
A liver microphysiological system of tumor cell dormancy and inflammatory responsiveness is affected by scaffold properties†
A. M.
Clark
a,
S. E.
Wheeler
a,
C. L.
Young‡
c,
L.
Stockdale
c,
J.
Shepard Neiman
c,
W.
Zhao
d,
D. B.
Stolz
aegh,
R.
Venkataramanan
ad,
D.
Lauffenburger
c,
L.
Griffith
c and
A.
Wells
*abfg
aDepartment of Pathology, University of Pittsburgh, S711 Scaife Hall, 3550 Terrace St, Pittsburgh, PA 15261, USA. E-mail: wellsa@upmc.edu
bDepartment of Bioengineering, University of Pittsburgh, Pittsburgh, PA, USA
cDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
dDepartment of Pharmaceutical Sciences, University of Pittsburgh, Pittsburgh, PA, USA
eDepartment of Cell Biology, University of Pittsburgh, Pittsburgh, PA, USA
fPittsburgh VA Medical Center, VA Pittsburgh Healthcare System, Pittsburgh, PA, USA
gMcGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA, USA
hUniversity of Pittsburgh Cancer Center, Pittsburgh, PA, USA
First published on 24th November 2016
Abstract
Distant metastasis is the major cause of breast cancer-related mortality, commonly emerging clinically after 5 or more years of seeming ‘cure’ of the primary tumor, indicating a quiescent dormancy. The lack of relevant accessible model systems for metastasis that recreate this latent stage has hindered our understanding of the molecular basis and the development of therapies against these lethal outgrowths. We previously reported on the development of an all-human 3D ex vivo hepatic microphysiological system that reproduces several features of liver physiology and enables spontaneous dormancy in a subpopulation of breast cancer cells. However, we observed that the dormant cells were localized primarily within the 3D tissue, while the proliferative cells were in contact with the polystyrene scaffold. As matrix stiffness is known to drive inflammatory and malignant behaviors, we explored the occurrence of spontaneous tumor dormancy and inflammatory phenotype. The microphysiological system was retrofitted with PEGDa-SynKRGD hydrogel scaffolding, which is softer and differs in the interface with the tissue. The microphysiological system incorporated donor-matched primary human hepatocytes and non-parenchymal cells (NPCs), with MDA-MB-231 breast cancer cells. Hepatic tissue in hydrogel scaffolds secreted lower levels of pro-inflammatory analytes, and was more responsive to inflammatory stimuli. The proportion of tumor cells entering dormancy was markedly increased in the hydrogel-supported tissue compared to polystyrene. Interestingly, an unexpected differential response of dormant cells to varying chemotherapeutic doses was identified, which if reflective of patient pathophysiology, has important implications for patient dosing regimens. These findings highlight the metastatic microphysiological system fitted with hydrogel scaffolds as a critical tool in the assessment and development of therapeutic strategies to target dormant metastatic breast cancer.
Introduction
Distant metastasis is responsible for the majority of breast cancer-related deaths, with two-thirds of these lethal tumors being detected 5 or more years after a seeming ‘cure’ of the primary disease.1 Dormant cancer cells persist within distant foreign microenvironments, existing in a reversible growth arrested state that confers chemoresistance to anti-proliferative agents.2 Presently, our understanding of the fundamental biology underpinning the biology of dormant disseminated disease and the instigators that awaken these clinically-silent growths is limited. Unraveling the inherent signals and mechanisms behind this poorly understood step of metastasis biology is likely to profoundly impact cancer patients through the development of therapies against these lethal outgrowths.It is well accepted that the tumor microenvironment plays a critical role in regulating metastatic resistance and reoccurrence. The tumor microenvironment is complex being comprised of stromal, immune, extracellular matrix components (ECM) and signaling factors, with each component contributing to the tumor etiology, growth and therapeutic response.3 In recent years, the biomechanical factors of the tumor microenvironment have also emerged as a key element.4,5 Specifically, biophysical properties influence numerous key attributes governing metastasis – i.e. tumor migration, aggressiveness, proliferation, chemotherapeutic response and dormancy,6,7 with increasing mechanical stiffness correlated with the aforementioned behaviors.4,8–10 Thus, in order to advance discovery efforts for effective treatment regimens, it is imperative to develop disease models that accurately recapitulate both the cellular and biomechanical properties of the native metastatic niche, in particular, quiescent dormancy.
As a result, researchers are turning towards tissue engineered ex vivo biomimetic microphysiological systems, also known as ‘organs-on-a-chip’. Various models of cancer metastasis have recently been developed11–13 and are able to mimic the pathophysiology of native tumors more reliably than standard 2D cell culture settings.13,14 Notably, the tumor cells within microphysiological systems exhibit several phenotypes of tumors generally not found in vitro, forming physiologically relevant cell–cell and cell–ECM interactions that result in gene expression similar to that of human tumors.15–17
We previously reported on an all-human ex vivo hepatic microphysiological system to study breast cancer metastasis.13 Spontaneous dormancy was achieved, but only in a subpopulation of breast cancer cells within the liver-like tissue. In this model, cells are seeded into a scaffold comprising an array of 0.3 mm channels in a thin (0.25 mm) polymer disc where they attach to the walls of the scaffold and form 3D tissue-like structures adherent to the scaffold.13 The tissue is perfused with a microfluidic pump to produce a physiological oxygen gradient across the tissue.18,19 We speculate that the dormancy response observed in the previous work may have been influenced by the nature of the polystyrene scaffold support, as the subpopulation of cells in direct contact with the polystyrene scaffold experienced the type of stiff substrate environment that has been correlated with activation of liver stellate cells20–22 as well as many kinds of tumor cells.23,24 Indeed, proliferative tumor cells were typically observed to be in direct contact with the scaffold whereas dormant cells were localized primarily in the 3D tissue region. In order to refine the experimental system towards a dormancy-specific model, we developed and tested a soft synthetic hydrogel scaffold to better match the biomechanical environment in liver.
Hydrogels are commonly employed as synthetic ECM analogues as they capture numerous desirable features of the native ECM of soft tissues.25,26 Herein, we used a polyethylene glycol (PEG)-based hydrogel modified with a fibronectin-derived adhesion peptide mimic, SynKRGD, to engender integrin-mediated cell-scaffold interactions. The SynKRGD peptide “PHSRN-K-RGD” contains both the arginylglycylaspartic acid (RGD) motif and the PHSRN synergy site from the 9th fibronectin type III repeat in a branched configuration to mimic features of the biophysical presentation in fibronectin.27,28 We investigated the inflammatory phenotype and proportion of breast cancer cells entering dormancy as well as their sensitivity to standard chemotherapies and compared outcomes for the standard stiff polystyrene scaffold and the soft hydrogel scaffold. The hydrogel scaffolds significantly enhance breast cancer cell dormancy and maintained a healthier hepatic microenvironment, which in turn was more responsive and sensitive to inflammatory perturbations. Interestingly, an unexpected differential response of dormant cells to varying chemotherapeutic doses was identified, which if reflective of patient pathophysiology, has important implications for patient dosing regimens.
