
The biphasic dose effect of lychee (Litchi chinensis Sonn.) pulp phenolic extract on alcoholic liver disease in mice†
Juan
Xiao‡
a,
Ruifen
Zhang‡
a,
Fei
Huang
a,
Lei
Liu
a,
Yuanyuan
Deng
a,
Zhencheng
Wei
a,
Yan
Zhang
a,
Dong
Liu
*b and
Mingwei
Zhang
*a
aSericultural & Agri-Food Research Institute, Guangdong Academy of Agricultural Sciences/Key Laboratory of Functional Foods, Ministry of Agriculture/Guangdong Key Laboratory of Agricultural Products Processing, Guangzhou 510610, China. E-mail: mwzhh@vip.tom.com
bShenzhen Key Laboratory of Fermentation, Purification and Analysis, Shenzhen Polytechnic, Shenzhen 518055, China. E-mail: liudongsz@szpt.edu.cn
First published on 29th November 2016
Abstract
Lychee (Litchi chinensis Sonn.), a subtropical fruit grown widely in Southeast Asia, has been used as a traditional Chinese medicine for liver protection. Our previous study showed that phenolics from lychee pulp had hepatoprotective activity against restraint stress-induced liver injury. The present study investigated the protective effects of lychee pulp phenolic extract (LPPE) on ethanol-induced liver injury and the mechanism responsible for these effects. Mice were divided into four groups and subjected to either a control liquid diet (control group, CON), a 4% (w/v) ethanol-containing liquid diet (ethanol group, EtOH), a 4% (w/v) ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE (low dose LPPE-supplemented group, EtOH + L-LPPE), or a 4% (w/v) ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE (high dose LPPE-supplemented group, EtOH + H-LPPE) for 8 weeks. The ethanol-induced hepatic steatosis and increases of triglyceride levels in the serum and liver were ameliorated by L-LPPE supplementation but aggravated by H-LPPE supplementation. Moreover, L-LPPE supplementation improved the antioxidant status, reduced the nuclear translocation of nuclear erythroid 2-related factor 2 (Nrf2), and reduced the expression of Nrf2 target genes in the liver. H-LPPE supplementation resulted in the improved antioxidant status and reduced the expression of Nrf2 target genes. Furthermore, L-LPPE suppressed the expression of lipid synthesis genes and increased the expression of fatty acid β-oxidation genes. However, H-LPPE increased the expression of genes responsible for lipid synthesis and uptake and reduced the expression of fatty acid β-oxidation genes. Additionally, L-LPPE supplementation effectively decreased the serum endotoxin level and reversed ethanol-induced alterations in the intestinal microbiota composition. Collectively, LPPE showed biphasic effects on ethanol-induced liver injury in mice, indicating that a moderate intake of LPPE or Litchi pulp could be useful for the prevention and control of alcoholic liver disease.
1. Introduction
Long-term excessive consumption of alcohol inevitably leads to different levels of alcoholic liver disease (ALD), which is a major cause of morbidity and mortality worldwide, especially in China.1 In the initial stage of ALD, chronic alcohol consumption promotes fat accumulation in the liver, which subsequently leads to the development of fatty liver.2 Excessive fat accumulation increases the risk of developing progressive liver injuries, such as alcoholic steatohaepatitis and cirrhosis.1,3Alcohol metabolism induces a number of important physiological and biochemical changes in the liver, including overproduction of free radicals and increased NADH/NAD+ redox potential, which triggers oxidative stress and disturbs lipid metabolism.2,3 Nuclear erythroid 2-related factor 2 (Nrf2) is a key transcription factor that regulates the adaptive response to oxidative stress.4 Nuclear translocation of Nrf2 is induced in the livers of alcohol-fed mice, suggesting a protective role of Nrf2 against alcohol-induced liver injury.5 Moreover, alcohol consumption results in enhanced lipogenesis and impaired fatty acid oxidation predominantly by targeting key transcription factors controlling these metabolic processes, including sterol regulatory element binding protein 1c (SREBP-1c) and peroxisome proliferator-activated receptor α (PPARα).6–8 Alcohol consumption also has effects on hepatic lipoprotein absorption and secretion.2 Phenolics from fruit and vegetables have been demonstrated to slow the progression of ALD through inhibiting oxidative stress and regulating lipid metabolism.6,7
Lychee is a subtropical fruit with an attractive appearance, delicious taste and good nutritional value. Lychee has been reported to have hepatoprotective effects in traditional Chinese medicine.9 Recent studies have revealed that lychee pulp has an abundance of phenolic compounds.10–14 Lychee pulp extract has been demonstrated to offer hepatoprotection in CCl4-induced mice, which is closely associated with the antioxidant properties of phenolics.15 Phenolic-rich lychee pulp extract has shown antioxidant activity in vitro and hepatoprotective activity against restraint stress-induced liver injury in mice via inhibition of oxidative stress.12,14,16 Quercetin 3-O-rutinoside-7-O-a-L-rhamnosidase, rutin and (−)-epicatechin have been characterized as the main phenolic components of lychee pulp and have been demonstrated to possess potent antioxidant activity in vitro.12–14 The hypolipidaemic activity of rutin and (−)-epicatechin in animals has been elucidated.17–19 Quercetin 3-O-rutinoside-7-O-a-L-rhamnosidase was firstly reported to be the main antioxidant phenolic component of lychee pulp by our group.13 Few reports have shown the bioactivity of quercetin 3-O-rutinoside-7-O-a-L-rhamnosidase, although the ameliorative effects of quercetin and some of its glycosides, including rutin and isoquercitrin, on lipid metabolism have been demonstrated in many studies.20,21 Based on previous findings, we hypothesized that lychee pulp phenolic extract (LPPE) may offer beneficial effects on ALD via regulating oxidative stress and lipid metabolism.
Gut microbiota dysbiosis is a critical contributor to the development of ALD by affecting intestinal permeability that subsequently increases circulating endotoxins, which in turn trigger liver inflammation.22,23 The ameliorated liver injury in ALD patients and animals is accompanied by restored gut microbiota.22–24 Most dietary phenolics are not absorbed directly but are instead metabolized in the colon by intestinal bacteria.25 Research has found that some polyphenols and their metabolites can stimulate the growth of commensal and beneficial microbiota with the inhibition of pathogenic strains in vitro and in vivo, indicating that phenolics may play an important role in maintaining the balance of intestinal bacteria.26,27 Consumption of flavanol-enriched mango pulp can contribute to health by altering the intestinal microbiota composition in mice fed a high-fat diet.28 The effect of phenolics on gut microbiota dysbiosis in ALD patients and animals has not been reported until now, although phenolics from fruit and vegetables have been demonstrated to attenuate ALD.6,7
For these reasons, we investigated the potential protective effect of LPPE against ALD in C57BL/6 mice fed an ethanol-containing liquid diet. The potential hepatoprotective mechanisms related to oxidative stress and hepatic lipid metabolism were studied. Furthermore, we explored the effect of LPPE on the gut microbiota profile in ALD mice.
