
Improving the efficiency of the Diels–Alder process by using flow chemistry and zeolite catalysis†
S.
Seghers
a,
L.
Protasova
b,
S.
Mullens
b,
J. W.
Thybaut
c and
C. V.
Stevens
 *a
*a
aSynBioC Research Group, Department of Sustainable Organic Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, 9000 Ghent, Belgium. E-mail: chris.stevens@ugent.be
bVITO, Vlaamse Instelling voor Technologisch Onderzoek, Boeretang 200, 2400 Mol, Belgium
cLaboratory for Chemical Technology, Department of Chemical Engineering and Technical Chemistry, Faculty of Engineering and Architecture, Ghent University, Technologiepark 914, 9052 Ghent, Belgium
First published on 25th October 2016
Abstract
The industrial application of the Diels–Alder reaction for the atom-efficient synthesis of (hetero)cyclic compounds constitutes an important challenge. Safety and purity concerns, related to the instability of the polymerization prone diene and/or dienophile, limit the scalability of the production capacity of Diels–Alder products in a batch mode. To tackle these problems, the use of a high-pressure continuous microreactor process was considered. In order to increase the yields and the selectivity towards the endo-isomer, commercially available zeolites were used as a heterogeneous catalyst in a microscale packed bed reactor. As a result, a high conversion (≥95%) and endo-selectivity (89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11) were reached for the reaction of cyclopentadiene and methyl acrylate, using a 1:1 stoichiometry. A throughput of 0.87 g h−1 during at least 7 h was reached, corresponding to a 3.5 times higher catalytic productivity and a 14 times higher production of Diels–Alder adducts in comparison to the heterogeneous lab-scale batch process. Catalyst deactivation was hardly observed within this time frame. Moreover, complete regeneration of the zeolite was demonstrated using a straightforward calcination procedure.
:11) were reached for the reaction of cyclopentadiene and methyl acrylate, using a 1:1 stoichiometry. A throughput of 0.87 g h−1 during at least 7 h was reached, corresponding to a 3.5 times higher catalytic productivity and a 14 times higher production of Diels–Alder adducts in comparison to the heterogeneous lab-scale batch process. Catalyst deactivation was hardly observed within this time frame. Moreover, complete regeneration of the zeolite was demonstrated using a straightforward calcination procedure.
1 Introduction
The Diels–Alder (DA) reaction is universally acknowledged for its rapid, atom-efficient and clean access to molecular complexity on a small scale. This reaction involves a [4 + 2]-cycloaddition between a diene and a dienophile, introducing two new σ-bonds as indicated in red in Scheme 1.1 This enables one of the most efficient routes towards six-membered compounds. Controlling the stereo-, regio-, and enantioselectivity is essential for the synthesis of complex natural products and pharmaceuticals. Typically, a mixture of endo- and exo-isomers is obtained (Scheme 1). The endo products, however, are mostly obtained as the major products due to favorable secondary orbital interactions.1,2 Despite its wide use at the research level, problems associated with reactivity, selectivity and scale-up still have to be overcome for Diels–Alder reactions. The core problems are the instability of the reagents, the required stoichiometric excess of the reagent, the exothermic behavior of the main and side reactions, poor selectivity, extreme reaction times and temperatures, as well as the use of (toxic) catalysts.3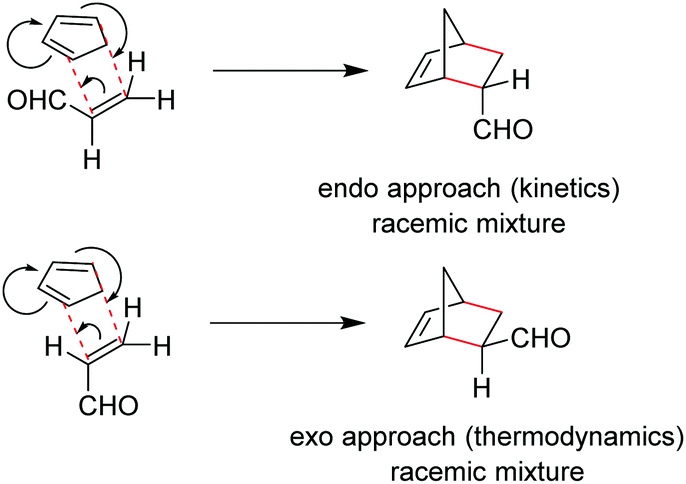 | ||
| Scheme 1 Stereoselectivity of the Diels–Alder reaction. | ||
Indeed, the industrial use of one of the most popular transformations for organic chemists is limited due to safety and purity concerns. Due to competing, rapid and uncontrolled polymerization with a potentially explosive character, the scale-up of the DA reaction constitutes an important challenge in view of industrial application. Process engineers tend to consider the DA reaction by definition in the field of non-scalable transformations and, in many cases, refuse chemical routes featuring the DA reaction as a key step.3,4 For instance, of the top 200 pharmaceutical products by US retail sales in 2010, only one marketed drug is produced industrially using the DA reaction.3,5,6 Buprenorphine is a potent narcotic and is additionally used to treat opioid addiction.7 The synthesis from thebaine or oripavine includes a DA reaction step with methyl vinyl ketone (MVK) as a dienophile (Scheme 2).8 Although little is known about DA reactions on the industrial scale, Funel and Abele recently published a review containing a limited number of industrial relevant DA reactions.3
 | ||
| Scheme 2 Buprenorphine synthesis from thebaine.8 | ||
In view of the aim of our research group to facilitate challenging reactions in flow,9 microreactor technology (MRT) is believed to be able to overcome the previously described problems. As a consequence of its small dimensions, the reaction environment can be controlled meticulously. The desired reaction time, temperature and, due to efficient mixing properties, stoichiometry can exactly be set. A fast, safe and low cost increase of the production capacity constitutes the key element to address the DA reaction in a continuous flow set-up. Moreover, the combination with heterogeneous catalysis tends to be a perfect match as the immobilization of a solid catalyst in a microreactor simplifies both the recuperation of the catalyst and the work-up of the product.10
DA reactions are known to be catalyzed by Lewis or Brønsted acids. The replacement of homogeneous catalysts, such as AlCl3, HF or H2SO4, by heterogeneous analogues, is of great interest. In addition to concerns about health, safety and corrosion, the use of homogeneous catalysts is also accompanied by high costs for work-up and disposal of waste streams. Being solid acids, zeolites, often metal-doped or -exchanged, and mesoporous silica derived catalysts are extensively investigated for DA reactions.11,12 Zeolites consist of SiO4 and AlO4 tetrahedra as elementary building blocks in well-ordered 3D structures. Within the cavities, both Brønsted and Lewis acid sites are present.13,14 The use of such solid acid catalysts enhances the conversion and the selectivity to the endo-isomer, as the interaction between the acid sites of the solid catalyst and the electron withdrawing group of the dienophile reduces the LUMO's energy. This provides a better overlap with the HOMO of the diene.1,11g,i Moreover, microporous or mesoporous structures also exhibit spatial confinement of the reagents, even directing the product selectivity in the one or the other direction.11e,f,12 The use of zeolites in combination with MRT is much less investigated.15 However, some research groups have performed reactions by combining microreactors and acid catalysis by zeolites: dehydration of methanol to dimethylether,16 benzylation of benzene with benzyl alcohol,17 Knoevenagel condensation and the epoxidation of 1-pentene.18,19
In this work, the reaction between cyclopentadiene (CPD), by far the most popular diene for DA reactions, and methyl acrylate (MA), both reactive and polymerization prone substrates, was chosen as the generic reaction to optimize the DA reaction under continuous flow conditions (Scheme 3). Among the commercially available zeolites (e.g. (US)Y, ZSM-5, Beta and Mordenite), zeolite Y and Beta type catalysts have the largest pore diameter and surface area.20 The subtype Ultrastable Y (USY) is obtained after a dedicated dealumination treatment.21 In general, the large pore zeolite types USY and Beta are most often used as a solid acid catalyst in organic synthesis.12,22 H-USY was selected as a starting point for this analysis, however, a batch screening of different commercially available zeolites was carried out.
