
Identification of the catalytically active component of Cu–Zr–O catalyst for the hydrogenation of levulinic acid to γ-valerolactone†
Satoshi
Ishikawa
ab,
Daniel R.
Jones
b,
Sarwat
Iqbal
b,
Christian
Reece
b,
David J.
Morgan
b,
David J.
Willock
*b,
Peter J.
Miedziak
b,
Jonathan K.
Bartley
b,
Jennifer K.
Edwards
b,
Toru
Murayama
a,
Wataru
Ueda
ac and
Graham J.
Hutchings
*b
aCatalysis Research Center, Hokkaido University, N-21, W-10, Sapporo, 001-0021, Japan
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: willockdj@cardiff.ac.uk; hutch@cardiff.ac.uk
cDepartment of Material and Life Chemistry, Faculty of Engineering, Kanagawa University, 3-27, Rokkakubashi, Kanagawa-ku, Yokohama, 221-8686, Japan
First published on 27th October 2016
Abstract
Cu–ZrO2 catalysts were synthesized by the methanothermal (Me) and oxalate gel precipitation (Og) methods. Detailed characterization of the catalysts synthesized by the Me method shows that these contain only Cu substituted into the tetragonal ZrO2 lattice. For catalysts prepared using the Og method Cu is found not only in the tetragonal ZrO2 lattice but also in the form of CuO particles on the zirconia surface. When these materials were tested for the hydrogenation of levulinic acid (LA) to γ-valerolactone (GVL) it was found that Me materials show no catalytic activity, whereas GVL was formed using Og catalysts. A reduction treatment of the Og catalysts prior to use resulted in a marked increase in the catalytic activity, however, no activity increase was observed when the Me material was exposed to a similar treatment before testing. Based on these results and characterization data, we conclude that the catalytically active component of Cu–ZrO2 catalysts for the hydrogenation of LA is reduced Cu particles dispersed on the catalyst surface with strong interaction with the Cu incorporated zirconia support, while the role of Cu in the zirconia lattice is to improve the adhesion of these particles and maintain their dispersion.
1. Introduction
Levulinic acid (LA) is one of the most important lignocellulosic derived materials because of its high chemical potential to be converted into value-added chemicals such as γ-valerolactone (GVL), 1,4-pentanediol, valeric acid and 2-methyltetrahydrofuran.1–7 Among these products, GVL has a wide range of applications. It can be used as a gasoline blend, a solvent in lacquers, a food additive and a precursor to a variety of monomers.2,8–11 Numerous papers have appeared focused on ruthenium based heterogeneous catalysts for hydrogenation of lignocellulose derived substrates, including LA to GVL.6,12–20 However, ruthenium is a relatively expensive metal whose supply is likely to limit its application in the large scale production of bio-derived materials that will be required for a sustainable future. For truly sustainable processes both the feedstock and the catalyst must be derived from renewable or recyclable materials therefore catalysts employing abundant elements are an important target.Copper–zirconia (Cu–ZrO2) catalyst systems have recently been reported as promising candidates for the LA to GVL reaction.21,22 These catalysts can work with water as a solvent and can decompose formic acid to produce hydrogen in situ (HCOOH → H2 + CO2). This means that they are able to operate under the conditions in which LA is produced (acidic hydrolysis of biomass)22 and so may be expected to offer great industrial potential for the production of GVL from lignocellulose. However, most of the literature has focused on improving the catalytic activity from a formulation point of view rather than on identifying and understanding the catalytically active component. Hengne and Rode have reported that Cu is the active component in Cu–ZrO2 catalyst acting in the same way as a traditional supported transition metal hydrogenation catalyst.21 However, it has also been suggested that Zr can also be responsible for hydrogenation23–25 and Chia and Dumesic have reported that ZrO2 itself can catalyze LA to GVL in various alcohol solvents by catalytic transfer hydrogenation through the Meerwein–Ponndorf–Verley (MPV) reaction pathway.25 The active Cu–ZrO2 catalyst is normally prepared by a co-precipitation method with a very high Cu loading.21,22 This synthesis method leads to the incorporation of Cu into the ZrO2 lattice as well as deposition of CuO particles on the zirconia surface. Sloczynski et al. have reported that the introduction of Cu into the tetragonal ZrO2 lattice increases both the Lewis and Brønsted acid site concentrations.26 Roman-Leshkov et al. pointed out that Lewis acids can also enhance transfer hydrogenation activity.27 This complex mixture of factors makes identification of the catalytically active component of Cu–ZrO2 catalysts for the hydrogenation of LA to GVL challenging but also suggests that there is scope to design a new class of catalysts based on these materials once an understanding of the active species has been achieved. In particular, the roles of Cu that is incorporated into the oxide lattice and that which forms a separate surface phase need to be differentiated.
In this paper, we attempt to identify the catalytically active component of Cu–ZrO2 catalysts by comparing materials having a consistent structure of the bulk oxide phase with and without surface Cu particles. We show how catalyst synthesis methodology and pre-treatment can dramatically influence performance. We believe this work provides useful information for designing more active catalysts composed of abundant elements for the LA to GVL reaction that will be economically and environmentally competitive with the state-of-the-art Ru/C formulations. This could also lead to new applications across the range of hydrogenation reactions that will underpin the future exploitation of biomass as a sustainable resource.
2. Experimental
2.1 Synthesis of pure tetragonal zirconia oxide
Synthesis of pure tetragonal zirconia (t-ZrO2) was carried out following the method reported by Li et al.28 In a typical synthesis, 8.137 g of ZrO(NO3)2·6H2O (Zr: 0.024 mol, Acros Organics, 99.5%) was dissolved in 60 ml of methanol with the help of ultrasonication. Then, 2.88 g of urea (0.048 mol, Fisher Scientific, >99%) was added and stirred for another 10 minutes at room temperature. The mixed solution was transferred to a 120 ml Teflon-lined autoclave and methanothermal synthesis was carried out at 175 °C for 20 h. The obtained powder was filtered and dried at 110 °C overnight under air. Finally, the dried material was calcined at 400 °C for 4 h with 10 °C min−1 ramp under static air. The obtained material is abbreviated as t-ZrO2.2.2 Synthesis of Cu–ZrO2 catalyst by methanothermal method
1.159 g of Cu(NO3)2·3H2O (Cu: 0.005 mol, Acros Organics, 99%) and 6.509 g of ZrO(NO3)2·6H2O (Zr: 0.019 mol) were dissolved in 60 ml of methanol with the help of ultrasonication. Then 2.88 g of urea (0.048 mol) was added and stirred for 10 min at room temperature before transferring into a 120 ml Teflon-lined autoclave. Methanothermal synthesis was carried out at 175 °C for 20 h. The obtained material was filtered and dried overnight at 110 °C under air, followed by the calcination at 500 °C for 2 h with 10 °C min−1 ramp under static air. The obtained material is abbreviated as 7.6Cu(Me), where the number represents the Cu molar ratio to overall metal content of the catalyst (100 × Cu/(Cu + Zr)). The exact amount of Cu metal was obtained from Inductively Coupled Plasma (ICP) analysis. (Me) in the material name indicates the methanothermal preparation method.2.3 Synthesis of Cu–ZrO2 catalyst by oxalate gel precipitation method
The oxalate gel precipitation method (Og) was based on the reports from Fan and co-workers.22,29–32x moles of Cu(NO3)2·3H2O (x = 0.002, 0.004, 0.006, 0.008, and 0.010) and (0.020 − x) mol of ZrO(NO3)2·6H2O were dissolved in 200 ml of ethanol at room temperature. Then, 0.024 mol of oxalic acid dihydrate (BDH Chemicals, 99.5%) was added and stirred for 2 h at room temperature. The formed gel was filtered out and dried at 110 °C under air. The dried material was calcined at 550 °C for 2 h with a 10 °C min−1 ramp under static air. Materials obtained via this route are referred to as xCu(Og) (x = 10.7, 21.0, 31.3, 41.6, and 51.8), where x corresponds to Cu molar ratio to the overall metal content of the catalyst (100 × Cu/(Cu + Zr)) as obtained from ICP analysis.2.4 Synthesis of Cu/support catalyst by deposition–precipitation method
To prepare catalyst samples by deposition–precipitation, the desired amount of Cu(NO3)2·3H2O was dissolved in 100 ml of water, followed by addition of corresponding amount of support (Cu weight percent: 2.5 wt%, 20 wt%). t-ZrO2 and 7.6Cu(Me) were used as the supports for these materials. The amounts of Cu and support were set so as to give 1.0 g of final product. 0.20 M of K2CO3 (2.80 g in 100 ml of water) was added slowly into the prepared solution until a pH of 9.0 was obtained (initial pH = 1.6). The resulting precipitates were aged for 6 h. The material was filtered and washed with over a liter of cold water. After drying at 110 °C under air, calcination was carried out at 300 °C for 2 h in air. Materials obtained by deposition–precipitation in this way will be referred to as y wt%Cu/support, where y represents the weight percent of Cu against the entire catalyst in the preparative conditions (y = 2.5, 20).2.5 Reduction treatment
Reduction treatment was carried out under continuous flow of 5%H2/Ar at 300 °C for 2 h with a 10 °C min−1 ramp. Heat treated catalysts are indicated by adding “-HR” to the abbreviations discussed above.2.6 Characterization
Powder X-ray diffraction (PXRD) was performed on an X'Pert Pro diffractometer with a monochromatic Cu-Kα source (λ = 0.154 nm) operated at 40 kV and 40 mA. The scans were recorded in the 2θ range between 10° and 80°.Raman analysis was performed on an inVia Raman Microscope (Renishaw) using both 514 nm and 785 nm lasers for powder samples.