2. Materials and methods
2.1. Chemicals and reagents
Procyanidin B2, (−)-epicatechin, rutin, and isorhamnetin-3-O-rutinoside were purchased from Sigma-Aldrich (St Louis, MO, USA). Quercetin 3-O-rutinoside-7-O-a-L-rhamnosidase (purity > 98%) was separated according to our previous research.13 HPLC-grade, LC-MS-grade acetic acid and acetonitrile were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), endotoxin, bile acid, triglyceride (TG), total cholesterol (TC), thiobarbituric acid reactive substances (TBARS), superoxide dismutase (SOD), glutathione peroxidase (GSH-Px) and catalase (CAT) kits were all obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The Lieber–DeCarli control liquid diet and ethanol-containing liquid diet were purchased from Trophic Animal Feed High-Tech Co. Ltd (Nantong, China).2.2. Preparation and analysis of LPPE
Fresh lychee (cv. Feizixiao) was purchased from a local fruit market in Guangzhou, China. Phenolic-rich lychee pulp extract was prepared as previously described.16 The extract (600 mg) was dissolved in 1% methanol and then applied onto a Toyopearl HW-40s column (250 mm × 50 mm I.D., Tosoh Chemical Co., Tokyo, Japan). The column was eluted using 1% methanol (900 mL) to remove low molecular mass impurities at a flow rate of 2 mL min−1. The eluant obtained by eluting with methanol (1500 mL) was concentrated and then lyophilized to recover LPPE (357 mg). The total flavonoid content of LPPE was measured on the basis of comparison with a calibration curve of rutin by the AlCl3-NaNO2 method.14 The phenolic composition and contents of the extract were determined by HPLC-DAD according to previously published methods.16The total flavonoid content of LPPE was 85.6 ± 3.98%, and its main phenolic components were procyanidin B2 (104.98 ± 3.11 mg g−1), (−)-epicatechin (34.91 ± 1.20 mg g−1), quercetin 3-O-rutinoside-7-O-a-L-rhamnosidase (260.49 ± 9.21 mg g−1), rutin (54.06 ± 1.52 mg g−1), and isorhamnetin-3-O-rutinoside (16.49 ± 0.50 mg g−1) (ESI Fig. S1†). Moreover, the other phenolic components were deduced as procyanidins and flavanone glycosides by comparing the relative retention time and maximum absorption wavelength with previous research on phenolic compositions of lychee pulp.11 These components were calculated as the (−)-epicatechin equivalent (mg EE per g) and the rutin equivalent (mg RE per g), respectively (ESI Table S1†).
2.3. Animals and experimental design
All experimental procedures involving animals were approved by the Animal Ethical and Welfare Committee of Sun Yat-Sen University (approval no. IACUC-DB-16-0302) and followed the Guiding Principles in the Care and Use of Animals. Eight-week-old pathogen-free male C57BL/6 mice (22 ± 2 g) were purchased from the Centre of Laboratory Animal Science Research of Sun Yat-Sen University (Guangzhou, China). Animals were housed in an environment maintained at 25 ± 1 °C, 55 ± 5% relative humidity, and diurnal light supply.During the one week acclimation, mice were supplied with a rodent chow diet and water ad libitum. The mice were then randomly divided into the following four groups (n = 10 per group): control group (CON), ethanol group (EtOH), low dose LPPE-supplemented group (EtOH + L-LPPE), and high dose LPPE-supplemented group (EtOH + H-LPPE). Animals were housed two per cage and given liquid diets. The liquid diet provided 1 kcal mL−1 based upon the Lieber–DeCarli formulation. The liquid diet provided 35% of calories from fat, 19% of calories from carbohydrates, 18% of calories from proteins, and 28% of calories from ethanol (4% (w/v) ethanol-containing liquid diet) or isocaloric maltose dextrin (control liquid diet). The CON group was pair-fed the control liquid diet with the EtOH group. The EtOH group, EtOH + L-LPPE group and EtOH + H-LPPE group were given the ethanol-containing liquid diet alone or supplemented with 0.4 and 0.8 g L−1 of LPPE, respectively, ad libitum for 8 weeks. The doses of LPPE in the EtOH + L-LPPE and EtOH + H-LPPE groups were equivalent to 150 and 300 mg per kg body weight, which was based on our previous studies.16 All diets were freshly prepared from powder and provided every day at 5:00 p.m. The body weight and food intake were recorded weekly and every day, respectively.
Mice were euthanized by inhalation with ether (Sinopharm Chemical Reagent Co., Ltd, Shanghai, China) after fasting for 12 h. Blood was collected and centrifuged at 3000g for 10 min at 4 °C to obtain the serum for biochemical tests. The mice were then sacrificed by cervical dislocation. The liver was immediately removed, washed with chilled normal saline, weighed and cut into many portions. One portion was immediately placed into 4% paraformaldehyde for haematoxylin and eosin (H&E) staining analysis. The other portions were flash frozen in liquid nitrogen and kept at −80 °C. Faeces were immediately collected from the ileum and rectum and stored at −80 °C.
2.4. Measurement of serum AST, ALT and ALP activities as well as the endotoxin level
The serum levels of ALT, AST and ALP were measured colourimetrically using commercial kits. The endotoxin level was measured with the Limulus Amebocyte Lysate test kit. All biochemical indices were determined on an Infinite® M200 PRO plate reader (Tecan Austria GmbH, Grodig, Austria).2.5. Measurement of serum bile acid, TG, and TC levels as well as hepatic TG and TC levels
Serum bile acid, TG and TC were measured colourimetrically using commercial kits. Total lipids were extracted from liver homogenates by using a chloroform/methanol mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v).29 The TG and TC levels in the total lipids were determined using commercial kits.
:1, v/v).29 The TG and TC levels in the total lipids were determined using commercial kits.
2.6. Liver histopathology
The histopathological alteration in the liver was determined by H&E staining and oil-red O staining using a standard procedure. For H&E staining, paraffin sections (5 μm thick) were cut, deparaffinized in xylene, rehydrated in alcohol gradients, and then stained with H&E. For oil-red O staining, frozen sections (5 μm thick) were cut, stained with oil-red O, and counterstained with haematoxylin. An image was captured using a light microscope (Leica DMI 4000B, Heidelberg, Germany) at 200× magnification.2.7. Measurement of lipid peroxidation and antioxidant enzymes
Liver samples were homogenized with chilled normal saline in an ice bath. The 10% (w/v) homogenate was centrifuged at 3000g for 10 min at 4 °C, and the supernatant was used for TBARS, SOD, GSH-Px and CAT tests. Moreover, as an important index of oxidative stress, the ratio of TBARS to SOD (TBARS/SOD ratio) was calculated.30 All parameters were determined using the respective kits. Lipid peroxidation was measured from the formation of malondialdehyde based on the amount of TBARS. SOD activity was measured according to the xanthine and xanthine oxidase method. GSH-Px activity was determined based on the ability of the enzyme to catalyse the oxidation of reduced glutathione to oxidized glutathione. CAT activity was determined based on the ability of the enzyme to break down H2O2.2.8. Quantitative real-time PCR (qRT-PCR)