 | ||
| Scheme 3 Target reaction to develop a continuous flow and heterogeneously catalyzed DA process. | ||
Being a hot topic in organic chemistry, the DA reaction has already been extensively studied considering a wide variety of (expensive) ionic liquids and Lewis acid catalysts, focusing on maximizing the yields and the endo/exo-selectivity.11j,23–25 Although some research aims at inversing this selectivity, e.g. through the use of a bulky Lewis acid catalyst,2a,26 in most cases the maximal formation of the typical major endo-adduct is desired. As the exo-isomer is generally obtained after isomerisation from an endo/exo-mixture under basic conditions,27 also this research project is directed towards the endo-isomer and thus the maximization of the inherent selectivity of a (catalyzed) Diels–Alder reaction. With regard to the reaction of cyclopentadiene and methyl acrylate, in many publications, yields are up to 90% and higher, albeit obtained on a small scale and often requiring an excess of CPD and long reaction times.11j,24,25 Mostly, the batch processes are limited to an endo/exo-selectivity of about 80:20.24,27c Still, some research groups report an excellent selectivity of 90:10 and even higher.11j,25 In conclusion, the objectives to be achieved can be set at a yield of at least 90% and a selectivity of 90:10 to be able to compete with the majority of the reported batch investigations.
The DA cycloaddition has been performed using MRT before, mainly taking advantage of the excellent heat transfer, pressurized conditions and the rapid optimization ability.28 In an attempt to produce aromatics from a renewable feedstock, Cheng and Huber reported the production of mixtures of aromatic compounds using gas-phase DA type reactions between different furanic species and olefins over a ZSM-5 catalyst in a continuous flow fixed-bed reactor in the temperature range of 450–600 °C.11b In another paper the DA reaction between cyclopentadiene and crotonaldehyde was performed using a solid Lewis acid in a microreactor set-up. This solid acid consisted of aluminium oxide functionalized silica monoliths. Dichloromethane was used as a solvent at a reactor temperature of 37 °C. Conversions of 15 to 80% were reached. The optimized contact time was determined to be 9.4 min, resulting in 72% conversion, an endo/exo-distribution of 90:10 and a productivity of 0.033 mmol g−1 min−1. Although this work shows promising results, there is still room for improvement in terms of conversion, productivity and scalability, especially using acrylates in view of their industrial importance.29 More recently, Chiroli et al. described a continuous flow Diels–Alder reaction using a chiral organocatalyst on a silica support. The yield varied from 55 to 95% and an endo/exo-selectivity of 45:55 was obtained at room temperature. However, an excess of 7 equivalents of cyclopentadiene was used and the need for long residence times (up to 25 h) leads to low space–time yields (0.0024 mmol g−1 min−1).30
2 Experimental
2.1 Materials
All reagents and solvents were purchased from Sigma-Aldrich Belgium, except for the zeolite materials CBV 500 (Si/Al-ratio 2.6), CBV 720 (Si/Al-ratio 15), CBV 760 (Si/Al-ratio 30), CBV 3024E (Si/Al-ratio 15), CBV 21A (Si/Al-ratio 10) and CP814E* (Si/Al-ratio 12.5), which were obtained from Zeolyst International.20 The zeolite powder was pelletized under a 10 ton press and sieved to obtain a 125–200 μm particle fraction. Dichloromethane was dried by distillation from calcium hydride, while tetrahydrofuran (THF) and toluene were distilled from sodium and benzophenone prior to use. Ethyl acetate and acetonitrile were dried over molecular sieves. Extra dry 2-methyl THF (stabilized, AcroSeal®) was purchased from ACROS Organics and was used without any further drying step. Cyclopentadiene was obtained by cracking and distillation of dicyclopentadiene before each experiment. The used zeolite material was characterised using N2-sorption and thermogravimetric analysis (Table 1). These results show a significantly higher specific surface area (SSA) and pore volume of H-Beta. The analysed H-USY and H-Beta species contain a comparable water content of about 10 to 15 wt%. Additionally, the acid properties of the commercially available zeolite powders were measured using NH3-TPD (Table 2).| Zeolite type | Product codea | Shape | SSAb,c (m2 g−1) | Pore volumec (cm3 g−1) | Pore radiic (Å) | Water contentd (wt%) |
|---|---|---|---|---|---|---|
| a Zeolyst International. b Specific surface area. c Determined using N2-sorption. d Determined using thermogravimetric analysis. | ||||||
| H-USY | CBV 720 | Powder | 104 | 0.24 | 19.5 | 14 |
| H-USY | CBV 720 | Pellets | 109 | 0.38 | 19.2 | 14 |
| H-Beta | CP814E* | Powder | 177 | 0.88 | 18.2 | 12 |
| H-Beta | CP814E* | Pellets | 155 | 0.87 | 18.1 | 12 |
| Zeolite type | Product codeb | Si/Al |
T
l,maxc (°C) |
T
h,maxc (°C) |
C
ASd (10−2 mol kg−1) |
ΔHde (kJ mol−1) |
|---|---|---|---|---|---|---|
| a Ammonia temperature programmed desorption. b Zeolyst International. c Low- and high-temperature maximum, respectively corresponding to the weak and strong acid sites. d Acid site concentration. e Heat of desorption of ammonia. | ||||||