Surface areas were determined by multi-point N2 adsorption at 77 K on a Micromeretics Gemini 2360 according to the Brauner Emmet Teller (BET) method. Prior to the analysis, samples were degassed at 120 °C for 1 h under N2 flow.
Temperature programmed reduction (TPR) was carried out on a Thermo 1100 series TPDRO (Quantachrome) equipped with a cold trap with 75 ml min−1 of 10%H2/Ar using a 10 °C min−1 ramp rate. Samples (0.050 g) were pre-treated at 110 °C under a flow of argon (20 ml min−1) for 20 min prior to reduction in order to clean the surface. Analysis was performed under 10%H2/Ar (BOC 99.99%, 20 ml min−1) flow with 5 °C min−1 ramp, thermal conductivity detector (TCD) current of 150 mA, and an attenuation setting level of 8.
N2O titration was performed on a ChemBet (Quantachrome) for catalysts after reduction. Prior to the analysis, the catalysts were treated in situ under 30 ml min−1 of 10%H2/Ar flow at 180 °C. Then the temperature was reduced to 65 °C with He purging in order to remove H located on the catalyst surface. N2O was pulsed until no signals were detected using a TCD detector. After titration, a known amount of N2 was fed for calibration. A total of 1.46 × 1019 Cu atom m−2 and a stoichiometry of 2Cu/N2 were used to calculate Cu surface area.31,32
Transmission electron microscopy (TEM) analysis was performed on a JEM-2100F (JEOL). Prior to TEM analysis, samples were dispersed with ethanol under ultrasonication. Supernatant liquid was dropped on the Ni-grid and dried overnight for analysis.
Elemental compositions in the bulk were determined by Inductively Coupled Plasma (ICP) (ICPE-9000, Shimadzu). 0.025 g of the materials were dissolved in the mixture solution of 0.5 ml of hydrogen fluoride (Kanto) and 3.0 ml of nitric acid (Kanto) at 80 °C overnight. Then, the obtained solutions were diluted with distilled water up to 50 ml (solution 1). 1 ml of solution 1 was transferred to another tube and again diluted by distilled water up to 50 ml (solution 2) so that the catalyst concentration in solution 2 was 10 ppm. The diluted solutions (2) were subjected to ICP analysis. To calibrate this analysis, standard solutions of 1000 ppm Zr (Wako) and 1000 ppm Cu (Wako) were mixed and diluted up to 0.2, 0.5, 1.0, 2.5, and 5.0 ppm (Cu and Zr, respectively).
X-ray Photoelectron Spectroscopy (XPS) was performed using a Kratos Axis Ultra-DLD photoelectron spectrometer, using monochromatic Al Kα radiation, at 144 W power.
2.7 Hydrogenation of LA to GVL
All experiments were carried out using a 50 mL capacity stainless steel autoclave (Model 5500HP, Parr Instruments). The reactor was equipped with a gas inlet valve for charging gas into the reactor and a gas release valve for releasing pressure and gas sampling. A stirring shaft was used to vigorously stir the mixture (2000 rpm) during the reactions, and the temperature of the liquid was measured using a thermocouple. A Parr Instruments Model 4836 controller was used to control temperature and stirring speed. Unless otherwise stated reactions were carried out under a set of standard conditions: reaction temperature 200 °C, catalyst amount 50 mg and 10 g of 5% levulinic acid (98%, Sigma Aldrich) diluted with distilled water was used as a substrate. Before the reaction, the reactor was purged three times with 5 bar of nitrogen gas, in order to remove residual air, followed by purging three times with 35 bar of hydrogen. The reactor was then pressurized to 35 bar of H2 at which point the reference zero time for the reaction was set. For the reusability experiments, first, a reaction was performed with fresh catalyst and then the catalyst was filtered off and transferred back to the reactor without drying and a new catalytic run was performed along with the fresh substrate under standard reaction conditions.Blank runs, without the catalyst present, showed no LA conversion. Acetonitrile (Acros Organics, 99.9%) was used an internal standard for analysis and the products were detected by Varian 450 GC equipped with CP-Sil 5CB (50 m, 0.32 mm, 5 μm) column and an FID detector. The calculated carbon balance was always in the range of 95–100%.
2.8 DFT calculations
Calculations were performed using the CASTEP33–41 package with the Perdew–Burke–Ernzerhof (PBE) functional and auto generated pseudopotentials. A plane wave basis set was used with a three dimensional periodic boundary, with a cut off value of 630 eV and finite basis set corrections were included for all calculations. The calculations included the G06 long range dispersion force correction by Grimme, and a Hubbard U value of 7 eV was used for the Cu d orbitals.42 The different Cu doping levels in ZrO2 were created using a 3 × 3 × 3 supercell (Zr52O104), in which the appropriate number of Zr4+ ions were replaced with Cu2+, along with oxygen vacancies to ensure the simulation cell remained charge neutral when all ions are considered in their formal oxidation states. The Cu atom positions were chosen to ensure the second neighbor shell of each Cu ion consisted only of Zr atoms. Tests in which Cu atoms were placed within second neighbor distances resulted in very high lattice energies and/or failure to converge the geometry optimization of the cell. Optimization of both the atomic positions and the unit cell dimensions were carried out in order to calculate lattice parameters for each level of Cu doping. The reported lattice parameters were taken as an average over 8 structures corresponding to charge compensating defects at each of the 8 oxygen atoms surrounding the Cu substituted cation site. To compare the likely stability of alternative doping levels we calculate the mixing energy, Emixing, based on the equation:| Emixing = ECuxZrn−xO2n−x − ((n − x)EZrO2 + xECuO) | (1) |
Here ECuxZrn−xO2n−x is the calculated energy for the unit cell with x-Cu atoms substituted on Zr sites, EZrO2 is the lattice energy per formula unit for the pure ZrO2 calculated using the same 3 × 3 × 3 supercell as used for the doped calculations and ECuO is the calculated energy for a reference CuO unit cell. Cu doping levels are quoted using the same percentage molar metal content as defined for experimental samples (100 × Cu/(Cu + Zr)).