Liver total RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed on a B960 real-time thermocycler (Hangzhou Jingle Scientific Instrument Co., Ltd, Hangzhou, China) using a Reverse Transcriptase M-MLV (RNase H-) (Vazyme Biotech Co., Ltd, Nanjing, China) at 42 °C for 60 min followed by 70 °C for 15 min. The synthesized cDNA was stored at −20 °C. qRT-PCR was conducted on an ABI ViiA 7 Detection System (Applied Biosystems, Foster City, CA, USA) using an AceQ® qPCR SYBR® Green Master Mix (Vazyme Biotech Co., Ltd, Nanjing, China) with a predenaturation step at 95 °C for 5 min followed by 45 cycles of 95 °C for 10 s and 60 °C for 30 s. Each sample was performed in triplicate and normalized to GAPDH. The relative expression levels of genes were calculated by the 2−ΔΔCT method as previously described and presented as a ratio of the treatment group to the CON group.30 The primers used (Sangon Biotech (Shanghai) Co., Ltd, Shanghai, China) are listed in Table 1.| Gene name and (abbreviation used) | Primer sequences |
|---|---|
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | F(5′-3′): GGAGAAACCTGCCAAGTATGATGAC |
| R(5′-3′): GAGACAACCTGGTCCTCAGTGTA | |
| Nuclear factor erythroid 2-related factor 2 (Nrf2) | F(5′-3′): CAGTGCTCCTATGCGTGAATCCC |
| R(5′-3′):TGCCCTAAGCTCATCTCGTGTGA | |
| NAD(P)H dehydrogenase quinone 1 (NQO1) | F(5′-3′): GTCTGGAAACCGTCTGGGAGGA |
| R(5′-3′):GCCCACAGAGAGGCCAAACTTG | |
| Heme oxygenase 1 (HO-1) | F(5′-3′): ACCGCCTTCCTGCTCAACATTG |
| R(5′-3′):CTCTGACGAAGTGACGCCATCTG | |
| Glutamate-cysteine ligase catalytic subunit (GCLC) | F(5′-3′): CACCTGGATGATGCCAACGAGTC |
| R(5′-3′):GCACCTCCATTGGTCGGAACTC | |
| Glutamate-cysteine ligase modifier subunit (GCLM) | F(5′-3′): TGTCTAAGAAGGCGGCTTGATGC |
| R(5′-3′):TGAGTCCAACTGAGCAGCAACAC | |
| Glutathione-S-transferase alpha 1 (GSTα1) | F(5′-3′): ACTACCTTGTGGGCAACAGGCT |
| R(5′-3′):GGAGGCTGCTGATTCTGCTCTTG | |
| Glutathione-S-transferase m 1 (GSTm1) | F(5′-3′): CAGCACCAGCACCATGCCTATG |
| R(5′-3′):CTGTCAAAGTCGGGAGCGTCAC | |
| Fatty acid synthase (FAS) | F(5′-3′): GAGAGAGCCTGCCACCCATGAT |
| R(5′-3′):CAGTCCCTGGAACCTGCTAGTC | |
| Acyl-CoA synthetase long-chain family member 1 (ACSL1) | F(5′-3′): TGATCTGGTGGAACGAGGCAAGA |
| R(5′-3′):TTCTGGAGGCTTGGGCTTCACT | |
| Acetyl-CoA carboxylase 1 (ACC1) | F(5′-3′): ACAGACCGTGGTAGTTGGCAGAG |
| R(5′-3′):GCTTCAGAATCCAGGTTCGCAGG | |
| 1-Acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) | F(5′-3′): GCCAGAACTGCCATGTCTGTGAT |
| R(5′-3′):CATTGTCGTTGCGTGTACCCTCT | |
| Diacylglycerol-O-acyltransferase 1 (DGAT1) | F(5′-3′): CTGCCTGCACACTCCTCCTAGT |
| R(5′-3′):GACAACACAGACTCCCAGCACTG | |
| Diacylglycerol-O-acyltransferase 2 (DGAT2) | F(5′-3′): AGCACCCGACCCAGAAAGACAT |
| R(5′-3′):TCAGTTCACCTCCAGCACCTCAG | |
| Carnitine palmitoyltransferase 1α (CPT1α) | F(5′-3′): CCGCCACCTCTTCTGCCTCTAT |
| R(5′-3′):GAGTCTGGCTCGTGGACAACCT | |
| Microsomal triglyceride transfer protein (MTP) | F(5′-3′): TGCTCAGACTCCAGACTCGCTAG |
| R(5′-3′):CTTCATCAGGGTGGGTGGCAAAG | |
| Very low density lipoprotein receptor (VLDLR) | F(5′-3′): GGATGGCAGCGACGAGAAGAAC |
| R(5′-3′):GGCACTGTTCAGGGCTTTCATCA |
2.9. Western blot analysis
Total protein lysates of the liver were extracted using RIPA buffer supplemented with 1% protease inhibitor cocktail and 1% phenylmethylsulphonyl fluoride. Liver nuclear protein extract was obtained by homogenizing the liver in ice-cold nuclear protein extraction buffer using a commercial kit (Beyotime Biotechnology, Shanghai, China). The protein concentration was measured by a BCA protein assay. The protein extract was loaded onto a 10% sodium dodecylsulphate-polyacrylamide gel (30 μg protein per lane), transferred onto a polyvinylidene difluoride membrane (0.45 μm, Merck Millipore, Darmstadt, Germany), and blocked with 5% skim milk before immunostaining with primary antibodies against Nrf2 (1:400, Santa Cruz Biotechnology, Santa Cruz, CA, USA), SREBP-1c, PPARα, fatty acid synthase (FAS), carnitine palmitoyltransferase 1α (CPT1α), very-low density lipoprotein receptor (VLDLR) or microsomal triglyceride transfer protein (MTP) (1:1000, Abcam, London, UK). The membrane was then incubated with horseradish peroxidase-conjugated goat anti-mouse IgG or rabbit anti-goat IgG (1:10000, Tianjin Sungene Biotech, Tianjin, China). The bound complexes were stained with an enhanced chemiluminescent substrate using the Fast Western Blot kit (Pierce, Rockford, IL, USA) and exposed to a film. The film was scanned using a Plustek SW500 scanner (Plustek, Taiwan, China), and the band intensities were measured using Quantity One 1-D analysis software (Bio-Rad, Hercules, CA, USA). Histone and β-actin (Tianjin Sungene Biotech) were detected as the loading controls for the nuclear protein and total protein extracts, respectively.
2.10. Determination of the faecal microbial composition by high-throughput sequencing analysis
Total bacteria DNA was extracted from faeces using the PowerFecal™ DNA Isolation kit (MO BIO Laboratories, Carlsbad, CA, USA) following the manufacturer's instructions. Sequencing and sequence analysis were performed at BGI-Shenzhen (Shenzhen, China) according to previously published methods.31 Briefly, the extracted DNA was subjected to Illumina MiSeq sequencing of the V4 hypervariable region of the 16S rRNA gene followed by analysis using QIIME 1.8.0 to assess faecal microbiota.2.11. Statistical analysis
Data were expressed as the means ± standard deviation (SD) and analysed by one-way ANOVA followed by a Duncan post hoc test using SPSS 16.0 software. For gut microbial composition analysis, data were expressed as the means ± standard error (SE) and compared using Metastats. p < 0.05 was regarded as indicating a significant difference.3. Results
3.1. Change in the body weight and energy intake
There were no significant differences in the initial body weight, final body weight or body weight gain among the four groups (p > 0.05) (Table 2). The total energy intake among the four groups was equivalent (p > 0.05).| CON | EtOH | EtOH + L-LPPE | EtOH + H-LPPE | |
|---|---|---|---|---|
| Data are means ± SD. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase. | ||||
| Initial body weight (g) | 21.63 ± 1.10a | 21.80 ± 0.75a | 21.95 ± 0.79a | 22.02 ± 1.06a |
| Final body weight (g) | 24.07 ± 1.35a | 24.05 ± 1.01a | 24.75 ± 0.95a | 23.90 ± 1.35a |
| Body weight gain (g) | 2.52 ± 0.55a | 2.35 ± 0.46a | 2.72 ± 0.42a | 2.20 ± 0.65a |
| Total caloric intake (kcal per mice) | 635.36 ± 20.21a | 631.15 ± 46.29a | 628.81 ± 40.69a | 611.93 ± 27.88a |
| Liver-to-body weight ratio (%) | 4.03 ± 0.22a | 4.72 ± 0.51b | 4.22 ± 0.31a | 5.40 ± 0.50c |