| H-Y | CBV 500 | 2.6 | 262 | 498 | 179 | 110 |
| H-USY | CBV 720 | 15 | 282 | 482 | 47 | 99 |
| H-USY | CBV 760 | 30 | 236 | 423 | 39 | 69 |
| H-Beta | CP814E* | 12.5 | 322 | 579 | 39 | 116 |
| H-ZSM-5 | CBV 3024E | 15 | 253 | 471 | 78 | 139 |
| H-Mordenite | CBV 21A | 10 | 264 | 638 | 96 | 176 |
2.2 Microreactor device
The X-Cube™ flow reactor is commercially available from ThalesNano and is shown in Fig. 1. The reagent solutions are pumped in the stainless steel tubing by means of two high pressure (HPLC) pumps. Each pump can deliver a 0.1–3 mL min−1 flow rate. In the employed set-up, a single reagent solution and the corresponding pump were used. The reagent solution was placed under a N2-pressure, using a pressurisation module of the commercially available Africa flow system (Syrris). The tubing was connected to the insulated CatCart® (catalyst cartridge). In the commercial system, two cartridges can be used in parallel. These cartridges can be mounted and dismounted easily and can be heated from 20 to 200 °C in 5 °C intervals, separately. For all experiments, only one CatCart® was used after a manual fill with the zeolite material (loading: 370 mg CBV 720; 350 mg CBV 760; 340 mg CP814E*). Finally, the mixture was pumped to the pressure sensor and the back pressure regulator, to which the outlet is connected. The reactor pressure can be set per 5 bar, up to a maximum of 150 bar. A schematic representation of the X-Cube™ flow reactor is given in Fig. 2. The reactor was rinsed with (dry) dichloromethane and subsequently with the reagent solution at 100 °C and 30 bar before start-up, as the catalyst initially showed a high activity towards cyclopentadiene polymerisation. A full experiment description can be found in the ESI.† | ||
| Fig. 1 Set-up for the continuous flow Diels–Alder process (left) using a zeolite filled CatCart® (right). | ||
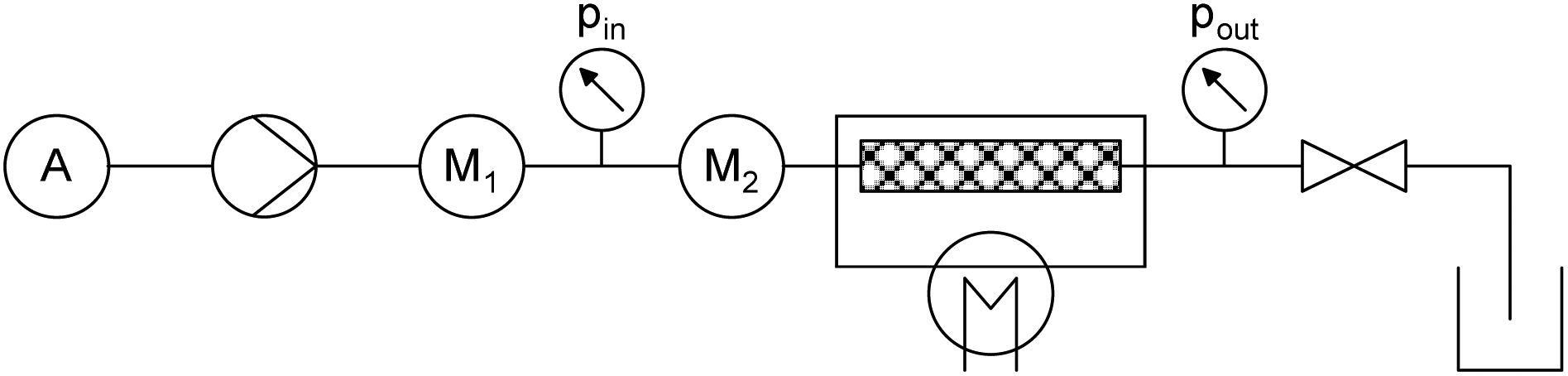 | ||
| Fig. 2 Schematic representation of the X-Cube™ flow reactor in the used set-up (M = Mixer). | ||
2.3 Analysis
The conversion of methyl acrylate to the end product was determined by integration of the methoxy-signals of MA (3.76 ppm), the endo-adduct (3.62 ppm) and the exo-adduct (3.69 ppm) in the 1H-NMR spectrum of the crude reaction mixtures. The space–time (ST) and site-time (SiT) were obtained as the ratio of the catalyst bed mass, respectively the molar amount of acid sites (AS), to the corresponding molar reagent flow rate.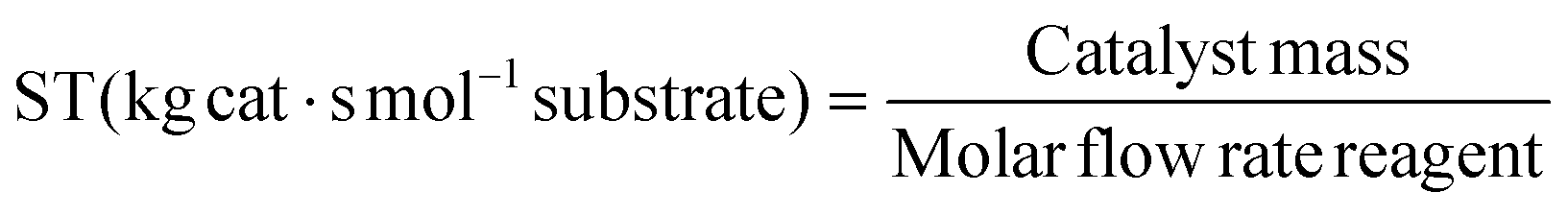

3 Results and discussion
3.1 Batch solvent screening
In order to develop a sustainable production process, a thoughtful solvent choice has to be made. A key example of a green solvent is without doubt water. The Diels–Alder reaction is an important example of an organic reaction which can be accelerated using water as a solvent.31–33 However, taking a look at the application of the Diels–Alder reaction in organic synthesis hardly shows any reaction that is not performed in either a chlorinated or an aromatic solvent.34,35 A quick database search demonstrates that dichloromethane is by far the utmost used solvent for the cycloaddition of CPD and MA. Toluene and hexane are located on respectively the second and third place. Additionally, this reaction has been evaluated in a wide variety of ionic liquids.36 To the best of our knowledge, only four papers have reported the performance of this reaction in water.33b,37 By stirring MA and 2 equiv. of CPD at room temperature in water for 72 h, the end product was obtained in 89% yield and a endo/exo-distribution of 87:13.37a Unfortunately, combining water as a solvent and a zeolite catalyst is not an option, as both reagents and, if any, end products accumulate inside the catalyst pore system.
Thus, the evaluation of the DA reaction between CPD and MA (Scheme 3) was initiated in batch in various organic solvents in order to determine the catalyst potential and the solvent of choice. Thermogravimetric analysis of the zeolites showed a water content of about 10 to 15 wt% (Table 1). In order to remove loosely bound water molecules, the zeolite material was kept for 24 h in an oven at 100 °C prior to its use. This constitutes a milder zeolite activation compared to calcinations or other high temperature and/or vacuum treatments found in the literature.11a–c,e–j Among the evaluated dry solvents, i.e. dichloromethane (DCM), THF, 2-methyl THF, ethyl acetate and acetonitrile, the highest conversion and endo/exo-selectivity were obtained in dry DCM (Table 3). Although the conversion was higher in dry toluene at reflux temperature, dichloromethane was chosen for optimization of the reaction in a continuous flow device. As a 3 times lower reflux temperature in dichloromethane still results in more than half of the conversion in toluene and a significantly higher endo-selectivity, it was envisaged that dichloromethane had more potential to speed up the reaction and to lead to high conversions and selectivity. In batch, however, even in the presence of a high catalyst loading (30 wt%), a mere 43% conversion was reached after 3.5 h.