Materials and methods
Cell sources
Human hepatocytes (Hep) and human nonparenchymal cells (NPC) are excess pathological specimens from therapeutic partial hepatectomies for metastatic colorectal carcinoma or other benign diseases, such as focal nodular hyperplasia. These cells are available from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)-funded Liver Tissue and Cell Distribution System (LTCDS) with the procurement core directed by Dr. David Geller at the University of Pittsburgh, which was funded by NIH contract # HHSN276201200017C. Through this distribution system the livers are perfused and separate isolations of hepatocytes and NPCs are provided to investigators. To eliminate contaminating debris, hepatocytes and red blood cells, the NPC fraction is further purified via Percoll gradients, as previously reported.29 All cells used in these investigations were approved as exempted by the University of Pittsburgh Institutional Review Board (IRB).Cell lines
The MDA-MB-231 breast cancer cell line was purchased from ATCC (Manassas, VA) and transfected with red fluorescent protein (RFP), as described previously.30 Cells were maintained in RPMI 1640 with 10% heat-inactivated fetal bovine serum and 25 IU ml−1 penicillin and streptomycin (Gibco, Life Technologies). For seeding into the ex vivo hepatic microphysiological system, cells were trypsinized, centrifuged and resuspended in hepatocyte maintenance medium (detailed below).Ex vivo hepatic microphysiological system
The ex vivo hepatic microphysiological system (LiverChip) is assembled as recommended by the manufacturer (CN Bio Innovations Limited, Oxford UK). The system is seeded with cells as described by Wheeler et al.13 Briefly, human hepatocytes and NPCs are seeded into the system at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (6 × 105 cells per well) in William's Medium E (Gibco, Life Technologies, Grand Island, NY) supplemented with the Hepatocyte Thawing and Plating Supplement Pack (Life Technologies), prepared as recommended by the manufacturer. Hepatocytes alone and in all co-culture conditions were cultured for approximately 16 hours before changing to William's Medium E supplemented with the Hepatocyte Maintenance Supplement Pack (Life Technologies), prepared as recommended by the manufacturer. Day 3 is considered the first day of complete tissue formation. Subsequently, the human breast cancer cells, MDA-MB-231, are introduced at a density of 500 cells per well into the formed hepatic tissue on this aforementioned day (experimental overview, Fig. S1†). For microenvironmental responsiveness assays, hepatic niche cultures (i.e. hepatocytes and NPCs only) were stimulated with 1 μg ml−1 lipopolysaccharide (LPS) (L2880, Sigma Aldrich) on day 14 (24 hours). For chemotherapy treatments, hepatic niche cultures containing MDA-MB-231 cells were treated with either doxorubicin (APP Pharmaceuticals LLC) or cisplatin (APP Pharmaceuticals LLC, Schaumburg, IL) on day 7 to 10 (72 hours).
:1 (6 × 105 cells per well) in William's Medium E (Gibco, Life Technologies, Grand Island, NY) supplemented with the Hepatocyte Thawing and Plating Supplement Pack (Life Technologies), prepared as recommended by the manufacturer. Hepatocytes alone and in all co-culture conditions were cultured for approximately 16 hours before changing to William's Medium E supplemented with the Hepatocyte Maintenance Supplement Pack (Life Technologies), prepared as recommended by the manufacturer. Day 3 is considered the first day of complete tissue formation. Subsequently, the human breast cancer cells, MDA-MB-231, are introduced at a density of 500 cells per well into the formed hepatic tissue on this aforementioned day (experimental overview, Fig. S1†). For microenvironmental responsiveness assays, hepatic niche cultures (i.e. hepatocytes and NPCs only) were stimulated with 1 μg ml−1 lipopolysaccharide (LPS) (L2880, Sigma Aldrich) on day 14 (24 hours). For chemotherapy treatments, hepatic niche cultures containing MDA-MB-231 cells were treated with either doxorubicin (APP Pharmaceuticals LLC) or cisplatin (APP Pharmaceuticals LLC, Schaumburg, IL) on day 7 to 10 (72 hours).
High impact polystyrene scaffolds
The high impact polystyrene scaffolds (CN Bio Innovations Limited, Oxford UK; Table 1) are freshly coated with 1% rat tail collagen type I (BD Biosciences, San Jose, CA) in phosphate buffered saline (PBS) for 1 hour at room temperature and washed with PBS before placement in the LiverChip.| Properties | Polystyrene | Hydrogel |
|---|---|---|
| Mechanical properties | ∼3 GPa | ∼300 kPa |
| Channels number | 301 | 41 |
| Channel dimensions | 300 μm | 700 × 300 μm |
| Channel volume | 0.018 mm3 | 0.041 mm3 |
| Total 3D tissue volume | 5.4 mm3 | 1.68 mm3 |
| 2D surface area | 21.3 mm2 (27%) | 7.79 mm2 (17.6%) |
| Depth | 250 μm | 250 μm |
| Cell adhesion | Coated with 1% collagen | SynKRGD, fibronectin mimic |

|

|
Hydrogel scaffolds
PEG-based hydrogel scaffolds (Table 1) were simultaneously produced and bonded to microporous support membranes by UV polymerization of PEG diacrylate precursor solutions in a micromolding process using polydimethylsiloxane (PDMS) mold with metalized posts. An aluminum mold was fabricated via rapid prototyping using SoldWorks. PDMS (Dow Corning Sylgard 184 Elastomer kit, Krayden, Glenview, IL) was cast and cured on an aluminum master mold by adding the curing agent to the base in a 1:9 mass ratio, degassing the PDMS prepolymer under vacuum, casting against the master, and curing at 60 °C overnight. The PDMS patterns consisted of raised features with a thickness of 250 μm and lengths and widths of 700 μm and 300 μm, respectively. The raised features of the PDMS mold were then metalized by overlaying a stainless steel mask and sputtering a 50 nm thick layer of chromium followed by a 200 nm thick layer of gold using electron beam sputtering (AJA International, ATC model). Metalization of these features blocks the UV radiation and prevents crosslinking of the PEG diacrylate (PEGDa) solution in select regions of the membrane.
PEGDa hydrogels were formed in a one-pot synthesis as follows. PEGDa (700 Mw, Sigma Aldrich) was dissolved 20% (w/w) in PBS. The cell adhesive peptide Ac-PHSRNGGGK-(Ac-GGGERCG)-GGRGDSPY-CONH2 (custom synthesis by Boston Open Labs, Cambridge, MA), incorporated through thiol–ene reaction, was added to the dissolved PEGDA to yield a final concentration of 0.1 mM. Photoinitiator 1-[4-(2-hydroxyethoxy)phenyl]-2-hydroxy-2-methyl-1-propanone-1-one photoinitiator (Irgacure 2959 or I2959, BASF, Chicago, IL) dissolved 50% (w/w) in dimethyl sulfoxide (DMSO, Fisher Scientific) was added to the PEG solution to yield a final concentration of 0.25% (w/w). The PDMS molds were treated in a Harrick Plasma Cleaner for 20 seconds to improve wetting of the mold. Next, 40 μL of precursor solution was added to each mold, then polyvinylidene fluoride (PVDF) membrane filters with 5 μm pore size (Millipore, Billerica, MA) were laid on top of the prepolymer and subsequently covered by a glass substrate. The precursor solution was exposed to UV light at an intensity of 180 mW cm−2 through the PDMS mold for 90 seconds. After polymerization, the scaffolds were demolded and punched to attain a final diameter of 10 mm.
Enzyme-linked immunosorbent assays
Alpha-1-antitrypsin (A1AT) and fibrinogen secretion from hepatic tissue was measured using Enzyme-linked immunosorbent assays (ELISAs) kits according to the manufacturer's instructions (Genway Biotech Inc, San Diego, CA). Supernatants were used at a 1:125 dilution for A1AT and a 1:25 dilution for fibrinogen.
Clinical chemistry assays
Assays for glucose (GLU), blood urea nitrogen (BUN), aspartate transaminase (AST) and alanine aminotransferase (ALT) were performed in the College of American Pathologists certified clinical laboratories at the University of Pittsburgh Medical Center (UPMC, Pittsburgh, PA) in accordance with all governmental regulations.Immunofluorescence microscopy
Polystyrene and hydrogel scaffolds were harvest, fixed, stained and imaged as previously described.13 The primary antibodies used were: mouse anti-actin, α-smooth muscle actin (Sigma C6198) and rabbit ant-Ki67 (Abcam, ab15580). Secondary antibodies used were goat anti-mouse or goat anti-rabbit Cy3, Cy5 (Jackson Immunoresearch, West Grove PA) or Alexa 488 (Invitrogen, Grand Island, NY). Counterstains used was 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) for nuclei.Cytochrome P450 assay
Four cytochrome P450 (CYP P450) enzyme activities were measured using a previously validated CYP cocktail assay.31 CYP cocktail substrates used in this assay were selected based on the FDA guidance over the preferred and acceptable CYP substrates (http://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm) for evaluating CYP activities in vitro. Further, the validated 4-probe CYP cocktail assay has no significant inter-substrate interactions.Signaling analyses – multiplex immunoassays
Determination of 77 unique cytokines, chemokines, and growth factors was performed using the human group 1 27-plex and 40-plex chemokine panels, cancer panels 1 and 2 (16- and 18-plex, respectively). Assays were completed according to the manufacturer's instructions (BioRad Laboratories, Hercules, CA, USA), with the exception that coupled beads, biotinylated detection antibodies, and streptavidin–phycoerythrin fluorescent reporters were diluted 2-fold. All analytes were evaluated in neat, undiluted samples; a total volume of 50 μL sample from coculture medium was analyzed per multiplex panel. To attain measurements within the working range of the assay, samples were diluted 2-, 4-, 8-, 16-, 32- and 100-fold specifically for 10 cytokines (Table S1†). Standard and sample diluents consisted of WEM (Life Technologies, Carlsbad, CA, USA) in the presence of 0.75% BSA (Sigma-Aldrich, St. Louis, MO, USA) as a final concentration.Assays were performed in parallel (unmixed) to avoid antibody cross-reactivity between groups. Prepared arrays were assessed by the 3D suspension array system (BioRad Laboratories, Hercules, CA, USA) utilizing xMAP technology licensed by Luminex. Data were collected with xPONENT for FLEXMAP 3D software, version 4.2 (Luminex Corporation, Austin, TX, USA) and results were evaluated initially in BioPlex Manager software version 6.1 (BioRad Laboratories, Hercules, CA, USA). Median fluorescence intensity (MFI) values were converted to absolute concentrations via calibration to fifteen-point standard series that implemented a 2-fold serial dilution. Assay performance metrics for each analyte are summarized in Table S1.†
Signaling data processing and statistical analyses
To quantify the concentration of each analyte, the five-parameter (5PL) logistic model was employed for the best curve fit of standards. Regression analysis minimized the weighted sum of squared errors (wSSE). In general, the weights are set equal to the inverse variance; however, for immunoassays, the high-response end of a curve approaches saturation of the detector thus variance is approximated more appropriately by a power function,| variance = A(response)B | (1) |
Cancer cell detection and enumeration
Images of the scaffolds sacrificed and fixed on day 15 were taken using an Olympus BX51 with a 2× (PlanApo NA = 0.08) objective. Digital images were obtained on an Olympus CCD camera using Magnafire image acquisition software. The proportional presence of RFP-expressing MDA-MB-231 cells was determined using MetaMorph software (Molecular Devices, LLC, Sunnyvale, CA). Images of the entire scaffolds were inclusively thresholded and the RFP positive portion measured as a percentage of total scaffold area.Statistics
Mann–Whitney non-parametric tests, Student t-tests or Wilcoxon rank sum tests were used and alpha p-values were set at <0.05. For these test, graphs and statistics were generated using GraphPad Prism version 7 (GraphPad Software, Inc, La Jolla, CA). For the multivariate statistical techniques, such as unsupervised hierarchical clustering, MATLAB version 2012b was used (Mathworks Inc, Natick, MA, USA).Results
Hepatic tissue function is maintained in scaffolds composed of different matrices
Following isolation and purification, hepatocytes undergo a brief adjustment to culture conditions. Inherently, some of the hepatocytes are injured during the initial procedures, however the viable cells recover and form tissue. This was observed using standard clinical injury markers of AST and ALT (Fig. 1A). For both polystyrene and hydrogel-supported hepatic tissue the levels of ALT (18.7 IU L−1 and 20.3 IU L−1, respectively) and AST (81.4 IU L−1 and 95.3 IU L−1, respectively) were initially high following introduction of hepatic cells into the system. However, as the hepatic tissue established, cells adjusted to the culture conditions while dying cells were eliminated, and injury-related markers rapidly decline to undetectable levels by day 15. Active protein catabolism was determined by measuring urea (BUN), a byproduct of protein catabolism. BUN levels were transiently elevated in samples collected on day 3, but declined and remained detectable through to day 15 (Fig. 1B). As expected, measurements of glucose in culture effluent at days 3, 7, and 15 remained at around the input level of 200 mg dL−1 (Fig. 1C), although consumption may be masked by the continual replacement of media every 2 days. Overall, no appreciable differences were observed in the clinical markers between the hydrogel and polystyrene scaffolds supporting hepatic tissue from 7 different donor-matched primary hepatocytes and NPCs. | ||
| Fig. 1 Hydrogel scaffolds maintain hepatocyte health and function similarly to polystyrene scaffolds. (A–C) Freshly isolated donor matched human hepatocytes and NPCs were seeded into the microphysiological system and cultured through 15 days. Effluent samples were obtained on day 3, 7 and 15 (n = 7 donors). Hepatocyte injury was evaluated via clinical analyses of (A) ALT and ALT. (B) BUN was analyzed with clinical tests to determine continued hepatocyte catabolism of proteins. (C) GLU consumption from the media (200 mg dL−1) was analyzed by measuring glucose levels. (D) Active CYP metabolism was assessed on day 3 and 13 by LC-MS/MS for drug metabolites (n = 5 donors) and normalized to tissue volume. Significant differences were determined via non-parametric pairwise Wilcoxon rank sum tests (*p < 0.05, **p < 0.01, ***p < 0.001). (E) Representative images of tissue formed within each scaffold type on day 15, depicted using phase contrast (top and bottom) and fluorescent nuclear staining with DAPI (middle). | ||
Active CYP activity, a key functional aspect of hepatocytes, was also maintained throughout the experimental period. Activities of four CYP enzymes (i.e. CYP1A2, CYP3A4/5, CYP2C9 and CYP2D6) were assessed on days 3 and 13 of culture (Fig. 1D). Hepatic metabolism was marginally higher overall in hydrogel-supported hepatic tissue and better maintained over the culture period. Comparison of individual CYP activities at days 3 and 13 for polystyrene-supported tissue found maintenance of CYP3A4, but a significant reduction in the activity of CYP1A2, CYP2C9 and CYP2D6. In contrast, only CYP2D6 was reduced in the tissue supported by hydrogel scaffolds.
The formation of 3D, multicellular tissue is fundamental to recapitulating organ physiology ex vivo. Tissue formation within the channels of each scaffold was assessed on day 15 (Fig. 1E). Both scaffolds were capable of forming intact tissue; however, hydrogel scaffolds were associated with a greater proportion of channels occupied with hepatic tissue, and fewer cells residing directly upon the scaffold surface.
Hydrogel matrices enhance the physiologic health and responsiveness of hepatic tissue
Stiff matrices can activate hepatic cells, altering the secretion of growth factors, cytokines, and matrix toward a pro-inflammatory, pro-fibrotic phenotype.20–22 Compared to polystyrene-supported tissue, we hypothesized that the more compliant and permeable hydrogel would diminish the production of pro-inflammatory factors within the hepatic microenvironment. To compare the signaling profiles within the microenvironment of polystyrene- and hydrogel-supported hepatic tissue, culture effluent was assessed at day 15 using multiplex bead-based assays in conjunction with xMAP technology (Table S1,† 27-plex pro-inflammatory and 40-plex chemokine panels). Differential signaling profiles of pro-inflammatory factors was observed between these two scaffolds types among donor-matched primary hepatocytes and NPCs (n = 5), as depicted in Fig. 2A. The levels of secreted cytokines, chemokines and growth factors were reduced in the effluent from hydrogel-supported tissue. | ||
| Fig. 2 Hydrogel scaffolds created a healthier physiological microenvironment and enhanced the responsiveness of hepatic tissue. (A and B) Levels of secreted analytes (pg mL−1) in day 15 effluent from hepatic tissue supported by either polystyrene or hydrogel scaffolds were measured; 53 analytes were above the LoD (n = 5 donors). (A) Scatter plot depicts levels of secreted analytes in hepatic tissue supported by polystyrene vs. hydrogel scaffolds. Significantly altered analytes (black circles) via non-parametric pairwise Wilcoxon rank sum tests (p < 0.05): IL-12p70, IL-13, IL-15, SDF-1α/β and VEGF-A, and those with a >1.5 ratio change (red circles). (B) Graphs summarize analytes exhibiting marked ratio changes >1.5 in polystyrene (top) or hydrogel (bottom) scaffolds. (C) Immunofluorescent images of hepatic tissue formed in polystyrene or hydrogel scaffolds on day 15, obtained using a 20× objective. Red – α-SMA; blue – DAPI. (D and E) Fold changes in acute phase protein production (A1AT and fibrinogen) following stimulation with LPS. Protein levels were measured by ELISA on day 15 following stimulation with 1 μg mL−1 LPS for 24 hours (n = 2 donors each in duplicate). Fold change was determined by for each patient donor by normalizing each stimulated group to its respective unstimulated control group. Significant differences were determined via non-parametric pairwise Wilcoxon rank sum tests (*p < 0.05). | ||
Hepatic tissue supported by polystyrene vs. hydrogel scaffolds showed >1.5-fold elevation in multiple signals including those noted to be produced by stimulated macrophages (macrophage inhibitory protein (MIP)-1β, interleukin (IL)-6, IL-1β, monocyte chemoattractant protein (MCP)-4 (CCL13), Gro-α (CXCL1), IL-12, IL-15, tumor necrosis factor (TNF)-α), hepatic stellate cells (MCP-1 (CCL2), eotaxin, stromal cell-derived factor (SDF)-1) and endothelial cells (TECK (CCL25), VEGF-A, MIP-1α, IL-7); these are involved in mediating pro-inflammatory responses (Fig. 2B). Key signals associated with fibrosis and chronic inflammation in the liver (basic fibroblast growth factor (b-FGF), IL-13, IL-17A, IL-5 and IL-1β) were markedly increased (>2-fold elevation) within the culture effluent of polystyrene-supported hepatic tissue as compared to hydrogel-supported. Conversely, only a few signals were elevated in effluent from hepatic tissues supported by hydrogel scaffolds. Marked increases were observed in IL-2, a key cytokine involved in immunotolerance via direct effects on T cells;33 and macrophage inhibitory factor (MIF), a cytokine with anti-fibrotic properties in the liver that inhibits the migration and proliferation of activated-hepatic stellate cells.34 Together these factors potentially increase cell survival, and the subsequent rebuilding and repair of hepatic tissue. Notably, monokine induced by interferon gamma (MIG or CXCL9), a pleiotropic chemokine reported to exert both pro- and anti-fibrotic activities was elevated in hydrogel-supported hepatic tissue; MIG and IP-10 (CXCL10) activate that same CXCR3 receptor that exerts potent anti-angiogenic activities.35–37 In general, our results demonstrate that polystyrene scaffolds predominantly created a more pro-inflammatory environment (26 pro-inflammatory signals elevated >1.5 fold-change). These aspects are further confirmed in Fig. 2C where α-smooth muscle actin (α-SMA) staining, a hepatic tissue activation marker, was increased in polystyrene-supported tissue.
Hepatic tissue capable of responding to stressors is essential in order to accurately compare ‘normal’ and ‘diseased’ conditions. NPCs are pivotal factors involved in the modulation of hepatocyte responses. Our results indicate that when hydrogel-supported hepatic tissue was exposed to inflammatory stimulus (LPS), both hepatocyte (p = 0.0286) and NPC function was markedly increased compared to polystyrene-supported tissue, as determined by elevated levels of hepatocyte specific acute phase reactant proteins in the culture effluent (A1AT and fibrinogen; Fig. S3†). Importantly, the responsiveness of hydrogel-supported tissue became considerably more evident when fold changes in acute phase protein production for stimulated groups were matched to donor controls (Fig. 2D and E). Consistent with signaling results in Fig. 2A, the basal level of acute phase protein production was lower in hydrogel-supported tissue compared to polystyrene.
Pro-inflammatory signaling profiles, immunofluorescence of hepatic tissue, and assessment of hepatic acute phase proteins in response to a physiologically-relevant inflammatory stimulus, highlights advantages of hydrogel scaffolds, in comparison to standard polystyrene scaffolds, in providing an environment conducive to recreating a more physiologically healthier and responsive hepatic tissue.
Spontaneous quiescence of breast cancer cells in the hepatic metastatic niche is enhanced by hydrogel matrices
In Wheeler et al.,13 we reported the LiverChip ex vivo hepatic microphysiological system to be a pertinent model to investigate the pathophysiology of breast cancer metastases. Distinctively, this system enables the recreation of spontaneous dormancy in a small sub-population of the aggressive and malignant MDA-MB-231 breast cancer cell line. In this current study, we queried whether a greater sub-population of breast cancer cells undergo spontaneous dormancy in a microenvironment supported by the hydrogel scaffold, which is inherently less stiff than the polystyrene scaffold used in previous investigations, and is also more permeable to nutrients and small signaling molecules. A significant reduction in the outgrowth of MDA-MB-231 cells was observed in hepatic tissue supported by hydrogel compared to polystyrene scaffolds after 12 days (Fig. 3A, >7-fold decrease, p = 0.0016). MDA-MB-231 cells established primarily as quiescent, non-proliferative populations as indicated by the absence of Ki67 staining (markers of proliferation) in Fig. 3B (bottom panel) and the very modest if any increase in cell number from day 7 to 15 (Fig. S4A†). Meanwhile, numerous MDA-MB-231 clusters formed in the polystyrene-supported hepatic tissue and primarily comprised the proliferating sub-population (Ki67+) (Fig. 3B, top and middles panels) and exhibited an increase in number consistent with exponential growth from day 7 to 15 (Fig. S4A†). Generally, proliferating cells (asterisks) clustered near the channel edges on the surface of the polystyrene scaffolds (i.e. the rigid surfaces of the scaffolds, Fig. 3B, top panel), while quiescent cells (arrows) were located within the hepatic tissue (Fig. 3B, middle panel). In contrast, MDA-MB-231 cells supported within hydrogel scaffolds predominantly colonized and remained as single cells with rounded epithelial-like morphology (Fig. 3B, bottom panel). Our results infer that the hydrogel-supported hepatic metastatic niche provides a microenvironment where the proliferative capacity of MDA-MB-231 cells is reduced or halted, even when the number of cancer cells seeded increased 10-fold (Fig. S4B†). Taken together these data demonstrate the significant effects that matrices properties have on recapitulating an actively growing or dormant metastatic microenvironment. | ||
| Fig. 3 Enhanced spontaneous quiescence in aggressive breast cancer cells in tissue supported hydrogel scaffolds. On day 3, MDA-MB-231 cells (500 cells per well) expressing RFP were seeded into the hepatic tissue and cultured through 15 days after which effluent samples were obtained and scaffolds sacrificed and fixed. (A) Complete scaffolds were imaged in the RFP channel (2× objective) and the percent of total area positive for RFP was calculated (n = 3 donors). Significant differences were determined via non-parametric pairwise Wilcoxon rank sum tests (**p < 0.01). (B) Representative immunofluorescent images of MDA-MB-231 cells in polystyrene and hydrogel scaffolds after 12 days in culture (day 15). Red – MDA-MB-231 cells; green – Ki67; blue – DAPI. Arrow: Attenuated cell, asterisk: proliferating cells. (C–F) Levels of secreted analytes (pg mL−1) in day 15 effluent from 4 groups were measured: polystyrene-supported hepatic tissue in the absence (n = 5 donors) or presence of MDA-MB-231 cells (n = 6 donors), and hydrogel-supported hepatic tissue in the absence (n = 5 donors) or presence of MDA-MB-231 cells (n = 3 donors); 30 analytes were above the LoD. Scatter plot depicts levels of secreted analytes in (C) hepatic tissue with MDA-MB-231 cells supported by polystyrene vs. hydrogel scaffolds, (D) polystyrene-supported hepatic tissue in the presence or absence of MDA-MB-231 cells, or (E) hydrogel-supported hepatic tissue in the presence or absence of MDA-MB-231 cells. Significantly altered analytes (black circles) via non-parametric pairwise Wilcoxon rank sum tests (p < 0.05): (C) TNF-α, (D) uPA, and those exhibiting marked ratio changes of >1.5 (red circles). (F) Unsupervised hierarchical clustering of 30 cancer-related signaling factors with the relative secretion of analytes averaged across the four experimental groups; concentrations were mean-centered. Gray – Hep/NPC; red – Hep/NPC + MDA-MB-231. | ||
To further evaluate the differences among the actively growing or dormant phenotype of breast cancer cells as a consequence of the metastatic microenvironment created within polystyrene- vs. hydrogel-supported hepatic tissue, 34 cancer markers were evaluated in the culture effluent at day 15 (Table S1,† cancer panels 1 and 2). Multiplex bead-based assays were employed to identify variations in signaling networks as a consequence of matrix type. First, we compared the difference in signals between the hepatic metastatic niche in the presence of MDA-MB-231 cells supported by polystyrene and hydrogel scaffolds. Overall, metastatic tissue from the polystyrene microenvironment secreted more cancer-associated signals than the hydrogel microenvironment (Fig. 3C). Greater than 1.5-fold increase was observed in breast cancer-associated signals (plasminogen activator inhibitor-1 (PAI-1), hepatocyte growth factor (HGF)), promoters of metastases, growth and survival (angiopontein-2, platelet derived growth factor (PLGF), vascular endothelial growth factor (VEGF)-A, TNF-α, epidermal growth factor (EGF)) as well as pro-inflammatory signals (insulin-like growth factor-binding protein (IGFBP)-1, IL-6) (Fig. 3C). In hydrogel-supported metastatic tissue, three breast cancer-associated markers (urokinase-type plasminogen activator (uPA), stem cell factor (SCF) and osteopontin) were elevated compared to polystyrene (Fig. 3C).
To determine the impact of cancer cells on signaling profiles within hepatic microenvironments, we compared each tissue in the presence or absence of MDA-MB-231 cells. The addition of MDA-MB-231 cells to the hepatic tissue supported by polystyrene scaffolds resulted in elevations in only a few signals greater than baseline values of the hepatic niche itself (Fig. 3D). PAI-1, a breast cancer-biomarker,38 was the only cytokine markedly elevated by the presence of MDA-MB-231 cells (Fig. 3D, 5.7-fold). Conversely, a greater impact on signal secretion was observed for the more compliant hydrogel scaffolds in the presence of MDA-MB-231 cells (Fig. 3E). Consistent with comparisons to the polystyrene-supported metastatic niche, elevations in uPA, SCF, osteopontin, VEGF-A, secreted VEGF receptor (sVEGFR)-2 and IL-6 were observed in the hydrogel-supported metastatic niche compared to the baseline values. Distinctively, promoters of dormancy were also increased – b-FGF can be a direct inducer of dormancy39 and while SCF is a cancer biomarker, it is also involved in dormant stem cells maintenance.40,41 Integration of the MDA-MB-231 cells may have been enhanced by the increased levels of osteopontin, which has been shown previously to significantly enhance heterotypic interactions with hepatocytes in colorectal cancer metastasis to liver.42
These data collectively indicate that the high baseline concentration of signaling factors present within the culture effluent of hepatic tissue supported by the polystyrene matrix masks important biological responses, such as the presence of metastatic cells. These findings are corroborated by multivariate analyses such as the unsupervised hierarchical clustergram depicted in Fig. 3F, where it is evident that elevated concentrations (red) of cytokines are secreted to the effluent of hepatic tissue supported by polystyrene scaffolds when compared to the more compliant and permeable hydrogels. The enhanced signaling response to MDA-MB-231 cells correlates with the enhanced sensitivity to stimuli in Fig. 2D and E. These results provide a critical assessment of the microenvironment, demonstrating that the selected matrix of the microphysiological system must closely align with the in vivo liver microenvironment in order to mimic the fundamental physiological effects. The implementation of a hydrogel-supported hepatic tissue appears a preferable protocol for anti-metastasis drug discovery, where exploratory biomarkers such as cytokines, growth factors, and their receptors can be identified as predictive markers of prognosis.
Efficacy of chemotherapeutic doses are dependent upon the relevance of the metastatic niche
The chemotherapies used to treat primary disease principally target actively proliferating cells; as a result, dormant metastases are resistant. Presently, current model systems do not recapitulate this scenario. We postulated that the enhanced dormancy provided by a hydrogel matrix would facilitate protection from chemotherapy as observed in patients. Polystyrene-supported metastatic tissue treated with standard chemotherapies, doxorubicin (Fig. 4A) and cisplatin (Fig. 4B), resulted in a dose-dependent response against MDA-MB-231 cells. However, within the more physiological metastatic niche created by more compliant hydrogel scaffolds, a differential response to chemotherapeutic treatment was observed. As predicted, high doses of chemotherapy were minimally effective against the predominantly dormant MDA-MB-231 cells within the hydrogel-supported metastatic tissue. Interestingly, proliferation (emergence) of MDA-MB-231 cells was observed following treatment with the lower doses of chemotherapy (doxorubicin, p = 0.0503; and cisplatin, p = 0.330), a finding that was not anticipated and may provide novel insights into therapeutic strategies. Clinical hepatic injury markers were unaffected by the chemotherapeutic doses tested (Fig. 4D).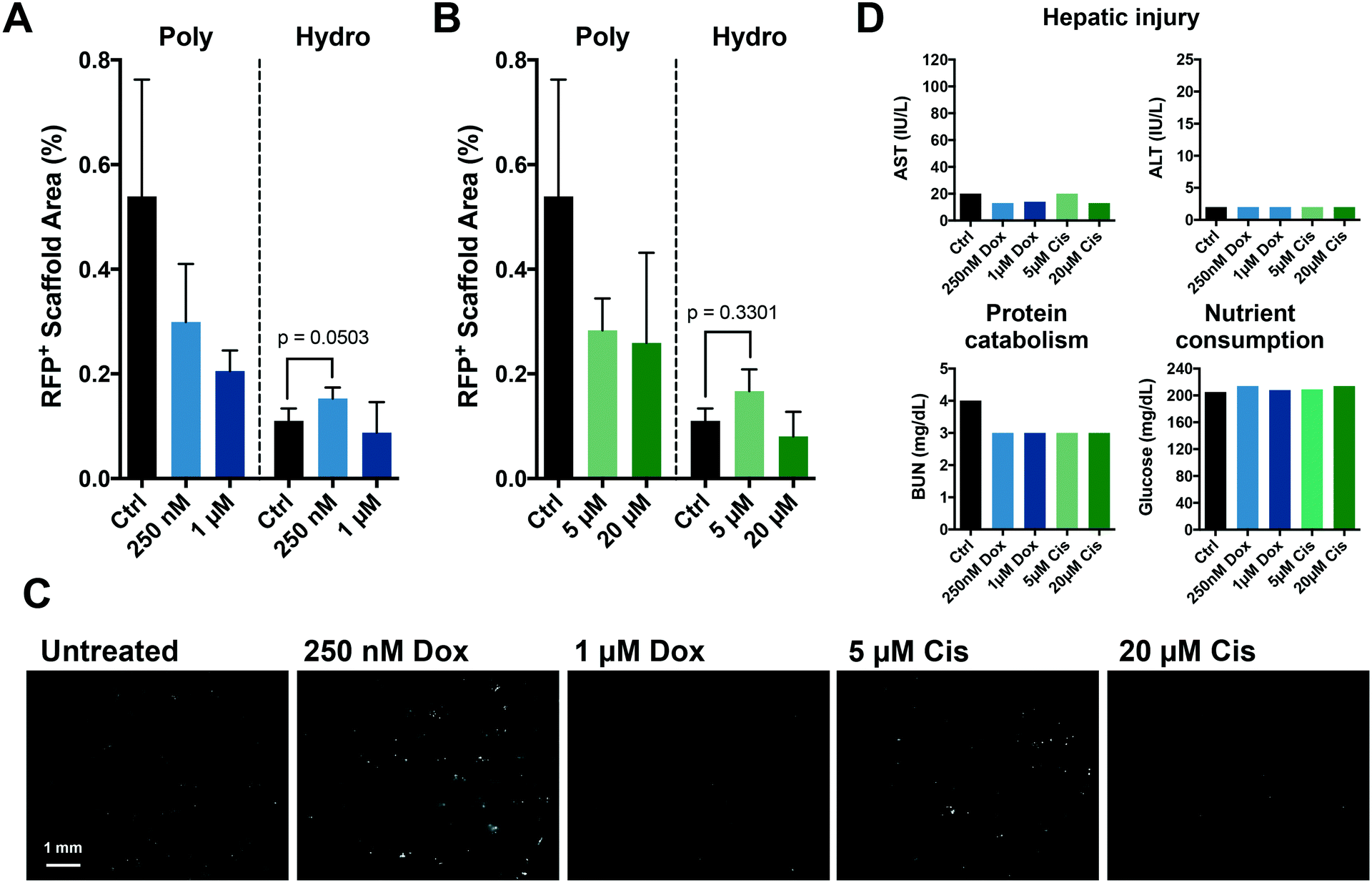 | ||
| Fig. 4 Differential response of breast cancer cells to standard chemotherapy in tissue supported by polystyrene and hydrogel scaffolds. Co-cultures of MDA-MB-231 cells with hepatocytes and NPCs were treated for 72 hours with doxorubicin (250 nM or 1 μM) or cisplatin (5 μM, 20 μM) from day 7 to 10. (A and B) Complete scaffolds were imaged in the RFP channel (2× objective) and the percent of total area positive for RFP was calculated following treatment with (A) doxorubicin or (B) cisplatin (n = 2 donors). (C) Representative images of MDA-MB-231 cells on day 15 after treatment with doxorubicin or cisplatin. White – RFP. (D) Effluent samples were from day 9 (after 48 hours of chemotherapy) were evaluated via clinical analyses of hepatic injury markers, ALT and ALT. BUN was analyzed with clinical tests to determine continued hepatocyte catabolism of proteins. GLU consumption from the media (200 mg dL−1) was analyzed by measuring glucose levels (n = 1 donor). | ||
Discussion
Accurately recapitulating the pathophysiology of dormant metastatic breast cancer is imperative in order to test and identify effective treatment regimes against this fatal stage of breast cancer. Recently, in vitro organ-on-a-chip models of cancer metastasis have been developed, each focusing on specific phases of metastasis.11,12 The challenge in these systems is not simply to grow tumor cells in the ectopic tissue microenvironment but to recapitulate the pathophysiology that blocks therapeutic efficacy in patients. Breast cancers are often dormant following implantation into the metastatic site, rendering standard cytotoxic therapies ineffective.We previously reported an all-human ex vivo hepatic model of metastases that could elicit spontaneous dormancy in a subpopulation of breast cancer cells within the liver.13 Although dormancy was achieved in some cells, the majority of the ectopically seeded cells grew out as a macrometastasis. We posited that these results were a direct consequence of using conventional polystyrene scaffolds as tissue stiffness is known to be a key driver of stellate and endothelial cell activation43 as well as tumor migration, aggressiveness, and proliferation.6 In order to enrich the dormant subpopulation within the LiverChip system, we employed a matrix scaffolding constructed from a more compliant synthetic hydrogel biomaterial, PEGDa which more closely approximates the physiological mechanical properties of the liver.44,45 Although gels synthesized from low molecular weight (700 kDa) PEGDa macromers are known to be at least moderately cell adhesive due to protein adsorption, we further enhanced cell interactions by functionalization with the integrin-binding peptide SynKRGD.27,28
Herein, we confirmed that using a scaffolding biomaterial that more closely approximates the physiologic and mechanical properties of the liver provides a microenvironment more conducive to recreating a physiologically relevant hepatic tissue. Clinical markers of hepatic injury were comparable between tissues supported by conventional polystyrene and hydrogel scaffolds over the culture period; however; the proportion of tissue formed within the channels of hydrogel scaffolds was enhanced (Fig. 1E, bottom panel) and active drug metabolism was better maintained (Fig. 1D). Reduced inflammation within the hepatic niche with hydrogel- over polystyrene-supported hepatic tissue was confirmed by investigations that quantified the baseline signaling microenvironment within the hepatic niche wherein lower levels of inflammatory signals were observed by hydrogel-supported tissue (Fig. 2A and B). Markers of fibrosis and inflammation (b-FGF, IL-13, IL-17A, IL-5 and IL-1β) as well as factors produced by activated NPCs (MIP-1β, IL-6, IL-1β, MCP-4, Gro-α, IL-12, IL-15, TNF-α, MCP-1, eotaxin, SDF-1, TECK, VEGF-A, MIP-1α, IL-7) dominated the microenvironment of polystyrene-supported hepatic tissue, whereas the three analytes that were increased in hydrogel-supported tissue were associated with anti-fibrotic properties (MIF, MIG)34,37 as well as immunotolerance (IL-2).33 Subsequently, the both hepatocytes and NPCs within the hydrogel-supported hepatic microenvironment were more sensitive and responsive to inflammatory perturbations (Fig. 2D and E), indicating that they maintained their native functionality in the ex vivo system. This was a particularly important outcome for modeling dormancy as the NPC populations of metastatic organs (i.e. immune and stromal cells) are well-known drivers of metastatic progression and emergence.43,46–49
Our study further supported the perspective that matrices more closely aligned with the native physiologic proprieties of metastatic organs allow for improved recapitulation of dormancy specifically and disease processes in general. MDA-MB-231 cells within the hepatic tissue of hydrogel scaffolds correlated with a significantly reduced growth potential and exhibited classic features of dormancy – non-cycling single cells with a rounded epithelial morphology (Fig. 3A). These results are consistent with findings by others indicating the importance of considering matrix rigidity in cellular models.50 Our studies demonstrated that proliferating, mesenchymal cells were proximal to the rigid surface of the polystyrene scaffold, while non-proliferating, epithelial-like cells resided within the hepatic tissue (Fig. 3B, top and middle panel). These observations inferred that matrix stiffness directed the phenotype of carcinoma cells. Within hydrogel-supported metastatic tissue, we found that the majority of breast cancer cells attained a dormant phenotype, regardless of their proximity to the channel edge (Fig. 3B, bottom panel). This observation suggested that despite the hydrogel matrix being more rigid than native tissue (∼300 kPa vs. ∼300–600 Pa, respectively), the closer approximation of the overall matrix properties (including both stiffness and cellular interactions with the matrix) was sufficient to produce a native phenotype. Furthermore, signaling in the in hydrogel-supported metastatic niche correlated with a microenvironment supportive of quiescence, being comprised of factors that maintain stem cell dormancy (SCF) as well as those that direct the induction of dormancy (b-FGF). Potent anti-angiogenic factors (MIG and IP-10)35,36 were also elevated in the hydrogel-supported hepatic niche, which may have contributed to promoting the dormant cell state upon MDA-MB-231 cell seeding. It should be noted that outgrowth from dormancy has been linked to activated endothelial cells in vitro43 and neo-angiogenesis in an animal model.47 Thus, the limitation of angiogenesis may protect the dormant cells from re-awakening. Although b-FGF has only been shown for indolent breast cancer cells lines (e.g. MCF-7 and T47D) and not for MDA-MB-231 cells in vitro,39 the tumor microenvironment can have profound effects upon cell phenotype which has been observed in 3D.51 In general, elevated levels of uPA38 is associated with poor prognosis of cancer patients; similarly, high levels of uPA were observed in effluent of hydrogel-supported metastatic tissue in this study. However, the downstream effect of increased uPA levels has not been determined. MDA-MB-231 cells are known to constitutively produce uPA52 and this may merely be a marker of their presence.
The more physiologically-relevant microenvironment created by employing compliant, synthetic hydrogel scaffolds facilitated compelling insights into the effect of different chemotherapeutic doses on dormant breast cancer cells. The dormant phenotype was predicted to correlate with increased resistance to chemotherapy, as noted in patients,53 which was recapitulated in this system for the high doses of both doxorubicin and cisplatin (Fig. 4A and B). However, this increased resistance was only observed following treatment with high chemotherapeutic doses. Conversely, for both doxorubicin (p = 0.0503) and cisplatin (p = 0.3301), the lower doses induced a slight outgrowth of the more attenuated breast cancer cell population (Fig. 4A and B). These lower chemotherapy doses may be causing clinically undetectable injury to the hepatic tissue, resulting in cellular stress and activation of the more responsive NPCs, which in turn secrete signals that promote the outgrowth and emergence of breast cancer cells. Regardless of chemotherapies used or dosage, clinical markers of hepatic injury were unaffected, suggesting that such activations may be relatively localized and paracrine, and thus occurred in the absence of clinically evident liver toxicity. Cancer cells seeded in hydrogel matrixes exhibit higher clonogenic capacity compared to their counterparts on polystyrene,54 which may also contribute to the increased outgrowth at low chemotherapy doses in hydrogel but not polystyrene matrices. Our findings advocate a previously unsuspected behavior of dormant micrometastases and, if reflective of the human pathophysiology, has significant implications for treatment regimens administered to patients. If doses are not administered at the necessary therapeutic dose, we may actually be portending metastatic-related mortality.
Our data highlight the all-human ex vivo hepatic model of metastasis fitted with synthetic hydrogel scaffolds as an essential tool in the assessment and development of therapeutic strategies targeting dormant metastatic breast cancer. This microphysiological system is still in its infancy and will likely require additional modifications and adaptations, especially as we gain mechanistic insights into dormancy. For example, the effect of SynKRGD upon enhancing hepatic tissue health and function as well as the dormant phenotype under hydrogel compositions of varying stiffness is yet to be evaluated. Currently, this microphysiological system is limited to assessments involving endpoint fixation and imaging as well as evaluation of effluent at multiple time points; however, efforts are underway to incorporate a degradable hydrogel in order to facilitate the isolation of individual, attenuated breast cancer cells for a more in-depth characterization of proteomic, genomic, epigenomic and transcriptomic analyses. Additionally, as with most other studies, immortalized breast cancer cell lines have been mainly used to-date within the current iteration of the metastatic microphysiological system, although limited studies have used primary tumor cells.17,55 The primary cells would be more representative but confound interpretations due to intrinsic heterogeneity and uncertainty as to outcome as these come mainly from primary tumors rather than metastatic sites. Other aggressive metastatic cell lines that demonstrated directed hepatic predilection (e.g. colon CR4 (ref. 56)) or those derived from liver metastasis and thus exhibiting liver specific tropism (e.g. pancreatic SU.86.86,57 lung DMS 15358 or stomach NCI-N8759) could be utilized to demonstrate widespread validation; though we have found prostate and colorectal carcinoma and melanoma cells to undergo similar behavior.17,30 Alternatively, seeding of circulating tumors cells derived from the patient blood to even more closely mimic the human in vivo situation in combination with the degradable hydrogel feature may lead to notable discoveries into previously unidentified mechanisms governing dormancy.
Despite more than a century of genetic, molecular biology, and animal research into cancer, it largely remains incurable once it has disseminated. Novel biological insights attained from microphysiological systems, such as the one presented herein, combined with traditional animal and 2D models is the most likely path towards furthering our understanding and facilitating the development of therapies to cure malignant metastatic diseases. These microphysiological systems are now gaining attention in the academia, government, and pharmaceutical industries; however, they are imperfect with several challenges to be addressed (e.g. scaffolding matrices). Nevertheless, these systems provide a more reasonable approximation of the mechanical responses in vivo and allow unprecedented access to the process of metastatic implantation and initiation of dormancy. Metastasis is exceedingly complex and highly variable even within a single patient, thus it is unrealistic to expect to capture every facet of the human in vivo situation. Alternatively, designing and optimizing each microphysiological system to focus upon particular aspects of metastasis (i.e. dormancy, invasion or progression etc.) offer a promising path forward in order to the unravel and elucidate mechanistic insights into mechanisms underlying metastatic tumor resistance, as well as entry and exit from dormancy in order to impact exploratory research, R&D, drug development, and clinical practice.
Abbreviations
| A1AT | Alpha-1-antitrypsin |
| ALT | Alanine aminotransferase |
| AST | Aspartate transaminase |
| b-FGF | Basic fibroblast growth factor |
| BUN | Blood urea nitrogen |
| CYP | Cytochrome P450 |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| GLU | Glucose |
| HGF | Hepatocyte growth factor |
| HN | Hepatic niche |
| IGFBP | Insulin growth factor binding protein |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MCP | Monocyte chemoattractant protein |
| MIF | Macrophage inhibitory factor |
| MIG | Monokine induced by gamma |
| MIP | Macrophage inhibitory protein |
| PDMS | Polydimethylsiloxane |
| PEGDa | Polyethylene glycol diacrylate |
| PLGF | Platelet derived growth factor |
| RGD | Arginylglycylaspartic acid |
| SCF | Stem cell factor |
| SDF | Stem cell-derived factor |
| sVEGFR | Secreted vascular endothelial growth factor receptor |
| TNF | Tumor necrosis factor |
| uPa | Urokinase-type plasminogen activator |
| VEGF | Vascular endothelial growth factor |
Acknowledgements
The authors thank other members of their laboratories for thoughtful discussions as well as Dr. Jonathan Coppeta (The Charles Stark Draper Laboratory, LLC) for his helpful suggestions with respect to the hydrogel scaffolds. The work described within is funded by grants from the NIH (1UH3TR000496), a VA Merit Award, and DARPA (BAA-11-73 Microphysiological Systems: W911NF-12-2-0039).References
- C. A. Klein, Curr. Opin. Genet. Dev., 2011, 21, 42–49 CrossRef CAS PubMed.
- R. A. Weinberg, APMIS, 2008, 116, 548–551 CrossRef PubMed.
- M. R. Junttila and F. J. de Sauvage, Nature, 2013, 501, 346–354 CrossRef CAS PubMed.
- M. J. Paszek, N. Zahir, K. R. Johnson, J. N. Lakins, G. I. Rozenberg, A. Gefen, C. A. Reinhart-King, S. S. Margulies, M. Dembo, D. Boettiger, D. A. Hammer and V. M. Weaver, Cancer Cell, 2005, 8, 241–254 CrossRef CAS PubMed.
- H. Yu, J. K. Mouw and V. M. Weaver, Trends Cell Biol., 2011, 21, 47–56 CrossRef PubMed.
- S. S. McAllister and R. A. Weinberg, Nat. Cell Biol., 2014, 16, 717–727 CrossRef CAS PubMed.
- D. E. Jaalouk and J. Lammerding, Nat. Rev. Mol. Cell Biol., 2009, 10, 63–73 CrossRef CAS PubMed.
- M. H. Zaman, L. M. Trapani, A. L. Sieminski, D. Mackellar, H. Gong, R. D. Kamm, A. Wells, D. A. Lauffenburger and P. Matsudaira, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10889–10894 CrossRef CAS PubMed.
- Y. A. Miroshnikova, D. M. Jorgens, L. Spirio, M. Auer, A. L. Sarang-Sieminski and V. M. Weaver, Phys. Biol., 2011, 8, 026013 CrossRef CAS PubMed.
- K. R. Levental, H. Yu, L. Kass, J. N. Lakins, M. Egeblad, J. T. Erler, S. F. Fong, K. Csiszar, A. Giaccia, W. Weninger, M. Yamauchi, D. L. Gasser and V. M. Weaver, Cell, 2009, 139, 891–906 CrossRef CAS PubMed.
- T. K. Schuessler, X. Y. Chan, H. J. Chen, K. Ji, K. M. Park, A. Roshan-Ghias, P. Sethi, A. Thakur, X. Tian, A. Villasante, I. K. Zervantonakis, N. M. Moore, L. A. Nagahara and N. Z. Kuhn, Cancer Res., 2014, 74, 5359–5363 CrossRef CAS PubMed.
- Z. Liu and G. Vunjak-Novakovic, Curr. Opin. Chem. Eng., 2016, 11, 94–105 CrossRef PubMed.
- S. E. Wheeler, A. M. Clark, D. P. Taylor, C. L. Young, V. C. Pillai, D. B. Stolz, R. Venkataramanan, D. Lauffenburger, L. Griffith and A. Wells, Br. J. Cancer, 2014, 111, 2342–2350 CrossRef CAS PubMed.
- A. M. Clark, B. Ma, D. L. Taylor, L. Griffith and A. Wells, Exp. Biol. Med., 2016, 241, 1639–1652 CrossRef PubMed.
- K. Chwalek, L. J. Bray and C. Werner, Adv. Drug Delivery Rev., 2014, 79–80, 30–39 CrossRef CAS PubMed.
- A. Birgersdotter, R. Sandberg and I. Ernberg, Semin. Cancer Biol., 2005, 15, 405–412 CrossRef PubMed.
- C. Yates, C. R. Shepard, G. Papworth, A. Dash, D. Beer Stolz, S. Tannenbaum, L. Griffith and A. Wells, Adv. Cancer Res., 2007, 97, 225–246 CrossRef PubMed.
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC.
- M. J. Powers, K. Domansky, M. R. Kaazempur-Mofrad, A. Kalezi, A. Capitano, A. Upadhyaya, P. Kurzawski, K. E. Wack, D. B. Stolz, R. Kamm and L. G. Griffith, Biotechnol. Bioeng., 2002, 78, 257–269 CrossRef CAS PubMed.
- W. Zhao, L. Zhang, Z. Yin, W. Su, G. Ren, C. Zhou, J. You, J. Fan and X. Wang, Int. J. Cancer, 2011, 129, 2651–2661 CrossRef CAS PubMed.
- E. Olaso, K. Ikeda, F. J. Eng, L. Xu, L. H. Wang, H. C. Lin and S. L. Friedman, J. Clin. Invest., 2001, 108, 1369–1378 CrossRef CAS PubMed.
- R. G. Wells, Hepatology, 2008, 47, 1394–1400 CrossRef CAS PubMed.
- P. Lu, V. M. Weaver and Z. Werb, J. Cell Biol., 2012, 196, 395–406 CrossRef CAS PubMed.
- M. S. Samuel, J. I. Lopez, E. J. McGhee, D. R. Croft, D. Strachan, P. Timpson, J. Munro, E. Schroder, J. Zhou, V. G. Brunton, N. Barker, H. Clevers, O. J. Sansom, K. I. Anderson, V. M. Weaver and M. F. Olson, Cancer Cell, 2011, 19, 776–791 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- K. Saha, J. F. Pollock, D. V. Schaffer and K. E. Healy, Curr. Opin. Chem. Biol., 2007, 11, 381–387 CrossRef CAS PubMed.
- W. Kuhlman, I. Taniguchi, L. G. Griffith and A. M. Mayes, Biomacromolecules, 2007, 8, 3206–3213 CrossRef CAS PubMed.
- E. Cambria, K. Renggli, C. C. Ahrens, C. D. Cook, C. Kroll, A. T. Krueger, B. Imperiali and L. G. Griffith, Biomacromolecules, 2015, 16, 2316–2326 CrossRef CAS PubMed.
- A. J. Hwa, R. C. Fry, A. Sivaraman, P. T. So, L. D. Samson, D. B. Stolz and L. G. Griffith, FASEB J., 2007, 21, 2564–2579 CrossRef CAS PubMed.
- Y. L. Chao, C. R. Shepard and A. Wells, Mol. Cancer Ther., 2010, 9, 179 CrossRef PubMed.
- V. C. Pillai, S. C. Strom, S. N. Caritis and R. Venkataramanan, J. Pharm. Biomed. Anal., 2013, 74, 126–132 CrossRef CAS PubMed.
- D. Finney, Biom. J., 1978, 21, 689–690 Search PubMed.
- W. Liao, J. X. Lin and W. J. Leonard, Curr. Opin. Immunol., 2011, 23, 598–604 CrossRef CAS PubMed.
- T. T. Thuy le and N. Kawada, Hepatology, 2012, 55, 1295–1297 CrossRef PubMed.
- R. J. Bodnar, M. E. Rodgers, W. C. Chen and A. Wells, Arterioscler., Thromb., Vasc. Biol., 2013, 33, 2818–2829 CrossRef CAS PubMed.
- R. J. Bodnar, C. C. Yates and A. Wells, Circ. Res., 2006, 98, 617–625 CrossRef CAS PubMed.
- H. E. Wasmuth, F. Lammert, M. M. Zaldivar, R. Weiskirchen, C. Hellerbrand, D. Scholten, M. L. Berres, H. Zimmermann, K. L. Streetz, F. Tacke, S. Hillebrandt, P. Schmitz, H. Keppeler, T. Berg, E. Dahl, N. Gassler, S. L. Friedman and C. Trautwein, Gastroenterology, 2009, 137, 309–319 CrossRef CAS PubMed e301-303.
- R. Hildenbrand, A. Schaaf, A. Dorn-Beineke, H. Allgayer, M. Sutterlin, A. Marx and P. Stroebel, Histol. Histopathol., 2009, 24, 869–877 CAS.
- J. Barrios and R. Wieder, Cancer Microenviron., 2009, 2, 33–47 CrossRef CAS PubMed.
- Y. Ma, D. Liang, J. Liu, K. Axcrona, G. Kvalheim, K. E. Giercksky, J. M. Nesland and Z. Suo, Tumor Biol., 2012, 33, 967–978 CrossRef CAS PubMed.
- P. E. Boulais and P. S. Frenette, Blood, 2015, 125, 2621–2629 CrossRef CAS PubMed.
- J. Huang, C. Pan, H. Hu, S. Zheng and L. Ding, PLoS One, 2012, 7, e47901 CAS.
- D. P. Taylor, A. Clark, S. Wheeler and A. Wells, Breast Cancer Res. Treat., 2014, 144, 551–560 CrossRef CAS PubMed.
- M. Yin, J. A. Talwalkar, K. J. Glaser, A. Manduca, R. C. Grimm, P. J. Rossman, J. L. Fidler and R. L. Ehman, Clin. Gastroenterol. Hepatol., 2007, 5, 1207–1213 CrossRef PubMed , e1202.
- P. C. Georges, J. J. Hui, Z. Gombos, M. E. McCormick, A. Y. Wang, M. Uemura, R. Mick, P. A. Janmey, E. E. Furth and R. G. Wells, Am. J. Physiol., 2007, 293, G1147–1154 CrossRef CAS PubMed.
- D. G. DeNardo, M. Johansson and L. M. Coussens, Cancer Metastasis Rev., 2008, 27, 11–18 CrossRef CAS PubMed.
- C. M. Ghajar, H. Peinado, H. Mori, I. R. Matei, K. J. Evason, H. Brazier, D. Almeida, A. Koller, K. A. Hajjar, D. Y. Stainier, E. I. Chen, D. Lyden and M. J. Bissell, Nat. Cell Biol., 2013, 15, 807–817 CrossRef CAS PubMed.
- F. G. Giancotti, Cell, 2013, 155, 750–764 CrossRef CAS PubMed.
- M. S. Sosa, P. Bragado and J. A. Aguirre-Ghiso, Nat. Rev. Cancer, 2014, 14, 611–622 CrossRef CAS PubMed.
- R. W. Tilghman, C. R. Cowan, J. D. Mih, Y. Koryakina, D. Gioeli, J. K. Slack-Davis, B. R. Blackman, D. J. Tschumperlin and J. T. Parsons, PLoS One, 2010, 5, e12905 Search PubMed.
- V. M. Weaver, O. W. Petersen, F. Wang, C. A. Larabell, P. Briand, C. Damsky and M. J. Bissell, J. Cell Biol., 1997, 137, 231–245 CrossRef CAS PubMed.
- Y. Guo, P. Pakneshan, J. Gladu, A. Slack, M. Szyf and S. A. Rabbani, J. Biolumin. Chemilumin., 2002, 277, 41571–41579 CAS.
- R. E. Hurst, A. Bastian, L. Bailey-Downs and M. A. Ihnat, Ther. Adv. Med. Oncol., 2016, 8, 126–137 CrossRef PubMed.
- J. Schrader, T. T. Gordon-Walker, R. L. Aucott, M. van Deemter, A. Quaas, S. Walsh, D. Benten, S. J. Forbes, R. G. Wells and J. P. Iredale, Hepatology, 2011, 53, 1192–1205 CrossRef CAS PubMed.
- E. S. Sokol, D. H. Miller, A. Breggia, K. C. Spencer, L. M. Arendt and P. B. Gupta, Breast Cancer Res., 2016, 18, 19 CrossRef PubMed.
- R. A. Rowehl, S. Burke, A. B. Bialkowska, D. W. Pettet III, L. Rowehl, E. Li, E. Antoniou, Y. Zhang, R. Bergamaschi, K. R. Shroyer, I. Ojima and G. I. Botchkina, PLoS One, 2014, 9, e99091 Search PubMed.
- B. J. Drucker, F. M. Marincola, D. Y. Siao, T. A. Donlon, C. D. Bangs and W. D. Holder, In Vitro Cell. Dev. Biol., 1988, 24, 1179–1187 CAS.
- O. S. Pettengil, G. D. Sorenson, D. H. Wurster-Hill, J. C. Thomas, W. W. Noll, C. C. Gate and L. H. Maurer, Cancer, 1980, 45, 906–918 CrossRef.
- J. Park, H. Frucht, R. V. LaRocca, D. P. Bliss Jr., Y. Kurita, T. Chen, J. G. Henslee, J. B. Trepel, R. T. Jensen, B. E. Johnson, Y. Bang, J. Kim and A. F. Gazdar, Cancer Res., 1990, 50, 2773–2780 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6lc01171c |
| ‡ Applied BioMath, LLC, Lincoln MA. |
| This journal is © The Royal Society of Chemistry 2017 |