| Serum AST (U L−1) | 26.89 ± 3.39a | 33.97 ± 3.93b | 28.04 ± 1.98a | 37.06 ± 7.01b |
| Serum ALT (U L−1) | 18.33 ± 2.96a | 25.82 ± 5.95bc | 23.08 ± 3.39ab | 31.31 ± 7.93c |
| Serum ALP (U L−1) | 73.17 ± 5.70a | 82.35 ± 9.61a | 71.73 ± 8.04a | 74.52 ± 8.97a |
3.2. Liver-to-body weight ratio and serum biomarkers of hepatic function
The liver-to-body weight ratio, and serum ALT and AST activities of the EtOH group were 1.17-fold (p < 0.05), 1.41-fold (p < 0.05) and 1.26-fold (p < 0.05), respectively, higher than those of the CON group (Table 1). L-LPPE supplementation normalized these indices. However, compared with the EtOH group, H-LPPE supplementation significantly increased the liver-to-body weight ratio (1.14-fold, p < 0.05). There was no significant difference in ALP activity among the four groups. The results indicated that L-LPPE significantly protected against ethanol-induced liver injury and that H-LPPE did not exhibit ameliorative effects on ethanol-induced liver injury.3.3. Liver histopathology
Histopathological images of representative liver samples are presented in Fig. 1. In contrast to the CON group, ethanol feeding resulted in liver damage characterized by an irregular arrangement of hepatocytes and extensive fat droplets in hepatocytes. Fat droplets in the hepatocytes were indicated by vacuoles in H&E staining sections and aggregation of the orange dye in oil-red O staining sections. L-LPPE supplementation alleviated hepatic steatosis as indicated by fewer and smaller fat droplets in hepatocytes than in those of the EtOH group. Conversely, more and larger fat droplets were observed in the EtOH + H-LPPE group than in the EtOH group, revealing the aggravative effect of H-LPPE on ethanol-induced hepatocyte steatosis. | ||
| Fig. 1 Effects of LPPE on liver histopathology in ethanol-induced liver injury in mice. (A) H&E staining, 200× magnification; (B) oil-red O staining, 200× magnification. Representative liver sections from mice subjected to a control liquid diet (CON), an ethanol-containing liquid diet (EtOH), and an ethanol-containing liquid diet supplemented with 0.4 or 0.8 g L−1 LPPE (EtOH + L-LPPE or EtOH + H-LPPE groups) for 8 weeks. Thin arrows indicate lipid droplets. | ||
3.4. Lipid-related parameters in the serum and liver
Ethanol feeding for eight weeks resulted in significant increases in the serum and hepatic TG levels as well as the serum bile acid level compared to those of the CON group (p < 0.05) (Table 3), and L-LPPE supplementation reversed these changes. In contrast, H-LPPE supplementation increased the serum and hepatic TG levels compared to those of the EtOH group (1.33- and 1.81-fold, respectively). There were no significant differences in the serum or hepatic TC levels among the four groups.| CON | EtOH | EtOH + L-LPPE | EtOH + H-LPPE | |
|---|---|---|---|---|
| Data are means ± SD. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. TG, triglyceride; TC, total cholesterol. | ||||
| Serum bile acid (μmol L−1) | 28.64 ± 5.41a | 41.67 ± 5.18b | 32.10 ± 3.49a | 39.38 ± 6.99b |
| Serum TG (mmol L−1) | 0.69 ± 0.10a | 0.86 ± 0.06b | 0.68 ± 0.12a | 1.14 ± 0.24c |
| Serum TC (mmol L−1) | 2.99 ± 0.36a | 2.74 ± 0.20a | 2.70 ± 0.21a | 2.73 ± 0.36a |
| Liver TG (mg per g liver) | 17.87 ± 3.86a | 27.30 ± 5.34b | 20.18 ± 2.73a | 49.36 ± 7.40c |
| Liver TC (mg per g liver) | 9.56 ± 2.36a | 13.81 ± 3.70a | 10.31 ± 2.47a | 10.06 ± 2.63a |
3.5. Effects of LPPE on the hepatic redox status in ethanol-induced liver injury in mice
As shown in Table 4, ethanol feeding resulted in significant enhancements of the hepatic TBARS level and TBARS/SOD ratio but significant reductions in hepatic SOD, GSH-Px and CAT activities compared to those of the CON group (p < 0.05). However, L-LPPE supplementation reversed the ethanol-induced changes in the hepatic TBARS level, TBARS/SOD ratio and CAT activity. Moreover, H-LPPE supplementation normalized hepatic CAT activity.| CON | EtOH | EtOH + L-LPPE | EtOH + H-LPPE | |
|---|---|---|---|---|
| Data are means ± SD. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. TBARS, thiobarbituric acid reactive substances; SOD, superoxide dismutase; TBARS/SOD ratio, ratio of TBARS to SOD; GSH-Px, glutathione peroxidase; CAT, catalase. | ||||
| TBARS (nmol MDA equivalent per mg prot.) | 0.72 ± 0.08a | 0.90 ± 0.18b | 0.70 ± 0.07a | 0.76 ± 0.08ab |
| SOD (U per mg prot.) | 444.34 ± 80.07b | 338.60 ± 34.81a | 356.67 ± 16.34a | 326.99 ± 30.90a |
| TBARS/SOD ratio (%) | 0.17 ± 0.03a | 0.26 ± 0.04b | 0.20 ± 0.02a | 0.23 ± 0.02b |
| GSH-Px (U per mg prot.) | 531.69 ± 92.52b | 403.46 ± 41.39a | 467.17 ± 32.23ab | 425.86 ± 35.93a |
| CAT (U per mg prot.) | 12.66 ± 1.52b | 8.74 ± 2.52a | 15.17 ± 4.22b | 13.58 ± 3.13b |
3.6. Effects of LPPE on hepatic Nrf2 nuclear translocation and expression of Nrf2 target genes in ethanol-induced liver injury in mice
As shown in Fig. 2, the ethanol-induced elevation of the nuclear Nrf2 protein level was accompanied by significant upregulation of the mRNA levels of Nrf2 target genes (p < 0.05), such as NAD(P)H:quinine oxidoreductase 1 (NQO1), heme oxygenase-1 (HO-1), glutamate-cysteine ligase catalytic subunit (GCLC), glutamate-cysteine ligase modifier subunit (GCLM) and glutathione-S-transferases (GSTα1 and GSTm1), indicating ethanol-induced hepatic Nrf2 nuclear translocation. Compared to the EtOH group, however, L-LPPE supplementation significantly reduced the nuclear Nrf2 protein levels by 22% (p < 0.05) with concomitant significant decreases in the Nrf2-regulated mRNA levels of NQO1, HO-1, GCLC, GCLM and GSTm1 (p < 0.05). Furthermore, H-LPPE supplementation had no significant effect on nuclear Nrf2 protein levels, but it reversed the ethanol-induced increases in the mRNA levels of HO-1 and GCLM to levels equivalent to those of the CON group. | ||
| Fig. 2 Effects of LPPE on hepatic Nrf2 nuclear translocation and expression of Nrf2 target genes in ethanol-induced liver injury in mice. (A) Representative western blot of the nuclear Nrf2 protein level in the liver (n = 4); (B) qRT-PCR analysis of the mRNA levels of NQO1, HO-1, GSTα1, GSTm1, GCLC and GCLM in the liver (n = 4), which are shown relative to Gapdh expression. Data are means ± SD. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. | ||
3.7. Effects of LPPE on hepatic fatty acid and TG synthesis in ethanol-induced liver injury in mice