| Solvent | Loading (wt%) | RTa (h) | 1H-NMR | endo (%) | |
|---|---|---|---|---|---|
| MA (%) | EPb (%) | ||||
| a Reaction time. b End product (both endo- and exo-isomers). c Percentage of the endo-isomer in the final DA product. | |||||
| Dry DCM | 0 | 2 | 94 | 6 | 79 |
| Dry DCM | 0 | 3.5 | 90 | 10 | 79 |
| Dry DCM | 30 | 2 | 64 | 36 | 92 |
| Dry DCM | 30 | 3.5 | 57 | 43 | 92 |
| Dry THF | 30 | 2 | 85 | 15 | 73 |
| Dry 2-methyl THF | 30 | 2 | 77 | 23 | 71 |
| Dry EtOAc | 30 | 2 | 87 | 13 | 74 |
| Dry MeCN | 30 | 2 | 82 | 18 | 81 |
| Dry toluene | 30 | 2 | 36 | 59 | 73 |
3.2 Batch catalyst screening
In the next phase of this study, different commercially available zeolite types were evaluated in the batch mode (Table 4). Among the H-(US)Y family, species with a varying Si/Al-ratio were evaluated. In this way, the best suited balance between the amount of acid sites and the acid site strength was investigated within a specific zeolite framework.38 CBV 720 (Si/Al 15) exhibited the highest conversion. An even higher Si/Al-ratio only resulted in a slightly lower conversion and a similar selectivity. Next, the influence of the zeolite structure was considered and other zeolite types with a similar Si/Al-ratio were evaluated. Although these materials possess a comparable amount of acid sites, their strength and accessibility can be influenced by the catalyst architecture. Using H-ZSM-5 and H-Mordenite catalysts, low conversions were obtained. Finally, the highest conversion was observed using a H-Beta catalyst with an analogous Si/Al-ratio, which could be ascribed to the higher specific surface area and pore volume, as well as to the acid strength of the sites (Tables 1 and 2).39 The high maximum temperature corresponding to the strong acid sites indicates that, despite the lower amount of acid sites, their strength is higher compared to the CBV 720 material. The most promising zeolite H-USY and H-Beta species were applied as a catalyst under continuous flow conditions.| Zeolite type | Product codea | Si/Al | 1H-NMR | endo (%) | |
|---|---|---|---|---|---|
| MA (%) | EPb (%) | ||||
| a Zeolyst International. b End product (both endo- and exo-isomers). c Percentage of the endo-isomer in the final DA product. | |||||
| H-Y | CBV 500 | 2.6 | 82 | 18 | 90 |
| H-USY | CBV 720 | 15 | 64 | 36 | 92 |
| H-USY | CBV 760 | 30 | 73 | 27 | 91 |
| H-ZSM-5 | CBV 3024E | 15 | 89 | 11 | 86 |
| H-Mordenite | CBV 21A | 10 | 95 | 5 | 83 |
| H-Beta | CP814E* | 12.5 | 42 | 58 | 90 |
3.3 Dilute continuous flow experiments
In addition to the problematic scalability, only a moderate conversion was reached in batch. Therefore, the development of a continuous flow process was considered. The proper use of a high catalyst loading, a high pressure environment and efficient radial mixing, as it occurs in an ideal plug flow, inherently related to the proposed microreactor device, is expected to lead to an improved result. The batch experiments revealed that dichloromethane was the solvent of choice. For continuous flow experiments, the pressure drop over the system depends on the solvent viscosity, catalyst, system set-up and pump speed. As indicated by the preliminary flow experiments, the generated pressure drop over the packed catalyst bed (CatCart®, see Section 2.2) was too high (>100 bar) when commercial zeolite powder was used. To avoid this, after a pelletizing, breaking and sieving pre-treatment, a 125–200 μm fraction was used for all flow experiments resulting in a maximum pressure drop below 5 bar. An overview of the presented flow configuration can be found in Scheme 4. A starting solution was prepared, containing 0.5 M of both reagents. This was stored at 0 °C and placed under a N2-pressure, to ensure a good pump stability and to avoid system pressure fluctuations. As it has been stated before that Diels–Alder reactions are known to benefit from pressurized conditions, the continuous flow experiments were performed in a high temperature and high pressure environment (100 °C, 30 bar). | ||
| Scheme 4 The continuous flow Diels–Alder process using a zeolite USY catalyst bed. | ||
Analogous to the preliminary batch experiments, the zeolite catalyst was conditioned prior to its use, however, in the continuous flow configuration this drying was performed in situ. The CatCart® was packed with zeolite USY, after physical pre-treatment, and subsequently dried by a N2 flush of 1 h over the heated cartridge at 200 °C. A conversion of MA to the end product of 85% was observed, which decreased to 70% after 6 h (Table 5, entry 1). Additionally, the catalyst was evaluated without any conditioning procedure (Table 5, entry 2). In this way, a conversion of 95% was reached, decreasing to 81% after 6 h. Quite remarkably, the USY zeolite was thus found to be more active towards acid catalysis without any drying step. Innumerable examples can be found in the literature of acid catalyzed reactions which require a calcination step or a high temperature and/or a vacuum step to activate these aluminosilicate catalysts.11a–c,e–j,40
| Entry | Zeolite typeb,c | Si/Al | FR (mL min−1) | CT (min) | Equiv. MA | [CPD] (M) | [MA] (M) | Timed (h) | 1H-NMR | endo (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| EP (%) | ||||||||||
| a Void fraction in the catalyst bed, estimated value: 40% of total volume. b The catalyst was used without any drying procedure, unless specified otherwise. c The corresponding zeolite product code, as obtained from Zeolyst International, can be seen in Table 2. d Time since start-up of the process. e The catalyst was conditioned by a 1 h N2 flush over the cartridge at 200 °C. f Dichloromethane was not dried before use. | ||||||||||
| 1e | H-USY | 15 | 0.1 | 3.5 | 1 | 0.5 | 0.5 | 1 | 85 | 88 |
| 6 | 70 | 86 | ||||||||
| 2 | H-USY | 15 | 0.1 | 3.5 | 1 | 0.5 | 0.5 | 1 | 95 | 89 |
| 1–5.5 | 88 | 89 | ||||||||
| 5.5 | 83 | 88 | ||||||||
| 5.5–6.5 | 81 | 87 | ||||||||
| 3 | H-USY | 15 | 0.1 | 3.5 | 0.8 | 0.6 | 0.5 | 1 | 92 | 89 |
| 1–4 | 81 | 88 | ||||||||
| 4 | 70 | 87 | ||||||||
| 4 | H-USY | 15 | 0.1 | 3.5 | 1.2 | 0.5 | 0.6 | 1 | 96 | 88 |
| 2 | 98 | 88 | ||||||||
| 3 | 99 | 88 | ||||||||
| 4 | 99 | 88 | ||||||||
| 5 | 98 | 88 | ||||||||
| 6 | 98 | 88 | ||||||||
| 7 | 96 | 88 | ||||||||
| 5 | H-USY | 15 | 0.2 | 1.8 | 1.2 | 0.5 | 0.6 | 1 | 95 | 89 |
| 2 | 95 | 90 | ||||||||
| 3 | 95 | 89 | ||||||||
| 4 | 95 | 89 | ||||||||
| 5 | 80 | 89 | ||||||||
| 6 | 68 | 88 | ||||||||
| 7 | 62 | 88 | ||||||||
| 6f | H-USY | 15 | 0.1 | 3.5 | 1.2 | 0.5 | 0.6 | 1 | 93 | 87 |
| 2 | 96 | 88 | ||||||||
| 3 | 99 | 87 | ||||||||
| 4 | 96 | 88 | ||||||||
| 5 | 93 | 88 | ||||||||
| 6 | 84 | 87 | ||||||||
| 7 | 80 | 87 | ||||||||
| 7 | H-USY | 30 | 0.1 | 3.5 | 1 | 0.5 | 0.5 | 1 | 93 | 88 |
| 2 | 93 | 88 | ||||||||
| 3 | 93 | 88 | ||||||||
| 4 | 93 | 89 | ||||||||
| 5 | 93 | 89 | ||||||||
| 6 | 93 | 89 | ||||||||
| 7 | 92 | 89 | ||||||||
| 8 | H-Beta | 12.5 | 0.1 | 3.5 | 1 | 0.5 | 0.5 | 1 | 93 | 87 |
| 2 | 94 | 88 | ||||||||
| 3 | 94 | 88 | ||||||||
| 4 | 94 | 88 | ||||||||
| 5 | 94 | 88 | ||||||||
| 6 | 94 | 89 | ||||||||
| 7 | 94 | 88 | ||||||||
| 9 | H-Beta | 12.5 | 0.2 | 1.8 | 1 | 0.5 | 0.5 | 1 | 95 | 88 |
| 2 | 95 | 89 | ||||||||
| 3 | 95 | 89 | ||||||||
| 4 | 95 | 89 | ||||||||
| 5 | 95 | 89 | ||||||||
| 6 | 93 | 89 | ||||||||
| 7 | 93 | 89 | ||||||||
| 10 | H-Beta | 12.5 | 0.3 | 1.2 | 1 | 0.5 | 0.5 | 0.7 | 95 | 88 |
| 1 | 95 | 89 | ||||||||
| 2 | 94 | 88 | ||||||||
| 3 | 91 | 89 | ||||||||
| 4 | 87 | 89 | ||||||||
| 5 | 85 | 89 | ||||||||
| 11f | H-Beta | 12.5 | 0.2 | 1.8 | 1 | 0.5 | 0.5 | 1 | 94 | 89 |
| 2 | 94 | 89 | ||||||||
| 3 | 95 | 89 | ||||||||
| 4 | 94 | 89 | ||||||||
| 5 | 94 | 89 | ||||||||
| 6 | 93 | 89 | ||||||||
| 7 | 92 | 89 | ||||||||
On adjusting the dienophile to diene ratio, it was found that a small excess of dienophile (1.2 equiv.) resulted in a nearly quantitative conversion and high selectivity to the endo product, as well as a high stability of the process, which were the main goals of the present work (Table 5, entry 4). The process was monitored for 7 h. Noteworthily, the contact time (CT), which equals the retention time in the catalyst bed, was barely 3.5 min. This corresponds to a space–time of 444 kg cat·s mol−1 CPD. Repetition of these process conditions showed that the conversion remained at least 94% during the first 7 hours after start-up. In the resulting DA mixture, a selectivity of 88% endo-isomer was observed. In comparison, when an excess of CPD was used, a fast deactivation of the catalyst bed was observed (70% after 4 h, see Table 5, entry 3). Doubling the flow rate, corresponding to a contact time of 1.8 min and a space–time of 222 kg cat·s mol−1 CPD, or using dichloromethane without pre-drying (Table 5, entries 5 and 6), both gave rise to a comparably high conversion initially, but a fast drop in conversion afterwards. It indicates that the performance achieved with the higher space–time corresponds to the establishment of thermodynamic equilibrium.