3. Results and discussion
3.1 Hydrogenation of LA to GVL
Catalysts synthesized by the Me and Og methods were tested for the hydrogenation of LA to GVL. For all the reactions, the selectivity to GVL was 100% and no byproducts were detected. Fig. 1 shows the GVL yield obtained from a reaction time of 2 h at 200 °C under 35 bar hydrogen pressure.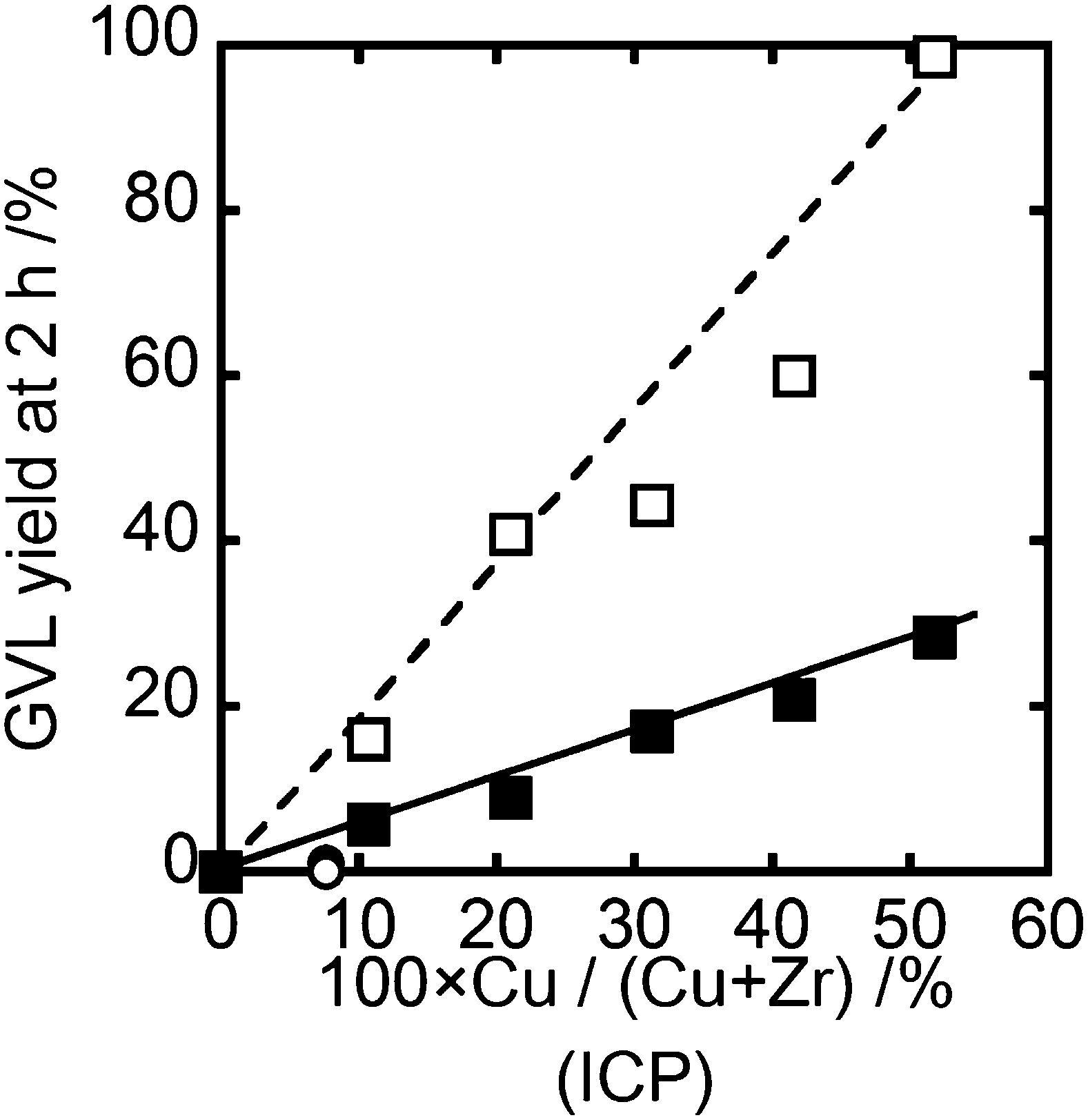 | ||
| Fig. 1 GVL yield as a function of Cu/(Cu + Zr) ratio determined by ICP. ○ Me-HR, ● Me, ■ Og, □ Og-HR. Reaction conditions: 200 °C, 35 bar H2, 0.050 g catalyst, 5 wt% LA in water, 2 h. | ||
Initial comparisons were carried out at relatively low Cu loadings (10Cu(Og) and 7.6Cu(Me)). The Me catalysts did not show any activity while the material prepared by the Og method gave a modest GVL yield of around 5% after a 2 h reaction time. Increasing the Cu loading of the Og catalyst lead to a linear increase in GVL yield rising to 25% for the 51.8Cu(Og) material. After reduction of the catalysts, the 7.6Cu(Me)-HR material was also found to be inactive. In contrast the catalytic activity of Og prepared materials was significantly increased for all loadings tested and a linear correlation between Cu loading and activity was maintained. This leads to almost 100% GVL yield being achieved for the case of the 51.8Cu(Og)-HR material. These observations clearly show that the Og preparation method produces catalytically active materials for LA hydrogenation and that a pre-reduction step before the catalysts are used greatly increases their activity.
A range of characterization methods were used to examine the physical properties that may lead to this stark difference between the catalytic performance of the 7.6Cu(Me) and xCu(Og) (x = 10.7, 21.0, 31.3, 41.6, and 51.8) samples and to establish the effect of the reduction step. Table 1 lists the catalyst compositions from ICP analysis (bulk) and XPS (surface), lattice parameter determined from PXRD, the BET surface area, CuO or Cu particle size (Scherrer equation), and Cu surface area (N2O titration). For catalysts synthesized by the methanothermal method (Me), 7.6% of the catalyst was found to be Cu by ICP, much lower than the theoretical Cu molar ratio (Cu/(Cu + Zr) = 20). For this reason, for all samples we quote the Cu content of the material from the ICP determined Cu amount. Furthermore the surface concentration of copper (determined by XPS) was reduced after reduction at 300 °C under a flow of 5%H2/Ar. This suggests that the reduction treatment caused more copper to be incorporated into the bulk structure. In contrast, catalysts prepared via oxalate gel precipitation (Og) showed good agreement between theoretical and measured ratio of Cu to total catalyst mass. For the Og materials, the measured Cu/(Cu + Zr) ratios were also unchanged by reduction treatment under a flow of 5%H2/Ar.
| Catalyst | Cu/(Cu + Zr) | Lattice parameter/Å | BET surface areac/m2 g−1 | CuO particle sized/nm | Cu particle sizee/nm | Cu surface areaf/m2 gcat−1 | |||
|---|---|---|---|---|---|---|---|---|---|
| Preparation | Entirea | Surfaceb | a | c | |||||
| a Determined by ICP. The value in bracket represents the bulk composition after the reduction. b Determined by XPS. The value in bracket represents the surface composition after the reduction. c Obtained by N2 adsorption at liq. N2 temperature. Surface area in bracket is the one of the reduced catalysts. d Obtained by PXRD of the oxidized catalysts and estimated from Scherrer equation. e Obtained by PXRD of the reduced catalysts and estimated from Scherrer equation. f Obtained by N2O titration. | |||||||||
| t-ZrO2 | 0 | 0.0 | 0.0 | 3.60 | 5.15 | 43.4 | — | — | — |
| 7.6Cu (Me) | 20 | 7.6 (5.3) | 5.7 (2.9) | 3.59 | 5.08 | 64.3 (—) | — | — | 0.2 |
| 10.7Cu (Og) | 10 | 10.7 (10.3) | 8.9 (12.7) | 3.60 | 5.13 | 62.6 (61.3) | — | 7.6 | 2.4 |
| 21.0Cu (Og) | 20 | 21.0 (20.7) | 17.0 (29.8) | 3.59 | 5.08 | 76.4 (76.1) | — | 16.0 | 2.0 |
| 31.3Cu (Og) | 30 | 31.3 (31.0) | 22.1 (26.1) | 3.59 | 5.08 | 66.7 (64.0) | 7.6 | 13.9 | 4.0 |
| 41.6Cu (Og) | 40 | 41.6 (41.0) | 32.0 (32.5) | 3.60 | 5.07 | 57.9 (—) | 8.5 | 17.1 | 1.9 |
| 51.8Cu (Og) | 50 | 51.8 (51.6) | 27.6 (42.8) | 3.60 | 5.08 | 61.1 (64.3) | 8.0 | 15.4 | 2.0 |
Powder X-ray diffraction patterns for all materials are shown in Fig. 2. Fig. 2(A) shows the patterns for the Me and Og catalysts before reduction. All the samples showed PXRD reflections at 30.3°, 35.2°, 50.3°, 60.1°, 63.0°, 73.2°, and 74.2°, ascribed to the tetragonal-zirconia phase (t-ZrO2). Fig. S1† shows the narrow region of same PXRD patterns to indicate slight change of the lattice parameter for the c-axis. Raman spectra of these catalysts also showed the bands attributed to t-ZrO2 (Fig. S2†), confirming that all catalysts synthesized have the bulk structure of t-ZrO2.29,43 Besides the PXRD reflections of t-ZrO2, an additional reflection attributable to CuO was found at 38.5° in xCu(Og) catalysts with x ≥ 31.3. So it appears that, at the higher loadings, segregation of CuO and Cu–ZrO2 phases leads to CuO particles that are large enough to observe by PXRD. The crystallite size of CuO on the basis of the Scherrer equation was, within error, around 8 nm (Table 1). For all samples in which CuO reflections are observed, the reflection position is the same as that of pure CuO, indicating no incorporation of Zr into the copper oxide particles. In addition, we observe an PXRD halo which implies the formation of an amorphous phase in the 14.8Cu(Me) catalyst (Fig. S3†).
 | ||
| Fig. 2 PXRD patterns of (A) calcined catalysts, (B) reduced catalysts; ● CuO, ○ Cu metal. HT = heat treated. | ||
The PXRD patterns of the Me and Og catalysts after reduction are shown in Fig. 2(B). The reduction treatment caused no changes in the PXRD peaks attributable to t-ZrO2 component in any of the materials. A new peak at 43.3° is also seen for the Og prepared materials at all Cu loadings that is attributable to Cu metal. At the same time the CuO peaks seen in Fig. 2(A) for Cu loadings of 31.3% and higher are absent after reduction, indicating that the reduction of CuO was complete under our reduction conditions (300 °C for 2 h under 5%H2/Ar flow). The crystallite size of Cu metal determined using the Scherrer equation (Table 1) was around 15 nm for all samples, except for the case of 10.7Cu(Og) which showed 7.6 nm Cu particles in agreement with previous reports.29 The increase of the particle size from 8 nm before the reduction to 15 nm after the reduction indicates some sintering has taken place. This is also seen from the XPS measurement of surface Cu species estimated from peak areas (Table 1). For example, the 51.8Cu(Og) sample has a surface Cu to total metals ratio of 27.6 before reduction compared with 42.8 for 51.8Cu(Og)-HR post-reduction, which we attribute to the increased thickness of the Cu particles. The appearance of the Cu metal peak in the PXRD of Fig. 2(B) for low Cu loadings suggests that phase segregation does occur for Og materials at all Cu loadings even if the CuO particle size is below that detectable by PXRD. No PXRD peak shifts of the Cu metal lines compared with pure Cu metal were observed, indicating no alloy formation. No change in the PXRD pattern was observed for the 7.6Cu(Me) sample following treatment in a reductive atmosphere to give 7.6Cu(Me)-HR, indicating there is no CuO surface phase for this material. Furthermore, the Cu ratio near the catalyst surface estimated by XPS (Table 1) is almost the same as that of entire sample estimated by ICP in 7.6Cu(Me) (XPS: 5.7; ICP: 7.6), indicating that Cu incorporated into the t-ZrO2 lattice has been uniformly dispersed.
Aside from the phase segregation for the Og prepared samples, the inclusion of Cu in both the Me and Og synthesis procedures leads to changes in the PXRD peaks seen for the t-ZrO2 phase. The diffraction peaks for t-ZrO2 in Cu–ZrO2 materials occur at higher diffraction angles than seen for pure t-ZrO2. These shifts have also been reported by Fan et al. for Cu–ZrO2 materials prepared by the Og method.29Table 1 also gives the a and c lattice parameters for the t-ZrO2 phase derived from the diffraction patterns of Fig. 2. For 7.6Cu(Me), although almost no change in the lattice parameter for the a-axis was observed (Table 1, 3.60 Å in pure t-ZrO2 and 3.59 Å in 7.6Cu(Me)), the lattice parameter for the c-axis is significantly lower at 5.08 Å for 7.6Cu(Me) compared to 5.15 Å in pure t-ZrO2. Catalysts prepared using the Og method also show almost no lattice parameter changes in the a axis (3.59–3.60 Å). However, for xCu(Og) with x below 21.0%, the measured c lattice parameter gradually decreases with the amount of Cu included in the synthesis (5.15 Å in pure t-ZrO2 and 5.13 Å in 10.7Cu(Og)) and becomes constant for x above 21.0% (5.07 Å–5.08 Å). This contraction in the c axis direction indicates that Cu is incorporated into the t-ZrO2 lattice.44 The observation that the c-lattice parameter does not change any further on the addition of Cu to xCu(Og), x ≥ 21.0 indicates the saturation level of Cu in the t-ZrO2 lattice. Additional Cu content will then be taken up by the separate CuO phase on the zirconia surface.
This interpretation is supported by the DFT calculation results presented in Table 2. We find good agreement between the DFT calculated and experimental a and c-lattice parameters for the pure t-ZrO2 phase (Table 1). The contraction seen on incorporation of Cu2+ cations into the bulk cell (Table 2 and Fig. S4†) is also reproduced as is the relative insensitivity of the a-parameter. The computer model also shows how, in the relaxed structures, the Cu ions move to adopt a square planar geometry in one of the faces of the cubic arrangement of O anions around the Zr cation site. We also find that, as the amount of Cu doping increases, so does the mixing energy (Table 2 and Fig. S5†), even when normalized to Cu content. Indeed, when a structure with Cu doping in excess of 20% in the ZrO2 lattice is relaxed with DFT, the calculation is difficult to converge and there is a large distortion of the unit cell suggesting that high levels of Cu doping cannot be accommodated by the t-ZrO2 lattice. At these levels of doping it is difficult to find an arrangement of Cu ions which do not share anion neighbors and so, presumably, the lattice strain introduced by Cu2+ moving to the square planar site cannot be accommodated. Since the lattice parameters of 7.6Cu(Me) and xCu(Og) (x ≥ 21.0) are almost the same, the amount of Cu incorporated into the t-ZrO2 lattice is expected to be the same for these materials. The saturation of the lattice parameter change observed in xCu(Og) (x ≥ 21.0) implies that both 7.6Cu(Me) and xCu(Og) (x ≥ 21.0) contain Cu inside the t-ZrO2 lattice at the maximum loading.
| Doping % | E mixing/eV | E mixing/x/eV | Lattice parameters (error) Å | ||
|---|---|---|---|---|---|
| a | b | c | |||
| Error estimated from standard deviation of the calculations using 8 alternative charge compensating O anion defect locations. | |||||
| 0% | 0.00 | 0.00 | 3.59 | 3.60 | 5.27 |
| 2% | 0.71 | 0.71 | 3.59 (0.02) | 3.59 (0.02) | 5.20 (0.03) |
| 4% | 3.29 | 1.65 | 3.58 (0.01) | 3.61 (0.02) | 5.20 (0.02) |
| 6% | 5.79 | 1.93 | 3.57 (0.02) | 3.61 (0.01) | 5.19 (0.03) |
| 8% | 7.61 | 1.90 | 3.58 (0.03) | 3.61 (0.03) | 5.18 (0.03) |
We have already shown in Fig. 1 that the Og prepared materials show a linear increase in GVL yield with Cu content of the catalysts. PXRD and modelling studies suggest an upper limit for Cu incorporation into the t-ZrO2 lattice with materials prepared by the Og method having surface CuO nano-particles at all Cu doping levels, which can be readily reduced to Cu metal.
To consider how Cu loading affects the catalytic activity we consider characterization data that will give information on the material surface area and likely proportion of Cu incorporated into zirconia vs. that present as surface nanoparticles. The BET surface areas (Table 1) of the Cu containing catalysts obtained using both Me and Og routes were not affected by Cu content with all samples having values in the narrow range of 57.9 to 76.4 m2 g−1. These values are in line with those obtained for catalysts obtained by co-precipitation from the aqueous metal nitrate salts for which we have found a dependence of GVL yield on measured surface area.45 However, here the relatively small difference in surface area for the 7.6Cu(Me) and 10.7Cu(Og) materials cannot explain the large difference in catalytic activity. In addition, no significant change in the surface areas was observed after reduction.
N2O titration was performed in order to assess the Cu metal surface area (2Cu(metal) + N2O → Cu2O + N2, Table 1). The Cu surface areas obtained were similar for all of the Og catalysts and were in the range of 1.9 to 4.0 m2 g−1, slightly lower than reported previously for Cu zirconia catalysts synthesized by the Og method.22,29 The detectable TCD signal obtained in N2O titration further confirmed the existence of Cu metal in the reduced Og catalysts while the reduced 7.6Cu(Me) material showed almost no TCD signal during N2O titration and the obtained Cu surface area was almost negligible (0.2 m2 g−1), confirming that there is no Cu metal on the catalyst surface after the reduction treatment. This suggests that Cu metal is important for the hydrogenation of LA to GVL for Cu–ZrO2 catalysts. The lack of a direct correlation of GVL yield with Cu surface area may be due to a different Cu surface area for the catalysts under reaction conditions (200 °C under 35 bar of pure H2 in the presence of water) compared to that evaluated by N2O titration condition (pre-reduced at 180 °C under 10%H2/Ar flow). However, the more strongly reducing conditions of the experimental system indicate that only a particular form of Cu metal is active for the reaction so that the total metal surface area need not correlate with activity.
The likely state of the Cu segregated phase under reaction conditions was investigated using TPR experiments (Fig. 3). It is notable that the 7.6Cu(Me) material showed no TPR signal across the entire temperature range. This is in-line with ICP and PXRD results which indicate that the Me catalyst contains Cu only incorporated into the t-ZrO2 lattice and with the N2O titration of the 7.6Cu(Me)-HR material which showed no Cu surface area. The TPR result for this Me material also demonstrates that Cu incorporated into the t-ZrO2 lattice is not reduced using the reduction conditions employed in this study.
 | ||
| Fig. 3 TPR spectra of Cu–ZrO2 catalysts after calcination. | ||
In contrast, for Og catalysts, clear TPR signals were observed with the signal area increasing with the amount of Cu used in the synthesis. The temperature of the TPR peak also increases with Cu loading which could be due to a change in the degree of interaction of CuO species with Cu doped t-ZrO2 or a change of CuO particle dispersion.29,46,47 Even so, all samples, except the highest 51.8Cu(Og) material, show a return of the signal to base line by the reaction temperature of 200 °C. We attribute these TPR signals obtained for the Og catalysts to the reduction of CuO particles dispersed on the catalyst surface which is likely to occur under our reaction conditions.
TEM images of the catalysts are shown in Fig. 4. As expected from ICP, PXRD, and TPR, no Cu particles were found on the 7.6Cu(Me) samples either before or after reduction. Lattice fringes were observed in the material with d-spacing of 2.84 Å which corresponds to the (111) planes of the t-ZrO2 lattice. The Og catalysts show the same fringe patterns but in addition CuO and Cu metal particles were observed. The size of the CuO and Cu metal particles were ca. 8 nm and ca. 13 nm, respectively, which match well with the size estimated from PXRD using the Scherrer equation. The number of CuO and Cu metal particles was found to increase with overall Cu content in the materials. Similar observations were made with the TEM images of calcined only catalysts as shown in Fig. S10.†
 | ||
| Fig. 4 TEM images of (a) 7.6Cu(Me), (b) 7.6Cu(Me)-HR, (c) 21.0Cu(Og), (d) 21.0Cu(Og)-HR, (e) 51.8Cu(Og), and (f) 51.8Cu(Og)-HR. | ||
Based on the above results, we conclude that the Me catalyst have no Cu particles, whereas the Og catalysts should be thought of as a Cu doped t-ZrO2 material supporting CuO or Cu nanoparticles depending on the reduction treatment and that both synthesis methods lead to the same bulk composition with the same crystal phase of zirconia. Hence, we can evaluate the contribution of bulk and Cu particles for catalysis separately. In the next section we will discuss the catalytic activity of these catalysts for levulinic acid hydrogenation.
Fig. 5(A) shows time online (TOL) data for 51.8Cu(Og) both reduced and unreduced. Reduced catalysts always show significantly higher catalytic activity than calcined catalysts. No induction period for the reaction was observed in 51.8Cu(Og)-HR catalyst. However, the calcined 51.8Cu (Og) did show an induction period of 30 min (Fig. 5(B)). The rate as judged by the initial slope of the GVL yield is also much higher for 51.8Cu(Og)-HR than for the 51.8Cu(Og) catalyst even when the induction period is taken into account (Fig. 5(A)).
 | ||
| Fig. 5 (A) TOL data for GVL yield. (B) GVL yield from 0 to 30 min. ■ 51.8Cu (Og); □ 51.8Cu (Og)-HR, ⊠ 51.8Cu (Og)-AR-HR. ◘ GVL yield after the removal of the catalyst. Reaction conditions: 200 °C, 35 bar H2, 0.050 g catalyst, 5 wt% LA in water. | ||
In order to reuse the catalyst a reaction was performed with 51.8Cu (Og) and 51.8Cu (Og)-HR catalysts under standard reaction conditions. After reaction, the catalyst was filtered, washed, dried at 110 °C overnight and retested. The activity of 51.8Cu (Og) was completely lost. On the other hand, the catalytic activity of the reused 51.8Cu (Og)-HR was ca. 10% after 30 minutes of reaction time vs. 42% with the fresh catalyst. In the next attempt, the catalyst (after first use) was dried at room temperature overnight, and the catalytic activity was found to be 18% after 30 minutes of reaction time. Fig. S6† shows the PXRD (A) patterns of the dried 51.8Cu (Og) catalysts both before and after the reaction (dried at 110 °C overnight). PXRD reflections attributable to t-ZrO2 were unchanged by the reaction. However, PXRD reflections of CuO (in 51.8Cu(Og)) disappeared after use for the reaction and new reflections attributable to Cu metal were observed. It is suggested that the observed decline in activity corresponds to the reduction of CuO to Cu under the reaction conditions. Fig. S6(B)† shows the XRD pattern of 51.8Cu (Og)-HR catalyst on reuse and it was found out that the Cu metal crystallite sizes for 51.8Cu(Og) and 51.8Cu(Og)-HR after the reaction for 30 min were 62.0 nm and 22.0 nm, respectively, based on the Scherrer equation.
The CuO particles on the 51.8Cu (Og) catalyst were unstable against the strong reduction condition (i.e. temperature and hydrogen pressure) and formed the much larger particles by sintering. On the other hand, the Cu particles of 51.8Cu (Og)-HR catalyst were relatively stable even under the reaction condition, possibly the reduction treatment strengthened the interaction between ZrO2 support and Cu metal, therefore, the degree of sintering after the reaction was moderate. This different stability between CuO particles over the non-reduced catalyst and Cu particles over reduced the catalyst are related to the different catalytic activity. The larger size of the Cu metal particles produced from the reduction of CuO under reaction conditions in 51.8Cu(Og) resulted in the lower catalytic activity compared with 51.8Cu(Og)-HR. In a third attempt, the catalyst was not dried after the reaction (filtrated and reused without drying), and the catalytic activity of the reused catalyst was 36% after 30 minutes of reaction time. The data for four reuse runs are presented in Fig. 7. The catalyst lost activity over number of uses and a steady decline in GVL yield was observed.
Our experiments clearly show that reduced Cu particles play a crucial role for the hydrogenation of LA and that there is probably a critical Cu particle size required for high catalytic activity. To investigate this further we considered the effect of removal of copper particles from the surface. For this purpose, 0.40 g of 51.8Cu(Og) catalyst was treated in 4.0 ml of aqua regia overnight. Then, the solution was made up to 100 ml with distilled water and the remaining solid was filtered off. The recovered material (0.19 g) was dried overnight at 110 °C, followed by calcination under static air at 550 °C for 2 h, then heat-treatment under 5%H2/Ar at 300 °C for 2 h. The catalysts obtained from this process are abbreviated as 51.8Cu(Og)-AR (after the aqua regia treatment and calcination) and 51.8Cu (Og)-AR-HR (after aqua regia, calcination and reduction), respectively. Fig. S7† shows the PXRD patterns of the catalysts after the aqua regia treatment, which confirm that Cu particles have been removed from the catalyst surface. The catalytic reaction using 51.8Cu (Og)-AR-HR was also performed and the results are shown in Fig. 5(A) (crossed squares). It was found that 51.8Cu (Og)-AR-HR showed no catalytic activity for this reaction, further demonstrating that reduced Cu particles on the catalyst surface are crucial for catalytic activity.
To evaluate the effects of any leaching of Cu species during the reaction, the catalyst was filtered out of the reaction mixture after 30 min and then the solution was monitored for a further 1.5 h without any solid catalyst. After the initial 30 min run time, no further increase in the GVL yield was observed (Fig. 5(A) circle-in-square symbol), indicating that any Cu species leached out during the reaction did not contribute towards catalytic activity. ICP analysis of the solution after the reaction showed that it contained less than 0.3% of the entire Cu content of the catalyst. From these experiments we conclude that the reaction is completely heterogeneous.
According to earlier reports which dealt with the hydrogenation of LA to GVL, the reaction proceeds via hydrogenation of LA to 4-hydroxypentanoic acid, followed by dehydration and ring closure.16,21 It has been reported that ZrO2 is amphoteric and so can adsorb LA during the reaction.24,25 Therefore, the role of reduced Cu particles is likely to be the dissociation of molecular hydrogen and hydrogenation of LA which is adsorbed on the oxide support material. Kinetic analysis was performed for the 51.8Cu(Og)-HR catalyst in order to evaluate the details of the reaction and test this hypothesis. Fig. 6(A) and (B) show the effect of LA concentration and H2 pressure on the reaction rate, respectively. From the slopes of the fitted straight lines, the reaction orders were calculated as practically zero (−0.1) with respect to LA and roughly first order (1.1) with respect to H2 pressure, respectively. These results indicate that the catalyst surface is saturated with LA (or their derivatives) and hydrogenation is the rate-limiting step.
 | ||
| Fig. 6 Effects of concentration of LA (A) and hydrogen pressure (B) on the hydrogenation of LA over 51.8Cu(Og)-HR. Reaction temperature: 200 °C; 30 min; 25–60 bar H2, 0.010–0.050 g catalyst, 2.5–15 wt% LA in water. | ||
We have also estimated an activation energy for this reaction, for these experiments the LA conversion was set below 10% by lowering the amount of catalyst used from 0.050 g (to between 0.010 g and 0.025 g). An estimated activation energy of 68 kJ mol−1 was obtained from the slope of the Arrhenius plot (Fig. S8†).
Based on the above results, we conclude that the catalytically active component of Cu–ZrO2 catalyst for the hydrogenation of LA to GVL are reduced Cu particles on the catalyst surface which are primarily responsible for the dissociation of hydrogen in the rate limiting step of the reaction under our conditions. It is also apparent that small Cu nanoparticles are required to achieve the highest rates of reaction.
3.2 The role of the support
We have shown in the previous section that reduced Cu particles deposited on the catalyst surface are the catalytically active component for the hydrogenation of LA to GVL. In this section, we will discuss the role of the t-ZrO2 support for this reaction. For this purpose, we have prepared the Cu–ZrO2 catalysts by a deposition–precipitation (Dp) method using pure t-ZrO2 or 7.6Cu(Me) as supports.The elemental composition determined by ICP, BET surface area, Cu particle size (Scherrer equation), and the amount of Cu particles estimated by TPR analysis of the synthesized catalysts are shown in Table S2.† After the deposition of Cu, only small variations in surface area are observed. Fig. S9† shows TPR spectra of 10.7Cu(Og), 41.6Cu(Og), 2.5 wt%Cu/7.6Cu(Me), 20 wt%Cu/t-ZrO2, and 20 wt%Cu/7.6Cu(Me). As shown in Table S1,† the estimated Cu particle amounts of 10.7Cu (Og), 2.5 wt%Cu/7.6Cu (Me), 41.6Cu (Og), 20 wt%Cu/t-ZrO2, and 20 wt%Cu/7.6Cu (Me) were 891, 671, 4130, 3831, and 3954 μmol g−1, respectively. This indicates that the amounts of Cu particles deposited by DP method are almost the same with those of Og catalysts. In addition, the reduction temperatures of CuO particles were almost the same in these catalysts. Since the reduction temperature relates to the dispersion degree of Cu particles on the catalyst surface, the Cu dispersion degree is considered to be very similar in these catalysts.46,47
Fig. 8 shows the PXRD patterns of 10.7Cu(Og), 41.6Cu(Og), 2.5 wt%Cu/7.6Cu(Me), 20 wt%Cu/t-ZrO2, and 20 wt%Cu/7.6Cu(Me) before and after reduction at 300 °C for 2 h under 5%H2/Ar flow. All catalysts show PXRD patterns consistent with t-ZrO2. No peak shifts between t-ZrO2 and 20 wt%Cu/t-ZrO2 were observed (Fig. 8, inset), indicating that Cu particles were only deposited on the catalyst surface by the Dp method and no Cu was introduced into the lattice. In the same manner, the PXRD peak positions in 7.6Cu(Me), 2.5 wt%Cu/7.6Cu(Me), and 20 wt%Cu/7.6Cu(Me) were the same and were consistent with those of 41.6Cu(Og).
 | ||
| Fig. 7 Reusability data for 51.8Cu (Og)-HR catalyst. Reaction temperature: 200 °C; 30 min; 35 bar H2, 0.050 g catalyst, 5 wt% LA in water. | ||
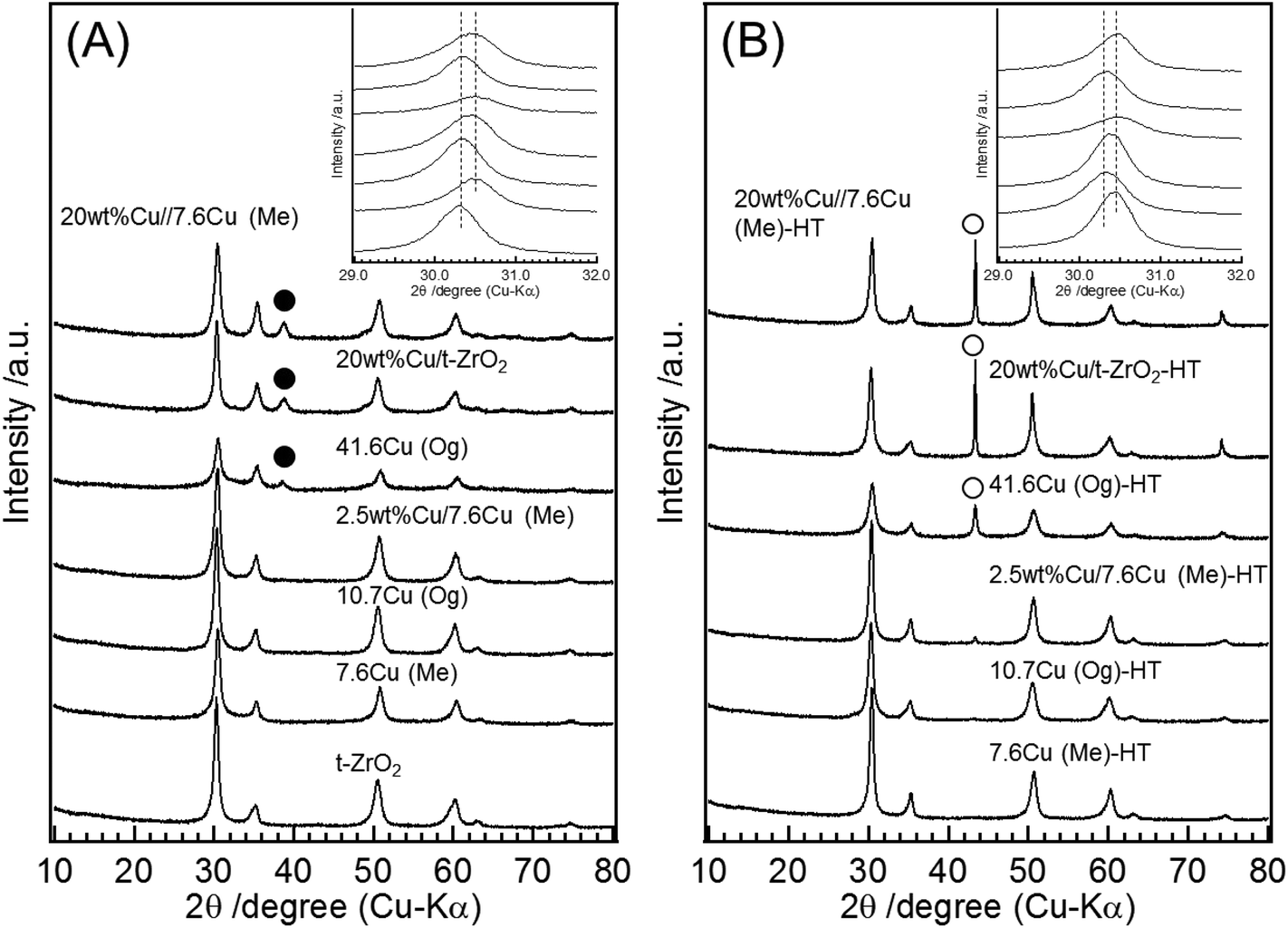 | ||
| Fig. 8 PXRD patterns of supports and catalysts. (A) Before reduction, (B) after reduction. ● CuO, ○ Cu metal. Insets are the enlarged PXRD patterns between 29–32°. | ||
As shown in Table S2,† the CuO particle sizes calculated using the Scherrer equation for 41.6Cu(Og), 20 wt%Cu/t-ZrO2, and 20 wt%Cu/7.6Cu(Me) were 8.5 nm, 9.1 nm, and 8.5 nm, respectively, showing that similarly sized surface particles are produced by the Og and Dp methods. It is interesting to note that after reduction, the Cu metal particle sizes were 17.1 nm, 50.2 nm, and 46.3 nm for 41.6Cu(Og), 20 wt%Cu/t-ZrO2, and 20 wt%Cu/7.6Cu(Me), respectively (Table S2†). This indicates that the Cu particles formed on 41.6Cu(Og) are more stable against sintering during the reduction process than those produced by Dp in 20 wt%Cu/t-ZrO2 and 20 wt%Cu/7.6Cu(Me), we attribute this to the different degree of interaction between Cu particles and the t-ZrO2 depending on the synthesis method to produce Cu–ZrO2 catalyst. For 10.7Cu(Og) and 2.5 wt%Cu/7.6Cu(Me), PXRD peaks of CuO were not seen before the reduction procedure was carried out. After the reduction, the PXRD peaks attributed to Cu metal were observed. Estimated Cu metal particle sizes were 7.6 nm and 19.1 nm, respectively (Table S1†).
Based on PXRD and TPR, we conclude that the catalysts synthesized by Dp method have the same bulk properties and the same amount of Cu particles on the supports with the similar dispersion. Here, we carried out the reaction using these catalysts. Fig. 9 shows the GVL yield of obtained catalysts at 2 h (reaction time). The supports (t-ZrO2 and 7.6Cu(Me)) were inactive for the reaction. The deposition of Cu lead to a slight increase in the catalytic activity as measured by GVL yield (2.5 wt%Cu/7.6Cu(Me), 5.3%; 20 wt%Cu/t-ZrO2, 2.7%; 20 wt%Cu/7.6Cu(Me), 3.7%). However, the activity was still lower than that obtained for the Og catalysts discussed earlier (10.7Cu(Og), 5.7%; 41.6Cu(Og), 20.7%). The difference between the Dp prepared materials and Og catalysts was particularly notable from the comparison of 41.6Cu(Og), 20 wt%Cu/t-ZrO2, and 20 wt%Cu/7.6Cu(Me). The Dp prepared materials in this group have almost the same amount of surface Cu with almost the same particle size as the Og catalysts and yet the Og catalyst gave more than 5 times the GVL yield after 2 h.
 | ||
| Fig. 9 GVL yield using Og and DP catalysts before (solid bar) and after the reduction (shadow bar). Reaction conditions: 200 °C; 35 bar H2, 0.050 g catalyst, 2 h, 5 wt% LA in water. | ||
After a pre-reaction reduction treatment, a significant increase in the catalytic activity was observed for Og catalysts as shown in Fig. 9 (10.7Cu (Og), 16.0%; 41.6Cu (Og), 59.9%). It is interesting to note, however, that the catalytic activity of the Dp catalysts was increased only slightly by the reduction step (2.5 wt%Cu/7.6Cu (Me), 7.5%; 20 wt%Cu/t-ZrO2, 6.6%; 20 wt%Cu/7.6Cu (Me), 7.7%).
As implied in Fig. 8, the Cu particles of Dp catalysts were unstable compared with those of Og catalysts, which we attribute to the different interaction degree between Cu particle and t-ZrO2 lattice. The Cu particles of Og catalysts are considered to be strongly interacted with Cu incorporated t-ZrO2 support and be stable even in the severe reaction conditions, which would explain the high catalytic activity compared with Dp catalysts. However we cannot prove this with the characterization techniques available to us. It was found that the support of Og catalysts stabilizes Cu particles and therefore is a key factor for the catalysis.
Based on these discussions, we suggest that reduced Cu particles, strongly interacted with Cu incorporated t-ZrO2 support, dissociate molecular hydrogen and the dissociated hydrogen reacts with adsorbed LA to form GVL.
4. Conclusions
Cu–Zr–O materials synthesized by the methanothermal method and by the oxalate gel precipitation method (Og method) have been used as catalysts for the hydrogenation of LA to GVL. All synthesized catalysts have the same crystal phase (tetragonal zirconia (t-ZrO2)) and similar BET surface areas. The material synthesized by the Me method contains Cu substituted within the t-ZrO2 lattice, but no evidence a separate CuO phase or Cu particle formation was found. This material was practically inactive as a catalyst for the reaction even after a pre-reduction step was included in the reaction procedure.Materials obtained using the Og method also contain Cu substituted into the t-ZrO2 lattice but in addition CuO particles were observed on the surface of these catalysts in the as prepared state and Cu metal nano-particles after a reduction in H2. The Og catalysts also showed promising activity for the hydrogenation of LA to GVL. A pre-reaction reduction treatment also led to a significant increase in the catalytic activity for the Og catalysts. Since the bulk composition, crystal structure, and surface area were almost the same for the Me and Og materials, we concluded that reduced Cu particles supported on Cu doped t-ZrO2 are responsible for the observed catalytic activity. We also used experiments based on DP prepared catalysts to suggest that the reduced Cu particles, strongly interact with Cu incorporated into the t-ZrO2 support allowing the material to maintain the dispersion of the active nanoparticles. The orders of reaction with respect to H2 and LA have been measured and suggest that the rate limiting step is the dissociate adsorption of molecular hydrogen.
The currently preferred catalyst of Ru/C has been optimized by Yan et al.48 Testing a 5%Ru/C material for the hydrogenation of LA to GVL they were able to achieve 92% conversion with close to 100% selectivity after 160 minutes of reaction at 130 °C under 12 bar of H2. The catalyst was dosed at 5% mass fraction against the LA reagent with methanol as solvent. In this work we have (Fig. 5) found that 51.8Cu(Og)-HR can achieve over 90% conversion at 60 minutes of reaction at 200 °C under 35 bar of H2. This catalyst was dosed at 10% mass fraction against the LA reagent with water as solvent.
The analysis presented here shows that surface supported Cu nanoparticles can show similarly high conversion and selectivity to GVL as the Ru/C system, albeit under more harsh reaction conditions. The higher temperature and H2 pressure reflect lower ability of Cu to activate hydrogen when compared to Ru. Our analysis also demonstrates that the active component of the Cu–ZrO2 system is present in relatively small amounts implying scope to reduce the Cu loading without loss of performance if the synthesis procedure can be refined further. The present work successfully demonstrates the catalytically active component of Cu–Zr–O catalyst for the hydrogenation of LA and will provide a way to design more effective catalysts, using abundant materials, to improve the catalytic activity for liquid phase hydrogenation chemistry.
Acknowledgements
This work was financially supported by the European Union FP7 NMP project NOVACAM (Novel cheap and abundant materials for catalytic biomass conversion, FP7-NMP-2013-EU-Japan-604319). Satoshi Ishikawa is currently a Research Fellow of Japan Society for the Promotion of Science. Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk). Computing resource was also provided by Advanced Re-search Computing at Cardiff (ARCCA) and the HPC-Wales supercomputer facilities.References
- Y. Yang, C.-J. Sun, D. E. Brown, L. Zhang, F. Yang, H. Zhao, Y. Wang, X. Ma, X. Zhang and Y. Ren, Green Chem., 2016, 18, 3558–3566 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- K. Kon, W. Onodera and K.-I. Shimizu, Catal. Sci. Technol., 2014, 4, 3227–3234 CAS.
- K.-I. Shimizu, S. Kanno and K. Kon, Green Chem., 2014, 16, 3899–3903 RSC.
- P. P. Upare, J.-M. Lee, Y. K. Hwang, D. W. Hwang, J.-H. Lee, S. B. Halligudi, J.-S. Hwang and J.-S. Chang, ChemSusChem, 2011, 4, 1749–1752 CrossRef CAS PubMed.
- W. Luo, U. Deka, A. M. Beale, E. R. H. van Eck, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- M. Li, G. Li, N. Li, A. Wang, W. Dong, X. Wang and Y. Cong, Chem. Commun., 2014, 50, 1414–1416 RSC.
- J. Q. Bond, D. M. Alonso, R. M. West and J. A. Dumesic, Langmuir, 2010, 26, 16291–16298 CrossRef CAS PubMed.
- X.-L. Du, Q.-Y. Bi, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Green Chem., 2012, 14, 935–939 RSC.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- N. Scotti, M. Dangate, A. Gervasini, C. Evangelisti, N. Ravasio and F. Zaccheria, ACS Catal., 2014, 4, 2818–2826 CrossRef CAS.
- A. M. Hengne, A. V. Malawadkar, N. S. Biradar and C. V. Rode, RSC Adv., 2014, 4, 9730–9736 RSC.
- X.-L. Du, L. He, S. Zhao, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815–7819 CrossRef CAS PubMed.
- P. P. Upare, J.-M. Lee, D.-W. Hwang, S. B. Halligudi, Y.-K. Hwang and J.-S. Chang, J. Ind. Eng. Chem., 2011, 17, 287–292 CrossRef CAS.
- Y. Yao, Z. Wang, S. Zhao, D. Wang, Z. Wu and M. Zhang, Catal. Today, 2014, 234, 245–250 CrossRef CAS.
- O. A. Abdelrahman, A. Heyden and J. Q. Bond, ACS Catal., 2014, 4, 1171–1181 CrossRef CAS.
- P. A. Son, S. Nishimura and K. Ebitani, RSC Adv., 2014, 4, 10525–10530 RSC.
- S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS.
- B. S. Akpa, C. D'Agostino, L. F. Gladden, K. Hindle, H. Manyar, J. McGregor, R. Li, M. Neurock, N. Sinha, E. H. Stitt, D. Weber, J. A. Zeitler and D. W. Rooney, J. Catal., 2012, 289, 30–41 CrossRef CAS.
- R. Luque and J. H. Clark, Catal. Commun., 2010, 11, 928–931 CrossRef CAS.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- J. Yuan, S.-S. Li, L. Yu, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Energy Environ. Sci., 2013, 6, 3308–3313 CAS.
- J. Kondo, Y. Sakata, K. Domen, K. Maruya and T. Onishi, J. Chem. Soc., Faraday Trans., 1990, 86, 397–401 RSC.
- J. Wang, S. Jaenicke and G.-K. Chuah, RSC Adv., 2014, 4, 13481–13489 RSC.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233–12235 RSC.
- K. Samson, M. Sliwa, R. P. Socha, K. Gora-Marek, D. Mucha, D. Rutkowska-Zbik, J. F. Paul, M. Ruggiero-Mikolajczyk, R. Grabowski and J. Sloczynski, ACS Catal., 2014, 4, 3730–3741 CrossRef CAS.
- L. Bui, H. Luo, W. R. Gunther and Y. Roman-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022–8025 CrossRef CAS PubMed.
- W. Li, H. Huang, H. Li, W. Zhang and H. Liu, Langmuir, 2008, 24, 8358–8366 CrossRef CAS PubMed.
- L.-C. Wang, Q. Liu, M. Chen, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Phys. Chem. C, 2007, 111, 16549–16557 CAS.
- Y. Ma, Q. Sun, D. Wu, W.-H. Fan, Y.-L. Zhang and J.-F. Deng, Appl. Catal., A, 1998, 171, 45–55 CrossRef CAS.
- X.-R. Zhang, L.-C. Wang, C.-Z. Yao, Y. Cao, W.-L. Dai, H.-Y. He and K.-N. Fan, Catal. Lett., 2005, 102, 183–190 CrossRef CAS.
- C.-Z. Yao, L.-C. Wang, Y.-M. Liu, G.-S. Wu, Y. Cao, W.-L. Dai, H.-Y. He and K.-N. Fan, Appl. Catal., A, 2006, 297, 151–158 CrossRef CAS.
- B. G. Pfrommer, M. Côté, S. G. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233–240 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. – Cryst. Mater., 2005, 220, 567–570 CAS.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- G. P. Francis and M. C. Payne, J. Phys.: Condens. Matter, 1990, 2, 4395 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Solid State, 2009, 80, 205414 CrossRef.
- A. K. Mishra, A. Roldan and N. H. de Leeuw, J. Phys. Chem. C, 2016, 120, 2198–2214 CAS.
- M. D. Rhodes and A. T. Bell, J. Catal., 2005, 233, 198–209 CrossRef CAS.
- C. Qiu, C. Chen, S. Ishikawa, T. Murayama and W. Ueda, Top. Catal., 2014, 57, 1163–1170 CrossRef CAS.
- D. R. Jones, S. Iqbal, S. Ishikawa, C. Reece, L. M. Thomas, P. J. Miedziak, D. J. Morgan, J. K. Edwards, J. K. Bartley, D. J. Willock and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 6022–6030 CAS.
- D. Mrabet, A. Abassi, R. Cherizol and T.-O. Do, Appl. Catal., A, 2012, 447–448, 60–66 CrossRef CAS.
- P. B. Sanguineti, M. A. Baltanas and A. L. Bonivardi, Appl. Catal., A, 2015, 504, 476–481 CrossRef CAS.
- Z.-P. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02598f |
| This journal is © The Royal Society of Chemistry 2017 |