As expected, a significantly higher expression level of nuclear SREBP-1c protein was found in the EtOH group than in the CON group. Moreover, increased mRNA and protein levels of FAS and increased mRNA levels of 1-acylglycerol-3-phosphate O-acyltransferase 2 (AGPAT2) and diacylglycerol acyltransferase 1 (DGAT1) were also found in the EtOH group (p < 0.05) (Fig. 3). The nuclear SREBP-1c protein level and FAS mRNA and protein levels in the L-LPPE-supplemented group were markedly decreased by 54%, 32% and 35%, respectively, compared to those of the EtOH group (p < 0.05). L-LPPE supplementation normalized the AGPAT2 and DGAT1 mRNA levels. Although ethanol intake had no apparent effects on the mRNA levels of acyl-CoA synthetase long-chain family member 1 (ACSL1), acetyl-CoA carboxylase 1 (ACC1) and DGAT2 (diacylglycerol acyltransferase 2), L-LPPE supplementation significantly decreased the ACSL1 and ACC1 mRNA levels (p < 0.05). Furthermore, H-LPPE supplementation significantly increased the FAS mRNA, FAS protein and DGAT1 mRNA levels by 22%, 22% and 33% (p < 0.05), respectively, compared to those of the EtOH group. | ||
| Fig. 3 Effects of LPPE on the expression of hepatic fatty acid and TG synthesis-related genes in ethanol-induced liver injury in mice. (A) Representative western blot of the nuclear SREBP-1c protein level and FAS protein level in the liver (n = 4). (B) qRT-PCR analysis of the mRNA expression levels of FAS, ACC1, ACSL1, AGPAT2, DGAT1 and DGAT2 in the liver (n = 4), which are shown relative to Gapdh expression. Data are means ± SD. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + l-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. | ||
3.8. Effects of LPPE on hepatic fatty acid β-oxidation in ethanol-induced liver injury in mice
As shown in Fig. 4, the marked ethanol-induced reduction in the hepatic nuclear PPARα protein level was accompanied by significant decreases in the mRNA and protein levels of CPT1α (p < 0.05). However, the ethanol-induced decrease of nuclear PPARα proteins was significantly increased by L-LPPE (1.62-fold) and H-LPPE (1.17-fold) supplementation. The ethanol-induced decreased mRNA and protein levels of CPT1α were elevated by L-LPPE supplementation but further decreased by H-LPPE supplementation. Moreover, the PPARα protein, CPT1α mRNA, and CPT1α protein levels were significantly higher in the L-LPPE-supplemented group than in the CON group (p < 0.05). | ||
| Fig. 4 Effects of LPPE on the expression of hepatic fatty acid β-oxidation-related genes in ethanol-induced liver injury in mice. (A) Representative western blot of the nuclear PPARα protein level and CPT1α protein level in the liver (n = 4). (B) qRT-PCR analysis of the mRNA expression level of CPT1α in the liver (n = 4), which are shown relative to Gapdh expression. Data are means ± SD. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. | ||
3.9. Effects of LPPE on hepatic TG-containing lipoprotein absorption and secretion in ethanol-induced liver injury in mice
As shown in Fig. 5, hepatic VLDLR mRNA and protein levels were significantly increased in the EtOH group by 43% and 54%, respectively, compared to those in the CON group (p < 0.05). H-LPPE but not L-LPPE supplementation notably increased the VLDLR mRNA and protein levels (p < 0.05). There was no significant difference in MTP expression among the four groups.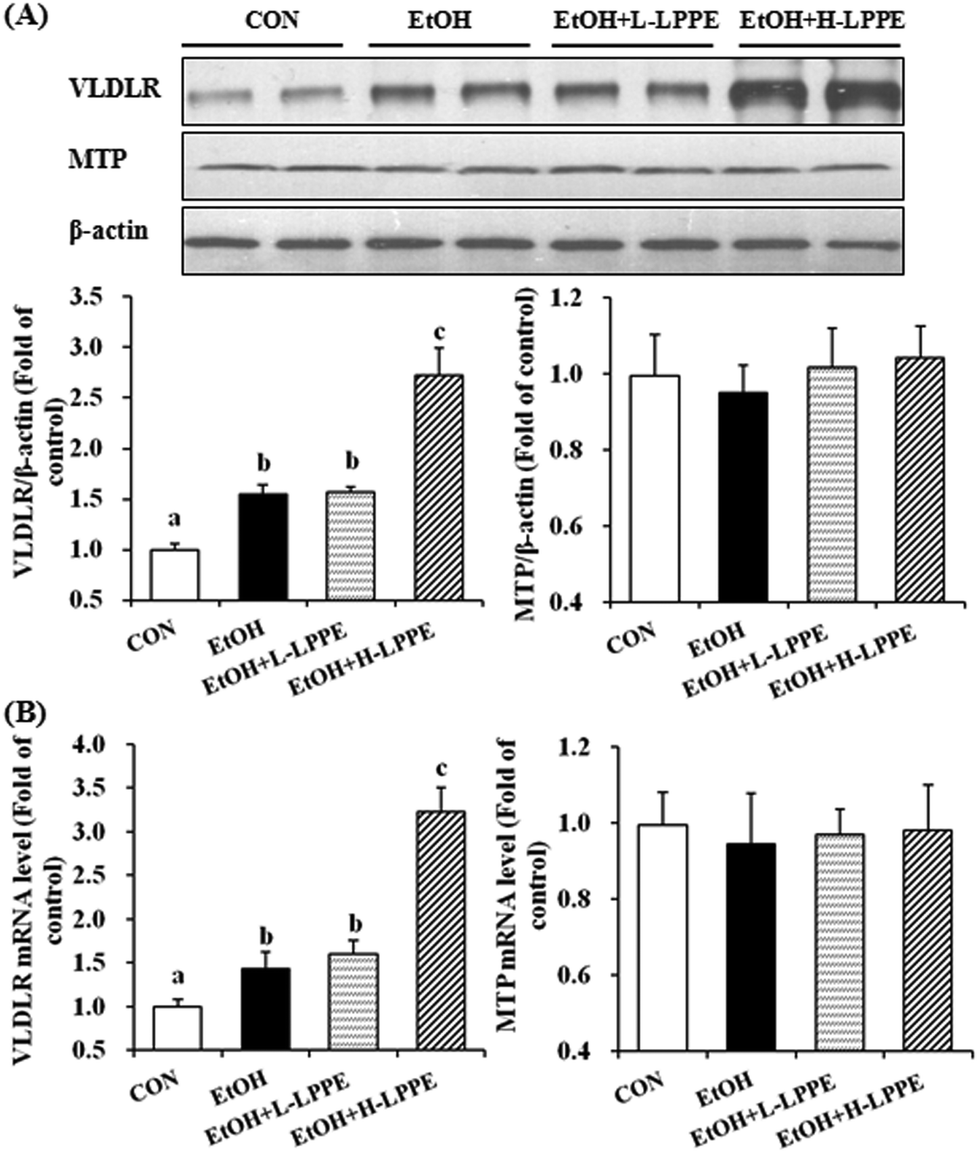 | ||
| Fig. 5 Effects of LPPE on the expression of hepatic TG-containing lipoprotein absorption and secretion-related genes in ethanol-induced liver injury in mice. (A) Representative western blot of VLDLR and MTP protein levels in the liver (n = 4). (B) qRT-PCR analysis of the mRNA expression levels of VLDLR and MTP in the liver (n = 4), which are shown relative to Gapdh expression. Different superscript letters indicate significant differences (p < 0.05). CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. | ||
3.10. Effects of LPPE on the intestinal microbial composition
We assessed the relative abundance of faecal microbiota in mice (Table 5). A total of eight different bacterial phyla were observed, in which Bacteroidetes, Proteobacteria, Firmicutes and Deferribacteres phyla represented 94–98% of the total sequences. At the phylum level, only the relative abundance of Cyanobacteria significantly declined in response to ethanol feeding. Compared to the EtOH group, the relative abundances of the Actinobacteria phylum and Coriobacteriaceae family were significantly higher in the L-LPPE-supplemented group but not in the H-LPPE-supplemented group, and the alcohol intake did not change their relative abundance compared to the CON group. Within the Firmicutes phylum, the ethanol-induced decrease of the Lactobacillus genus and the ethanol-induced expansion of the Dehalobacteriaceae family, Dehalobacterium and Ruminococcus genera were reversed by L-LPPE but not by H-LPPE supplementation. Parabacteroides distasonis and Bacteroides acidifaciens species (Bacteroidetes phylum), as well as the Sutterella genus (Proteobacteria phylum), were significantly decreased in response to ethanol feeding. L-LPPE supplementation reversed the ethanol-induced changes in Bacteroides acidifaciens.| Bacterial targets | Relative abundance (%) | |||
|---|---|---|---|---|
| CON | EtOH | EtOH + L-LPPE | EtOH + H-LPPE | |
| Data are means ± SE (n = 5). * Signifies significant differences (p < 0.05) when compared with the CON group; # signifies significant differences (p < 0.05) when compared with the EtOH group. n.d.: no detection. CON, mice fed a control liquid diet; EtOH, mice fed an ethanol-containing liquid diet; EtOH + L-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.4 g L−1 LPPE; and EtOH + H-LPPE, mice fed an ethanol-containing liquid diet supplemented with 0.8 g L−1 LPPE. | ||||
| Cyanobacteria phylum | 1.035 ± 0.43 | 0.004 ± 0.004* | 0.006 ± 0.006* | n.d. |
| Actinobacteria phylum | 0.23 ± 0.08 | 0.14 ± 0.01 | 0.31 ± 0.06# | 0.13 ± 0.08 |
| Coriobacteriaceae family | 0.23 ± 0.08 | 0.14 ± 0.01 | 0.31 ± 0.06# | 0.13 ± 0.08 |
| Firmicutes phylum | ||||
| Dehalobacteriaceae family | 0.046 ± 0.014 | 0.156 ± 0.011* | 0.089 ± 0.019# | 0.159 ± 0.024* |
| Dehalobacterium genus | 0.046 ± 0.014 | 0.156 ± 0.011* | 0.089 ± 0.019# | 0.159 ± 0.024* |
| Ruminococcus genus | 0.121 ± 0.071 | 0.300 ± 0.037* | 0.134 ± 0.028# | 0.266 ± 0.058* |
| Lactobacillus genus | 0.25 ± 0.03 | 0.12 ± 0.02* | 0.26 ± 0.02# | 0.07 ± 0.02* |
| Proteobacteria phylum | ||||
| Sutterella genus | 3.17 ± 0.40 | 1.27 ± 0.64* | 1.05 ± 0.60* | 1.25 ± 0.63* |
| Bacteroidetes phylum | ||||
| Parabacteroides distasonis species | 0.48 ± 0.14 | 0.11 ± 0.04* | 0.16 ± 0.09* | 0.13 ± 0.06* |
| Bacteroides acidifaciens species | 1.62 ± 0.35 | 0.62 ± 0.20* | 1.64 ± 0.08# | 0.44 ± 0.08* |
3.11. Effects of LPPE on the serum endotoxin level
Ethanol feeding resulted in a significant increase of the serum endotoxin level (75.94 ± 4.49 EU L−1) compared to the CON group (65.4 ± 6.66 EU L−1) (p < 0.05). L-LPPE (68.91 ± 3.42 EU L−1) but not H-LPPE (74.40 ± 5.19 EU L−1) supplementation significantly reduced the ethanol-induced increase of the serum endotoxin level.4. Discussion and conclusions