Based on the batch screening of catalysts, other promising zeolite catalysts were identified for evaluation under continuous flow conditions. A 1:1 CPD:MA stoichiometry could be applied to reach a high and stable conversion using a H-Beta catalyst or a H-USY species with a doubled Si/Al-ratio, respectively with a space–time of 390 and 420 kg cat·s mol−1 CPD (Table 5, entries 7 and 8). The better performance of the CBV 760 catalyst in comparison to CBV 720 in flow is different from the results obtained in batch (Tables 4 and 5, entries 2 and 7). This can be ascribed to the improvements made in the condition procedure of the catalyst. Next, attempts were made to increase the flow rate without affecting the high conversion (Table 5, entries 9 and 10). The optimal balance between conversion and throughput was found at a contact time of 1.8 min, corresponding to 195 kg cat·s mol−1 CPD (Table 5, entry 9). Moreover, in contrast to the H-USY catalyst, the H-Beta catalyzed continuous flow process in dichloromethane without pre-drying did not show a drop in conversion within a 7 h time frame (Table 5 entries 6 and 11). These reaction conditions were selected for the optimal Diels–Alder continuous flow process. Based on the experimental results at a flow rate of 0.3 mL min−1, deactivation is expected to be observed starting 9 h after the process start-up (Table 5, entry 10).
Because of the volatility of the reagents and the solvent, the end products were isolated as a mixture of endo and exo products in a nearly quantitative yield via mild rotary evaporation. Upon evaporation with mild heating, a product loss, mainly of the endo-isomer, was observed. The purity of the obtained mixture of DA products was verified using GC(-MS) and was found to be ≥95%. Under the optimal process conditions, the Diels–Alder products were isolated in a 94% yield, a 89% selectivity towards the endo-isomer and a GC-purity of 97% (Table 5, entry 11). A calculated throughput of 0.87 g h−1 over 7 h was reached, corresponding to a space–time yield of 0.29 mmol g−1 min−1. This implies a respectively 9 and 121 times higher space–time yield in comparison to the published continuous flow procedures of Sachse et al. and Chiroli et al.29,30 Noteworthily, our results were obtained using just a 1:1 stoichiometric ratio of diene and dienophile and a contact time of 1.8 min with the catalyst bed.
In an attempt to avoid the use of harmful dichloromethane, the second and third best solvents, as it was determined using a batch solvent screening (Table 3), were also evaluated under the optimized continuous flow conditions (Table 6). Unfortunately, in the case of toluene or acetonitrile as a solvent, both the conversion and the selectivity towards the endo-isomer were significantly lower in comparison to the use of dichloromethane.
| Entry | Solvent | FR (mL min−1) | CT (min) | Equiv. MA | [CPD] (M) | [MA] (M) | Timeb (h) | 1H-NMR | endo (%) |
|---|---|---|---|---|---|---|---|---|---|
| EP (%) | |||||||||
| a Void fraction in the catalyst bed, estimated value: 40% of total volume. b Time since start-up of the process. | |||||||||
| 1 | Acetonitrile | 0.1 | 3.5 | 1 | 0.5 | 0.5 | 1 | 66 | 83 |
| 2 | 64 | 84 | |||||||
| 3 | 62 | 83 | |||||||
| 4 | 61 | 84 | |||||||
| 5 | 59 | 84 | |||||||
| 6 | 57 | 83 | |||||||
| 7 | 55 | 83 | |||||||
| 2 | Toluene | 0.1 | 3.5 | 1 | 0.5 | 0.5 | 1 | 84 | 86 |
| 2 | 87 | 84 | |||||||
| 3 | 86 | 86 | |||||||
| 4 | 85 | 85 | |||||||
| 5 | 83 | 86 | |||||||
| 6 | 81 | 84 | |||||||
| 7 | 79 | 85 | |||||||
3.4 Neat continuous flow experiments
Finally, an effort was made to eliminate the need for solvent and thus the use of undiluted reagents was evaluated. To avoid any uncontrollable reaction in the reagent reservoir, the choice was made to handle two separate reagent streams. This configuration entails a slight excess of cyclopentadiene (1.1 equiv.). Despite the higher throughput, a lower conversion and selectivity were obtained (Table 7). Moreover, a small fraction of the cyclopentadiene dimer (about 6%) was observed on 1H-NMR as a side-product.Presumably, the neat DA reaction mainly takes place in the bulk phase and not on the catalyst surface. This was easily verified by calculating the acid site concentration in the catalyst bed. This was easily verified by calculating the known acidity of the catalyst. The acid site concentration of CBV 720 was determined using NH3-TPD to be 0.47 mmol H+ per g. Taking into account the internal concentration of MA, the limiting reagent, of 5.5 M, it appears that there is a catalyst loading of 9 mol% in the CatCart® or a site-time of 9.5 mol AS·s mol−1 MA. Therefore, knowing that the reaction also proceeds, albeit non-selective, without the catalyst, the catalyst loading is too low, leading to a non-catalyzed and thus non-selective background reaction. As a result, the high conversion but low endo-selectivity can be explained by the high temperature and pressure conditions. In comparison, under diluted continuous flow conditions (Table 5, entry 4), this catalyst loading is 99 mol%, corresponding to a site-time of 208.7 mol AS·s mol−1 CPD. These calculations were performed under the assumption that the physical pre-treatment does not affect the amount of acid sites, nor their accessibility.