Chronic alcohol consumption can change lipid profiles and result in ALD.6,7,29 Bile acid has long been known to be essential in dietary lipid absorption and cholesterol catabolism, and increased serum bile acid is observed in all stages of ALD.32 In the present study, chronic alcohol intake led to a noticeable increase in the liver-to-body weight ratio, AST activity and the bile acid level in the serum and the TG level in the serum and liver, which was consistent with previous studies.6,7,32 LPPE showed biphasic effects on ethanol-induced liver injury in mice in the present study. L-LPPE protected against ALD as reflected in the recovery of the liver-to-body weight ratio, serum AST activity, bile acid level, serum TG level and liver TG level. In contrast, H-LPPE notably aggravated ALD as evidenced by the markedly increased liver-to-body weight ratio, serum TG level and liver TG level.4.1. Potential mechanisms of the ameliorative effect of L-LPPE on ALD
Ethanol-induced oxidative stress has been implicated in the initiation and progression of ALD.2,3 Consistent with previous reports,5,7 ethanol intake induced significant oxidative stress in the EtOH group as evidenced by the elevated hepatic TBARS level and reduced antioxidant enzyme activity. Moreover, Nrf2 is a master regulator for transcriptional activation of a wide spectrum of genes involved in antioxidant response and detoxification.4 Ethanol has been reported to promote hepatic Nrf2 activation and further induce the expression of its target genes, including NQO1, HO-1, GCLC, GCLM, GSTα1 and GSTm1, suggesting a protective role of Nrf2 against alcohol-induced liver damage.5 The present study showed that L-LPPE supplementation diminished ethanol-induced oxidative stress, which was accompanied by decreased Nrf2 nuclear translocation and Nrf2-regulated gene expression, indicating that L-LPPE alleviated ethanol-induced oxidative stress.Dysregulation of lipid metabolism, which includes increased lipogenesis and/or inhibited fatty acid β-oxidation, is observed in ALD.6,7,29 SREBP-1c is an important nuclear receptor that regulates the expression of many lipogenesis genes. In response to ethanol consumption, the SREBP-1c precursor is cleaved into small fragments, and the mature SREBP-1c protein translocates into the nucleus where it upregulates the expression of lipogenesis-related genes.8 Similarly to previous research, our results showed that ethanol consumption promoted hepatic SREBP-1c nuclear translocation and upregulated FAS expression and AGPAT2 mRNA levels. FAS and AGPAT2 are the key enzymes involved in fatty acid and triglyceride synthesis.6–8 The abovementioned changes were reversed by L-LPPE supplementation, indicating that L-LPPE had an inhibitory effect on ethanol-induced lipogenesis. CPT1α is the rate-limiting enzyme of fatty acid β-oxidation, which plays a pivotal regulatory role in the transport of long-chain fatty acids from the cytoplasm into the mitochondria. CPT1α expression is mainly regulated by PPARα, a ligand-activated transcription factor.2,6,7 In the present study, the hepatic nuclear PPARα protein level and CPT1α expression levels were decreased in the EtOH group, and L-LPPE significantly increased PPARα nuclear translocation and CPT1α expression. Therefore, the ameliorative effect of L-LPPE on ALD was, at least in part, attributed to diminished lipogenesis and increased fatty acid β-oxidation in the liver through regulating nuclear receptor SREBP-1c and PPARα activation and downstream gene expression.
As a member of the low density lipoprotein (LDL) receptor superfamily, VLDLR is responsible for the hepatic uptake of LDL and intermediate density lipoprotein (IDL).33 The effect of VLDLR on maintaining the hepatic lipid content has been shown in previous research.5,34 Increased expression of hepatic VLDLR plays a critical role in the pathogenesis of ALD. Ethanol consumption leads to a 40-fold increase in VLDLR expression compared to the vehicle control group. In contrast, VLDLR-deficient mice are protected from the ethanol-induced fatty liver.5 Ethanol consumption also increased the hepatic VLDLR expression in the present study but to a much lower extent than the abovementioned study. However, L-LPPE supplementation showed no effect on ethanol-induced VLDLR overexpression. Thus, the ameliorative effect of L-LPPE on ALD was not associated with the alteration of hepatic VLDLR-mediated lipoprotein absorption.
Gut microbiota dysbiosis results in dysfunction of the intestinal barrier and translocation of bacteria and bacterial products, which eventually contribute to the progression of ALD.22,23 As bacterial products, systemic endotoxins mediate the liver inflammatory response by interacting with toll-like receptor 4 and other receptors in the liver.35 In the present study, the serum endotoxin level was reduced by L-LPPE supplementation. These data suggested that L-LPPE supplementation may influence the gut microbiota composition. We found an increase in the Lactobacillus genus in the EtOH + L-LPPE group compared to that in the EtOH group. The commensal probiotic Lactobacillus spp. chemically and competitively excludes pathogens and stimulates mucin secretion from intestinal L-cells, along with the modulation of inflammatory responses by stimulating Treg cells.36 L-LPPE supplementation also reversed the ethanol-induced increases of the Dehalobacteriaceae family, Dehalobacterium and Ruminococcus genera. The Dehalobacteriaceae family and Ruminococcus genus are more abundant in animals with gastrointestinal inflammation than those without, indicating that their abundances are positively associated with inflammation.37,38 Moreover, L-LPPE supplementation normalized the ethanol-induced decrease in Bacteroides acidifaciens species. Expansion of gut commensal Bacteroides acidifaciens is responsible for the higher insulin sensitivity and lower body weight and fat mass in lean mice than those in obese mice. Feeding mice with Bacteroides acidifaciens results in improved insulin sensitivity, less weight gain and less fat mass than feeding mice with phosphate buffered saline. These results suggest that the abundance of Bacteroides acidifaciens species is positively associated with the homeostasis of glucose and lipid metabolism.39 In addition, the relative abundances of the Actinobacteria phylum and Coriobacteriaceae family were significantly increased by L-LPPE supplementation. In our study, Coriobacterium was the only family identified from the Actinobacteria phylum. The potential health-related effects of the Coriobacterium family are still unclear. Harmsen and his colleagues indicated that Coriobacterium plays a role in the early development of the gut microflora of newborn infants.40 This study provided evidence that L-LPPE supplementation modulated beneficial changes in the gut microbiota in ethanol-fed mice, which may be important for gut homeostasis. Our study firstly indicated that the moderate intake of phenolics positively influenced the gut microbiota in ethanol-fed mice, which brings some hints for further investigation.
4.2. Potential mechanisms of the detrimental effect of H-LPPE on ALD
It is widely accepted that phenolics induce hormetic (biphasic) dose responses in a wide range of biological models.41–44 Hormetic dose response is characterized by a beneficial effect at a low dose but a harmful effect at a high dose.41,44 Green tea polyphenols (GTPs) and phenolic-rich ethanol extracts of red mould rice result in biphasic effects on hepatotoxicity and hypertension, respectively.43,44 Although the biphasic effects of phenolics have been widely reported in previous studies, the harmful effect of phenolics is seldom found in ALD animals. To our knowledge, only curcumin has been reported to accelerate ethanol-induced liver injury at a high dose but exhibits a protective effect at a low dose.45,46 For the first time, we found a biphasic dose response of LPPE on ALD in mice in this study.Researchers have suggested that the toxicity of phenolics at high doses is presumably due to their pro-oxidative properties.42,43 Overdoses of curcumin and epigallocatechin-3-gallate (EGCG) result in hepatotoxicity in healthy mice due to suppressed antioxidant enzymes and compensatory activation of Nrf2 and upregulation of its target genes.42,47 However, H-LPPE normalized hepatic CAT activity and the expression of HO-1 and GCLM, indicating that H-LPPE exerts antioxidant effects on ethanol-treated mice. Thus, H-LPPE aggravated ethanol-induced liver injury was not due to its pro-oxidant effects.
The present study found that H-LPPE notably aggravated ALD. The increased hepatic steatosis in the H-LPPE-supplemented group was attributed to the increased expression of hepatic FAS, DGAT1 and VLDLR as well as the decreased expression of CPT1α compared to those in the EtOH group, which led to increased lipid synthesis and uptake and decreased fatty acid β-oxidation. However, in our previous study, the equivalent phenolic dose of H-LPPE exerted hypolipidemic activity in high-fat diet fed mice (data not published). Why did H-LPPE show contradictory effects on two different animal models? As reported in a previous study, the hepatotoxicity caused by a high dose of GTPs was observed in dextran sodium sulphate-induced intestinal inflamed mice but not in healthy mice.43 Therefore, it can be concluded that H-LPPE aggravated ethanol-induced liver injury may be due to the common effect of H-LPPE and ethanol but not due to the direct toxic effects of H-LPPE.
Previous research has reported that ethanol–xenobiotic metabolism interactions increase individual susceptibility to the toxic effects of xenobiotics.48 A high dose of GTPs has adverse effects in dextran sodium sulphate-induced intestinal inflamed mice but not in healthy mice,43 indicating that the adverse effects of a high dose of GTPs may be due to the drug–polyphenol metabolism interactions. As mentioned above, H-LPPE showed a detrimental effect on ALD, but the equivalent phenolic dose of H-LPPE did not show any toxic effect in high-fat diet fed mice. Thus, in our opinion, the hyperlipidemic effect of H-LPPE in ethanol-treated mice was due to the ethanol–polyphenol metabolism interactions. Cytochrome P450 2E1 (CYP2E1) is mainly responsible for the detoxification of ethanol, which is induced by ethanol.3 CYP2E1 is also involved in the demethylation and hydroxylation of flavonoid.49,50 Moreover, UDP-glucuronosyltransferases and sulphotransferases are involved in the minor metabolic pathways of ethanol and catalyse an acetaldehyde metabolite, which plays an important role in the detoxification of ethanol.51 Detoxification of polyphenols is also catalysed by these enzymes in the liver.25,52 Goodin and Rosengren reported that a high dose of EGCG, but not a low dose of EGCG, increases CYP2E1 activity.53 Thus, co-administration of ethanol and H-LPPE may overload their common metabolizing enzymes, induce ethanol–polyphenol metabolism interactions, and finally lead to adverse effects. In the present study, we mainly focused on ALD related changes in LPPE-treated mice. The exact mechanism for H-LPPE aggravating liver injury needs further study.
The health benefits of phenolics have been reported to be related to their effect on maintaining the balance of intestinal bacteria.26–28 However, the effect of phenolics on gut microbiota dysbiosis in ALD patients and animals has not been reported previously. In the present study, H-LPPE supplementation exhibited no obvious effect on the gut microbiota composition or the serum endotoxin level compared to the EtOH group, although beneficial changes were found in the L-LPPE supplemented group. Thus, the detrimental effect of H-LPPE on ALD may be not associated with the changes of the gut microbiota. However, no available data help us to analyze why LPPE showed different effects on the gut microbiota of ALD mice at low and high doses.
In conclusion, we demonstrated the biphasic dose response of LPPE on ALD in mice. The potential mechanism for the ameliorative effect of L-LPPE on ALD included reducing oxidative stress and improving dyslipidaemia. L-LPPE supplementation modulated beneficial changes in gut microbiota in ethanol-fed mice. H-LPPE aggravated ethanol-induced liver injury partly as evidenced by increased expression of genes responsible for lipid synthesis and uptake as well as reduced fatty acid β-oxidation gene expression. These findings suggest that the moderate intake of LPPE or lychee pulp could be useful for the prevention of ALD.
Acknowledgements
This work was supported by a Joint Fund from the NSFC and Guangdong Provincial Government (U1301211); the Basic Research Program of Shenzhen (JCYJ20140901162541286); the National Natural Science Foundation of China (31571828; 31501478); the China Postdoctoral Science Foundation (2016M590764); the PhD Start-up Fund of Natural Science Foundation of Guangdong (2014A030310328) and the Guangdong Provincial Science and Technology Project (2016B070701012).References
- R. S. O'Shea, S. Dasarathy and A. J. McCullough, Hepatology, 2010, 51, 307–328 CrossRef PubMed.
- V. Purohit, B. Gao and B. J. Song, Alcohol. Clin. Exp. Res., 2009, 33, 191–205 CrossRef CAS PubMed.
- F. Bardag-Gorce, B. A. French, L. Nan, H. Song, S. K. Nguyen, H. Yong, J. Dede and S. W. French, Exp. Mol. Pathol., 2006, 81, 191–201 CrossRef CAS PubMed.
- W. Tang, Y.-F. Jiang, M. Ponnusamy and M. Diallo, World J. Gastroenterol., 2014, 20, 13079–13087 CrossRef CAS PubMed.
- Z. Wang, X. Dou, S. Li, X. Zhang, X. Sun, Z. Zhou and Z. Song, Hepatology, 2014, 59, 1381–1392 CrossRef CAS PubMed.
- C.-C. Tang, W.-L. Lin, Y.-J. Lee, Y.-C. Tang and C.-J. Wang, Food Funct., 2014, 5, 678–687 CAS.
- Z. Wang, B. Su, S. Fan, H. Fei and W. Zhao, Biochem. Bioph. Res. Co., 2015, 458, 757–762 CrossRef CAS PubMed.
- M. You, M. Fischer, M. A. Deeg and D. W. Crabb, J. Biol. Chem., 2002, 277, 29342–29347 CrossRef CAS PubMed.
- S. R. M. Ibrahim and G. A. Mohamed, J. Ethnopharmacol., 2015, 174, 492–513 CrossRef CAS PubMed.
- Q. Lv, F. Luo, X. Zhao, Y. Liu, G. Hu, C. Sun, X. Li and K. Chen, PLoS One, 2015, 10, e0120480 Search PubMed.
- Q. Lv, M. Si, Y. Yan, F. Luo, G. Hu, H. Wu, C. Sun, X. Li and K. Chen, J. Funct. Foods, 2014, 7, 621–629 CrossRef CAS.
- R. Zhang, Q. Zeng, Y. Deng, M. Zhang, Z. Wei, Y. Zhang and X. Tang, Food Chem., 2013, 136, 1169–1176 CrossRef CAS PubMed.
- D. Su, H. Ti, R. Zhang, M. Zhang, Z. Wei, Y. Deng and J. Guo, Food Chem., 2014, 158, 385–391 CrossRef CAS PubMed.
- D. Su, R. Zhang, F. Hou, M. Zhang, J. Guo, F. Huang, Y. Deng and Z. Wei, BMC Complementary Altern. Med., 2014, 14, 9–18 CrossRef PubMed.
- L. Bhoopat, S. Srichairatanakool, D. Kanjanapothi, T. Taesotikul, H. Thananchai and T. Bhoopat, J. Ethnopharmacol., 2011, 136, 55–66 CrossRef CAS PubMed.