3.5 Batch confirmation of the observed trends
In order to obtain a full comparison, the batch experiments were repeated using the non-dried zeolite material, which lead to an increase of the conversion (Table 8). To gain insight into the influence of the pelletizing pre-treatment, comparative batch experiments were carried out. For both the H-USY and the H-Beta catalyst, the powder and the pelletized form showed a comparable conversion and selectivity, although in the case of the H-Beta species a larger difference was observed in favour of the powder form (Table 8, entries 2, 3, 5 and 6). The deposition of cyclopentadiene on the catalyst surface was determined from the changing CPD:MA ratio, which should theoretically equal one at every moment of the experiment. The deviation of this ratio from one can be ascribed to the deposition of cyclopentadiene (polymers) on the catalyst bed. This process leads to a biased view of the conversion of CPD to the end products from 1H-NMR and is accompanied by a decreased CPD:MA ratio. In each batch experiment, the deposition of CPD amounted to 17–30% approximately. For comparison, also in flow a continuous deposition of CPD on the catalyst surface was observed (1–6%). In batch, it appears that the conversion benefits from the drying of the solvent (Table 8, entries 2, 4, 5 and 7). The continuous flow experiment using the H-USY catalyst indicates that without the pre-drying of dichloromethane a sudden accelerated decline in conversion can be observed (Table 5, entry 6). However, this was not seen within a 7 h time frame when the H-Beta catalyst was used (Table 5, entry 11).
| Entry | Zeolite typea | Si/Al | Catalyst form | Solvent | 1H-NMR | endo (%) | CPD deposition (mmol g−1) | |
|---|---|---|---|---|---|---|---|---|
| MA (%) | EP (%) | |||||||
| a The corresponding zeolite product code, as obtained from Zeolyst International, can be seen in Table 1. | ||||||||
| 1 | No | — | — | Dry DCM | 90 | 10 | 79 | — |
| 2 | H-USY | 15 | Pelletized | Dry DCM | 47 | 53 | 92 | 13 (26%) |
| 3 | H-USY | 15 | Powder | Dry DCM | 46 | 54 | 92 | 15 (29%) |
| 4 | H-USY | 15 | Pelletized | DCM | 56 | 44 | 92 | 15 (30%) |
| 5 | H-Beta | 12.5 | Pelletized | Dry DCM | 36 | 64 | 92 | 11 (23%) |
| 6 | H-Beta | 12.5 | Powder | Dry DCM | 30 | 70 | 89 | 11 (21%) |
| 7 | H-Beta | 12.5 | Pelletized | DCM | 38 | 62 | 91 | 9 (17%) |
In flow, a high loading and a continuous recuperation of the catalyst results in a high catalytic productivity. Assuming that a batch process is stopped at 70% conversion after 3.5 h (Table 8, entry 6) as from then on hardly any change in conversion was observed, a catalytic productivity of 5.3 g EP per g H-Beta could be obtained. In contrast, the 7 h flow process, as presented in Table 5, entry 11, has a catalytic productivity of 18.7 g EP per g H-Beta. Moreover, this productivity is a minimal value, as the catalyst deactivation was not monitored to its boundaries. Evaluating both the lab-scale batch and continuous processes at 3.5 h, the flow process has a 14 times higher production of Diels–Alder end products (0.87 g h−1).
3.6 Spent catalyst characterisation and regeneration
Although hardly any deactivation of the used zeolite was observed within a 7 h time frame under the optimized conditions, the regeneration advantage of zeolite catalysis was considered. Therefore, to demonstrate the reactivation of the catalyst, an excess of cyclopentadiene was used to accelerate the deposition (Fig. 3, process 1). As can be seen from Table 5, entry 3 and Table 9, process 1, a small excess of CPD leads to a fast drop in conversion. Next, after the loss of conversion and selectivity, the challenge remained to regenerate the catalyst's activity. In the first attempt, a 4 h high vacuum treatment at 150 °C was carried out, after a continuous flow ethanol rinse. Subsequently, a continuous flow process under the optimized conditions was evaluated (Fig. 3, process 2). The obtained conversion and selectivity, however, were unsatisfying (Table 9, process 2). Thus, a more severe reactivation procedure was performed. Again a continuous flow rinse of ethanol was performed after which the zeolite was calcined for 5 h at 500 °C. In this way, the catalyst was reactivated and a high conversion and endo-selectivity was observed again under the optimized continuous flow conditions (Fig. 3 and Table 9, process 3). A mixture of end products was isolated in a 92% yield, with a GC-purity of 99%. | ||
| Fig. 3 Process conditions of the regeneration experiments of the used zeolite USY. | ||
In addition to our work, sieve analysis was carried out on the used pelletized zeolite, which revealed attrition of small zeolite particles upon reaction (Table 10).
3.7 Expansion to other derivatives
To demonstrate the versatile performance of this process, alternative dienophiles were evaluated (Table 11). Isolation was performed using mild rotary evaporation. Methyl vinyl ketone (MVK) gave rise to an excellent yield and selectivity towards the endo-isomer. Although this selectivity was slightly lower in case acrolein (AL) was used, a high yield of Diels–Alder products was obtained. In order to obtain a high conversion with acrylonitrile (AN) as a dienophile, the temperature was raised to 200 °C. Finally, dimethyl acetylene dicarboxylate (DMAD) also lead to an excellent conversion for 7 hours.| Dienophile | Timea (h) | Conversionb (%) | Yieldc (%) | endo (%) |
|---|---|---|---|---|
| a Time since start-up of the process. b The conversion was determined by integration of the signals of dienophile and end product(s) in the 1H-NMR spectrum of the crude reaction mixtures, unless specified otherwise. c GC-purity is indicated between brackets. d The conversion was determined by integration of the signals of diene and end product(s) in the 1H-NMR spectrum of the crude reaction mixtures. e T = 200 °C. f After distillation. | ||||
|
1 | 94 | 94 (97) | 89 |
| 7 | 92 | |||

|
1 | 100 | 99 (97) | 89 |
| 7 | 100 | |||

|
1 | 98d | 92 (100) | 78 |
| 7 | 98d | |||

|
1 | 84e | 84 (96) | 54 |
| 7 | 82e | |||

|
1 | 86 | 60f | — |
| 7 | 90 | |||
4 Conclusions
An efficient continuous flow process was developed for the Diels–Alder reaction of cyclopentadiene and methyl acrylate. Using zeolite H-Beta catalysis and high pressure conditions, a high conversion (≥92%) and selectivity towards the endo-isomer (89:11) were obtained in just 1.8 min contact time in the packed catalyst bed. Moreover, a 1:1 stoichiometry diene to dienophile was applied. These values are similar to some of the best small scale batch procedures and thus the safely scalable continuous flow process can compete with previously developed batch processes, as these often require an excess of cyclopentadiene, long reaction times, a (toxic) Lewis acid catalyst or expensive ionic liquids. The process was monitored for 7 h and a calculated throughput of 0.87 g h−1 was reached, corresponding to a 3.5 times higher catalytic productivity and a 14 times higher production of Diels–Alder adducts in comparison to the small scale batch process. In contrast to various literature examples, the zeolite material was found to be more active in acid catalysis without any drying step. Afterwards, the end products could be isolated in a straightforward manner in high yields. The use of neat reagent streams was also evaluated, but resulted in a slightly decreased conversion and a significant loss of endo-selectivity. The regeneration of the used zeolite catalyst was performed using a 5 h calcination step at 500 °C. Finally, the versatility of this process was demonstrated as alternative dienophiles were evaluated, leading to high yields.
Acknowledgements
Financial support for this research from the Fund for Scientific Research Flanders (FWO Vlaanderen) and VITO (Vlaamse Instelling voor Technologisch Onderzoek) and BOF (Bijzonder Onderzoeksfonds) of Ghent University is gratefully acknowledged.Notes and references
- P. Wothers, N. Greeves, S. Warren and J. Clayden, Organic Chemistry, Oxford University Press, 2001, pp. 907–924 Search PubMed.
- (a) M. Hatano, T. Mizuno, A. Izumiseki, R. Usami, T. Asai, M. Akakura and K. Ishihara, Angew. Chem., Int. Ed., 2011, 50, 12189–12192 CrossRef CAS PubMed; (b) M. K. Kesharwani and B. Ganguly, Croat. Chem. Acta, 2009, 82, 291–298 CAS; (c) J.-H. Zhou, B. Jiang, F.-F. Meng, Y.-H. Xu and T.-P. Loh, Org. Lett., 2015, 17, 4432–4435 CrossRef CAS PubMed.
- J. A. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822–3863 CrossRef CAS PubMed.
- V. Casson, T. Snee and G. Maschio, J. Hazard. Mater., 2014, 270, 45–52 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- J. T. Njardarson, http://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed 01/02/2016).
- N. D. Campbell and A. M. Lovell, in Addiction Reviews, ed. G. R. Uhl, Blackwell Science Publ, Oxford, 2012, vol. 1248, pp. 124–139 Search PubMed.
- (a) A. Machara, L. Werner, M. A. Endoma-Arias, D. P. Cox and T. Hudlicky, Adv. Synth. Catal., 2012, 354, 613–626 CrossRef CAS; (b) L. Werner, A. Machara, D. R. Adams, D. P. Cox and T. Hudlicky, J. Org. Chem., 2011, 76, 4628–4634 CrossRef CAS PubMed.
- (a) D. R. J. Acke and C. V. Stevens, Green Chem., 2007, 9, 386–390 RSC; (b) A. Cukalovic, J.-C. R. Monbaliu and C. Stevens, in Synthesis of Heterocycles via Multicomponent Reactions I, ed. R. V. A. Orru and E. Ruijter, Springer, Berlin Heidelberg, 2010, vol. 23, pp. 161–198 Search PubMed; (c) T. S. Heugebaert, C. V. Stevens and C. O. Kappe, ChemSusChem, 2015, 8, 1648–1651 CrossRef CAS PubMed; (d) F. E. A. Van Waes, J. Drabowicz, A. Cukalovic and C. V. Stevens, Green Chem., 2012, 14, 2776–2779 RSC; (e) F. E. A. Van Waes, S. Seghers, W. Dermaut, B. Cappuyns and C. V. Stevens, J. Flow Chem., 2014, 4, 118–124 CrossRef CAS.
- (a) A. Pohar and I. Plazl, Chem. Biochem. Eng. Q., 2009, 23, 537–544 CAS; (b) P. Watts and C. Wiles, J. Chem. Res., 2012, 181–193 CrossRef CAS; (c) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC.
- (a) C. Bernardon, B. Louis, V. Beneteau and P. Pale, ChemPlusChem, 2013, 78, 1134–1141 CrossRef CAS; (b) Y.-T. Cheng and G. W. Huber, Green Chem., 2012, 14, 3114–3125 RSC; (c) J. M. Fraile, J. I. Garcia, D. Gracia, J. A. Mayoral, T. Tarnai and F. Figueras, J. Mol. Catal. A: Chem., 1997, 121, 97–102 CrossRef CAS; (d) S. Ghosh and N. L. Bauld, J. Catal., 1985, 95, 300–304 CrossRef CAS; (e) M. V. Gomez, A. Cantin, A. Corma and A. de la Hoz, J. Mol. Catal. A: Chem., 2005, 240, 16–21 CrossRef CAS; (f) S. Imachi and M. Onaka, Tetrahedron Lett., 2004, 45, 4943–4946 CrossRef CAS; (g) T. Kugita, M. Ezawa, T. Owada, Y. Tomita, S. Namba, N. Hashimoto and M. Onaka, Microporous Mesoporous Mater., 2001, 44, 531–536 CrossRef; (h) T. Kugita, S. K. Jana, T. Owada, N. Hashimoto, M. Onaka and S. Namba, Appl. Catal., A, 2003, 245, 353–362 CrossRef CAS; (i) G. Mashayekhi, M. Ghandi, F. Farzaneh, M. Shahidzadeh and H. M. Najafi, J. Mol. Catal. A: Chem., 2007, 264, 220–226 CrossRef CAS; (j) M. Onaka, N. Hashimoto, Y. Kitabata and R. Yamasaki, Appl. Catal., A, 2003, 241, 307–317 CrossRef CAS; (k) J. K. Park, S. W. Kim, T. Hyeon and B. M. Kim, Tetrahedron: Asymmetry, 2001, 12, 2931–2935 CrossRef CAS; (l) M. Zendehdel, N. F. Far and Z. Gaykani, J. Inclusion Phenom. Macrocyclic Chem., 2005, 53, 47–49 CrossRef CAS.
- D. E. De Vos and P. A. Jacobs, Microporous Mesoporous Mater., 2005, 82, 293–304 CrossRef CAS.
- J. Weitkamp, Solid State Ionics, 2000, 131, 175–188 CrossRef CAS.
- A. F. Masters and T. Maschmeyer, Microporous Mesoporous Mater., 2011, 142, 423–438 CrossRef CAS.
- (a) A. Gavriilidis, P. Angeli, E. Cao, K. K. Yeong and Y. S. S. Wan, Chem. Eng. Res. Des., 2002, 80, 3–30 CrossRef CAS; (b) Y. S. S. Wan, J. L. H. Chau, A. Gavriilidis and K. L. Yeung, Microporous Mesoporous Mater., 2001, 42, 157–175 CrossRef CAS.
- (a) Y. Liu, S. Podila, D. L. Nguyen, D. Edouard, P. Nguyen, C. Pham, M. J. Ledoux and P. H. Cuong, Appl. Catal., A, 2011, 409, 113–121 CrossRef; (b) Q. Tang, H. Xu, Y. Y. Zheng, J. F. Wang, H. S. Li and J. Zhang, Appl. Catal., A, 2012, 413, 36–42 CrossRef.
- N. Candu, M. Florea, S. M. Coman and V. I. Parvulescu, Appl. Catal., A, 2011, 393, 206–214 CrossRef CAS.
- G. C. Zhang, X. F. Zhang, J. Lv, H. O. Liu, J. S. Qiu and K. L. Yeung, Catal. Today, 2012, 193, 221–225 CrossRef CAS.
- Y. S. S. Wan, J. L. H. Chau, A. Gavriilidis and K. L. Yeung, Chem. Commun., 2002, 878–879 RSC.
- http://zeolyst.com/our-products.aspx (accessed 20/11/2015).
- (a) W. Lutz, Adv. Mater. Sci. Eng., 2014, 2014(1), 1–20 CrossRef; (b) V. Patzelova, A. Zukal and J. Kloubek, Chem. Pap. – Chem. Zvesti, 1985, 39, 361–367 CAS; (c) Z. M. Yan, M. A. Ding, J. Q. Zhuang, X. C. Liu, X. M. Liu, X. W. Han, X. H. Bao, F. X. Chang, L. Xu and Z. M. Liu, J. Mol. Catal. A: Chem., 2003, 194, 153–167 CrossRef CAS.
- (a) A. Corma and H. Garcia, Catal. Today, 1997, 38, 257–308 CrossRef CAS; (b) S. E. Sen, S. M. Smith and K. A. Sullivan, Tetrahedron, 1999, 55, 12657–12698 CrossRef CAS.
- D. Nakashima and H. Yamamoto, Org. Lett., 2005, 7, 1251–1253 CrossRef CAS PubMed.
- (a) I. H. Chen, J. N. Young and S. J. Yu, Tetrahedron, 2004, 60, 11903–11909 CrossRef CAS; (b) J. R. Harjani, R. D. Singer, M. T. Garcia and P. J. Scammells, Green Chem., 2009, 11, 83–90 RSC; (c) G. Imperato, E. Eibler, J. Niedermaier and B. Konig, Chem. Commun., 2005, 1170–1172 RSC; (d) E. Janus, I. Goc-Maciejewska, M. Lozynski and J. Pernak, Tetrahedron Lett., 2006, 47, 4079–4083 CrossRef CAS; (e) I. Lopez, G. Silvero, M. J. Arevalo, R. Babiano, J. C. Palacios and J. L. Bravo, Tetrahedron, 2007, 63, 2901–2906 CrossRef CAS; (f) D. Sarma and A. Kumar, Appl. Catal., A, 2008, 335, 1–6 CrossRef CAS; (g) G. H. Tao, L. He, W. S. Liu, L. Xu, W. Xiong, T. Wang and Y. Kou, Green Chem., 2006, 8, 639–646 RSC; (h) X. P. Xu, M. T. Ma, Y. M. Yao, Y. Zhang and Q. Shen, Eur. J. Inorg. Chem., 2005, 676–684 CrossRef.
- (a) K. Erfurt, I. Wandzik, K. Walczak, K. Matuszek and A. Chrobok, Green Chem., 2014, 16, 3508–3514 RSC; (b) V. Escande, T. K. Olszewski and C. Grison, C. R. Chim., 2014, 17, 731–737 CrossRef CAS; (c) A. Kumar and S. S. Pawar, J. Org. Chem., 2007, 72, 8111–8114 CrossRef CAS PubMed; (d) B. Mathieu, L. de Fays and L. Ghosez, Tetrahedron Lett., 2000, 41, 9561–9564 CrossRef CAS; (e) K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2003, 125, 11460–11461 CrossRef CAS PubMed; (f) W. Stefaniak, E. Janus and E. Milchert, Catal. Lett., 2011, 141, 742–747 CrossRef CAS; (g) Z. Tang, B. Mathieu, B. Tinant, G. Dive and L. Ghosez, Tetrahedron, 2007, 63, 8449–8462 CrossRef CAS.
- (a) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS; (b) A. Barba, S. Barroso, G. Blay, L. Cardona, M. Melegari and J. R. Pedro, Synlett, 2011, 1592–1596 CAS; (c) S. Erol and I. Dogan, Tetrahedron, 2013, 69, 1337–1344 CrossRef CAS; (d) M. Hatano and K. Ishihara, Chem. Commun., 2012, 48, 4273–4283 RSC; (e) K. Ishihara and K. Nakano, J. Am. Chem. Soc., 2005, 127, 10504–10505 CrossRef CAS PubMed; (f) Z. J. Jia, Q. Zhou, Q. Q. Zhou, P. Q. Chen and Y. C. Chen, Angew. Chem., Int. Ed., 2011, 50, 8638–8641 CrossRef CAS PubMed; (g) T. Kano, Y. Tanaka and K. Maruoka, Org. Lett., 2006, 8, 2687–2689 CrossRef CAS PubMed; (h) K. Maruoka, H. Imoto and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 12115–12116 CrossRef CAS; (i) S. C. Pellegrinet and R. A. Spanevello, Org. Lett., 2000, 2, 1073–1076 CrossRef CAS PubMed; (j) W. R. Roush and B. B. Brown, J. Org. Chem., 1992, 57, 3380–3387 CrossRef CAS; (k) J. H. Zhou, B. Jiang, F. F. Meng, Y. H. Xu and T. P. Loh, Org. Lett., 2015, 17, 4432–4435 CrossRef CAS PubMed.
- (a) M. Kanao, A. Otake, K. Tsuchiya and K. Ogino, Int. J. Org. Chem., 2012, 2, 26–30 CrossRef CAS; (b) P. Klein, M. Thyes, M. Grosse and K. M. Weber, Preparation of biperiden used for treating Parkinson's disease, by reacting phenylmagnesium compound and bicycloheptenenyl-substituted propanone of high exo/endo ratio, DE10124451 (A1), 2002; (c) A. Okamoto, M. S. Snow and D. R. Arnold, Tetrahedron, 1986, 42, 6175–6187 CrossRef CAS.
- (a) S. Abele, S. Hock, G. Schmidt, J. A. Funel and R. Marti, Org. Process Res. Dev., 2012, 16, 1114–1120 CrossRef CAS; (b) F. Benito-Lopez, A. J. Mettelalj, R. J. M. Egberink, A. H. Velders, R. M. Tiggelaar, H. Gardeniers, D. N. Reinhoudt and W. Verboom, Chim. Oggi, 2010, 28, 56–59 CAS; (c) M. Rasheed and T. Wirth, Chim. Oggi, 2011, 29, 53–55 Search PubMed.
- A. Sachse, V. Hulea, A. Finiels, B. Coq, F. Fajula and A. Galarneau, J. Catal., 2012, 287, 62–67 CrossRef.
- (a) V. Chiroli, M. Benaglia, F. Cozzi, A. Puglisi, R. Annunziata and G. Celentano, Org. Lett., 2013, 15, 3590–3593 CrossRef CAS PubMed; (b) V. Chiroli, M. Benaglia, A. Puglisi, R. Porta, R. P. Jumde and A. Mandoli, Green Chem., 2014, 16, 2798–2806 RSC.
- S. W. Breeden, J. H. Clark, D. J. Macquarrie and J. R. Sherwood, in Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue, Wiley, 2012, pp. 243–262 Search PubMed.
- J. Clayden, N. Greeves and S. Warren, in Organic Chemistry, Oxford University Press, 2012, pp. 877–908 Search PubMed.
- (a) R. Breslow and U. Maitra, Tetrahedron Lett., 1984, 25, 1239–1240 CrossRef CAS; (b) R. Breslow, U. Maitra and D. Rideout, Tetrahedron Lett., 1983, 24, 1901–1904 CrossRef CAS; (c) P. A. Grieco, P. Garner and Z.-m. He, Tetrahedron Lett., 1983, 24, 1897–1900 CrossRef CAS; (d) D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS; (e) R. B. Woodward and H. Baer, J. Am. Chem. Soc., 1948, 70, 1161–1166 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- K.-i. Takao, R. Munakata and K.-i. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
- https://scifinder.cas.org/ (accessed 08/08/2016).
- (a) M. J. Diego-Castro and H. C. Hailes, Tetrahedron Lett., 1998, 39, 2211–2214 CrossRef CAS; (b) D. A. Jaeger and C. E. Tucker, Tetrahedron Lett., 1989, 30, 1785–1788 CrossRef CAS; (c) A. Manna and A. Kumar, J. Phys. Chem. A, 2013, 117, 2446–2454 CrossRef CAS PubMed.
- M. J. Remy, D. Stanica, G. Poncelet, E. J. P. Feijen, P. J. Grobet, J. A. Martens and P. A. Jacobs, J. Phys. Chem., 1996, 100, 12440–12447 CrossRef CAS.
- A. Funez, J. W. Thybaut, G. B. Marin, P. Sanchez, A. De Lucas and J. L. Valverde, Appl. Catal., A, 2008, 349, 29–39 CrossRef CAS.
- (a) P. Botella, A. Corma, M. T. Navarro, F. Rey and G. Sastre, J. Catal., 2003, 217, 406–416 CrossRef CAS; (b) L. Cerveny, K. Mikulcova and J. Cejka, Appl. Catal., A, 2002, 223, 65–72 CrossRef CAS; (c) M. G. Cutrufello, I. Ferino, R. Monaci, E. Rombi, V. Solinas, R. Magnoux and A. Guisnet, Appl. Catal., A, 2003, 241, 91–111 CrossRef CAS; (d) X. C. Hu, G. K. Chuah and S. Jaenicke, Microporous Mesoporous Mater., 2002, 53, 153–161 CrossRef CAS; (e) N. Srinivas, A. P. Singh, A. V. Ramaswamy, A. Finiels and P. Moreau, Catal. Lett., 2002, 80, 181–186 CrossRef CAS; (f) G. Takeuchi, Y. Shimoura and T. Hara, Appl. Catal., A, 1996, 137, 87–91 CrossRef CAS; (g) J. Wang, J.-N. Park, Y.-K. Park and C. W. Lee, J. Catal., 2003, 220, 265–272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6gc02334g |
| This journal is © The Royal Society of Chemistry 2017 |