- D. Su, R. Zhang, C. Zhang, F. Huang, J. Xiao, Y. Deng, Z. Wei, Y. Zhang, J. Chi and M. Zhang, Food Funct., 2016, 7, 508–515 CAS.
- V. Umarani, S. Muvvala, A. Ramesh, B. V. Lakshmi and N. Sravanthi, Toxicol. Mech. Methods, 2015, 25, 1–24 CrossRef PubMed.
- M. Sinha, D. K. Das, K. Manna, S. Datta, T. Ray, A. K. Sil and S. Dey, Free Radical Res., 2012, 46, 842–849 CrossRef CAS PubMed.
- N. Sathaporn, M. Kittana, T. Thavaree and E. Ekk, Molecules, 2011, 16, 5054–5061 CrossRef PubMed.
- M. Kobori, S. Masumoto, Y. Akimoto and H. Oike, Mol. Nutr. Food Res., 2011, 55, 530–540 CAS.
- W. Hassan, G. Rongyin, A. Daoud, L. Ding, L. Wang, J. Liu and J. Shang, Oxid. Med. Cell. Longevity, 2014, 313602 Search PubMed.
- L. Bull-Otterson, W. Feng, I. Kirpich, Y. Wang, X. Qin, Y. Liu, L. Gobejishvili, S. Joshi-Barve, T. Ayvaz, J. Petrosino, M. Kong, D. Barker, C. McClain and S. Barve, PLoS One, 2013, 8, e53028 CAS.
- A. W. Yan, D. E. Fouts, J. Brandl, P. Stärkel, M. Torralba, E. Schott, H. Tsukamoto, K. E. Nelson, D. A. Brenner and B. Schnabl, Hepatology, 2011, 53, 96–105 CrossRef CAS PubMed.
- I. A. Kirpich, N. V. Solovieva, S. N. Leikhter, N. A. Shidakova, O. V. Lebedeva, P. I. Sidorov, T. A. Bazhukova, A. G. Soloviev, S. S. Barve, C. J. McClain and M. Cave, Alcohol, 2008, 42, 675–682 CrossRef CAS PubMed.
- A. Scalbert and G. Williamson, J. Nutr., 2000, 130, 2073S–2085S CAS.
- H. C. Lee, A. M. Jenner, C. S. Low and Y. K. Lee, Res. Microbiol., 2006, 157, 876–884 CrossRef CAS PubMed.
- S. Jang, J. Sun, P. Chen, S. Lakshman, A. Molokin, J. M. Harnly, B. T. Vinyard, J. F. Urban, C. D. Davis and G. Solano-Aguilar, J. Nutr., 2016, 146, 673–680 CrossRef CAS PubMed.
- B. Ojo, G. D. El-Rassi, M. E. Payton, P. erkins-Veazie, S. Clarke, B. J. Smith and E. A. Lucas, J. Nutr., 2016, 146, 1483–1491 CrossRef CAS PubMed.
- H.-Q. Yin, Y.-C. Kim, Y.-S. Chung, Y.-C. Kim, Y.-K. Shin and B.-H. Lee, Toxicol. Appl. Pharmacol., 2009, 236, 124–130 CrossRef CAS PubMed.
- J. Xiao, S. Li, Y. Sui, Q. Wu, X. Li, B. Xie, M. Zhang and Z. Sun, PLoS One, 2014, 9, e112773 Search PubMed.
- E. Yu, J. Xie, J. Wang, H. Ako, G. Wang, Z. Chen and Y. Liu, World J. Microbiol. Biotechnol., 2016, 32, 116 CrossRef PubMed.
- G. Xie, W. Zhong, H. Li, Q. Li, Y. Qiu, X. Zheng, H. Chen, X. Zhao, S. Zhang, Z. Zhou, S. H. Zeise and W. Jia, FASEB J., 2013, 27, 3582–3593 CrossRef PubMed.
- S. Takahashi, J. Suzuki, M. Kohno, K. Oida, T. Tamaim, S. Miyabom, T. Yamamoto and T. Nakai, J. Biol. Chem., 1995, 270, 15747–15754 CrossRef CAS PubMed.
- H. Jo, S. S. Choe, K. C. Shin, H. Jang, J. H. Lee, J. K. Seong, S. H. Back and J. B. Kim, Hepatology, 2013, 57, 1366–1377 CrossRef CAS PubMed.
- W. Zhong and Z. Zhou, World J. Gastrointest. Pathophysiol., 2014, 5, 514–522 CrossRef PubMed.
- T. A. Oelschlaeger, Int. J. Med. Microbiol., 2010, 300, 57–62 CrossRef CAS PubMed.
- C. W. Png, S. K. Lindén, K. S. Gilshenan, E. G. Zoetendal, C. S. McSweeney, L. I. Sly, M. A. McGuckin and T. H. J. Florin, Am. J. Gastroenterol., 2010, 105, 2420–2428 CrossRef CAS PubMed.
- R. Arrazuria, N. Elguezabal, R. A. Juste1, H. Derakhshani and E. Khafipour, Front. Microbiol., 2016, 7, 446 Search PubMed.
- J. Y. Yang, Y. S. Lee, Y. Kim, S. H. Lee, S. Ryu, S. Fukuda, K. Hase, C. S. Yang, H. S. Lim, M. S. Kim, H. M. Kim, S. H. Ahn, B. E. Kwon, H. J. Ko and M. N. Kweon, Mucosal Immunol., 2016 DOI:10.1038/mi.2016.42.
- H. J. M. Harmsen, A. C. M. Wildeboer-Veloo, J. Grijpstra, J. Knol, J. E. Degener and G. W. Welling, Appl. Environ. Microbiol., 2000, 66, 4523–4527 CrossRef CAS PubMed.
- E. J. Calabrese, M. P. Mattson and V. Calabrese, Hum. Exp. Toxicol., 2010, 29, 1034–1037 CAS.
- D. Wang, Y. Wang, X. Wan, C. S. Yang and J. Zhang, Toxicol. Appl. Pharmacol., 2015, 283, 65–74 CrossRef CAS PubMed.
- H. Inoue, M. Maeda-Yamamoto, A. Nesumi, T. Tanaka and A. Murakami, Biosci. Biotechnol., Biochem., 2013, 77, 1223–1228 CrossRef CAS PubMed.
- J.-J. Wang, H.-Y. Wang and C.-D. Shih, J. Agric. Food Chem., 2010, 58, 7940–7948 CrossRef CAS PubMed.
- H.-L. Zhao, C. H. Song and O. H. Chai, Phytother. Res., 2012, 26, 1857–1863 CrossRef CAS PubMed.
- H.-I. Lee, R. A. McGregor, M.-S. Choi, K.-I. Seo, U. J. Jung, J. Yeo, M.-J. Kim and M.-K. Lee, Life Sci., 2013, 93, 693–699 CrossRef CAS PubMed.
- P. Qiu, S. Man, J. Li, J. Liu, L. Zhang, P. Yu and W. Gao, J. Agric. Food Chem., 2016, 64, 2765–2771 CrossRef CAS PubMed.
- A. Meskar, E. Plee-Gautier, Y. Amet, F. Berthou and D. Lucas, Pathol. Biol., 2001, 49, 696–702 CrossRef CAS PubMed.
- S. E. Nielsen, V. Breinhol, U. Justesenoe, C. Cornett and L. O. Dragsted, Xenobiotica, 1998, 28, 389–401 CrossRef CAS PubMed.
- J. Xiao and P. Högger, Curr. Drug Metab., 2013, 14, 381–391 CrossRef CAS PubMed.
- X. Shi, D. Yao and C. Chen, J. Biol. Chem., 2012, 287, 6336–6349 CrossRef CAS PubMed.
- C. Manach, A. Scalbert, C. Morand, C. Remesy and L. Jimenez, Am. J. Clin. Nutr., 2005, 81, 230S–242S CAS.
- M. G. Goodin and R. J. Rosengren, Toxicol. Sci., 2003, 76, 262–270 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6fo01166g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |