
Recent advances in well-defined, late transition metal complexes that make and/or break C–N, C–O and C–S bonds
Addison N.
Desnoyer
and
Jennifer A.
Love
*
Department of Chemistry, The University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: jenlove@chem.ubc.ca
First published on 16th November 2016
Abstract
Chemical transformations that result in either the formation or cleavage of carbon–heteroatom bonds are among the most important processes in the chemical sciences. Herein, we present a review on the reactivity of well-defined, late-transition metal complexes that result in the making and breaking of C–N, C–O and C–S bonds via fundamental organometallic reactions, i.e. oxidative addition, reductive elimination, insertion and elimination reactions. When appropriate, emphasis is placed on structural and spectroscopic characterization techniques, as well as mechanistic data.
 Addison N. Desnoyer | Addison N. Desnoyer studied chemistry at the University of British Columbia Okanagan, working in the research group of Prof. Kevin M. Smith, where he obtained an inordinate fondness for first-row transition metals. He graduated with his Bachelor of Science (Hons.) degree in 2012, then joined the group of Prof. Jennifer A. Love at the University of British Columbia in Vancouver to pursue his PhD. He mainly studies the reactivity of low-valent nickel complexes with small molecules, but can occasionally be convinced to work with rhodium. Addison is the recipient of an NSERC CGS-D3 scholarship, an Izaak Walton Killam doctoral fellowship and a Michael Smith Foreign Study Supplement, which allowed him to venture to Berlin in 2016 to work for a term in the labs of Prof. Dr Thomas Braun at the Humboldt-Universität zu Berlin. |
 Jennifer A. Love | Prof. Jennifer Love began at UBC as an assistant professor in 2003, following her doctoral studies with Prof. Paul Wender at Stanford University and NIH-funded postdoctoral studies with Prof. Robert Grubbs at Caltech. She was promoted to associate professor in 2009 and professor in 2016. She has won a number of awards, including an Alexander von Humboldt Research Fellowship for Experienced Researchers in 2012, the AstraZeneca Canada Award in Chemistry in 2008, the UBC Science Killam Teaching Award in 2009 and a CNC IUPAC travel award in 2011. She became a Fellow of the Royal Society of Chemistry (UK) in 2016. She is an Associate Editor for Catalysis Science & Technology and has served on both the editorial board and editorial advisory board of Dalton Transactions. In 2016, she became the Senior Advisor to the Provost on Women Faculty. Love's program spans the traditional boundaries of organic and inorganic chemistry. Her group aims to develop fundamentally new, cost-effective and low-waste reactions for the synthesis of medicinally important compounds by exploring the mechanism of reactions at late transition metal centres. |
Introduction
The development of transition metal-catalyzed reactions that can selectively form C–C bonds has dominated the fields of organic and inorganic chemistry for decades, and the literature is replete with examples of systems that are efficient, atom economical and functional group tolerant.1 In contrast, transition metal complexes that form C–X (X = N, O, S) bonds have only recently been developed and studied in detail.2–8Given the synthetic utility of C–heteroatom bond formation and functionalization, it is not surprising that much effort has recently been put forth towards the development of new transformations that focus on C–X bonds, and several pertinent reviews have emerged.9–15 However, these reviews tend to focus on catalytic reactions, and mechanistic data is often speculative. Herein, we present an overview of well-defined, often stoichiometric reactivity of late transition metals with organic compounds that form C–N, C–O and C–S bonds, with an emphasis on the characterization techniques used and mechanistic experiments that provide valuable insight into these reactions.
The focus of our research program is the rational design of new chemical transformations that rely on intimate knowledge of the fundamental organometallic reactivity of transition metal complexes. In particular, our group focuses on the reactivity of late transition metals, as these tend to be catalytically active, yet tolerant of a wider range of functional groups and experimental condition (i.e. air and water) than early metal catalysts. Indeed, presented here are several elegant examples of how these fundamental transformations (i.e. oxidative addition, reductive elimination, insertion, elimination etc.) can be harnessed to form valuable organic products that are often difficult to prepare using traditional synthetic methods. We hope that our summary of recent work in this field will provide continued inspiration for synthetic chemists to explore new avenues in the chemistry of transition metals interacting with C–X bonds.
This Review is divided into three sections: one each for C–N, C–O and C–S bond formation and cleavage reactions. Each section is divided into subsections based on the group number of the transition metal. Broadly, we have attempted to present the discussion in chronological order. However, compounds that share similar reactivity within a given subsection have also been grouped together. Given the wide scope of this topic, we have focused on advances in C–X (X = N, O, S) bond formation where the organometallic products have been characterized experimentally rather than simply invoked as reactive intermediates, especially those cases that occur by unusual mechanisms. While we have focused on examples from the past two decades, we have also included seminal, early advances in the field.
C–N bond cleavage and formation
Group 8 metals
Hartwig et al. have described the synthesis and reactivity of a ruthenium benzyne complex 1 (Scheme 1), and found that with mild heating it reacted with both benzaldehyde and benzonitrile via an insertion reaction to give the ruthenium alkoxide 2 or amide 3, respectively.16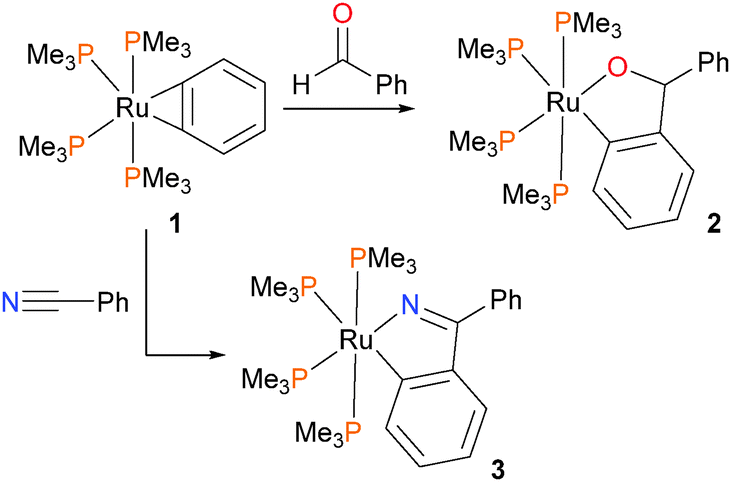 | ||
| Scheme 1 | ||
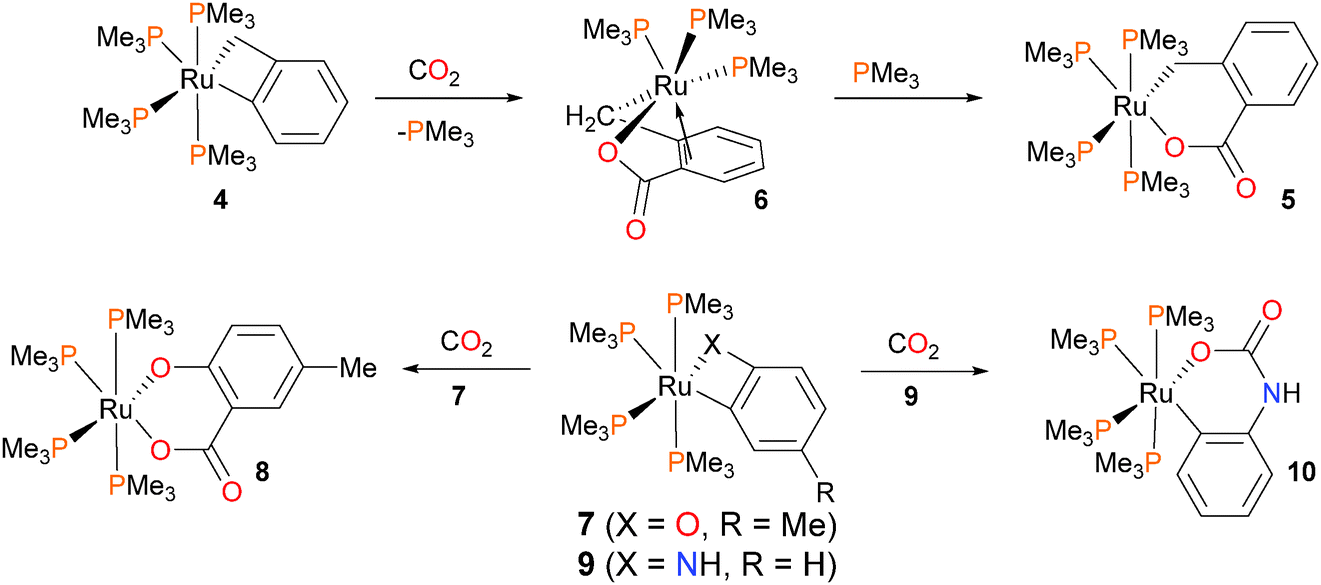
Shortly thereafter, the same group examined the reactivity of ruthenium benzyl complexes towards organic electrophiles (Scheme 2). Treatment of the benzylruthenium complex 4 with CO2 in toluene for 8 hours at 85 °C resulted in the formation of complex 5 in high yield (90–95%), which is the product of CO2 insertion into the Ru–Caryl rather than the Ru–Calkyl bond.17 If the reaction is performed in pentanes instead of arene solvents, an intermediate complex 6 precipitates from the reaction mixture, which can be characterized by NMR spectroscopic techniques as well as X-ray diffraction studies. Complex 6 contains three PMe3 ligands, and notably demonstrates η2-bonding of the aromatic ring with the metal centre. This highlights the necessity for phosphine dissociation and later re-coordination during this transformation. Analogous reactivity was observed with o-metallated cresolate complex 7, where insertion was found to occur exclusively into the Ru–Caryl bond to yield 8. Dissociation of phosphine is likely required here as well, as kinetic experiments demonstrated that 3 equivalents of added PMe3 to the reaction of 7 with CO2 resulted in a lengthening of the half-life of the reaction from 45 minutes to 18 hours. In contrast, treating anilide complex 9 with CO2 resulted in insertion into the Ru–N bond at much lower temperatures. In addition, performing the reaction with added PMe3-d9 at 0 °C revealed full conversion to product 10 without any incorporation of deuterated phosphine ligand into the product. Thus, the authors conclude that this insertion process likely occurs via direct attack of the nitrogen onto the carbon of CO2 and does not require dissociation of phosphine. Although at least five intermediates were observed by NMR spectroscopy at low temperatures, the authors were unable to definitively assign their structures.
 | ||
| Scheme 2 | ||
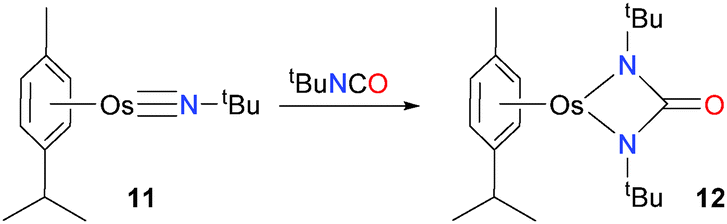
Osmium imido complex 11 has been shown to undergo cyclization with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of tBuNCO, giving the blue-green complex 12 in high yield (Scheme 3).18,19 Complex 12 displays a sharp absorbance band at 1656 cm−1 in the IR spectrum, clearly showing the presence on an amide group. In addition, the carbonyl carbon resonance at 174.4 ppm in the 13C NMR spectrum is also particularly diagnostic for an amide.
N bond of tBuNCO, giving the blue-green complex 12 in high yield (Scheme 3).18,19 Complex 12 displays a sharp absorbance band at 1656 cm−1 in the IR spectrum, clearly showing the presence on an amide group. In addition, the carbonyl carbon resonance at 174.4 ppm in the 13C NMR spectrum is also particularly diagnostic for an amide.
 | ||
| Scheme 3 | ||
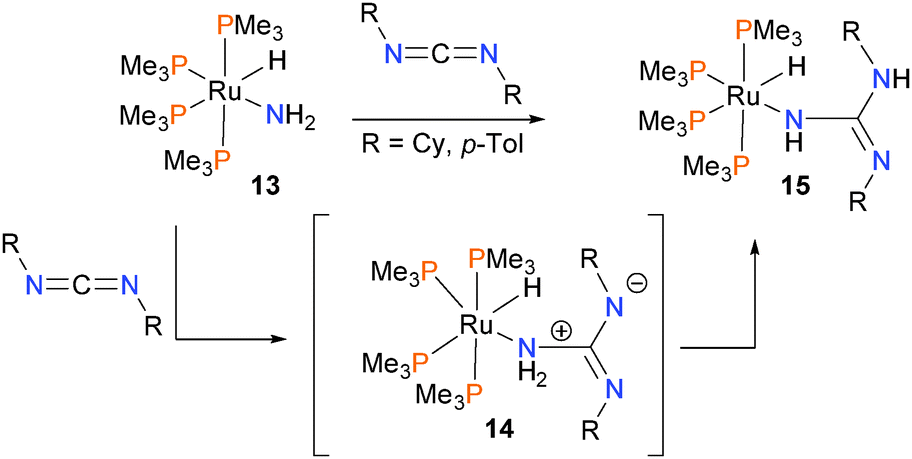
Insertion of carbodiimides into the N–H bond of the simple ruthenium amido complex 13 was found to proceed via direct attack of the nitrogen atom at the electrophilic carbon of the diimide to give intermediate 14, followed by proton transfer to yield final product 15 (Scheme 4).20 Evidence for this mechanism is that the reaction proceeds rapidly at low temperatures (for R = p-tolyl, at −80 °C; for R = Cy, rt) and that the rate is independent of added amounts of phosphine ligand.
 | ||
| Scheme 4 | ||
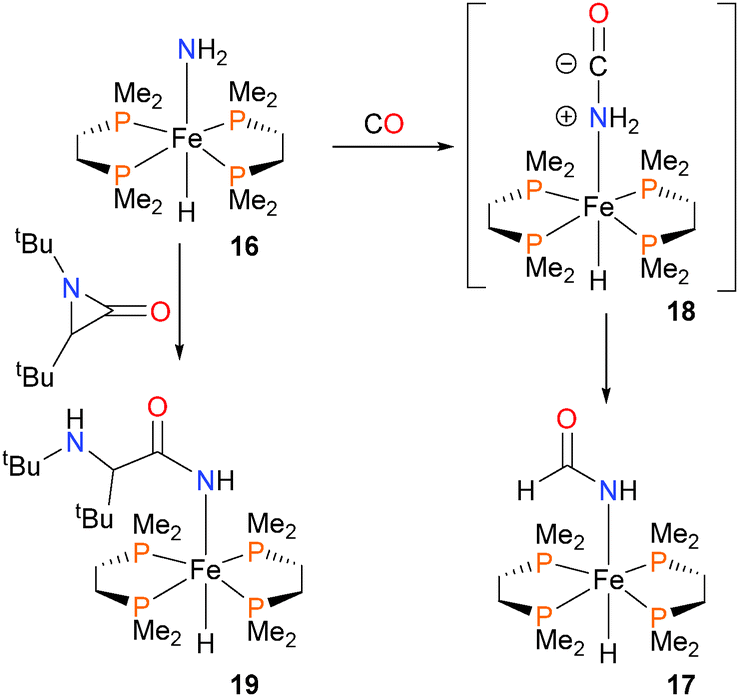
In 2003, Fox and Bergman described the synthesis of the first parent amido complex of iron,21 which exists as a mixture of cis and trans isomers in solution, as determined by NMR spectroscopy. Treatment of amido 16 with CO results not in the expected insertion of CO into the Fe–N bond, but rather complex 17via C–N bond formation that is formally the insertion of CO into one of the N–H bonds of the amido group (Scheme 5). The authors performed a variety of mechanistic experiments, and determined that the simple mechanism of nucleophilic attack of the amido directly onto the carbon atom of CO to yield intermediate 18, followed by a proton transfer, best fits the experimental data. A similar mechanism was found for the insertion of carbodiimides into the N–H bond of ruthenium amidos.20 Iron complex 16 was also found to be nucleophilic enough to induce ring-opening when reacted with aziridinone to form 19, presumably via a similar reaction pathway for the formation of 17.22 The Gunnoe group has also described insertion reactions of amido complexes of ruthenium with nitriles.23
 | ||
| Scheme 5 | ||
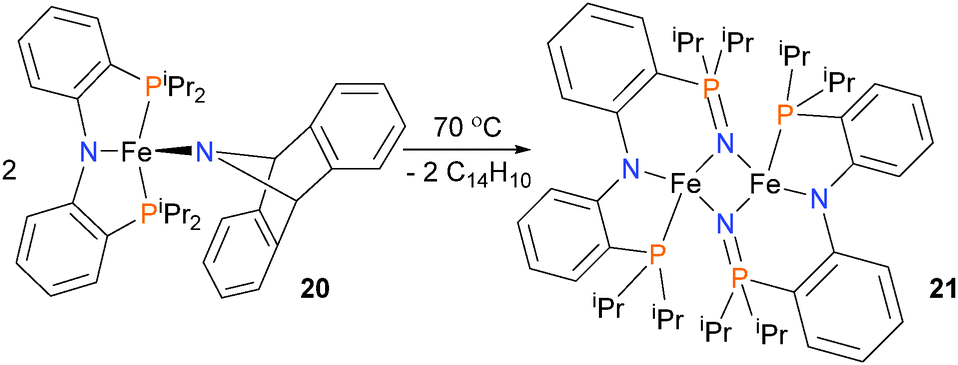
In an effort to prepare a nitride complex of iron, Mindiola and co-workers have prepared iron(II) amido complex 20, outfitted with a PNP-type pincer ligand. Thermolysis of complex 20 at 70 °C for 48 hours yields complex 21 in 69% yield, as determined by 1H NMR spectroscopy, as shown in Scheme 6. The authors propose several possible mechanisms for the formation of the final product 21,24 although all feature the elimination of anthracene and insertion of the nitrogen atom into the Fe–P bond of the pincer ligand. Iron nitrene complexes have been used for the preparation of carbodiimides.25
 | ||
| Scheme 6 | ||
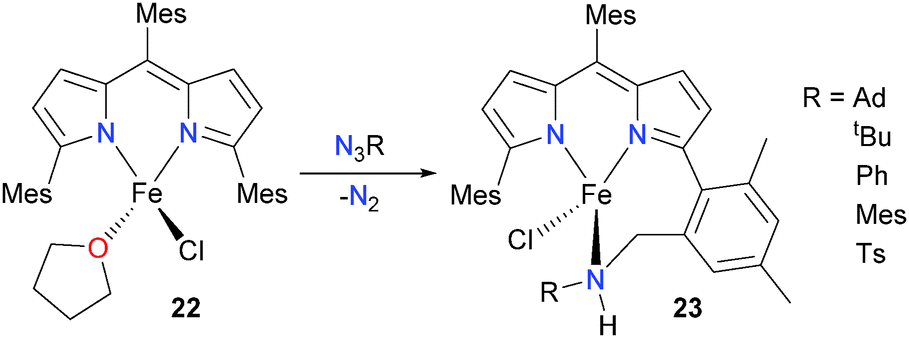
King and Betley have synthesized the iron dipyrromethene complex 22 and reacted it with organic azides in order to explore the possibility of preparing high-valent iron imidos.26 It was found that, for a variety of organic azides (Scheme 7), the resulting products 23 showed net insertion of the NR moiety into a C–H bond of the methyl group of the adjacent mesityl ring. The authors speculate that this process could proceed via H-atom abstraction by an Fe(IV) imido complex, akin to the typically accepted mechanism for hydroxylase enzymes such as cytochrome P450.27 Performing the reaction at low temperatures in an NMR tube did not reveal any appreciable amounts of observable intermediates during this amination reaction. Similarly, monitoring the reaction by UV/Vis spectrophotometry revealed only the decrease in the absorbance due to 22 with a concomitant increase in the absorbance band due to product 23 with an isosbestic point at 516 nm, again indicating no buildup of intermediates during the overall transformation.
 | ||
| Scheme 7 | ||
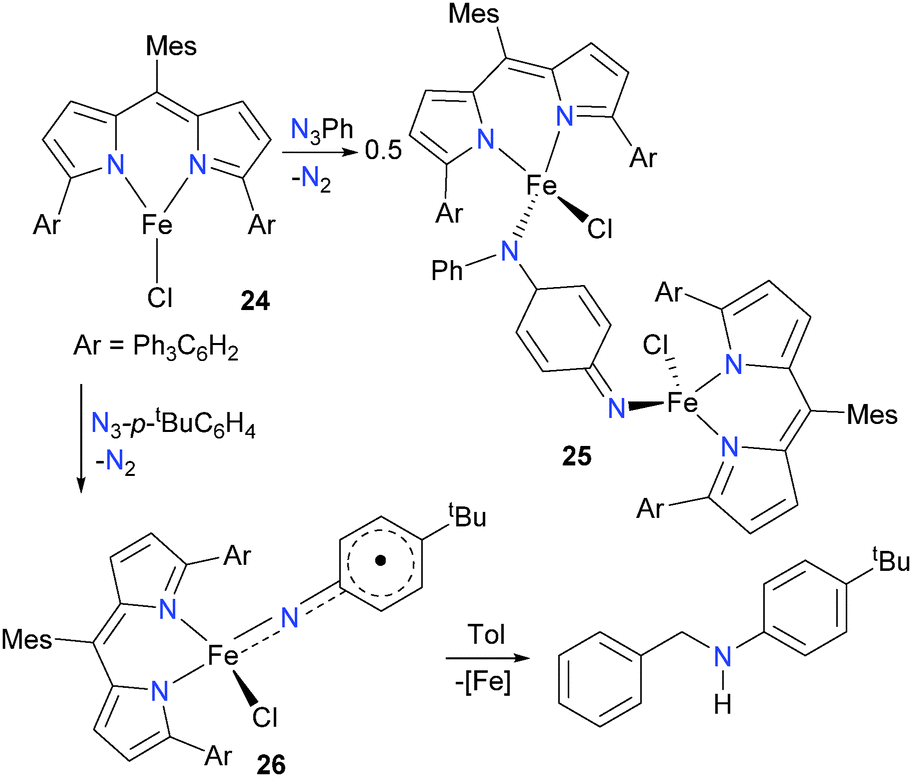
Several years later, the same group published a more extensive study featuring a modified ligand scaffold designed to remove the weak, easily abstractable C–H bonds of the flanking mesityl groups.28 Thus, a variety of analogues of 22 were prepared and fully characterized. The bulky complex 24 in particular (Scheme 8) proved adept at stabilizing the imidos generated from organic azides. If 24 is reacted with phenylazide, for example, the resulting product is the bimetallic 25, formed via C–N coupling of the transiently generated imido with the para-carbon of another equivalent of imido. This coupling could be prevented by instilling sufficient bulk at the para position of the aromatic ring (i.e. using p-tBuC6H4N3 instead of PhN3), resulting in monomeric complex 26, which was fully characterized. Complex 26 is best described as a high-spin iron(III) complex, antiferromagnetically coupled to an imido-based radical which is delocalized into the aromatic ring. This assignment of the electronic structure of 26 is based on the bond lengths determined from single crystal diffraction methods, as well as Mössbauer spectroscopy and broken-symmetry DFT calculations. Complex 26 was found to quantitatively react with toluene, resulting in the amination of the benzylic C–H bond.
 | ||
| Scheme 8 | ||
Use of complex 27, which features slight modifications to the dipyrromethene ligand allow the reaction of the imidyl radical to occur intramolecularly, generating a series of bound pyrrolidine complex of iron, complexes 28, that have been fully characterized.29 Importantly, the N-heterocycle can then be removed from the metal centre by protecting it with Boc2O, allowing for the regeneration of complexes 27. Thus, 27 was found to be a viable catalyst for the transformation of organic azides into a wide array of N-heterocycles, including azetidines and piperidines (not shown, see Scheme 9) in addition to pyrrolidines. A related methodology has also been applied to the preparation of a pyrrolidine using a palladium complex outfitted with a redox non-innocent ligand, in an unusual example of using a noble metal to facilitate single-electron chemistry.30 Further work from the Betley group demonstrated that complexes 27 were good catalysts for benzylic and allylic amination reactions, and detailed mechanistic studies found that the reaction was occurring via an H-atom abstraction and radical recombination pathway.31
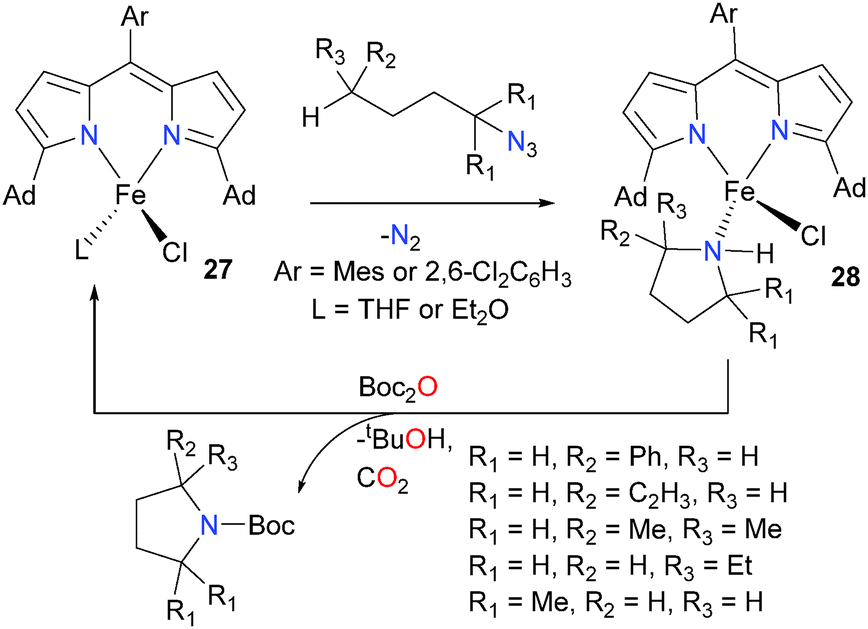 | ||
| Scheme 9 | ||
In 2009, the group of Kakiuchi published a paper on the reactivity of ruthenium complex 29 with o-acylanilines.32 In particular, they found that when the aniline featured a bulky pivalyl substituent, protonolysis of a hydride ligand occurred at elevated temperatures to generate chelated complex 30. Subsequent heating of 30 in the presence of an additional equivalent of the same aniline, however, resulted not in another protonolysis, but rather C–N bond cleavage to generate five-membered metallacycle 31 (see Scheme 10). This report was the first example of the C–N bond cleavage of unactivated anilines by a late transition metal. Ammonia was detected as a byproduct of this reaction by 1H NMR spectroscopy. The mechanism of this unusual transformation was not determined, although it was found that the addition of olefins increased the rate of formation of 31. Furthermore, 29 was found to be suitable for cross-coupling chemistry using a phenyl boronate ester. Indeed, Suzuki-type cross-coupling could be performed using catalytic amounts of 29, although additional phosphine ligand was required for optimal yields of biaryl product.
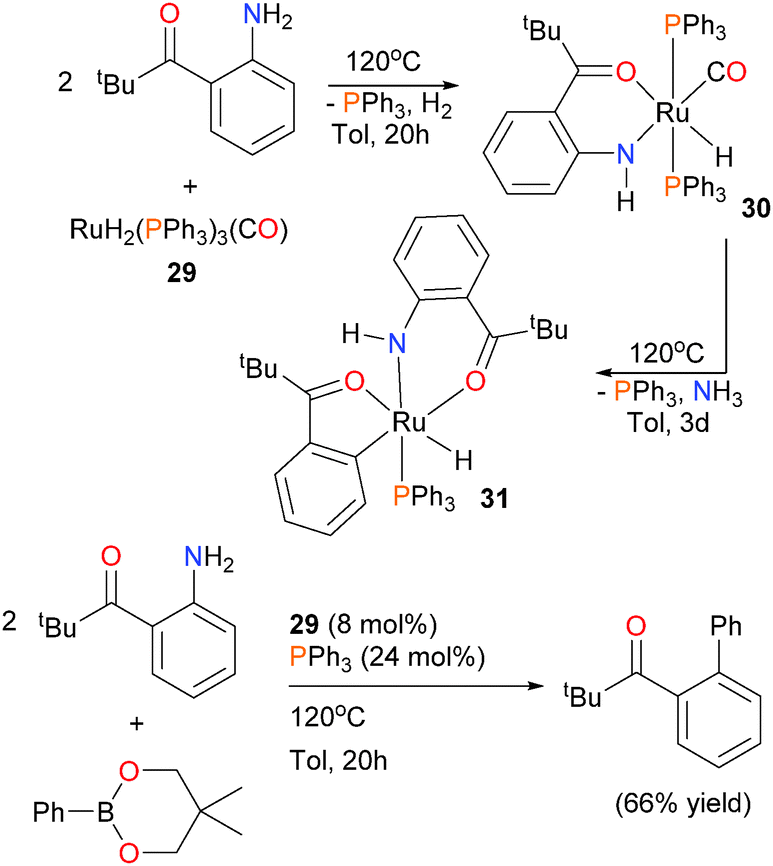 | ||
| Scheme 10 | ||
Thirty years after their first paper in the field of nitrosyl chemistry,33,34 the Bergman group reported an easily synthesized ruthenium dinitrosyl complex and its insertion chemistry with alkenes.35,36 Treatment of complex 32 with norbornadiene resulted in the formation of a new product, assigned as 33 based on NMR spectroscopic data. Unfortunately, 33 proved unstable in solution, and thus was difficult to definitively characterize. To circumvent this instability, a variety of neutral, chelating ligands were added. Tetramethylethylenediamine (TMEDA) proved to be the most convenient ligand for stabilizing the dinitrosoalkane product, and the structure of 34 was determined by an X-ray diffraction study and fully characterized (Scheme 11). In addition, the alkene scope of this transformation was found to be fairly broad, as a variety of higher alkenes could be used in place of norbornadiene. Related insertion of an alkyne37 and CO38 into the Ru–N bond of a binuclear ruthenium complex has been reported by Matsuzaka et al.
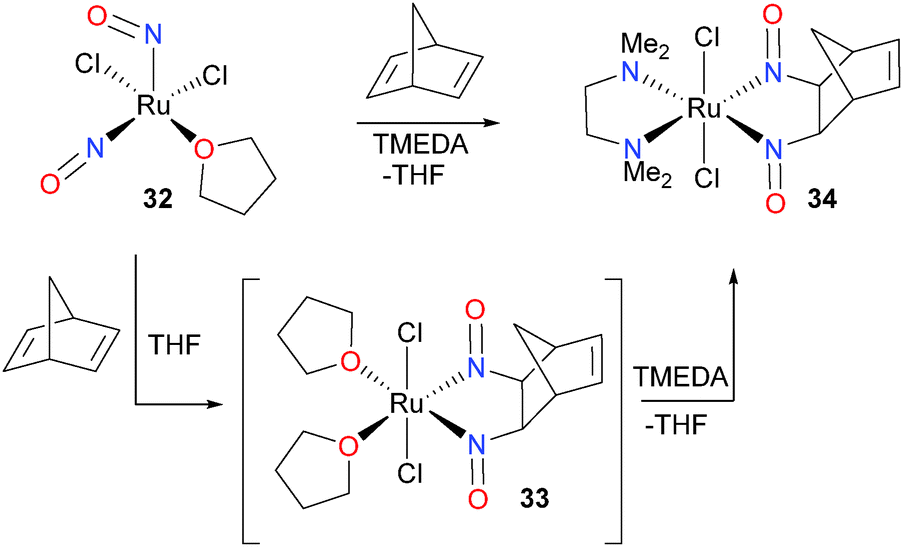 | ||
| Scheme 11 | ||
The Lau group has recently prepared a variety of ruthenium and osmium nitride complexes39 such as 35 (Scheme 12). They have shown them to be able to cleave the C–N bond of aniline via N–N coupling and H-atom transfer,40 to aziridinate alkenes (to yield 36),41 nitrogenate alkynes,42 to insert the nitride into the C–H bonds of xanthene (to yield 37) and other hydrocarbons43 and to couple to isocyanides to generate carbodiimides.44 Ruthenium nitrides have also been shown to insert into benzylic C–H bonds.45
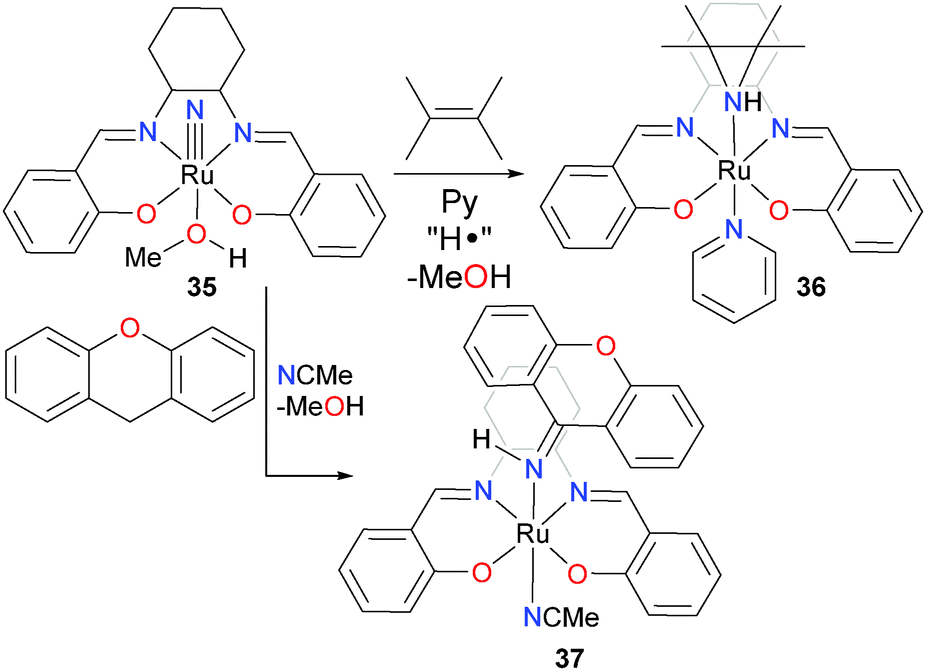 | ||
| Scheme 12 | ||
Brown has reported that osmium nitrides bearing a terpyridine (terpy) ligand, such as 38, react with alkenes to form rare azaallenium complexes 39via insertion of the nitride into the C–C bond of the alkene, as outlined in Scheme 13.46,47 Although this surprising transformation has been shown to be amenable to a wide variety of alkenes, mechanistic data has yet to be published.
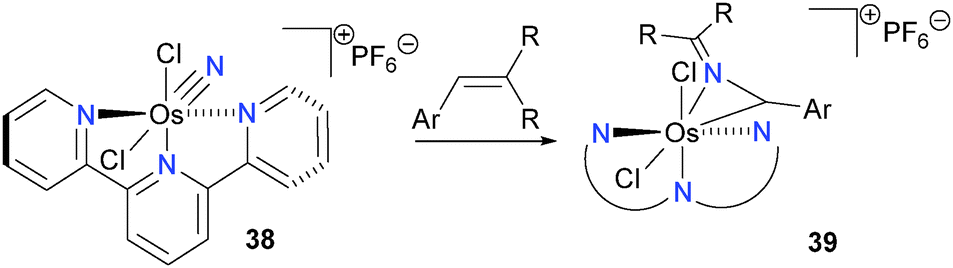 | ||
| Scheme 13 | ||
In contrast, osmium nitrides featuring tris(1-pyrazolyl)borate (Tp) as ancillary ligands react with substituted cyclohexadienes via a formal [4+1] cyclization to form bicyclic amido complexes.48 In the first reported example of the reaction of an electrophilic nitride ligand with a carbene, related osmium nitrides have also been shown to react with carbene precursors to form azavinylidene complexes.49 Moreover, these electrophilic nitrides can be arylated with a variety of carbanion equivalents, such as Grignards, zinc reagents and organoboranes.50 The resulting anilido complexes are themselves susceptible to nucleophilic aromatic substitution with amines such as pyrrolidine.51
In related work with iron, the Smith group has found that iron(IV) nitride complex 40 can react with styrene derivatives to form aziridinato complexes 4152 (Scheme 14), as well as undergoing cyclizations with dienes to give pyrrolides, amongst other coupling reactions.53–55 In addition, Powers et al. have shown that new C–N bonds can be formed by the alkylation of a bridging iron nitride with MeI.56
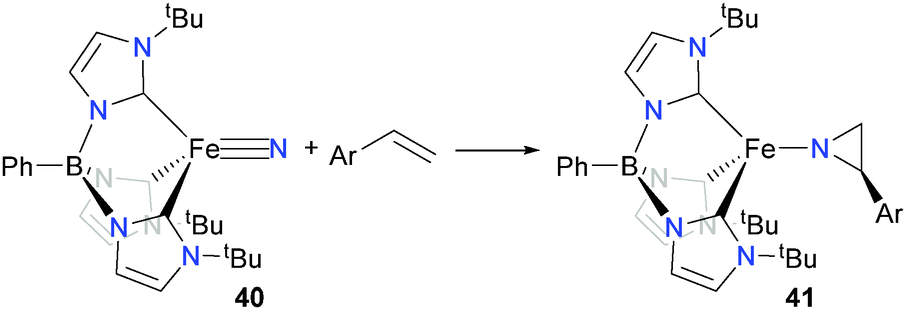 | ||
| Scheme 14 | ||
Group 9 metals
Becker et al. demonstrated that treatment of 42 with LiAlH4 at low temperatures, followed by aqueous work-up allowed for the isolation of diamines in good yields and with good stereoselectivity.57 The substrate scope was demonstrated to tolerate a wide range of alkenes, including 1-hexene and both trans and cis-β-methylstyrene, amongst others. The same group also found that dimer 43 could be reduced with sodium amalgam to generate 44, a pink, pyrophoric salt consistent with the formula CpCo(NO)Na. Alkylation with either MeI or EtI at −40 °C yielded the alkyl nitrosyl complex 45, which was characterized by NMR spectroscopic methods.58 When warmed to 0 °C in the presence of phosphines, complex 46 was formed, and was isolated in 80% yield. Further mechanistic experiments59 found that the rate of formation of 46 was independent of the concentration of phosphine, consistent with rate-limiting insertion of the alkyl group into the NO moiety to yield intermediate 47 prior to rapid phosphine coordination to give 46 as the ultimate product (Scheme 15). In addition, deuterium labeling experiments were performed, which demonstrated both that the insertion of the alkyl group into the NO moiety is intramolecular, and that β-hydride elimination, which is a common decomposition pathway for metal alkyls, does not occur to any appreciable extent in this cobalt system.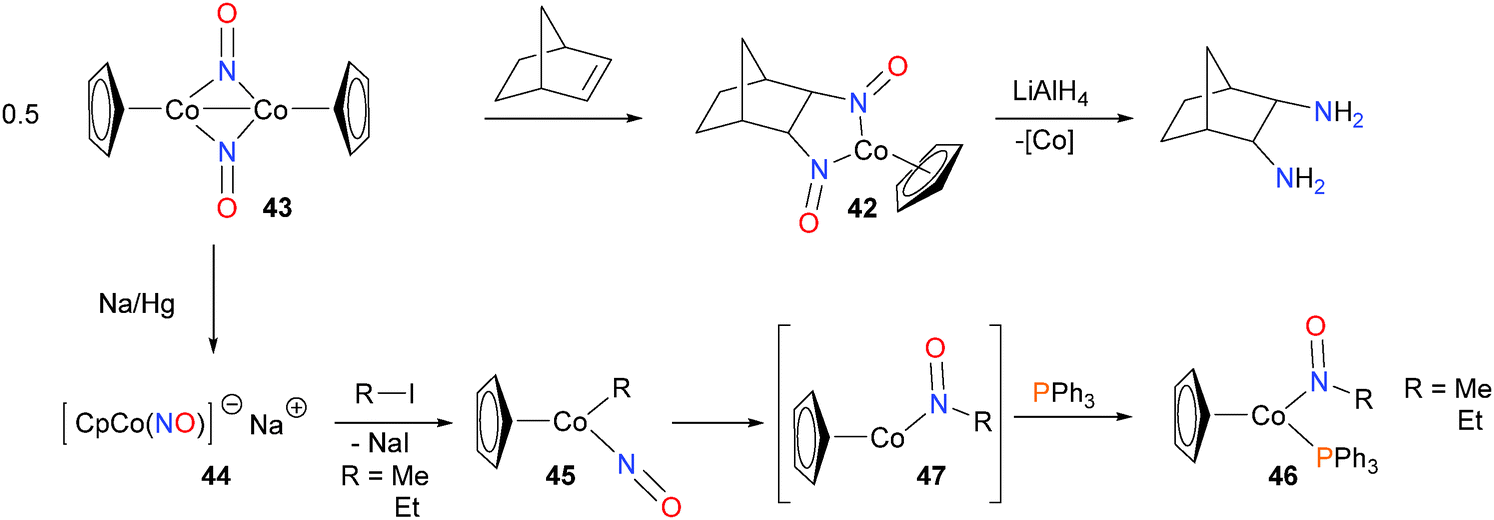 | ||
| Scheme 15 | ||
An early example of well-defined hydroamination was reported by Milstein and co-workers (Scheme 16).60 Oxidative addition of the N–H bond of aniline to iridium(I) complex 48 gave the iridium(III) hydride complex 49. Subsequent insertion of norbornene into the Ir–N bond results in azametallacyclobutane 50, which was fully characterized. Upon heating, C–H reductive elimination occurs, and dissociation liberates the product amine.
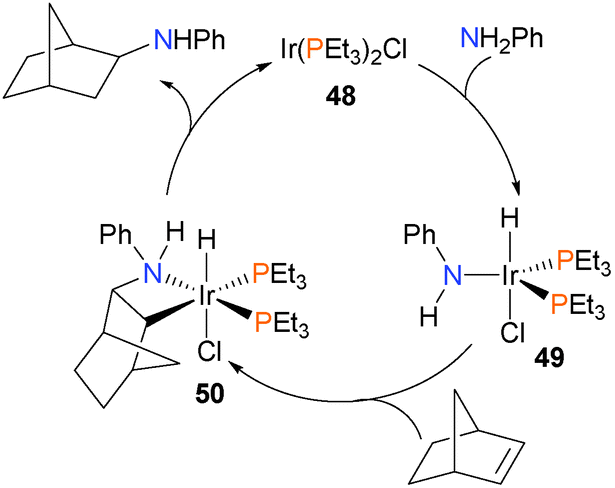 | ||
| Scheme 16 | ||
Heterocyclobutanes of iridium have also been shown to be capable of C–N bond formation (Scheme 17). Treating the Cp* complex 51 with either CO2 or tBuNCO results in attack of the amido nitrogen at the electrophilic carbon of the substrate, followed by proton transfer to yield either acid 52 or amide 53, respectively. In contrast, when the oxairidacyclobutane 54 is treated with CO2, ring expansion occurs to yield the six-membered iridaacetal 55.61 Interestingly, insertion of a single carbon unit into 51 can be achieved by treating 51 with tert-butylisocyanide, forming new C–X and C–Ir bonds in product 56.
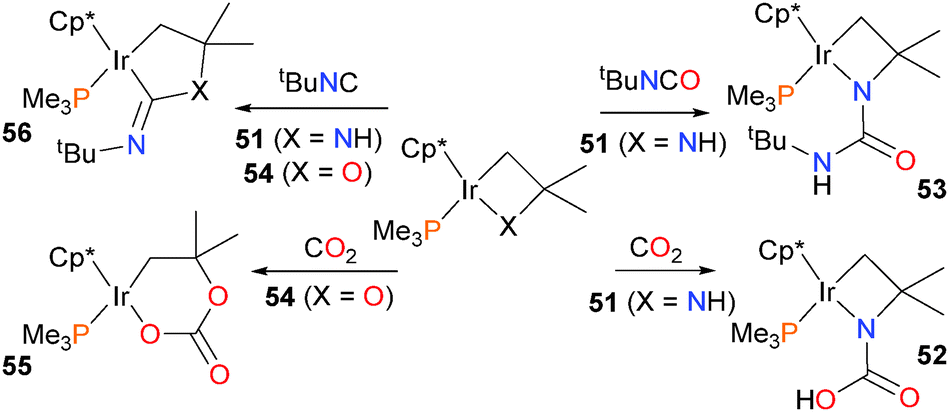 | ||
| Scheme 17 | ||
Iridium oxo complexes have also been shown to react with isocyanates.62 Treating iridium oxo dimer 57 with three equivalents of arylisocyanate gives the unusual, asymmetric product 58, which features two bridging imidos derived from the isocyanate as well as a bridging isocyanate (Scheme 18).
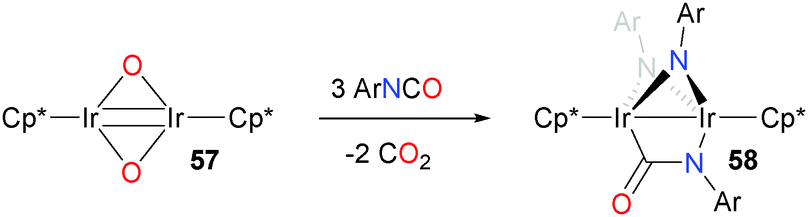 | ||
| Scheme 18 | ||
Glueck et al. have examined the reactivity of an unsaturated iridium imido towards a variety of C–N and C–O containing substrates (Scheme 19).63,64 When imido complex 59 was treated with two equivalents of CO or CNtBu, a three-membered metallacycle was formed via insertion of the Lewis basic ligand into the Ir![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond. Another equivalent of ligand also bonded to the metal centre to generate piano stool complexes 60 and 61, respectively. The imido ligand could also be cleaved from the metal centre via alkylation with an excess of MeI, generating the iodo dimer 62 and an equivalent of trimethylammonium iodide. Cyclization with CO2 produced the pseudo-two legged piano stool complex 63. Finally, imido 59 reacts with two equivalents of DMAD (dimethylacetylene dicarboxylate) to give an η4-pyrrole ligand in product 64. X-ray diffraction studies reveal that the iridium and nitrogen are no longer within bonding distance in this unusual sandwich complex. Interestingly, maleic anhydride reacts with 59 to yield 65, which was demonstrated by elemental analysis and mass spectrometry to be a 1
N bond. Another equivalent of ligand also bonded to the metal centre to generate piano stool complexes 60 and 61, respectively. The imido ligand could also be cleaved from the metal centre via alkylation with an excess of MeI, generating the iodo dimer 62 and an equivalent of trimethylammonium iodide. Cyclization with CO2 produced the pseudo-two legged piano stool complex 63. Finally, imido 59 reacts with two equivalents of DMAD (dimethylacetylene dicarboxylate) to give an η4-pyrrole ligand in product 64. X-ray diffraction studies reveal that the iridium and nitrogen are no longer within bonding distance in this unusual sandwich complex. Interestingly, maleic anhydride reacts with 59 to yield 65, which was demonstrated by elemental analysis and mass spectrometry to be a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct of 59 and the anhydride. Although NMR spectroscopy revealed that the olefin moiety was bound to the iridium in an η2-fashion, the authors conclude that 65 is likely an oligomer, based on the prohibitive strain of olefin binding in the monomeric form, as well as the low solubility of 65 in organic solvents. Reaction of 65 with CO yields complex 66, which is freely soluble and easily characterized by NMR spectroscopic methods.
:1 adduct of 59 and the anhydride. Although NMR spectroscopy revealed that the olefin moiety was bound to the iridium in an η2-fashion, the authors conclude that 65 is likely an oligomer, based on the prohibitive strain of olefin binding in the monomeric form, as well as the low solubility of 65 in organic solvents. Reaction of 65 with CO yields complex 66, which is freely soluble and easily characterized by NMR spectroscopic methods.
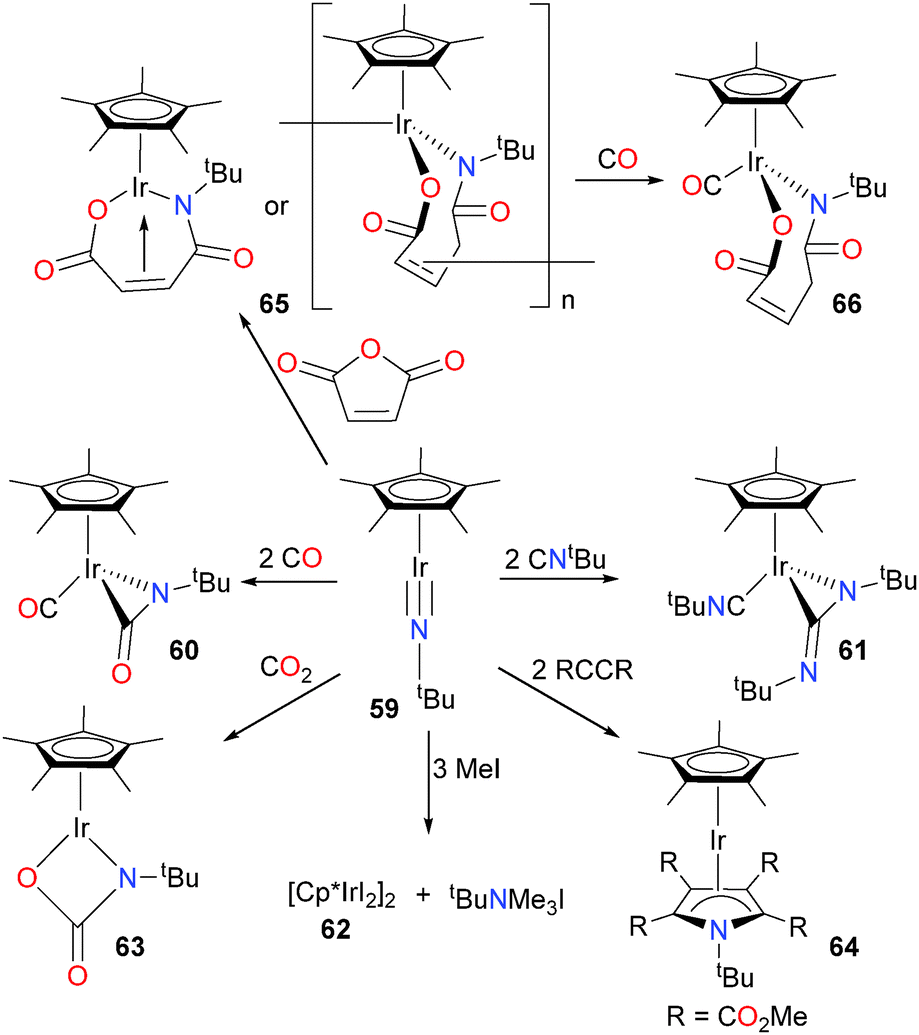 | ||
| Scheme 19 | ||
The Bergman group has also reported a more general synthetic route to the preparation of N-bound iridium carboxamide complexes 67via the reaction of arylnitriles with iridium hydroxide complex 68, as outlined in Scheme 20.65 This transformation was reliant on the use of an iridium triflate catalyst 69, which the authors propose serves to activate the nitrile towards nucleophilic attack from the iridium hydroxide complex (vide infra).66 The resulting carboxamide complexes can be protonolysed by strong acid to release the final amide product.
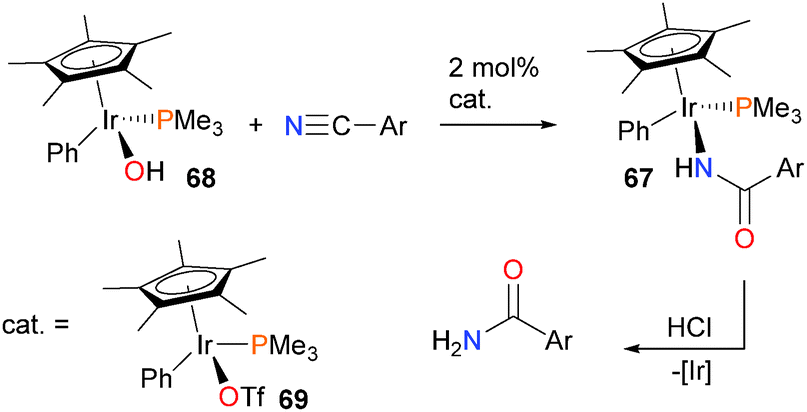 | ||
| Scheme 20 | ||
Iridium amido complex 70 has also been found to insert a variety of carbodiimides and isocyanates into an N–H bond rather than the Ir–N bond to form products of type 71 (Scheme 21).67
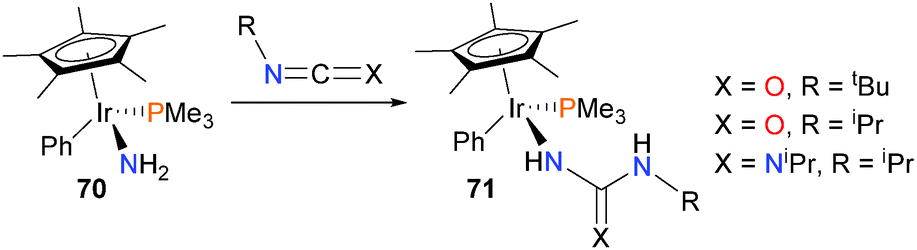 | ||
| Scheme 21 | ||
Holland and Bergman have reported that Cp*iridium guanidate complexes are excellent catalysts for carbodiimide metathesis,68 able to operate at much lower temperature than earlier reported systems. Initially, they noted that treating complex 72 with a large excess of carbodiimide led to the formation of iridium complex 73, as well as new carbodiimides (see Scheme 22). The authors also found that analogous reactivity could be achieved with isocyanates, and that stoichiometric experiments with p-tolNCO revealed the formation of six-membered metallacycle 74 in an equilibrium with starting material 72. Ring-expanded intermediate 74 could also be trapped with added PMe3. Ring-contraction from 74 results in the formation of iridium guanidate 75 and the release of carbodiimide. This mechanism of reversible, sequential ring-expansion and -contraction steps is notably different from traditional carbodiimide metathesis mechanisms, which typically proceed via [2+2] cycloaddition and cycloreversion steps.
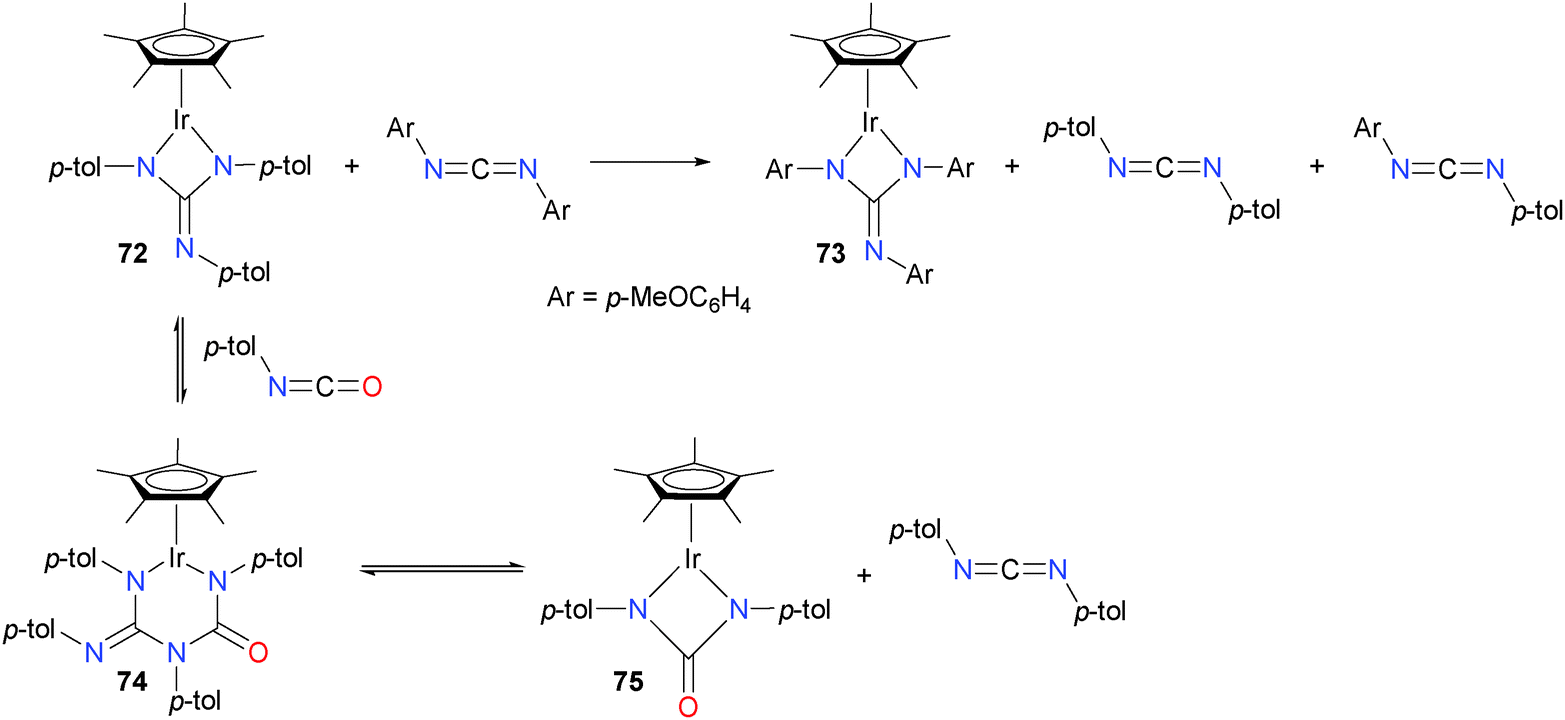 | ||
| Scheme 22 | ||
Zhao and Hartwig have shown that rhodium(I) iminyl complexes 76 undergo rare β-aryl eliminations when gently heated to yield nitriles and arylrhodium complexes such as 77.69 Mechanistic experiments found that the formation of products was inverse first-order in added phosphine, and that product conversion and rates of formation were unaffected by added nitrile. This is most consistent with a mechanism that requires dissociation of a PEt3 ligand from complex 76 to form intermediate 78 prior to β-aryl elimination to form 79. Coordination of phosphine would then form the observed arylrhodium complex 77 (Scheme 23). Notably, the authors found that both the steric and electronic features of the iminyl ligand dictated the reactivity towards β-aryl elimination. In a related paper, the same group also reported that rhodium(I) amidos can react with unactivated olefins to form imines, likely via olefin insertion into the Rh–N bond followed by β-H elimination.70
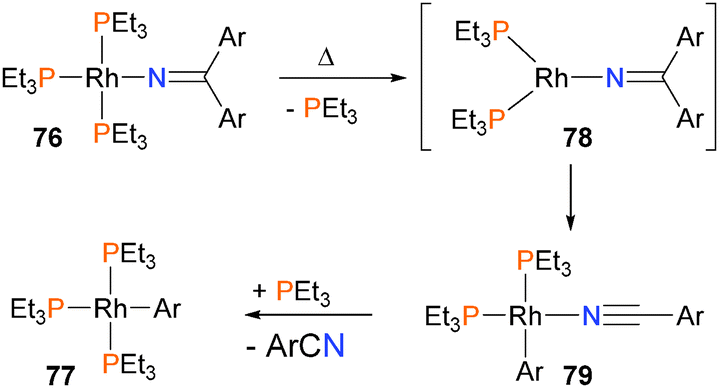 | ||
| Scheme 23 | ||
Tejel et al. have reported the preparation of binuclear rhodium imidos of the type 80, as outlined in Scheme 24.71 Treating 80 with CO results in an quick colour change from blue to green. The product of this reaction, complex 81, can be isolated as a crystalline solid and was fully characterized. It can be seen that two C–N bond forming events have occurred, likely via intermediate 82, resulting in the formation of a bridging ureylene ligand. In addition, another equivalent of CO has become ligated to one rhodium centre as a terminal ligand. Continued exposure of complex 81 to an atmosphere of CO results in a colour change from green to orange, with formation of both rhodium(I) complex 83 as well as ureylene-ligated rhodium(III) complex 84, likely formed via coordination of CO to the rhodium that already features a CO ligand followed by asymmetric cleavage of the Rh–Rh bond. Other insertions of CO into group 9 M–N bonds have been reported as well.72–75
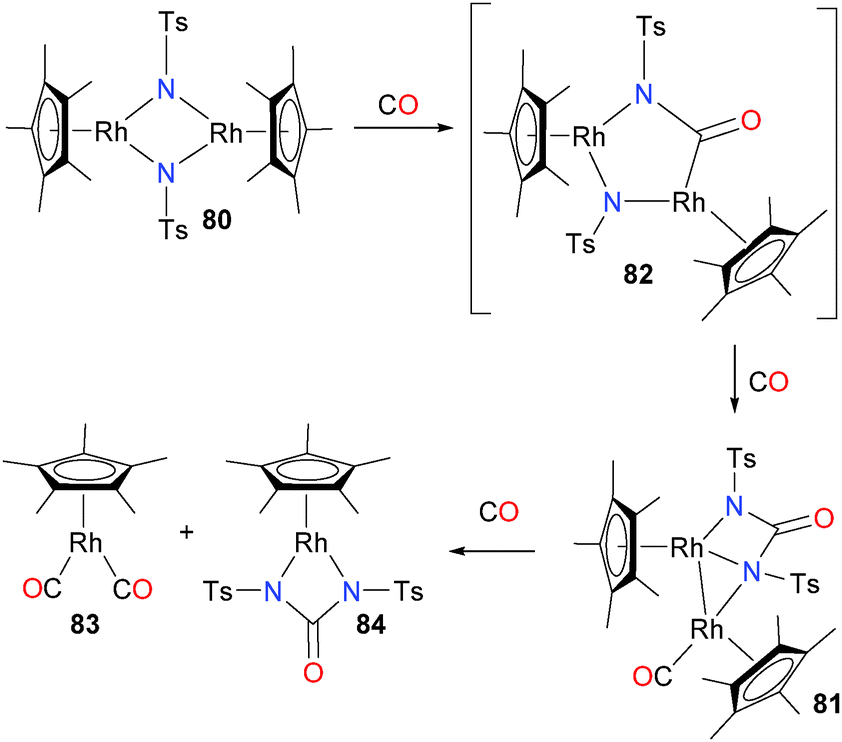 | ||
| Scheme 24 | ||
The group of Oro have prepared the amido-bridged trinuclear complex 85, shown in Scheme 25, and found that in the presence of added phosphine ligands, insertion of one of the olefin groups into the Ir–N bond occurs, giving three equivalents of mononuclear 86 as the final product in good yields.76 Theoretical calculations found that the barrier to intramolecular insertion via a four-membered transition state had a prohibitively high barrier of 30.9 kcal mol−1, inconsistent with the observed rapid reaction at room temperature. Thus, the authors examined an alternate intermolecular mechanism of insertion, where the nucleophilic amido stereoselectively attacks the olefin of another molecule of 85. This step occurs with a barrier of 14.4 kcal mol−1, which is more in line with the observed reaction rate. Subsequent insertion of the second olefin into the other amido occurs with a smaller barrier, and finally phosphine coordination to each metal gives two equivalents of the final product 86.
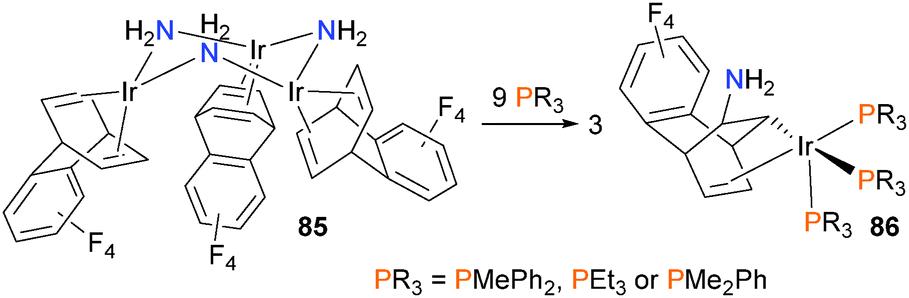 | ||
| Scheme 25 | ||
The Chang group has recently reported the catalytic amination of phenylpyridine77 and related compounds using Cp*rhodium complex 87 in the presence of a silver additive.78 Mechanistic studies, including the isolation and full characterization of intermediate 88, were performed, and they favour a pathway that involves formation of a discrete rhodium–nitrene complex (Scheme 26) that subsequently inserts into the Rh–C bond of the metalated phenylpyridine ligand. Complex 88 can be protonated with strong acids to release the amine product. Moreover, if additional phenylpyridine is used as the proton source, catalytic turnover can be achieved in good TONs. More broadly, it was found that this system is amenable to a wide variety of azides as well as phenylpyridine derivatives, making it synthetically appealing as an amination reaction.
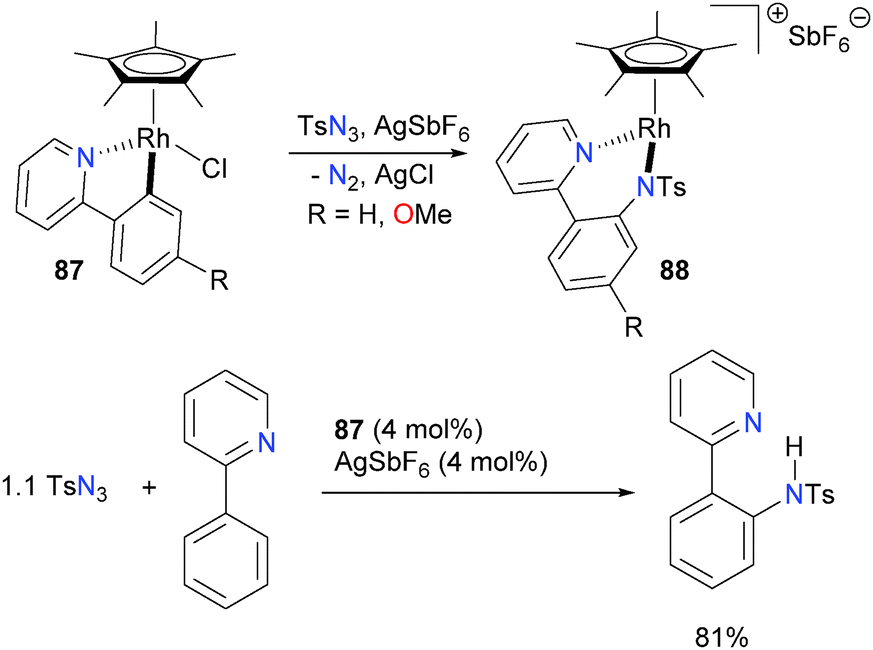 | ||
| Scheme 26 | ||
In 2012, the Love group found that azametallacyclobutanes 89 and 90 could be prepared by reacting ethylene complex 91 with the N-based oxidant PhINTs, as shown in Scheme 27.79 Interestingly, both isomers of the azametallocyclobutane were found to form on treatment of 91 with PhINTs in dichloromethane as solvent. This is in contrast to analogous work on the synthesis of rhodaoxetanes from 91 (vide infra).80 Separation of the two isomers was possible by HPLC, and although the azametallacyclobutanes were also found to resistant to crystallization, they could be rigorously characterized using 2D NMR techniques as well as mass spectrometry. Notably, it was found that the ratio of isomer formation was found to be heavily dependent on solvent choice, as use of protic solvents (MeOD, CD2Cl2/MeOD or D2O) resulted in the formation of only isomer 89.81 In addition, it was also found that the two isomers do not interconvert, even upon heating in DMSO for 3 days. A later study by the same group found that altering the nitrene precursor used for the oxidation also affected the distribution of isomers formed, as use of p-toluenesulfonyl azide in place of PhINTs formed exclusively the isomer 90, albeit in low yield (18% by 1H NMR spectroscopy).81
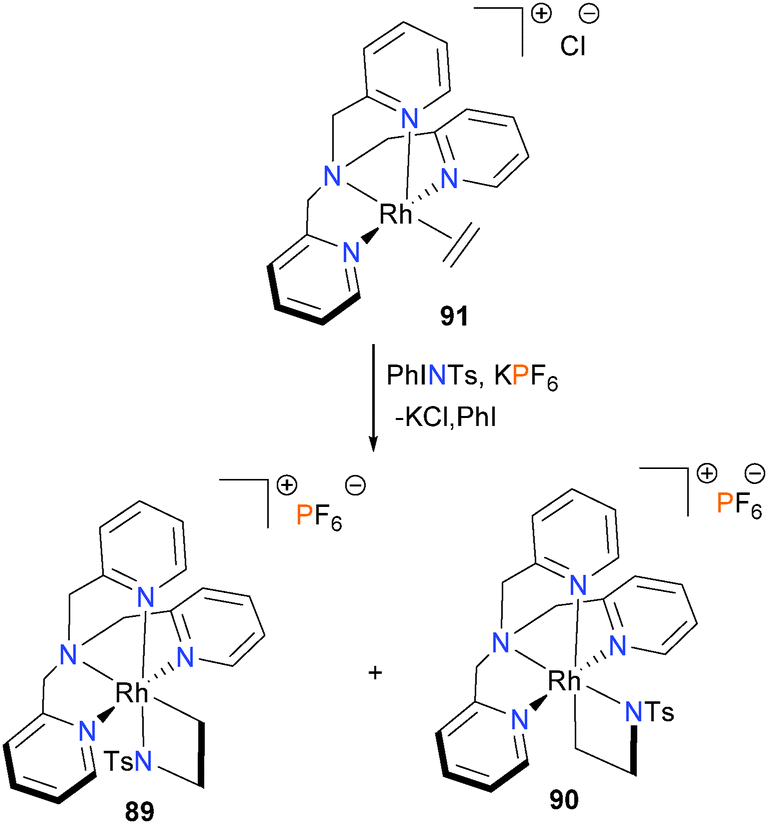 | ||
| Scheme 27 | ||
Shortly thereafter, Love and co-workers described the formation of substituted oxa- and azametallacyclobutane complexes of rhodium, prepared by substituting the ethylene ligand of complex 91 with higher olefins prior to oxidation with either H2O2 or PhINTs.82 Not surprisingly, multiple possible isomers were formed, with steric factors playing an important role in both the distribution of olefin complexes and their oxidation to form rhodaheterocyclobutanes.
Rhodium–ethylene complex 91 was also found to be able to undergo insertion with diazo compounds to form five-membered 3-rhoda-1,2-diazacyclopentanes such as 92.83 These unusual five-membered metallacycles were characterized by detailed NMR spectroscopic experiments, mass spectrometry and density functional theory (DFT) modeling. In most of the cases examined, high barriers to metallacycle inversion led to complexes that displayed low (i.e. C1) symmetry in solution, which rendered NMR spectral analysis non-trivial. However, the rhodacycle connectivity was unambiguously determined with the use of long-range 1H–15N HMBC experiments. Attempts to deprotect 92 with HOTs led to protonation of first the Rh–N bond, followed by a second, slower protonation of the Rh–C bond. In contrast, the Boc-substituted complex could be deprotected using an excess of trifluoroacetic acid to yield complex 93. Subsequent acylation with excess acetyl chloride yielded metallacycle 94, as outlined in Scheme 28.
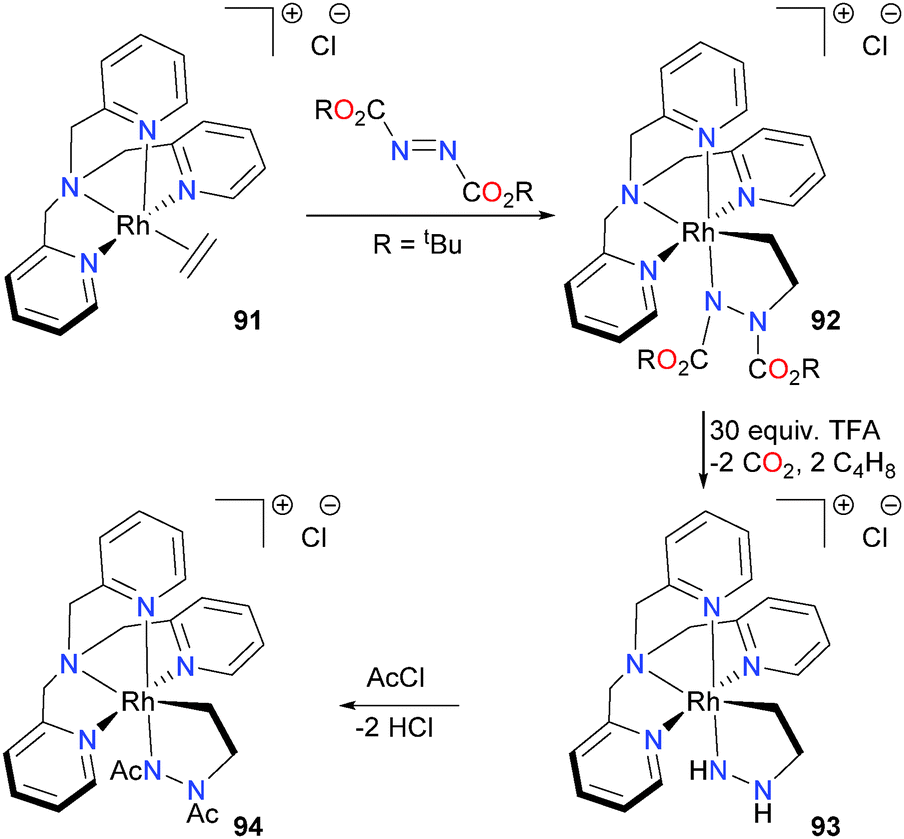 | ||
| Scheme 28 | ||
Braun and co-workers have recently disclosed that a rhodium(I) boryl complex 95, as shown in Scheme 29, is capable of inducing C–N bond cleavage of phenylisocyanate,84 resulting in the formation of rhodium–carbonyl complex 96 in 90% yield by NMR spectroscopy. Complex 96 was also characterized by X-ray diffraction studies and IR analysis. Complex 95 has also been shown to interact with both ketones and imines to generate reduced η3-type anionic ligands.85
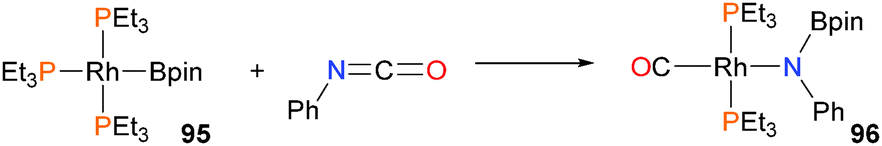 | ||
| Scheme 29 | ||
Insertions of CO86 and CO287 into M–N bonds have also been reported recently with rhodium–silylamide complexes. Treatment of complex 97 with CO led to the formation of the rhodium–cyanide complex 98. Labelling experiments showed that the carbon of the CN ligand is derived from CO. In addition, hexamethyldisiloxane was identified in the reaction mixture. Reacting 97 with COS resulted in a rapid colour change from dark green to red, and an X-ray diffraction experiment on the crystalline product obtained revealed the formation of complex 99, which features an unusual S-containing derivative of the common acetylacetonate ligand. Reducing the pressure of COS during this reaction allowed for the identification of an intermediate species by NMR, assigned as the O,S-chelating complex 100. Subsequent rapid silyl migration, insertion of another equivalent of COS and a second silyl migration then afford the final product, as shown in Scheme 30.
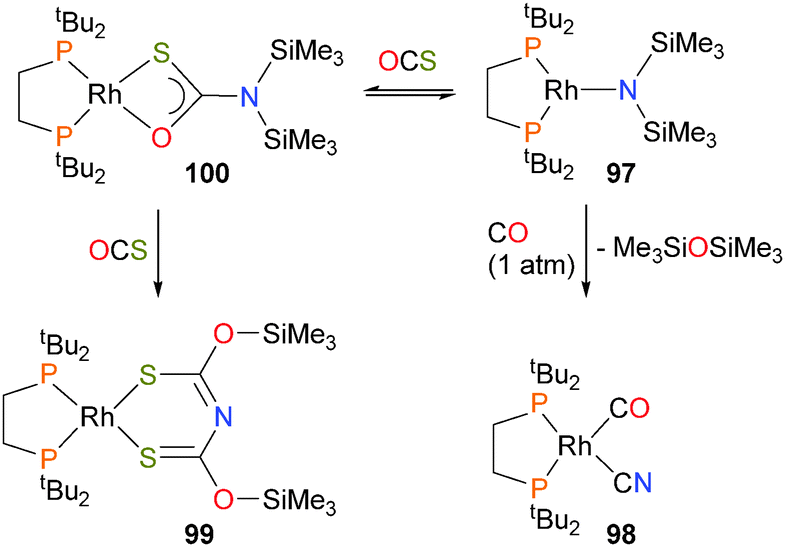 | ||
| Scheme 30 | ||
The Ozerov group has found that rhodium and iridium complexes are capable of reacting with pincer ligand precursors via oxidative addition of the N–Me bond to form complex 101 (Scheme 31).88 When rhodium is used as the metal centre, an intermediate rhodium(I) complex 102 can be observed by NMR spectroscopy prior to oxidative addition. Subsequent work found that tethering the aromatic rings of the PNP ligand precursor could be used to improve the C–N oxidative addition step.89,90
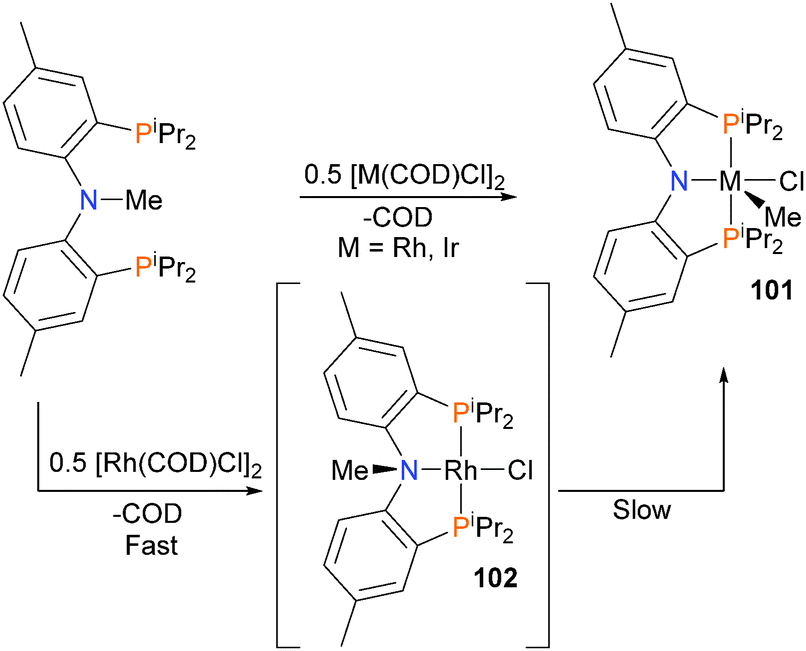 | ||
| Scheme 31 | ||
Group 10 metals
Hartwig–Buchwald amination is arguably one of the most useful synthetic developments of the last two decades, and as such, its numerous applications have been reviewed many times.91 Here, we wish to highlight early studies from the Hartwig group that unequivocally demonstrated that C–N reductive elimination from palladium(II) amido complexes.Inspired by a report from Boncella and co-workers,92 in a seminal early contribution, Driver and Hartwig describe the synthesis and characterization of monomeric and dimeric palladium(II) amido complexes such as 103, and observed C–N bond formation via reductive elimination at room temperature in high yield (92% yield after 1 hour) when 103 was treated with triphenylphosphine (Scheme 32).93
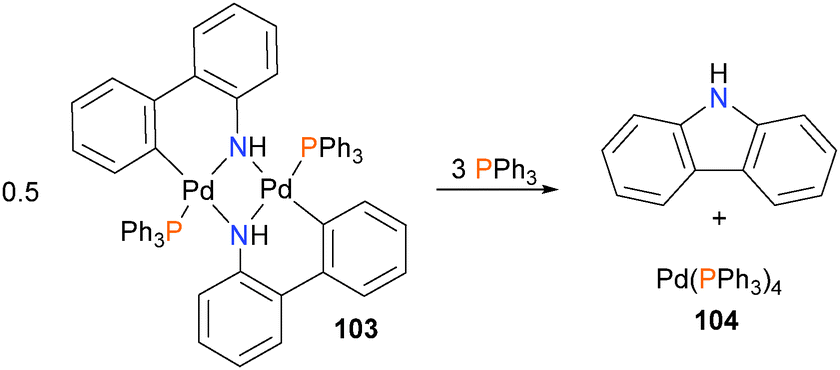 | ||
| Scheme 32 | ||
Subsequent catalyst optimization found that bidentate phosphines, such as DPPF94–96 and DPPBz97 are excellent ligands for both stoichiometric and catalytic studies of C–N bond formation. The scope of C–N reductive elimination from palladium(II) was later extended to iminyl,98 hydrazonato,99 and amidate complexes.100,101 Csp3–N bond formation was also found to be catalytically feasible with bisphosphine ligands.102
The Hartwig group has also recently reported Csp3–N reductive elimination from NHC complexes of palladium (Scheme 33).103 Treating norbornylpalladium complex 105 with free SIPr and heating the reaction mixture results in formation of arylamine and bisNHC complex 106 following reductive elimination. The stereochemistry of the product, as well as DFT calculations, indicate that the reductive elimination is concerted.
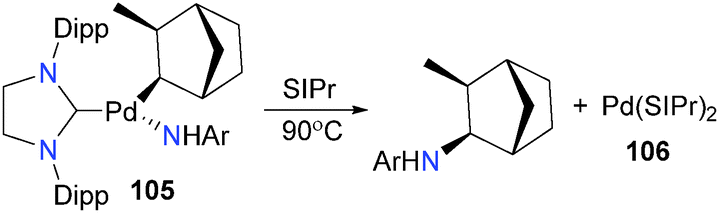 | ||
| Scheme 33 | ||
Oxidatively-induced reductive elimination has been observed for nickel alkyl–amido complexes, as shown in Scheme 34.104 For example, treatment of complex 107 with phenylazide gave complex 108. Complex 108 was found to be thermally unstable, reductively eliminating to form a C–N bond when stored at room temperature over the course of 2 days. This process could be greatly accelerated by adding an oxidant, either in the form of I2, O2, or the single-electron oxidant acetyl ferrocenium [(AcCp)2Fe+]. Koo and Hillhouse demonstrated that this process could be used for the nickel-mediated synthesis of a variety of indolines via the coupling of phenethyl Grignard reagents and arylazides.105 These reactions were not limited to cyclic species, as untethered alkyl–amido complexes of nickel were found to undergo similar reactivity when exposed to the same group of oxidants.104 In the following year, the Hillhouse group also communicated the analogous reactivity of bipyridyl-ligated complexes of nickel with arylazides.106
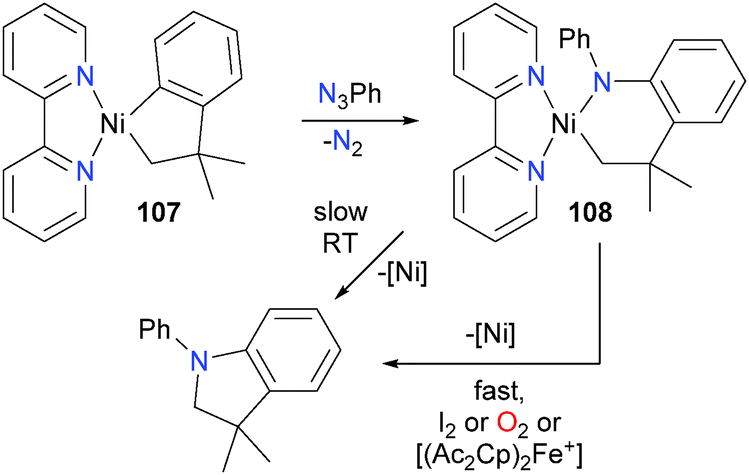 | ||
| Scheme 34 | ||
Boncella and co-workers have disclosed the synthesis of mesitylnickel(II) amido complexes, as well as their reactivity with organic electrophiles, as shown in Scheme 35.107 Complex 109 displays preferential reactivity at the Ni–N bond rather than the Ni–C bond in all the cases examined. Reacting amido 109 with CO2 results in the formation of carbamate complex 110. Interestingly, when allowed to react with diphenylketene, on the other hand, addition of the N–H bond is observed to occur across the CC moiety rather than the CO bond, yielding product 111. Similarly, treating 109 with isocyanates gives 112via preferential addition of the N–H bond across the CN rather than the CO group. Finally, insertion into the Ni–N bond can be induced by treating 109 with DMAD, yielding alkenyl complex 113, the Z-stereochemistry of which was confirmed by selective NOE experiments. The reactivity observed is analogous to that for the palladium congener of 109.108,109
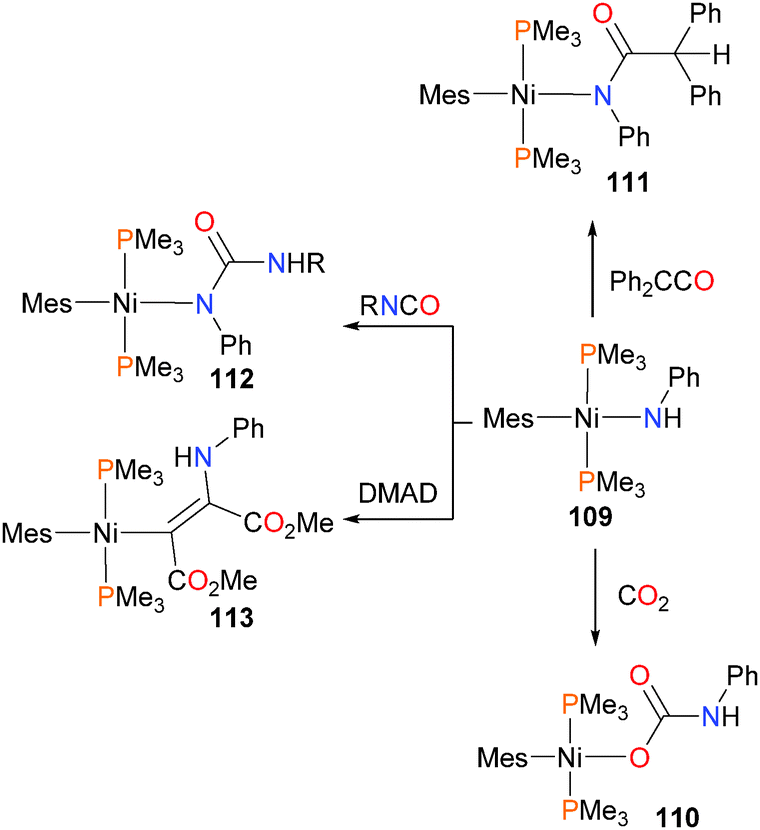 | ||
| Scheme 35 | ||
Holland et al. have also described the synthesis of nickel(II) amido complexes. They report that treatment of Cp*Ni(acac) and related derivatives with LiNH-p-tol is a good synthetic route to bimetallic complex 114.110 Reacting 114 with an excess of either CO or tBuNC breaks up the dimeric structure, resulting in carboxamide complex 115 or amidinyl complex 116, respectively. Both products also feature an additional equivalent of Lewis basic ligand coordinated to the nickel centre. Carboxamide 115, although it could be isolated pure in moderate yield, was found to readily decompose at room temperature in both solution and the solid-state to generate bridging carbonyl complex 117 as the main nickel-containing species, as well as a urea as the main organic product (see Scheme 36). CO insertion into group 10 amido complexes have been reported by others.111
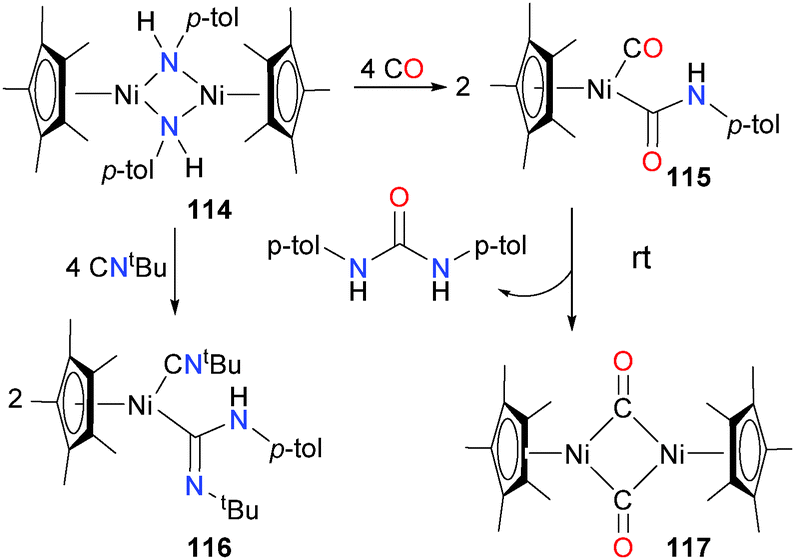 | ||
| Scheme 36 | ||
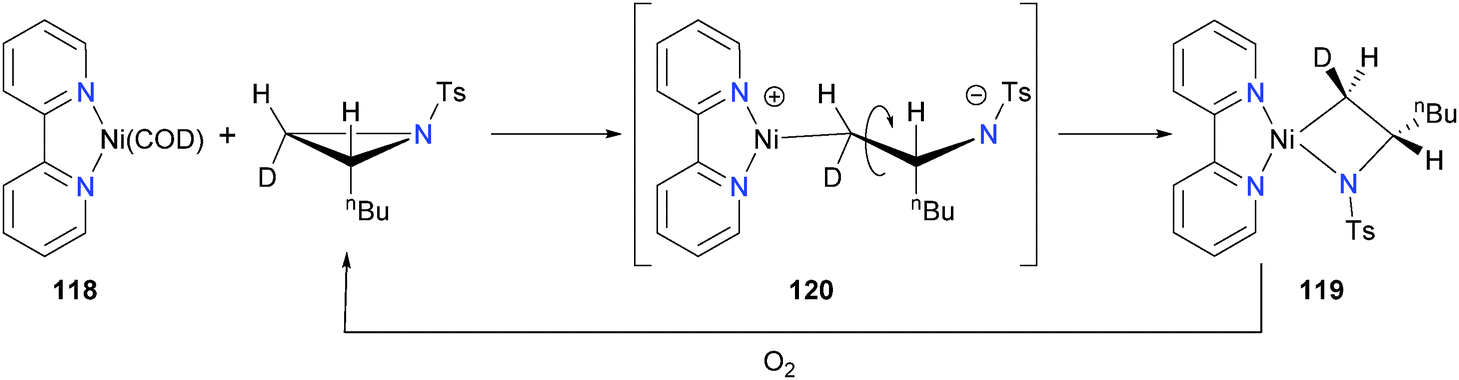
In 2002, Lin et al. reported a study of 118 with aziridines to yield azanickelacyclobutanes 119.112 Elegant use of deuterium labeling on the aziridine substrate demonstrated that this oxidative addition occurred with inversion of the stereochemical configuration at the methylene carbon, consistent with an SN2-type oxidative addition mechanism. Thus, the authors proposed that the azanickelacyclobutane product is formed via SN2-type ring-opening to give zwitterionic intermediate 120, which upon C–C rotation can close via attack on the tosyl-stabilized amide on the cationic metal centre (see Scheme 37). Curiously, upon oxidatively-induced reductive elimination from complex 119, the syn-aziridine is regenerated in high yield, indicating that the reductive elimination also proceeds with inversion of stereochemistry. The nickel species after reductive elimination was not characterized. Notably, the groups of Doyle113 and Jamison114 have since built on this fundamental stoichiometric study to develop new catalytic cross-coupling reactions. More recently, Xi and co-workers have prepared a related azanickelacyclobutane by reaction of IPr and Ni(COD)2 with 2,6-diazasemibulvalene.115
 | ||
| Scheme 37 | ||
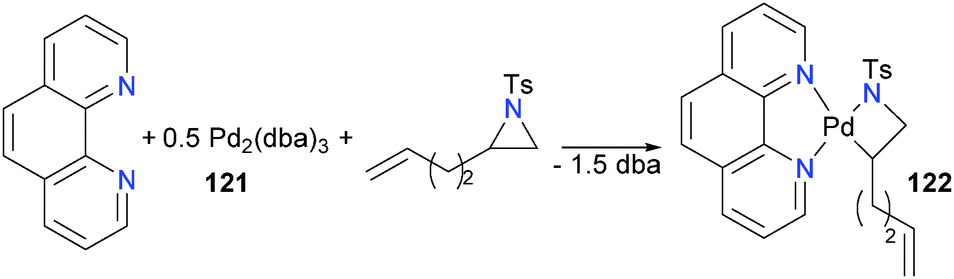
Ney and Wolfe have also reported the synthesis of a group 10 azametallacyclobutane. Mixing Pd2(dba)3 (121), phenanthroline and an aziridine formed palladacycle 122 in 45% isolated yield (Scheme 38).116 The presence of the pendant olefin was found to be important, with the authors proposing it serves as a directing group for the palladium(0) prior to oxidative addition. It should be noted, however, that using a more reactive nosyl-substituted aziridine obviated the need for the olefin tail. Similar to Hillhouse's system, deuterium labelling studies found that the oxidative addition proceeds with inversion of stereochemical configuration.
 | ||
| Scheme 38 | ||
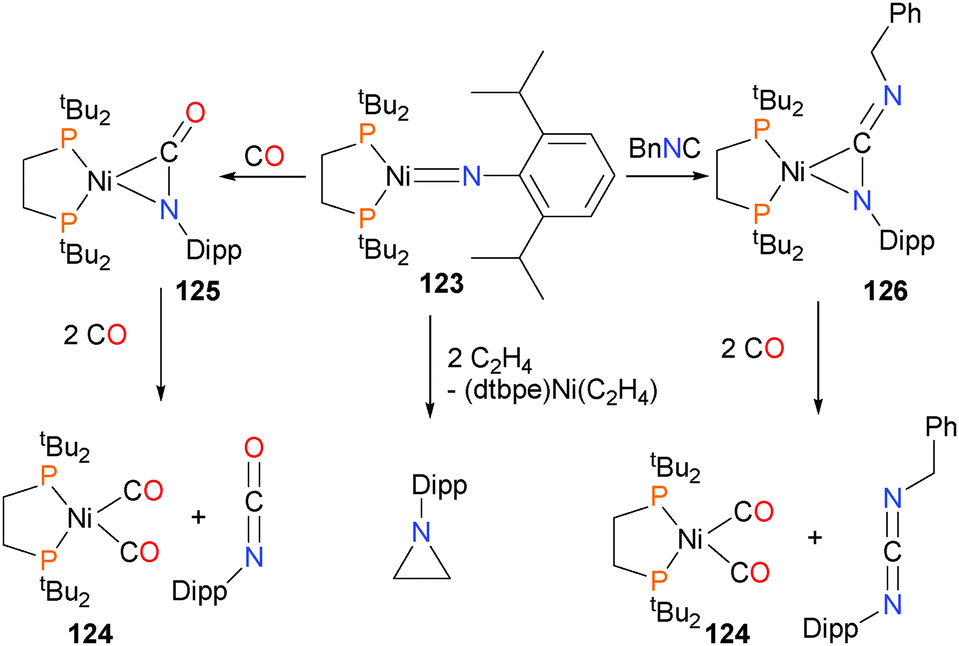
Mindiola and Hillhouse were the first to report the synthesis and structure of a nickel imido complex, 123.117 Shortly thereafter, reactivity studies found that the imido group could be coupled to CO to generate an isocyanate and carbonyl complex 124via complex 125. In addition, the imido could also be coupled to benzyl isocyanide to generate the η2-bound carbodiimide complex 126, which was characterized by X-ray diffraction studies. Exposure of 126 to an atmosphere of CO gas caused the release of the organic carbodiimide fragment, concomitant with the formation of complex 124.118 Shortly thereafter, Waterman et al. reported that complex 123 could also be used to aziridinate ethylene (Scheme 39). Interestingly, deuterium labeling studies demonstrated that this aziridination reaction occurred with retention of stereochemistry.119 A more detailed study on the reactivity of 123 was published by Mindiola et al. in 2014.120
 | ||
| Scheme 39 | ||
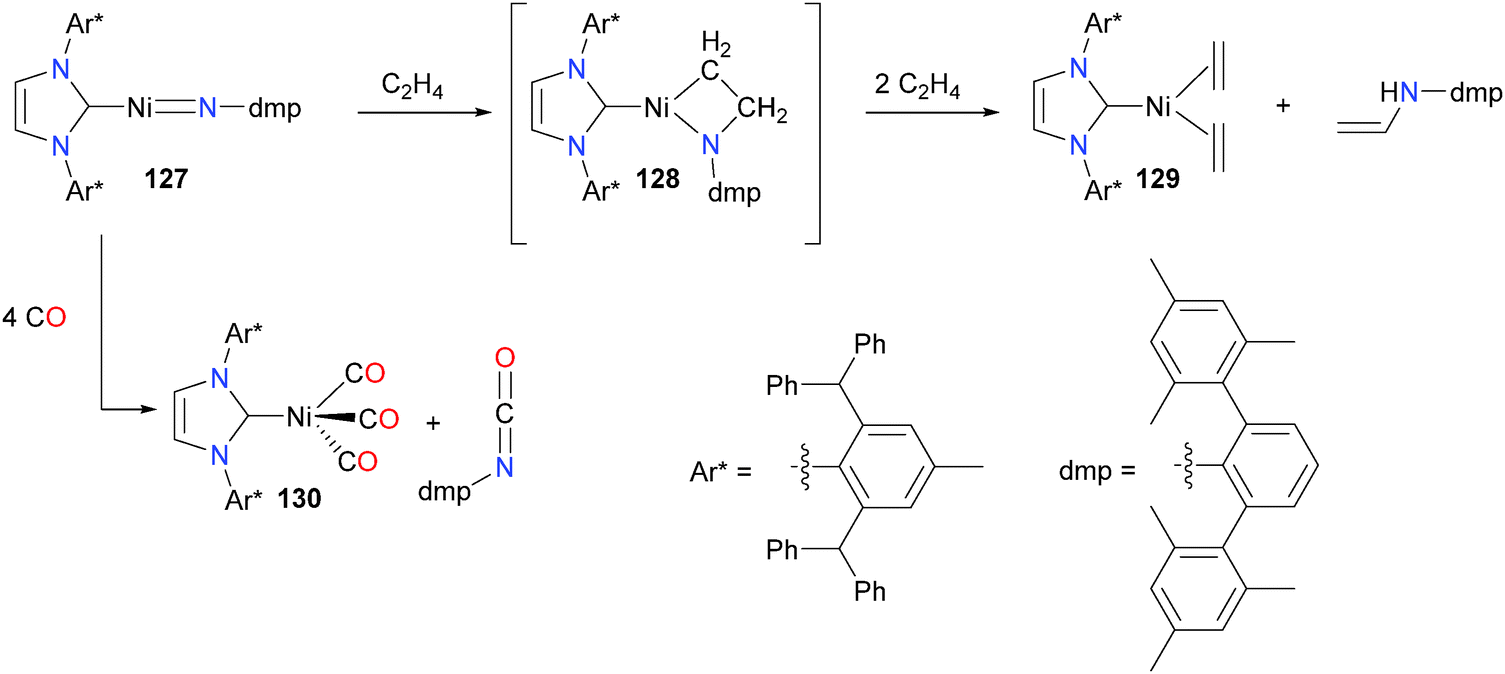
Extremely bulky NHCs have recently received much attention due to their ability to kinetically stabilize otherwise reactive moieties. An elegant example of this is the preparation of nickel nitrene complex 127, which features both very bulky Ar* groups on the N atoms of the NHC as well as large mesityl substituents at the ortho positions of the imido aryl group (Scheme 40).121 Treatment of 127 with ethylene at room temperature yielded the azametallacyclobutane 128, which was characterized by NMR spectroscopic experiments but could not be isolated. Over the course of 12 hours under an atmosphere of ethylene, 128 yielded complex 129 and a vinylamine. The authors propose that the amine is formed via either a 1,2-hydride shift or β-H elimination from 128 and subsequent N–H reductive elimination. The imido could also be coupled to CO to generate an isocyanate and nickel(0) complex 130.
 | ||
| Scheme 40 | ||
Other NHC ligands can also be useful for the preparation of isocyanates via the coupling of a imido group and CO.122 For instance, treating nickel complex 131 with MesN3 yields the bridging imido complex 132, which releases mesityl isocyanate when reacted with CO. Attempts to render this transformation catalytic were hampered by the rapid disproportionation of 131 under an atmosphere of CO. This decomposition pathway can be circumvented somewhat via chloride abstraction from 132 with NaBArF4 to yield the bridging chloride dimer 133, which is a competent catalyst for the formation of MesNCO from MesN3 and CO. Using 10 mol% 133 as a catalyst gave MesNCO in 52% yield after 6 hours at room temperature (Scheme 41).
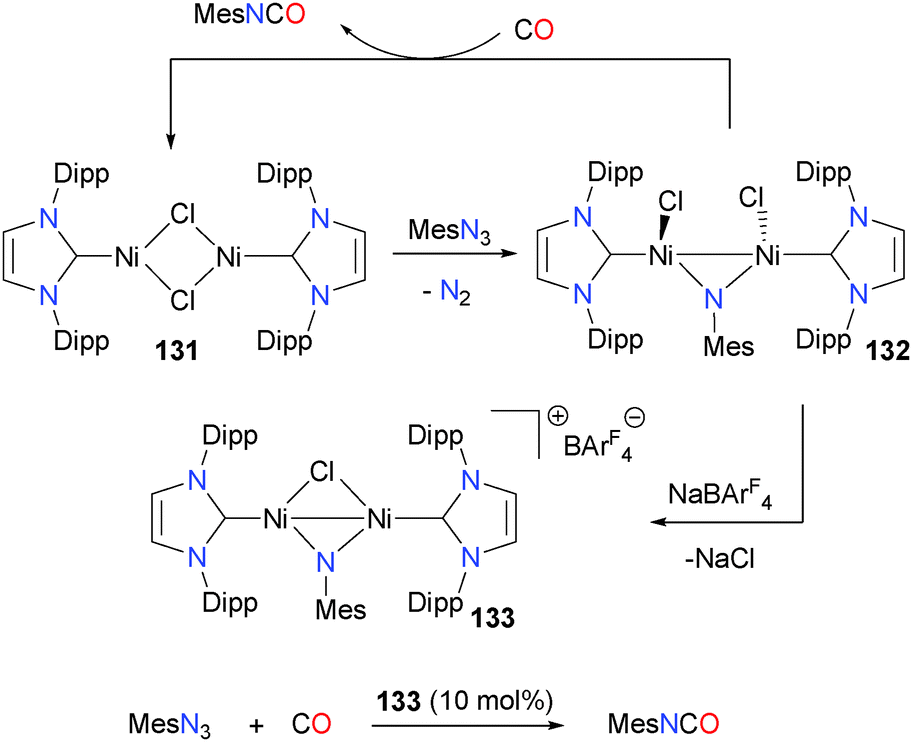 | ||
| Scheme 41 | ||
Bidentate NHCs have also been reported to stabilize nickel imidos.123 In fact, treatment of the chelating NHC complex 134 with Cp2FeB(C6F5)4 results in ligand-based oxidation to generate a carbon-centred radical species 135, which can undergo C–C coupling with another equivalent of itself to form binuclear quinoneimine complex 136, as outlined in Scheme 42. These Gomberg-type couplings of nitrogen-based radicals that are delocalized across an aromatic ring have also been reported for copper124–126 and rhodium.127 Bai and Stephan have also published related chemistry using a nickel(I) synthon with aryl azides.128 Other groups have also observed C–N bond formation upon treating group 10 complexes with nitrene precursors.129
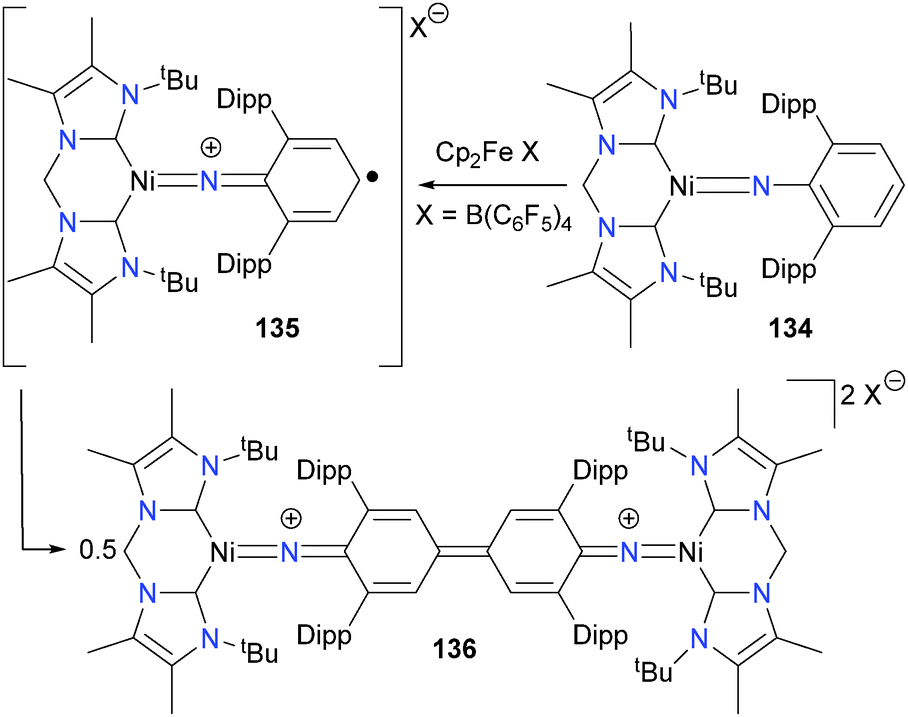 | ||
| Scheme 42 | ||
The Goldberg group reported the first example of directly-observed Csp3–N reductive elimination from a high-valent, late transition metal complex in 2007.130 Mechanistic studies indicate that upon heating, complex 137 undergoes sulphonamide dissociation to generate cationic, five-coordinate intermediate 138 (Scheme 43). Nucleophilic attack of sulphonamide anion on the electrophilic methyl group is the key C–N bond forming event, which reduces the platinum and releases the amide product along with complex 139. An analogous mechanism for C–O bond formation has also been proposed (vide infra). Others have since reported related studies of C–N bond formation.131,132
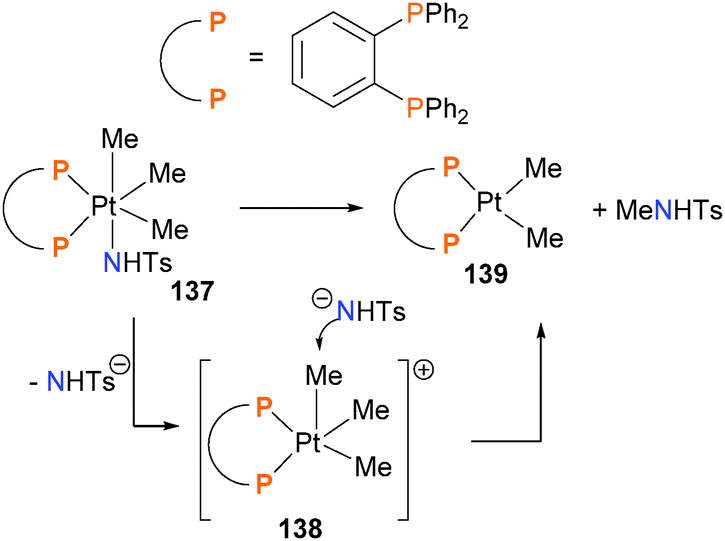 | ||
| Scheme 43 | ||
Ohashi et al. have explored the scope of nickel(0)-catalyzed dehydrogenative coupling of nitriles and 1,3-diynes to give substituted pyridines (see Scheme 44).133 Interestingly, they isolated an intermediate during catalysis that was identified by spectroscopic and X-ray diffraction methods as the η3-allyl complex 140, derived from oxidative cyclization of dienes and a nitrile from intermediate 141. Proton transfer yields intermediate 142, and subsequent reductive elimination was found to give a mixture of 1,2- and 2,5-dihydropyridines (not shown), which upon heating released dihydrogen to generate the final aromatic product. Related work from the same group has recently been reviewed.134
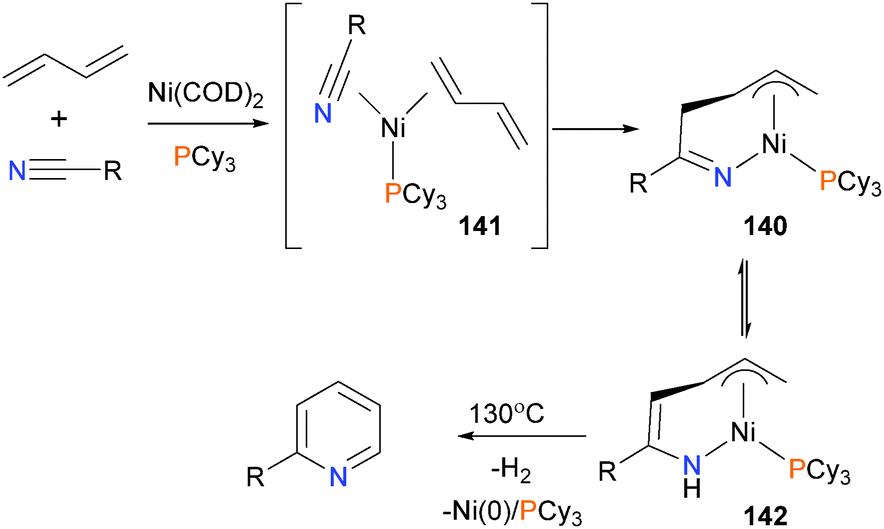 | ||
| Scheme 44 | ||
The surprisingly thermally robust azide complex 143, when treated with 5 bars of CO, gave the isocyanate complex 144 in good yield (Scheme 45).135 Complex 144 could also be prepared by independent synthesis. The reaction occurred both in the presence and absence of light, which implies the mechanism proceeds via a direct nucleophilic attack of the CO (likely first coordinated to the nickel centre) onto the azide nitrogen rather than the in situ generation of a nitrene, which would subsequently couple with CO.
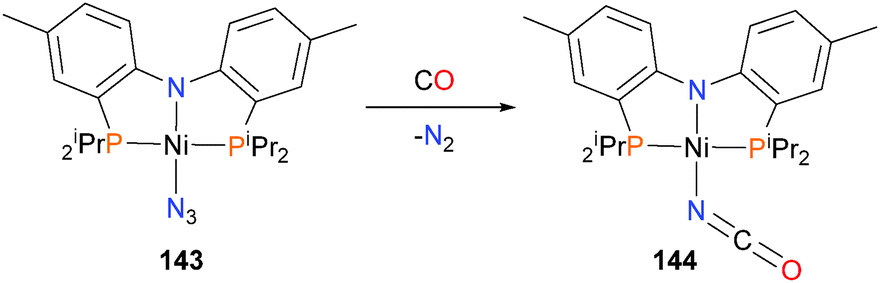 | ||
| Scheme 45 | ||
The Huang group has published work on the catalytic vinylation of aminals using various palladium catalysts.136 After catalyst optimization, they found that complex 145 provided the best yields of product under catalytic conditions. Reacting the catalyst precursor with the aminal substrate in stoichiometric amounts resulted in the formation of palladacycle 146 in high (94%) yield (Scheme 46). Complex 146 was fully characterized, and is the result of oxidative addition of the aminal C–N bond onto palladium(0), in this case likely formed by reduction of the palladium precursor with the solvent iPrOH. Oxidative addition of C–N bonds has been reported with other diphosphine complexes of group 10 metals.137,138
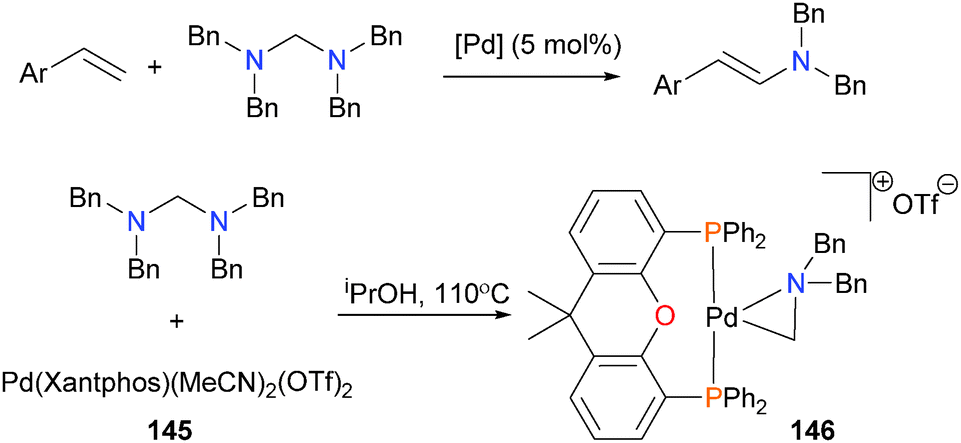 | ||
| Scheme 46 | ||
Miyazaki et al. have shown the oxidative addition of palladium(0) onto the N–C bond of cyanoformamides in the presence of Lewis acidic boranes, and found that the resulting palladium(II) complex can be used for catalytic aminocyanation of alkenes (Scheme 47).139 Complex 147 was characterized by NMR spectroscopy and X-ray diffraction studies. Borane-assisted oxidative addition reactions with nickel have also been reported.140
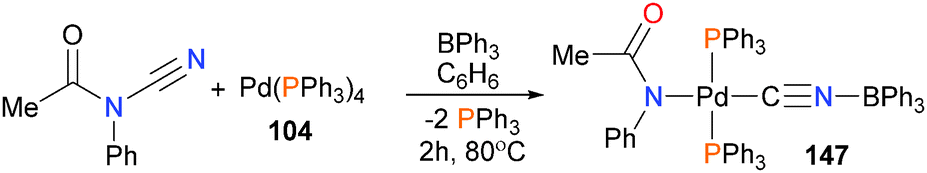 | ||
| Scheme 47 | ||
In 2006, Forrest and Michael reported an example of room temperature hydroamination of unactivated alkenes using PNP-bound palladium complex 148 as a catalyst.141 Several years later, the same authors reported more detailed mechanistic studies, including the X-ray crystal structure of acid-sensitive alkyl complex 149, which is a catalytic intermediate (Scheme 48).142 Related PNP–olefin complexes have also been demonstrated to be susceptible to C–N bond formation when treated with amines as nucleophiles.143,144 Later, intra-145 and intermolecular146,147 hydroamination was reported using platinum complexes.
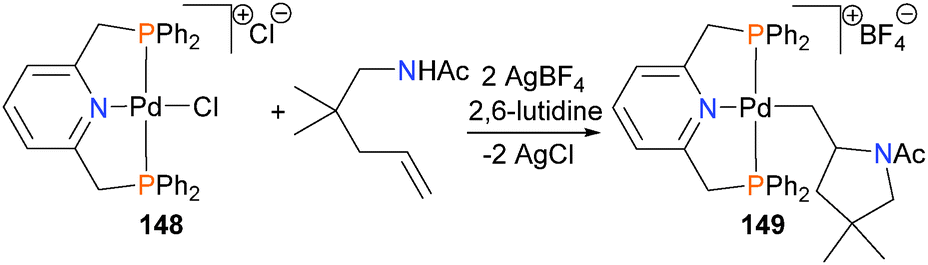 | ||
| Scheme 48 | ||
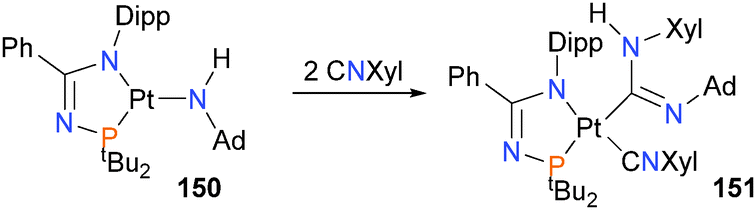
Kelly et al. have recently published a report on the reactivity of a three-coordinate platinum(II) amido complex 150,148 describing its insertion reactivity with an isocyanide to form complex 151, as shown in Scheme 49. Palladium(II) imidoyl complexes have also been found to react similarly with isocyanates.149
 | ||
| Scheme 49 | ||
Group 11 and 12 metals
Gunnoe and co-workers have prepared monomeric amido complexes of copper(I), such as complex 152, and found that they can be used to form new C–N bonds when treated with acrylonitrile,150 either EtBr or EtI as alkylating agents or with the trityl cation (Scheme 50).151,152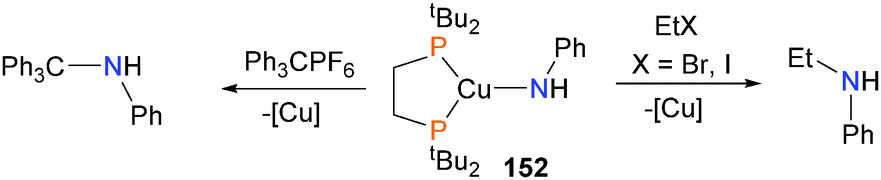 | ||
| Scheme 50 | ||
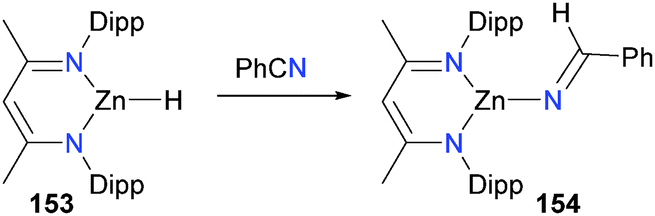
In a study examining the hydrosilation of ketones and nitriles, Boone et al. found that zinc hydride 153 reacted with benzonitrile to yield iminyl complex 154, formed via hydride insertion into the CN triple bond (Scheme 51).153 Curiously, the reaction was found to be second order in concentration of 153, leading the authors to propose an unusual mechanism of insertion whereby nitrile first coordinates to the metal centre of 153, and then another molecule of 153 attacks the bound nitrile, releasing 153 and generating the observed product via a six-membered transition state.
 | ||
| Scheme 51 | ||
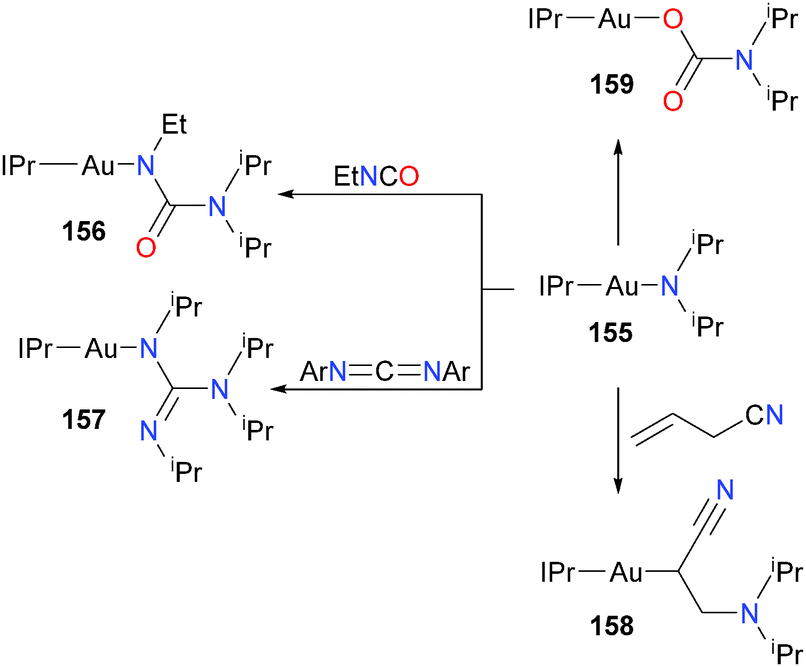
Gold amidos have been proposed as intermediates in hydroamination of alkenes and alkynes. In the first report of well-defined gold(I) amido complexes, Johnson et al. describe the reactivity of complex 155 with an array of organic electrophiles, as outlined in Scheme 52.154 Notably, 155 did not effect the amination of either diphenylacetylene or 2-hexyne, arguing against the involvement of gold amidos in hydroamination catalysis.
 | ||
| Scheme 52 | ||
Summary
Group 8 have been shown to undergo a variety of insertions reactions to form ring-expanded metallacycles. Iron in particular is an excellent platform for the formation of C–N bonds via radical processes, while ruthenium has been shown to be amenable to classic two-electron oxidative addition chemistry with aromatic Csp2–N bonds. While both iron and ruthenium nitride complexes tend to add to alkenes to form aziridines, osmium nitride complexes tend to undergo either Csp2–Csp2 bond cleavage (i.e. the conversion of complex 38 to complex 39) or [4+1] cyclizations.Iridium and rhodium amido complexes rapidly undergo insertion of π-bonds, typically via nucleophilic attack of the donor atom onto the electrophilic unsaturated compound. Both π-bound olefin and cyclometallated complexes of rhodium form Csp2–N and Csp3–N bonds when treated with nitrene reagents such as PhINTs and azides. In some cases, Csp2–N cleavage can be induced by reaction with a boryl group (i.e. the conversion of 95 to 96) or by tethering the amine to the metal using directing groups.
Hartwig–Buchwald amination has been proven to be an incredibly versatile method for the formation of Csp2–N bonds via reductive elimination from palladium(II) aryl–amido complexes. In contrast, related nickel complexes typically require oxidation to nickel(III) before undergoing Csp2–N or Csp3–N reductive elimination. In many cases, C–N bond formation proceeds through attack of an external nucleophile at the electrophilic donor atom.
Lastly, group 11 and 12 amido complexes are rare, and while they can be alkylated with alkyl halides or can insert heterocumulenes, they tend not to add directly to alkenes and alkynes, as demonstrated by Bergman and co-workers.154
C–O bond cleavage and formation
Group 8 metals
Hartwig et al. synthesized the ruthenaoxetane 160155,156 and reported its contrasting reactivity157 compared to known, related iridium compounds.61 For instance, it was found that 160 extruded α-methylstyrene when treated with either CO2 or benzaldehyde (Scheme 53). In the case of the former, the bicarbonate complex 161 was obtained in 83% isolated yield. In the case of the latter, the hydrido benzoate complex 162 was formed in 53% yield by 1H NMR spectroscopy.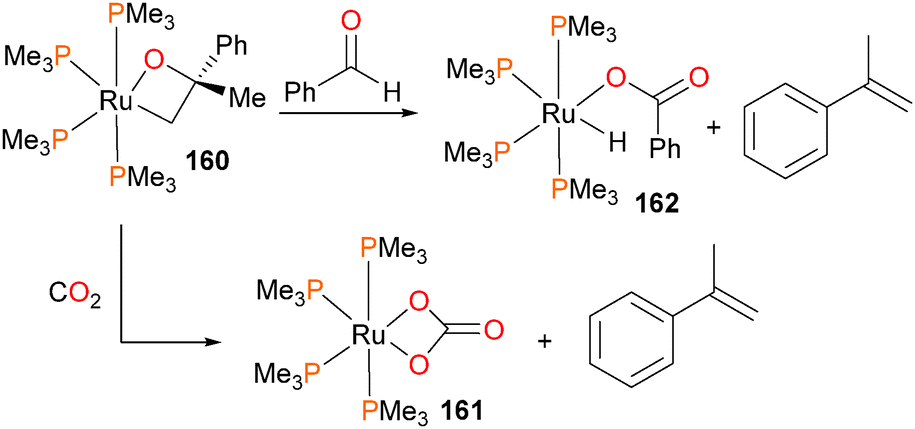 | ||
| Scheme 53 | ||
Unusual reactivity is observed when ruthenium–hydroxide complex 163 is reacted with either p-tolualdehyde or hexafluoroacetone to form complexes 164 and 165, respectively (Scheme 54).158 In both cases, a carboxylate ligand is formed. Isotopic labeling in the case of the former shows that the OH group is retained in the carboxylate, and that the carboxylate is fluxionally bound to the ruthenium (i.e. the 17O label derived from 163 is scrambled equally between both positions in the carboxylate). In the case of the latter, C–C bond cleavage occurs, and trifluoromethane is detected as a byproduct.
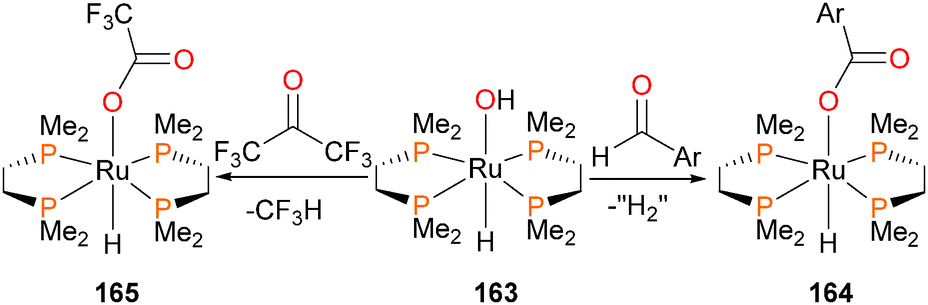 | ||
| Scheme 54 | ||
Sgro and Stephan have reported the synthesis and reactivity of a frustrated Lewis pair-like ruthenium system,159 capable of catalytically reducing CO2 in the presence of HBpin to MeOBpin and O(Bpin)2. Deprotonation of complex 166 with the strong base KHMDS yields 167, where the ruthenium is bound to the newly-formed imine in a π-fashion and also features a three-membered N–P–Ru ring. Reacting 167 with CO2 results in the rapid formation of complex 168, characterized by multinuclear NMR spectroscopic methods and X-ray crystallography. Reacting 168 with excess HBpin at slightly elevated temperatures results in slow but steady reduction of the bound CO2 moiety. Furthermore, treating 167 with benzaldehyde gives complex 169via a similar insertion pathway (Scheme 55).
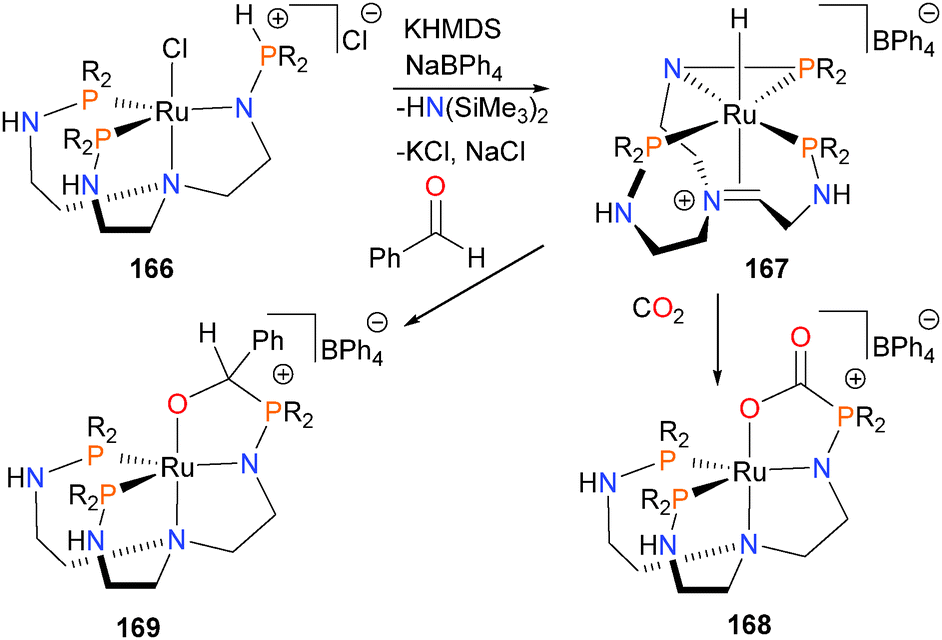 | ||
| Scheme 55 | ||
The terminal ruthenium carbide complex 170 can be oxidized to the carbonyl complex 171 with dimethyldixoirane or to the thiocarbonyl complex 172 with elemental sulfur.160,161 In the case of the thiocarbonyl, the oxidation can be reversed by treating 172 with the esoteric S-atom abstracting reagent 173, aided by the formation of a strong MoS bond, as shown in Scheme 56. Analogous reactivity has also been reported with osmium.162 A bridging carbide complex of ruthenium has also been shown to bind CO2.163
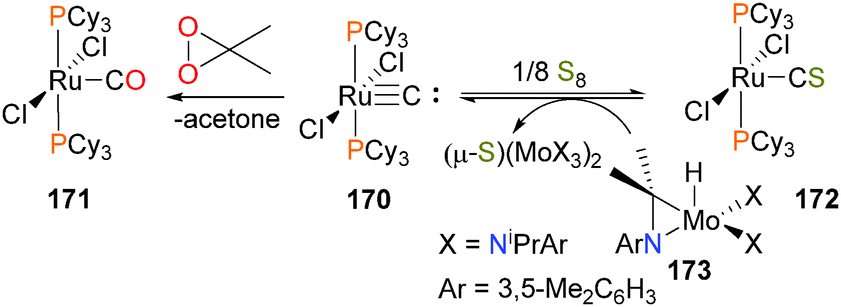 | ||
| Scheme 56 | ||
Oxidative addition of Caryl–O bonds has been demonstrated by Ueno et al. in 2007, when they reported that reacting 174 with an aryl ether in refluxing toluene formed the dark red ruthenium complex 175 (Scheme 57).164 This was the first example of directly observed Caryl–O oxidative addition of an unactivated ether to a transition metal.
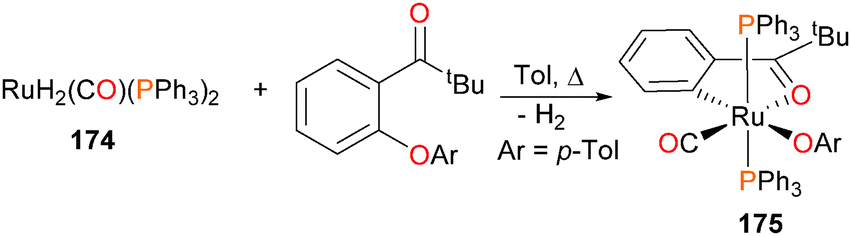 | ||
| Scheme 57 | ||
The group of Chirik has reported that bis(imino)pyridine complexes of iron, such as 176, can activate C–O bonds in both esters and ethers.165 Esters were found to undergo competing Cacyl–O and Cester–O bond activation (yielding mixtures of complexes such as 177 and 178, see Scheme 58).
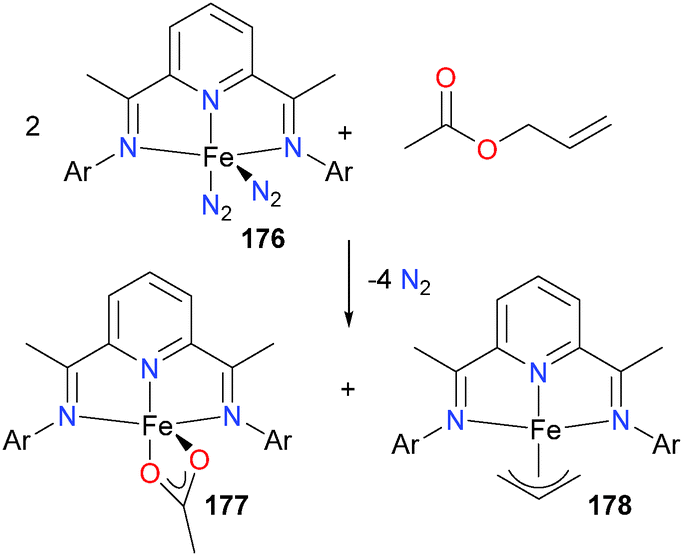 | ||
| Scheme 58 | ||
Goldberg and co-workers have developed a tetradentate ligand scaffold (see Scheme 59) that controls both the first- and second-coordination spheres of an iron complex in an effort to mimic the oxidation of organic substrates by non-heme iron enzymes.166,167 Reacting acetonitrile complex 179 with a hypervalent iodine reagent at low temperature results in the clean formation of iron oxo complex 180. Layering the reaction mixture with Et2O at −70 °C results in the growth of X-ray quality crystals, providing a rare example of a non-heme iron oxo complex that can be characterized by diffraction studies. On warming to room temperature, C–F hydroxylation of the ancillary ligand occurs rapidly, yielding complex 181. EPR analysis of the reaction mixture indicates that the activated fluorine may be bound to an unidentified, high-spin iron byproduct. The Agapie group has also recently shown related C–F activation of a fluorinated ancillary ligand by non-heme iron models.168
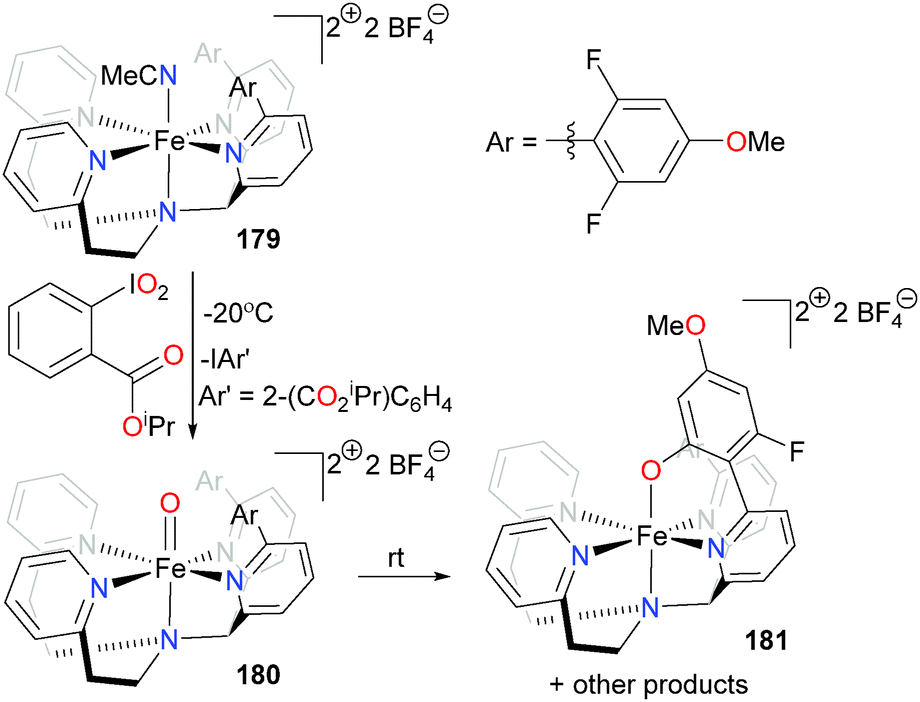 | ||
| Scheme 59 | ||
Group 9 metals
Milstein reported the first stable, isolable hydridoalkyl complex of rhodium in 1982,169 when it was reported that rhodium(I) complex 182 reacted with substituted epoxides (R = Me or Ph) to generate the complex 183, through either a 1,2-hydride shift or β-hydride elimination from zwitterionic intermediate 184 (or, alternatively, the ring-closed oxetane isomer). Complex 183 was characterized by NMR and elemental analysis, and the presence of the ketone can be gleaned by the strong absorbance bands in the IR spectra at 1650 cm−1 and 1625 for cm−1 for R = Me and Ph, respectively. Gentle heating resulted in reductive elimination of the hydride and alkyl groups to generate ketones and close the catalytic cycle. Crossover experiments indicated that reductive elimination is intramolecular, and kinetic analyses determined that the rate determining step was phosphine dissociation from 183 prior to rapid reductive elimination to generate rhodium(I) complex 185, which would subsequently re-coordinate phosphine (see Scheme 60). Shortly thereafter, related oxidative addition chemistry to form the iridium congener of 182 was reported.170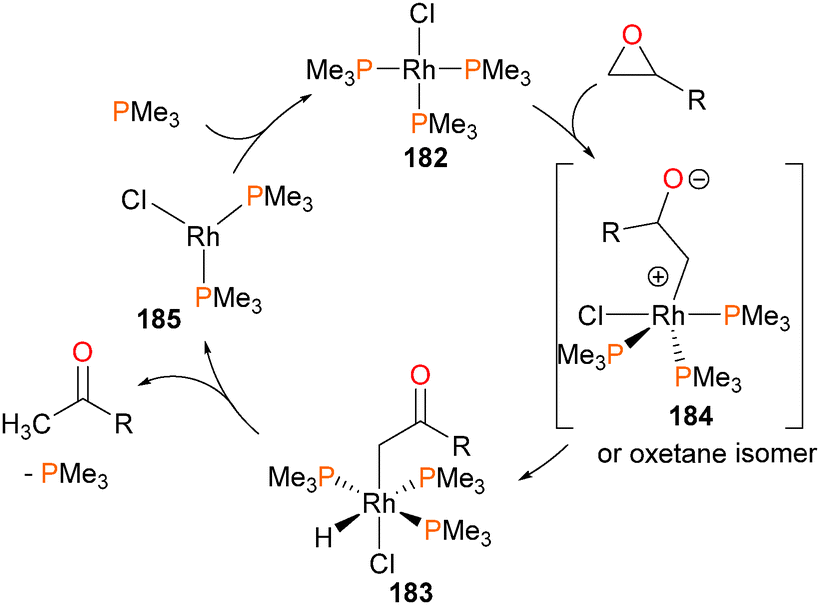 | ||
| Scheme 60 | ||
The authors proposed that complex 183 might also be formed via a rhodaoxetane isomer of intermediate 184,171 which could undergo β-hydride elimination to yield the observed hydrido product. To shut down this β-elimination pathway, rhodium(I) complex 186 was reacted with isobutylene oxide, as demonstrated in Scheme 61. The product of this reaction was found to be rhodaoxetane 187, which was formed in ca. 30% yield and characterized by NMR studies and X-ray diffraction experiments. Complex 187 could also be prepared independently via C–Br oxidative addition of complex 186 with an α-bromoalcohol, followed by deprotonation of the product 188 with the strong, hindered base KHMDS.
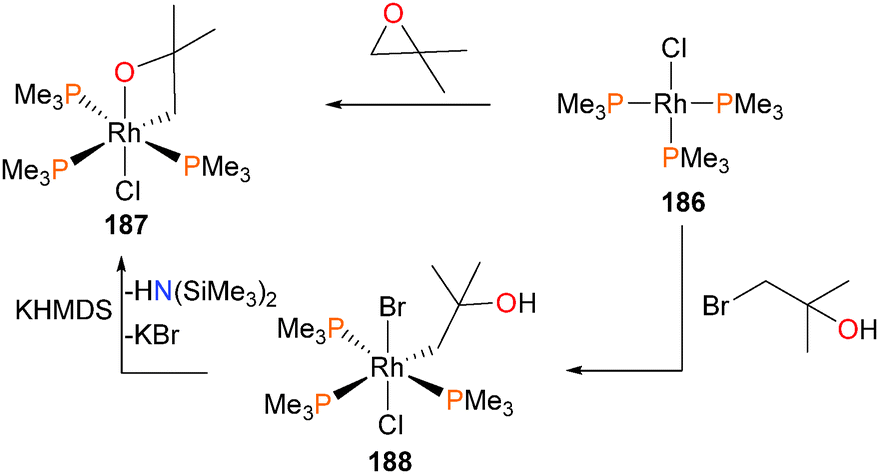 | ||
| Scheme 61 | ||
Han et al. have reported that anionic rhodium(I) porphyrin complexes such as 189 can induce ring-opening of epoxides to form β-hydroxyalkyl complexes such as 190 (Scheme 62). Deuterium labelling experiments reveal an inversion of stereochemical configuration at the α-carbon, consistent with an SN2-type mechanism. While complex 190 is stable under neutral conditions, addition of 18-crown-6/KOtBu results in rapid regeneration of 189 and the epoxide with overall retention of stereochemistry, consistent with 191 as an intermediate in this transformation.172
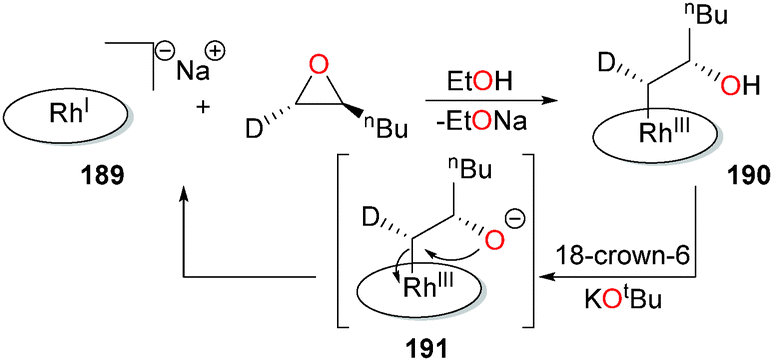 | ||
| Scheme 62 | ||
Newman and Bergman reported complex 192, one of the earliest examples of a mononuclear, late transition metal alkoxides.173 The authors found that 192 reacted with CO2, CS2 or a variety of isocyanates to selectively insert into the Ir–O bond over the Ir–H bond, producing complexes of type 193 (see Scheme 63).66,174,175 A later example of a tethered alkoxide was found to behave analogously.61 Glueck et al. have also reported on the synthesis and reactivity of iridium alkoxide complexes.175 The authors report that the alkoxide is fairly labile, able to be replaced by amines to yield iridium amido complexes (vide infra). Notably, other alkoxides such as 194 can be prepared by treating 192 with esters, such as phenyl acetate. The ethoxide ligand is suitably nucleophilic enough to attack anhydrides, resulting in ring-opened products 195.
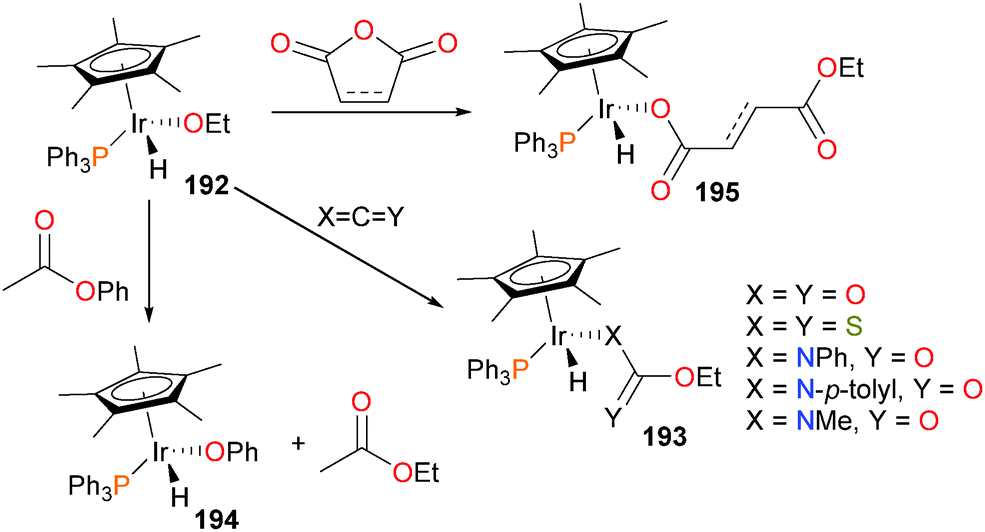 | ||
| Scheme 63 | ||
Iridium cabyne complex 196176 has been shown to be susceptible to O-atom transfer when treated with pyridine-N-oxide, resulting in scission of the C–C bond to form phenyl–carbonyl complex 197. Carbyne 196 also reacts with methanol to form a Fischer carbene complex, 198 (Scheme 64). This addition of methanol across the carbyne unit is effectively irreversible, as heating 198 at 95 °C for extended periods of time in the presence of 4 Å molecular sieves does not result in the reformation of complex 196.
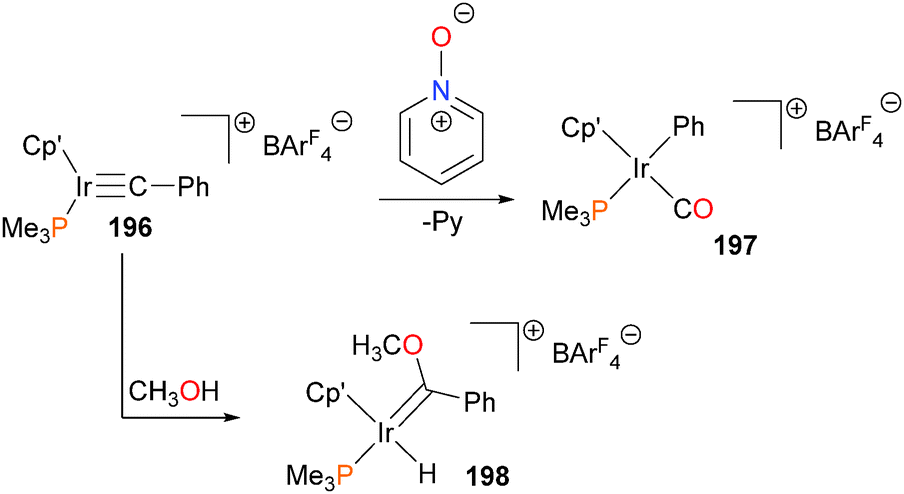 | ||
| Scheme 64 | ||
The first example of the insertion of an unactivated olefin into a late metal–oxygen bond was a report by Woerpel and Bergman, and is outlined in Scheme 65.177 Iridium hydroxide complex 199 reacts slowly under an atmosphere of ethylene to form the hydroxyethyl complex 200, which is not stable in the absence of ethylene. Over the course of 1 week at room temperature (or 2 days at 45 °C), 200 was found to decompose to give the aldehyde complex 201. Hydroxide complex 199 could also react with DMAD to generate the alkenyl complex 202, formed as solely the Z-stereoisomer, and maleic anhydride to product the maleate complex 203. IR analysis reveals two strong absorbance bands at 1726 cm−1 and 1550 cm−1, which the authors cite as evidence of the hydrogen-bonded maleate ligand as shown.
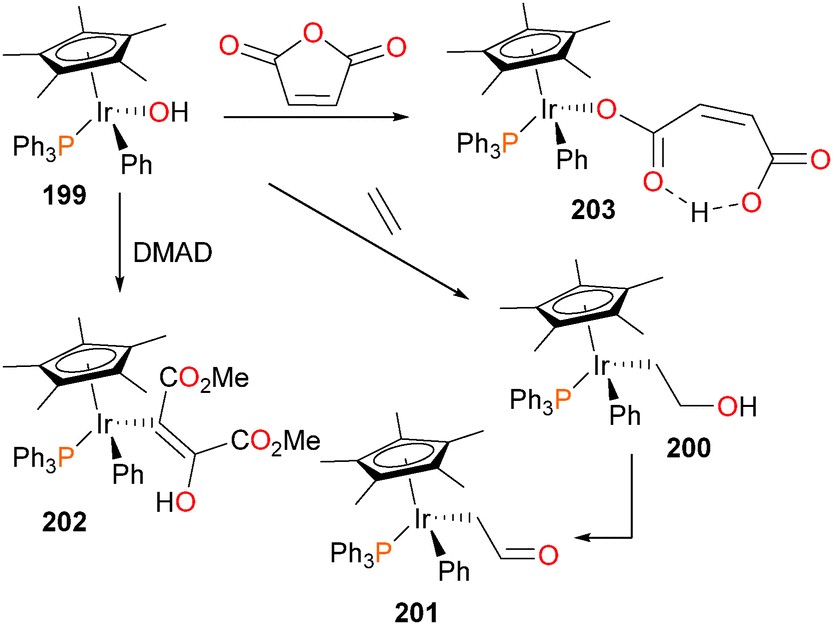 | ||
| Scheme 65 | ||
The authors were especially intrigued by the mechanisms of formation178 of complexes 200 and 201, given that the insertion of ethylene into the Ir–O bond, as well as the surprising formal dehydrogenation of the resulting hydroxyethyl complex to give formylmethyl complex 201 are relevant to the Wacker process for ethylene oxidation. It was quickly noticed that the rate of ethylene insertion into complex 199 was “batch-dependent”, with the authors concluding that some trace impurity in 199 must be catalyzing the insertion reaction. Indeed, addition of either PMe3 or PPh3 to 199 resulted in a dramatic decrease in the rate of ethylene insertion. Iridium triflate complex 69, which is the synthetic precursor to iridium hydroxide 199, was then added to these “inactivated” reactions, resulting in a reacceleration of the insertion rate. Clearly, the presence of a catalytic amount of triflate 69, remaining from the synthesis of hydroxide 199, was catalyzing the insertion chemistry, and could be quenched by the addition of phosphine. Further experiments revealed that ethylene is in fact reacting first with complex 69 to generate cationic olefin complex 204, which is then attacked by the nucleophilic OH group of 199. This potentially forms binuclear intermediate 205, which cleaves to generate the “insertion” product 200, as well as cationic 206, which could rapidly rebind either triflate to form 69 or ethylene to form 204. In cases where the reactions were allowed to continue, the complete conversion of hydroxyethyl 200 to formylmethyl 201 was observed. In addition, another iridium species, the hydride complex 207, was also detected. The authors propose that binuclear intermediate 205 can be deprotonated by Lewis basic hydroxide 199 to generate water and trinuclear intermediate 208, which could collapse as outlined in Scheme 66 to yield the formylmethyl complex 201, the hydride complex 207 and another equivalent of the cationic complex 206, which would once again either bind ethylene to generate 204 or triflate to form 69. Subsequent work found that this “cooperative” mechanism of β-elimination is also operative for the synthesis of aldehydes179 and carboxamides.65
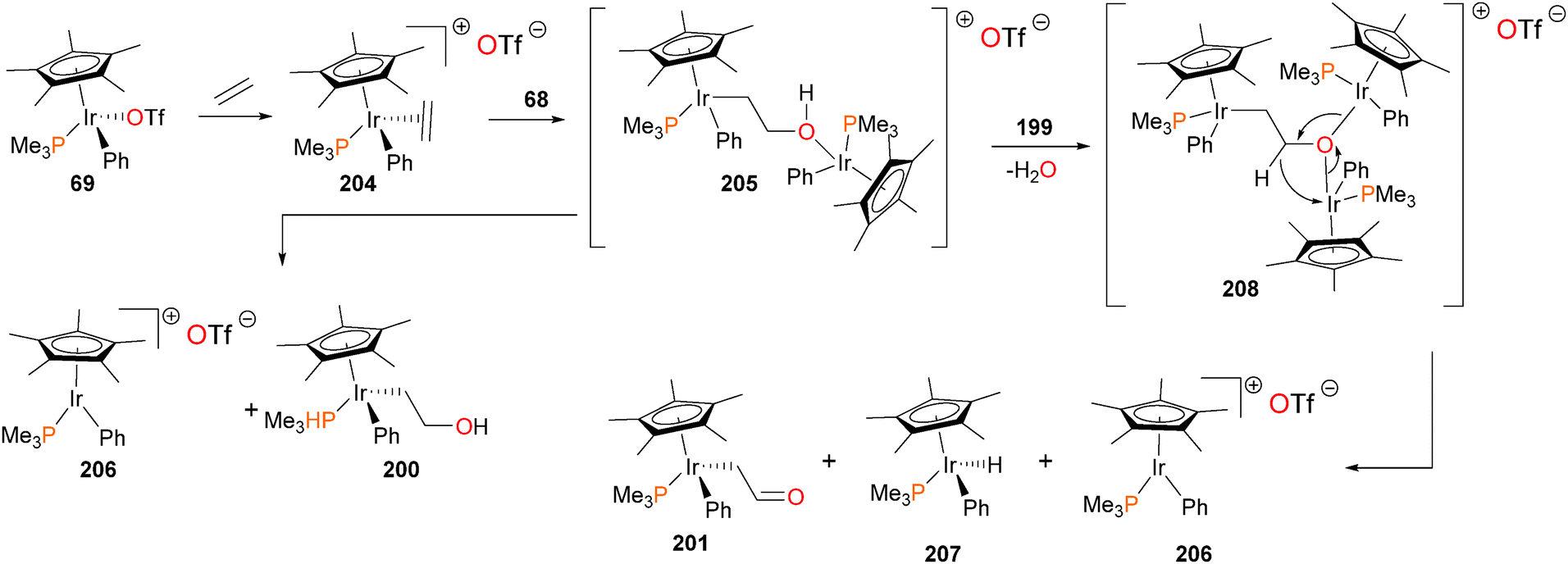 | ||
| Scheme 66 | ||
Interestingly, iridium methylidene complex 209 was found to react slowly with CO2 to generate the four-membered iridalactone 210 (shown in Scheme 67) in 45% isolated yield as a yellow crystalline solid.180 Complex 210 was characterized by NMR spectroscopy, and featured a strong absorbance band at 1662 cm−1 in the IR spectrum, indicative of the lactone functional group. Protonolysis of 210 with dry HCl results in the quantitative formation of acetic acid and the known Cp*Ir(PMe3)Cl2. More recently, Whited and Grubbs have also demonstrated that Fischer carbene complexes of iridium can react with CO2181 and other heterocumulenes182–185 to form formates and other carbonyl compounds.
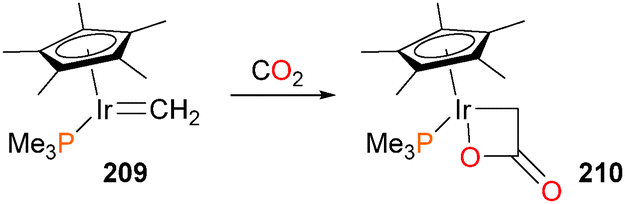 | ||
| Scheme 67 | ||
Photolysis of rhodium and cobalt pinacolato complexes 211186 were found to cleanly release 2 equivalents of acetone, and the resulting reduced metal species could be trapped by 2,3-dimethyl-1,3-butene to form 212, opening a new avenue into the preparation of M(I) synthons (Scheme 68). These pinacolato complexes were also found to be prone to insertion chemistry with a variety of heterocumulenes.187
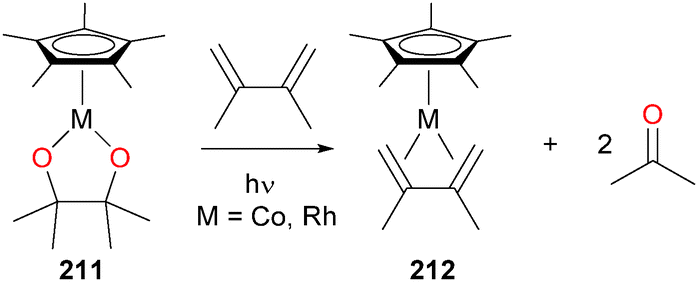 | ||
| Scheme 68 | ||
The Caulton group has reported a study on the nucleophilic attack of alkoxides on iridium- and rhodium-bound olefins such as 213188 and found that while the additions give predominantly the exo isomer of OR addition to the olefin (i.e. complex 214), the C–O bond formation can be reversed by the addition of the hydrogen-bonding solvent MeOH or by an acid (Scheme 69).
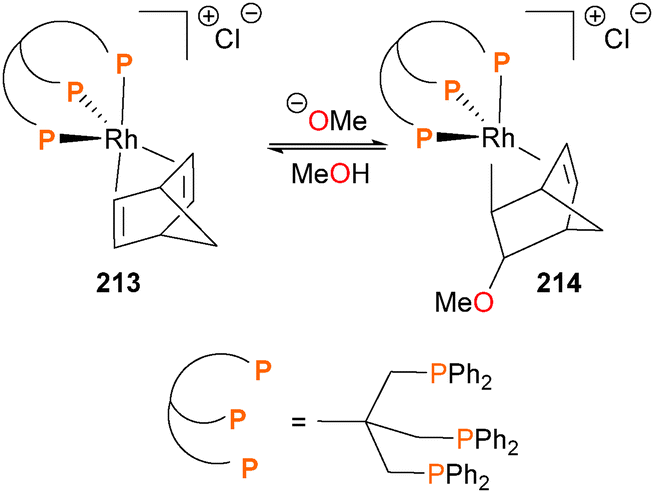 | ||
| Scheme 69 | ||
Rhodium–norbornadiene complex 215 was found to undergo smooth oxidation with ferrocenium or silver to generate the rhodium(II) complex 216,189 which was characterized by EPR spectroscopy and elemental analysis. In the absence of suitable crystals for diffraction studies, DFT optimizations were performed which suggest a square pyramidal geometry of 216, with the diene occupying the basal plane and the dipicolylamine ligand adopting a fac-orientation. Curiously, attempts to prepare 216via oxidation with FeCl3 in a mixture of acetone and either water or methanol led not to rhodium(II) complex 216 but to the new rhodium(III) complex 217, albeit in low yields (Scheme 70). Stoichiometric experiments revealed that complex 216 was undergoing a disproportionation reaction to generate equimolar amounts of 217 and 215, and cyclic voltammetry experiments revealed that this reaction is triggered by the presence of chloride. Due to the asymmetry of the pyridyl groups on the dipicolylamine ligand, 217 was formed as mixtures of diastereomers. The related formation of C–O bonds via the addition of alcohols190 or water191 to iridium complexes, respectively, have also been reported. Redox-triggered C–O reductive elimination has also been reported with iridium complexes.192
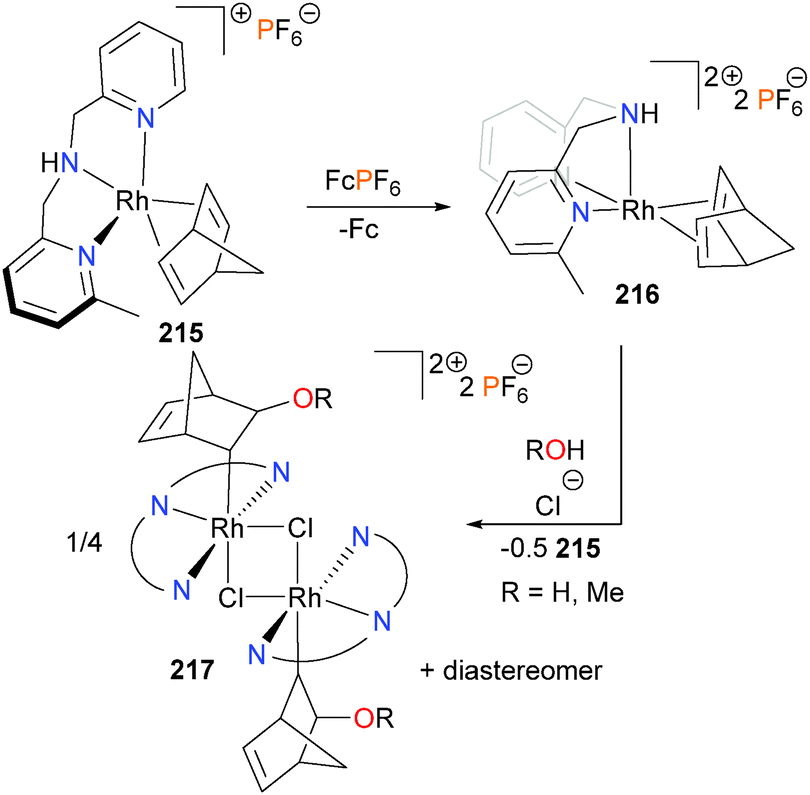 | ||
| Scheme 70 | ||
Rhodium-mediated oxygenation of COD with dioxygen has been reported by Tejel et al.193 to proceed via a 2-rhodaoxetane intermediate,194 which has been characterized by X-ray crystallography. Oxidation of starting material 218 with O2 forms binuclear 219, and addition of PMe3 to this complex results in the formation of a mononuclear rhodaoxetane 220, which then releases the ketone product (Scheme 71) upon coordination of another phosphine ligand. The authors propose that β-hydride elimination and reductive elimination occur prior to coordination of the final phosphine ligand to form the ultimate rhodium product 221. Blum and Milstein have previously observed related β-hydride eliminations from iridium complexes.195
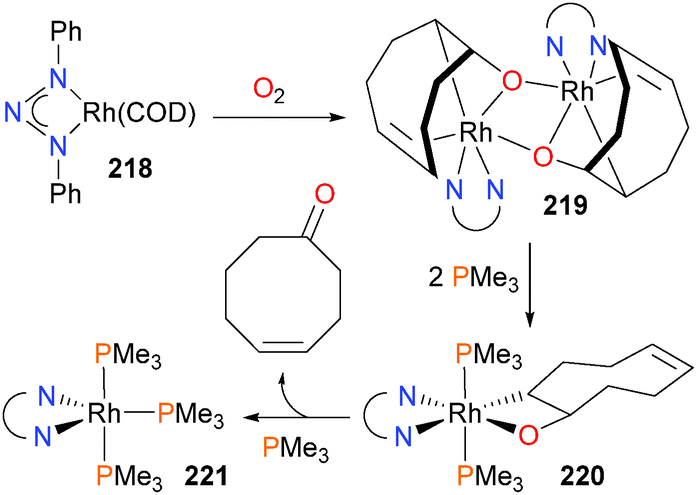 | ||
| Scheme 71 | ||
The research groups of Gal and others196–198 have reported considerable work on the oxygenation of metal-bound olefins using H2O2 and O2 as the oxidants. This work has been reviewed elsewhere,80 and so will not be repeated here except for select examples. For instance, it was found that treating Cramer's dimer 222 with tris(2-pyridylmethyl)amine gave rhodium–ethylene complex 91. While reacting 91 with aqueous H2O2 yielded rhodaoxetane 223 as the sole product,199 exposure of solid 91 to air results in the formation of a mixture of rhodadioxolanes 224 and 225, respectively (see Scheme 72).200 Notably, the nature of the counterion in these oxygenations was found to be quite important, as mixtures of 224 and 225 were obtained when using PF6, but only isomer 224 was formed when using BPh4 as the counterion.
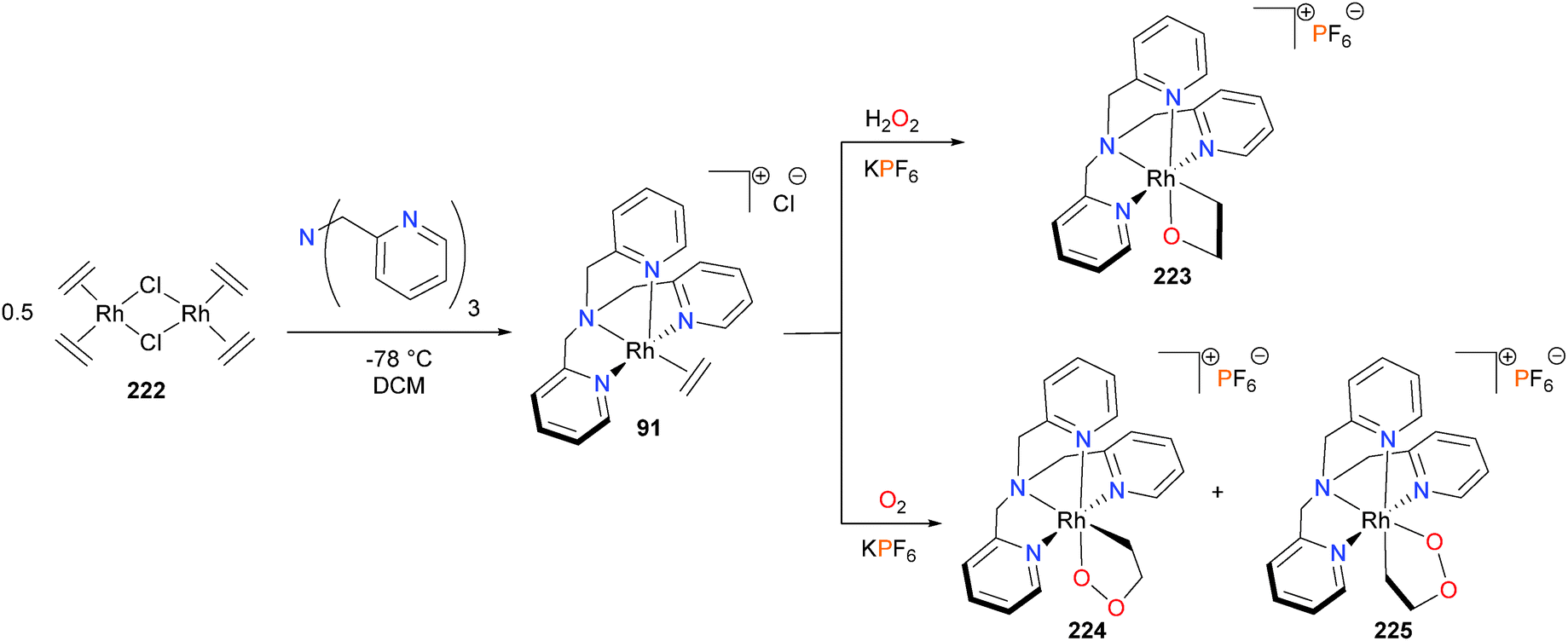 | ||
| Scheme 72 | ||
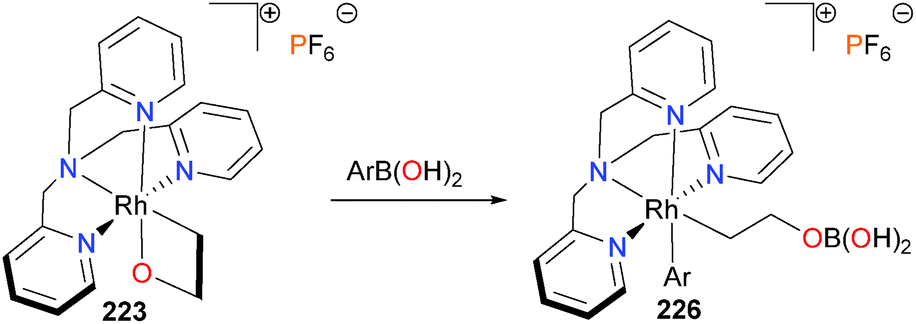
In 2010, the Love group reported that rhodaoxetane 223 can react with aryl- and alkenylboronic acids to yield ring-opened rhodium complexes 226, as outlined in Scheme 73.201 The products were characterized by a wide array of NMR spectroscopic techniques and mass spectrometry, as the products were resistant to crystallization. The transmetalation was observed to be general for arylboronic acids, but alkylboronic acids were reluctant to undergo comparable reactivity. Initial mechanistic experiments indicate that coordination of the rhodaoxetane oxygen to the sp2-hybridized boron of the boronic acid yields an ate-complex, which undergoes subsequent transmetalation to the observed product.
 | ||
| Scheme 73 | ||
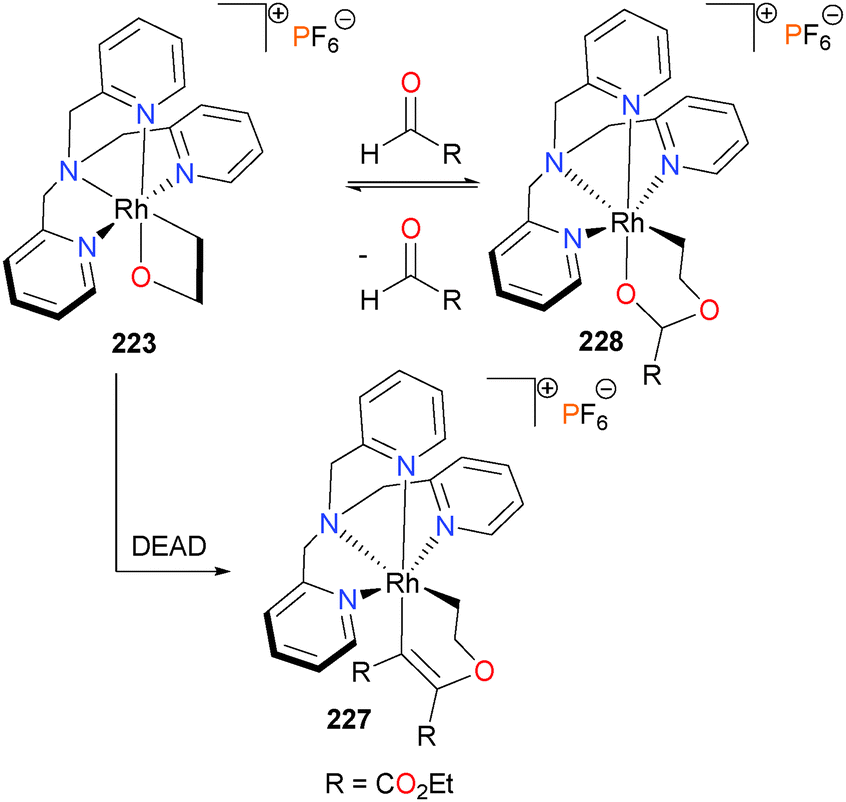
In 2014, Love and co-workers published a study on the reactivity of rhodaoxetane 223 with a variety of unsaturated electrophiles.202 Gal and co-workers had previously reported that 223 could insert acetone over the course of several weeks into the Rh–O bond to generate a ring-expanded rhodaketal complex.203 Later, it was found that 223 could also insert electron-deficient alkynes such as DMAD or DEAD to form 227. Insertions of these alkynes into Rh–O bonds had previously been reported by Yamamoto.204,205 Other small molecules, such as CO2, CS2 and aldehydes were also found to undergo insertion into the Rh–O bond of 223 (Scheme 74). Several of these complexes were characterized crystallographically.206 Notably, the insertion of aldehydes to form 228 was found to be reversible. Tejel et al. have also observed related aldehyde insertion reactions in rhodium-catalyzed Tischenko coupling,207 and several other examples of reversible aldehyde insertions have been reported with other late metals.208,209
 | ||
| Scheme 74 | ||
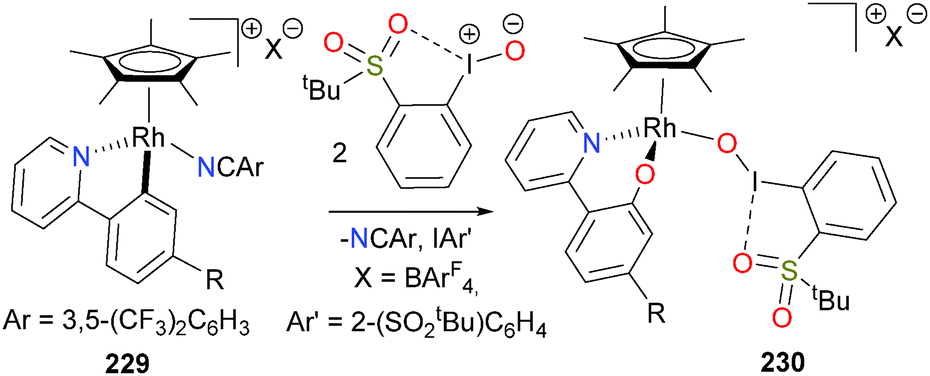
Related to the work of the Chang group (vide supra), Turlington et al.163,164 have reported that 2-tert-butylsulfonyliodosylbenzene can oxidize cationic rhodium and iridium complexes such as 229 (Scheme 75). In the case of rhodium congener, aryloxide 230 was generated, which was fully characterized. Treatment of 230 with acids results in cleavage of the Rh–O bond and release of 2-pyridylphenol.
 | ||
| Scheme 75 | ||
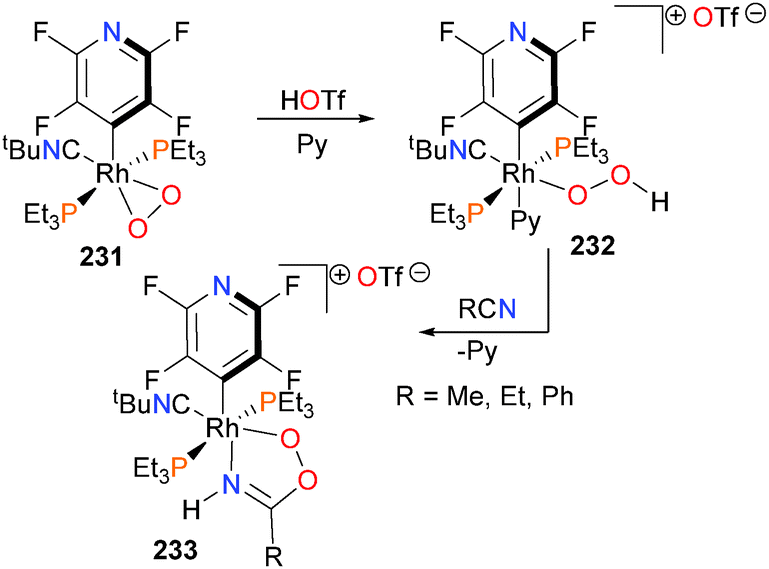
A recent report by Bittner et al. describes the synthesis and isolation of a transition metal stabilized analogues of peroxyimidic acid (Scheme 76).210 Protonation of rhodium–dioxygen complex 231 with triflic acid yields the end-on bound hydroperoxide complex 232, which is stable at room temperature in solution but decomposes when exposed to vacuum. Further reaction of 232 with nitriles furnishes the peroximidate complex 233via C–O bond formation and subsequent proton transfer. Related peroxo complexes have also been prepared by alkylation of rhodium-bound dioxygen.211 Examples of C–O bond formation upon treatment of rhodium complexes with O2 have also been reported.212
 | ||
| Scheme 76 | ||
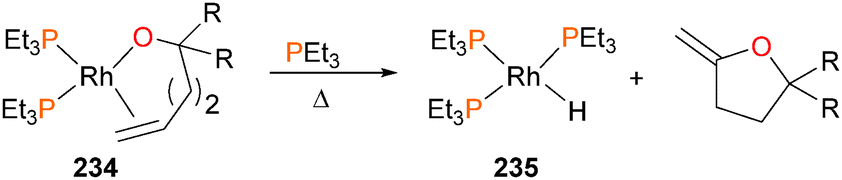
Zhao et al. have prepared rhodium(I) complexes that feature tethered alkoxo and olefin co-ligands.213 Treating complex 234 with additional PEt3 ligand at slightly elevated temperatures results in the clean formation of 2,2-disubstituted 5-methylenetetrahydrofuran (Scheme 77) and hydride 235. Kinetic analysis of the formation of product 235 indicates that the reaction is unimolecular, and a similar rate of formation of 235 in cyclohexane, benzene and THF indicate that the reaction likely does not proceed through ionic intermediates. Deuterium labelling studies found the product formed as a single isomer, consistent with syn addition of the metal and alkoxo across the CC double bond.
 | ||
| Scheme 77 | ||
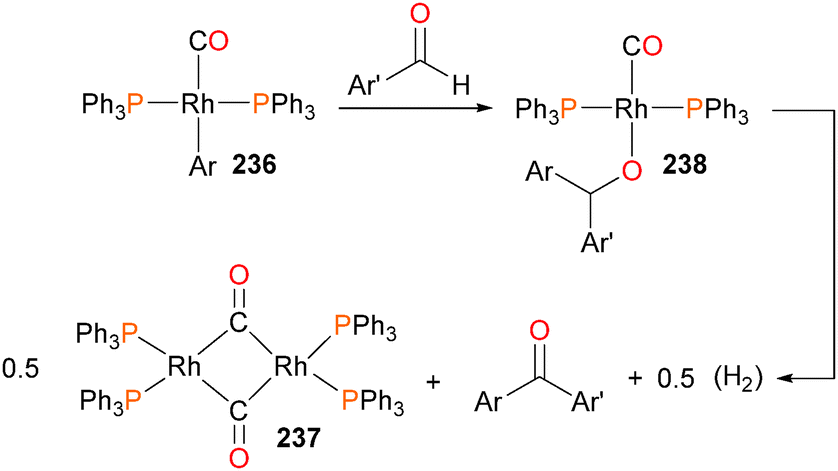
Krug and Hartwig presented the first example of aldehyde insertion into metal–aryl bonds with a paper in 2002, describing the reactivity of rhodium(I) aryl complexes with aldehydes.214 Heating aryl complex 236 with aldehydes at 85 °C gave ketones as the final organic product, along with rhodium dimer 237. If the reaction was conducted at room temperature with naphthaldehyde as the substrate, an intermediate complex 238 was observed by NMR spectroscopy (Scheme 78). This intermediate was identified by independent synthesis, fully characterized, and indeed, gave the expected ketone product when heated under the same reaction conditions as above. In addition, it was found that intermediate 238 could be hydrolysed to give diarylmethanols, as well as react with additional aldehyde to generate esters via Tischenko-like coupling. Later work indicated that this reactivity could be extended to imines and alkynones.215 In related work, β-aryl elimination has also been observed for other rhodium(I) complexes.216
 | ||
| Scheme 78 | ||
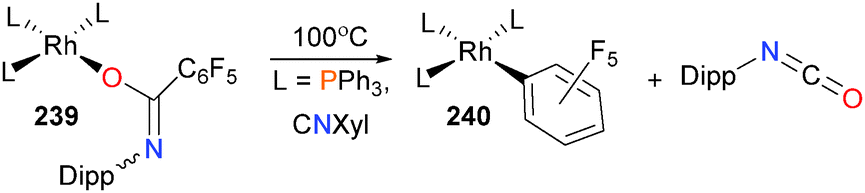
Drover et al. have found that when rhodium(I) amidate complexes bearing electron-withdrawing C6F5 substituents such as 239 will eliminate isocyanate when heated in toluene to form rhodium(I) aryl complexes 240 (Scheme 79).217
 | ||
| Scheme 79 | ||
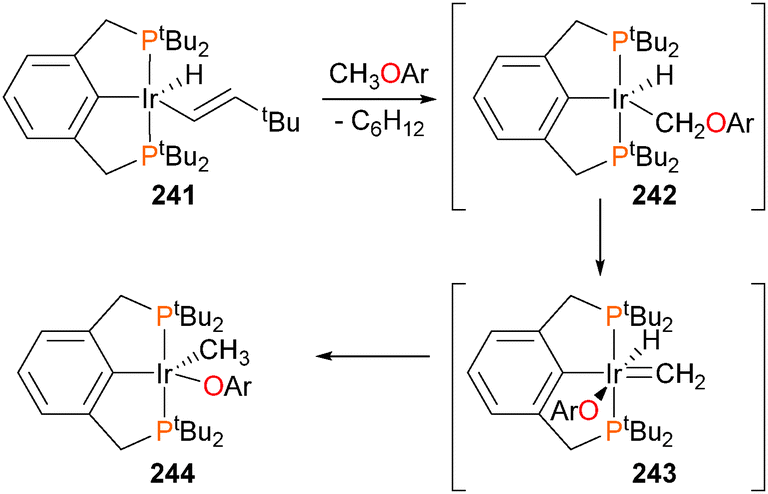
The Goldman group first reported in 2009 that iridium pincer complex 241 was capable of activating the C–O bond of methyl and ethyl ethers (Scheme 80).218 Subsequent isotope labelling experiments and DFT studies indicated that the reaction likely does not proceed via a simple direct oxidative addition of the C–O bond, but rather through reductive elimination of C6H12 followed by C–H oxidative addition to form 242, α-aryloxide elimination to give 243, and finally hydride insertion into the methylidene to generate the observed product 244.219 Similar C–O and C–H activations have been recently reported by Santos et al. using Tp*-ligated iridium complexes.220–222 Notably, the groups of Ozerov223 and Milstein224,225 have also observed related C–O activation chemistry with rhodium pincer complexes.
 | ||
| Scheme 80 | ||
Doyle et al. have reported oxidation of the iridium pincer complex 245 with N2O to generate the iridacycle 246 (Scheme 81).226 Based on the X-ray crystallographic data and NMR spectroscopic data, the authors propose that 246 is best described as a hybrid between the π-bound ketone and iridaepoxide resonance forms. Reactivity studies227 showed that the O atom could be removed via reaction with hydrogen to regenerate carbene complex 245 with concomitant formation of water.
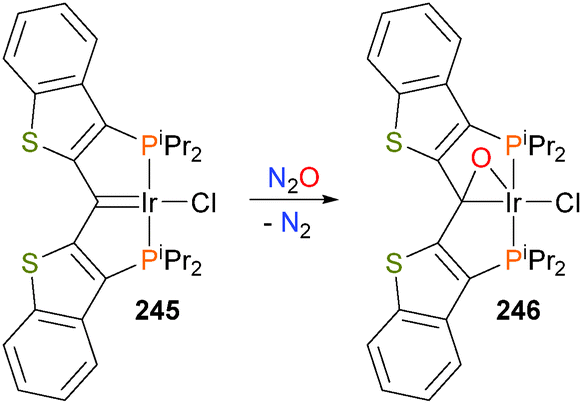 | ||
| Scheme 81 | ||
The Love and Schafer groups have recently reported an elegant example of the coupling of terminal alkynes and a phosphoramidate mediated by a Cp*Ir(III) complex.228 Addition of alkyne to coordinatively unsaturated complex 247 gives intermediate 248. Following rearrangement to form transient vinylidene 249, nucleophilic attack of the O atom of the phosphoramidate ligand results in the formation of dark green iridacycle 250 in high yield (Scheme 82). Although catalytic turnover was not possible, the functionalized phosphoramidate could be removed from the metal centre via protonolysis with TFA and subsequent hydrolysis.
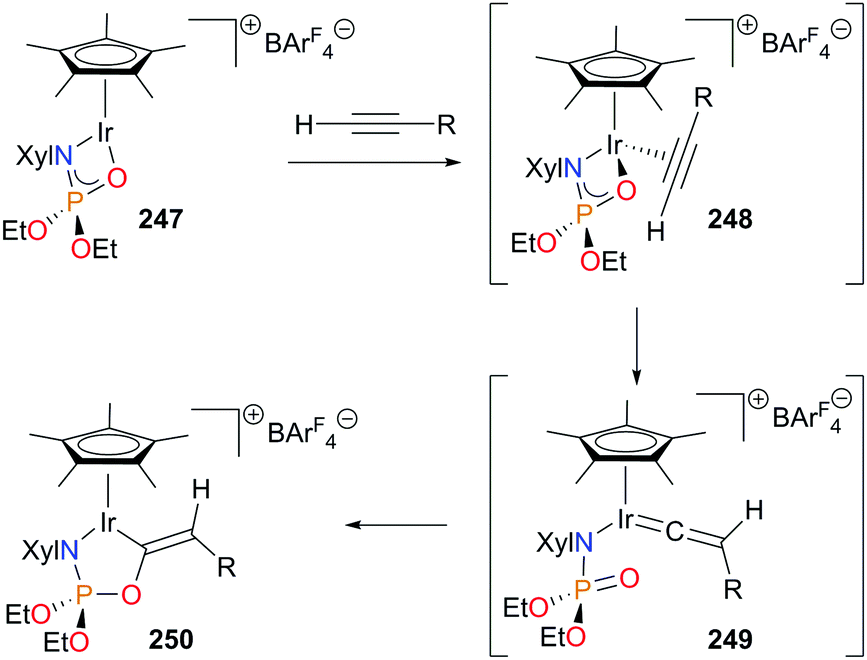 | ||
| Scheme 82 | ||
Group 10 metals
Although the insertion of an olefin into a late transition metal alkoxide had been previously proposed to be an intermediate in the Wacker process, the first well-defined example of this reactivity was reported in 1984, when Brynzda et al. published a communication on the insertion of tetrafluoroethylene into the Pt–O bond of platinum(II) alkoxide complex 251.229 Subsequent mechanistic studies found that a rapid pre-equilibrium occurs between complex 251 and olefin, generating a five-coordinate species 252 that undergoes slow insertion into the Pt–O bond to form 253, as outlined in Scheme 83.230 Crossover experiments revealed that the methoxide does not dissociate from the metal centre during this transformation. More recently, a report by Lohr et al. described related insertion reactions of CO2 into Pt–OH bonds.231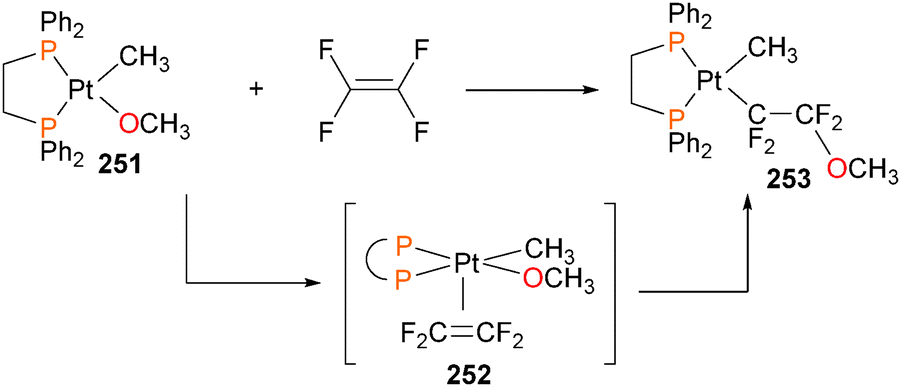 | ||
| Scheme 83 | ||
Carmona et al. reported the synthesis and small molecule reactivity of cyclometalated nickel complex 254, as shown in Scheme 84.232 Stirring complex 254 with polymeric paraformaldehyde yields complex 255, which is the product of insertion into the Calkyl–Ni bond. While complex 254 was characterized definitively by an X-ray diffraction study, the structure of 255 was assigned based on NMR data, particularly the strong coupling to a 31P nucleus (J = 88 Hz) for the ipso aromatic carbon resonance in the 13C NMR spectrum. The structure was found to be binuclear on the basis of elemental analysis (which showed loss of one equivalent of PMe3) as well as by molecular weight determinations. Reacting 254 with CO2 results in insertion into the Caryl–Ni bond, generating nickelalactone 256. Exposing 256 to an atmosphere of CO results in insertion of CO and reductive elimination to generate the cyclic anhydride, which could be isolated after workup as colourless crystals. Similarly, treating 255 with CO results in insertion and reductive elimination results in lactone formation.
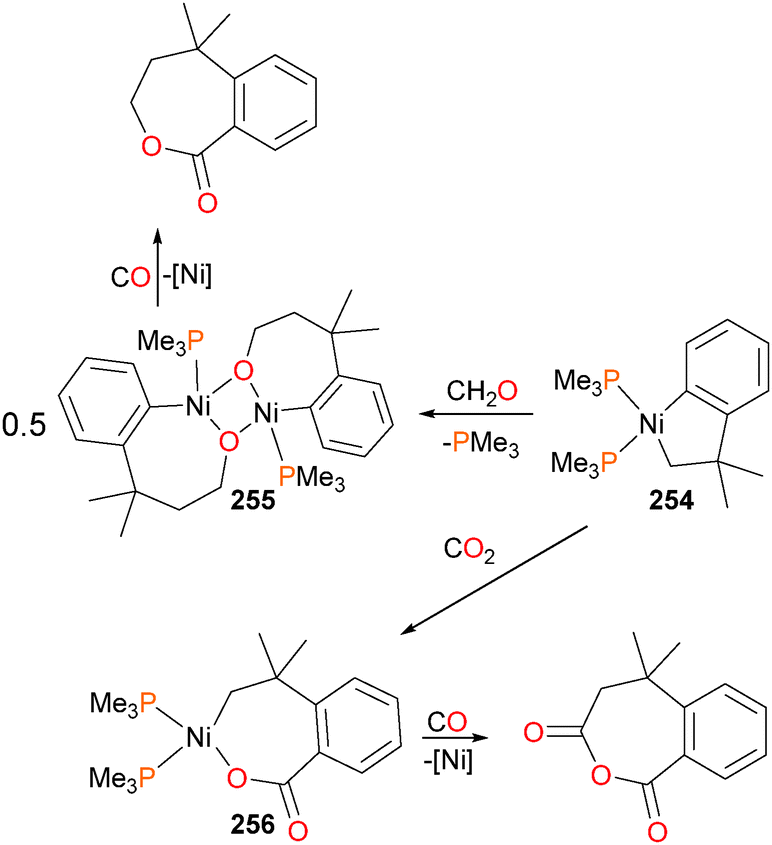 | ||
| Scheme 84 | ||
Seminal advancements in the formation of C–O, C–N and C–S bonds have been reported from the Hillhouse group over the last two decades. Early work in the Hillhouse group was spent developing the use of nitrous oxide (N2O) as an O-atom transfer reagent on late transition metals. For instance, in 1993 Hillhouse and co-workers reported that the metallacycle 257 reacts with 1 atm of N2O in benzene at 50 °C to produce the ring-expanded mixed alkyl–alkoxide complex 258, as shown in Scheme 85.233 Later reactivity studies of 258 found that it was susceptible to protonolysis with HCl to release n-butanol, oxidatively-induced reductive elimination when treated with one half of an equivalent of iodine to generate THF, as well as insertion and reductive elimination when treated with an excess of CO to generate a lactone.234 Related formation of acyclic esters from the treatment of (bpy)nickel(II) complexes with CO have also been reported by the group of Yamamoto.235,236
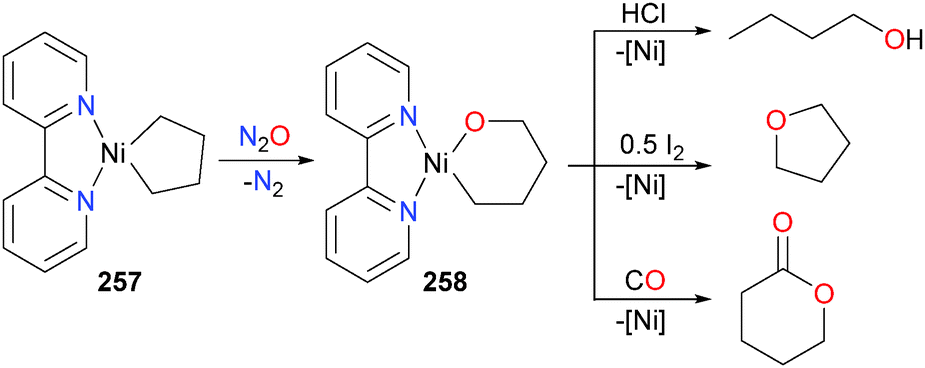 | ||
| Scheme 85 | ||
In related work, it was demonstrated that 118 could be used to effect the synthesis of more complex tetrahydrofuran derivatives (see Scheme 86). The mechanism involves olefin cyclodimerization to give 259, followed by O-atom insertion with N2O to generate nickelacycle 260, followed by oxidatively-induced reductive elimination237 with I2 to generate the cyclic ether.238
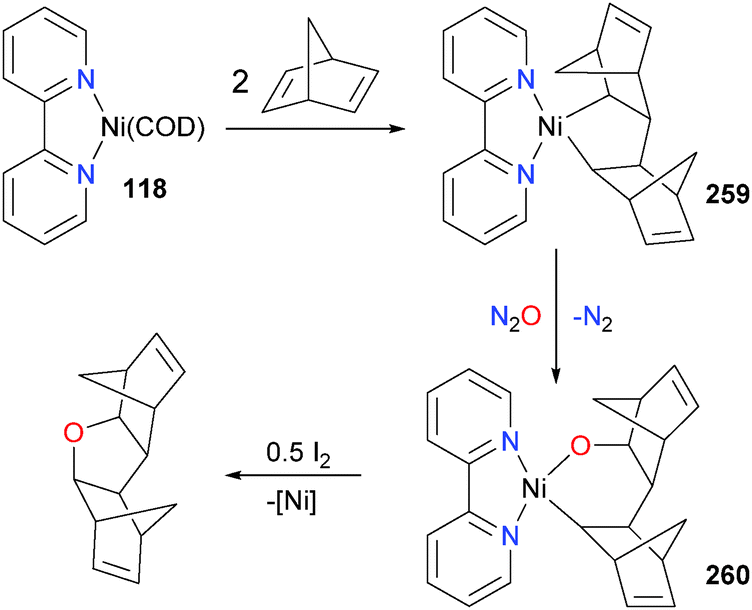 | ||
| Scheme 86 | ||
Perhaps not surprisingly, the use of smaller, more labile phosphines as ancillary ligands led to the formation of binuclear species, bridged by the alkoxide groups (Scheme 87).239 Oxidation of 254 with N2O yields 261. Once again, these dimeric species were found to be susceptible to protonolyses with strong acids and insertion and reductive elimination when treated with CO. The addition of chelating ligands, such as 1,2-bis(dimethylphosphino)ethane (dmpe), 1,10-phenanthroline (phen) or bpy, after oxidation with N2O resulted in the formation of mononuclear complexes such as 262.
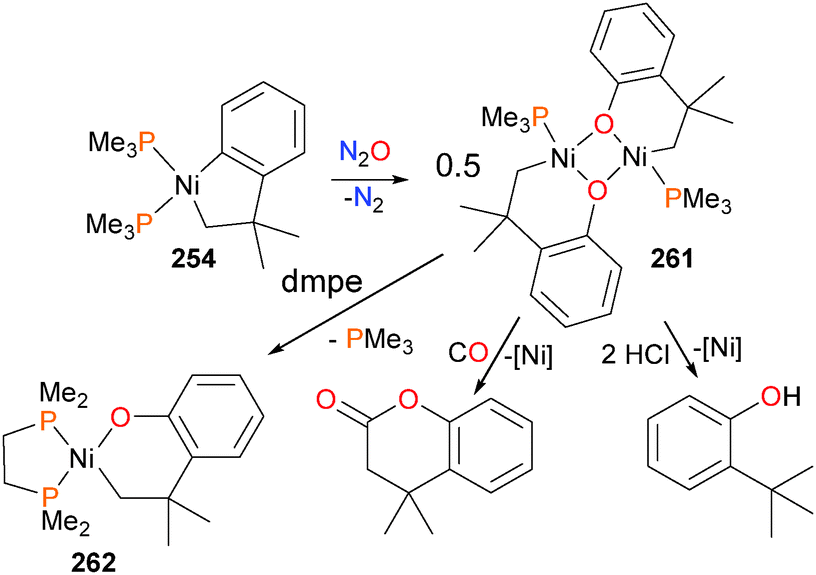 | ||
| Scheme 87 | ||
Subsequently, Hillhouse demonstrated that 255 could react with dry O2 to yield a chroman in modest yield (see Scheme 88).240 In contrast, when complex 255 was heated in the absence of an oxidant, the main organic product isolated was found to be an aldehyde, which the authors assigned as being formed via β-H elimination from 255, followed by reductive elimination of the aryl and hydrido groups of 263. In addition, the aldehyde product was also found to be a suitable candidate for insertion chemistry, as it could react with another equivalent of 255 to undergo Tischenko-like coupling to generate an ester via intermediate 264.
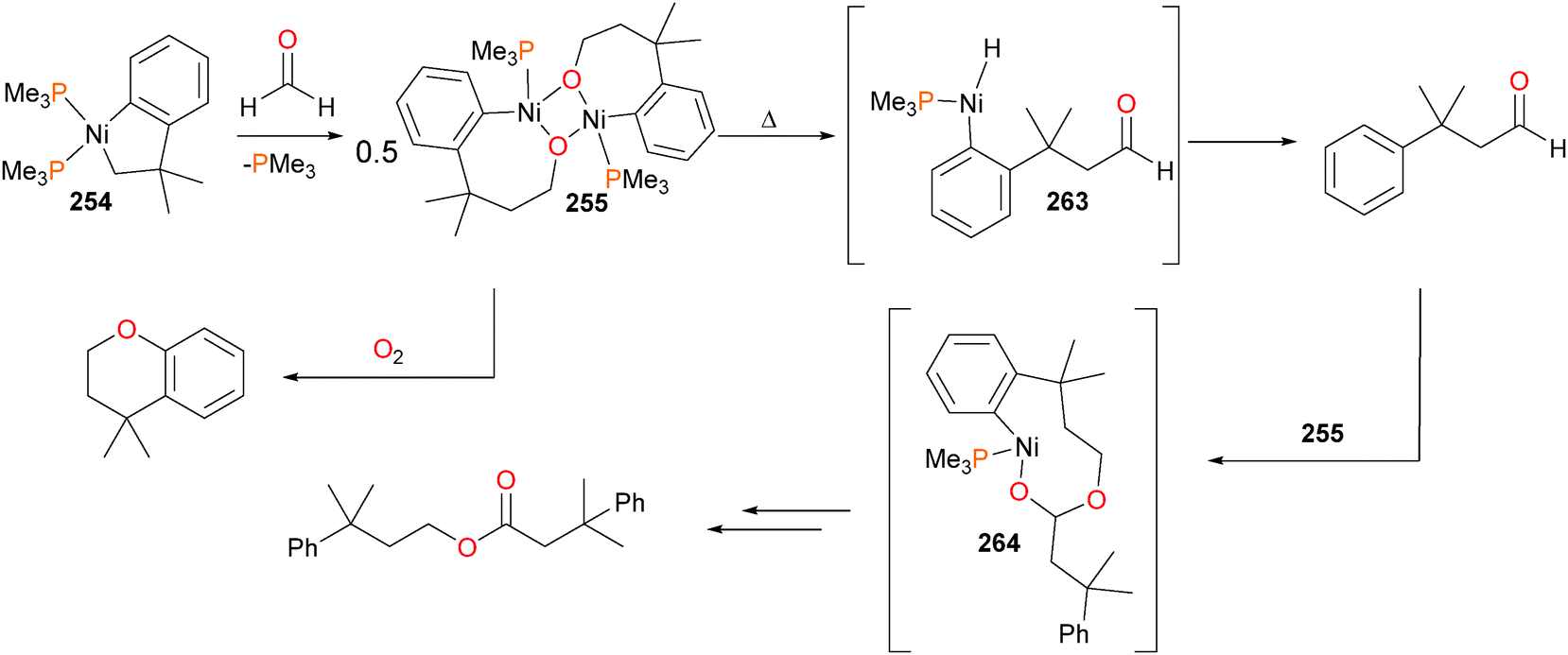 | ||
| Scheme 88 | ||
Mindiola and Hillhouse also reported that nickel alkylidene complex 265 could react with a variety of electrophiles to form cyclic products, as demonstrated in Scheme 89.241 For example, alkylidene 265 reacts with diphenylketene via a formal [2+2] cyclization to generate the first example of a structurally well-defined nickela(II)oxetane, complex 266. Two equivalents of smaller electrophiles (i.e. CO2 in this case) were found to react at the metal centre to yield the six-membered nickelacycle 267, presumably via insertion into the Ni–C bond of intermediate 268. In addition, SO2 was found to react at the Ni–C bond of 265 to generate a three-membered nickelacycle 269via formation of a C–S bond.
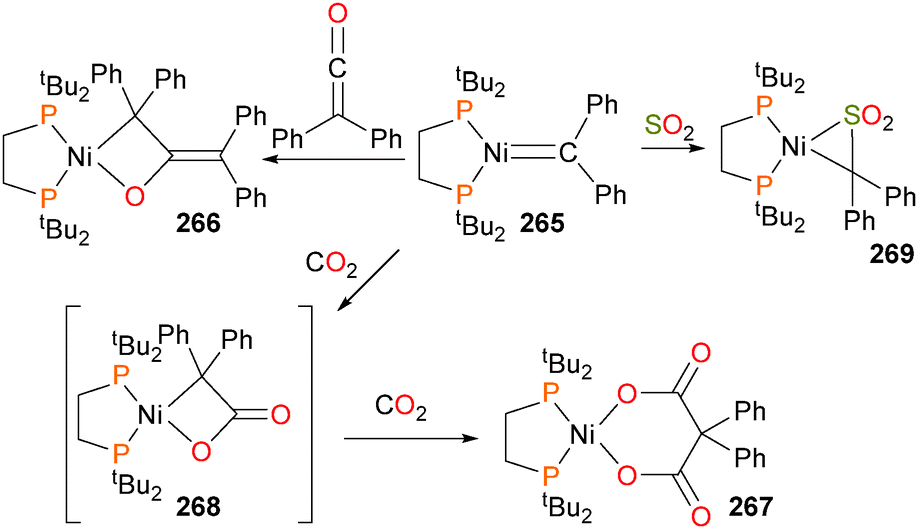 | ||
| Scheme 89 | ||
Indeed, alkylidene complex 265 demonstrates rich cyclization chemistry, as it reacts with N2O to yield benzophenone adduct 270242via O-atom transfer to the alkylidene group. This transformation likely occurs via the formation of five-membered intermediate 271, which eliminates N2 to generate the observed product, as shown in Scheme 90. Analogously, organic azides can also react with alkylidene 265 to give N2 and imines via C–N coupling. This transformation also likely occurs via a five-membered intermediate 272, as indicated by DFT studies.243
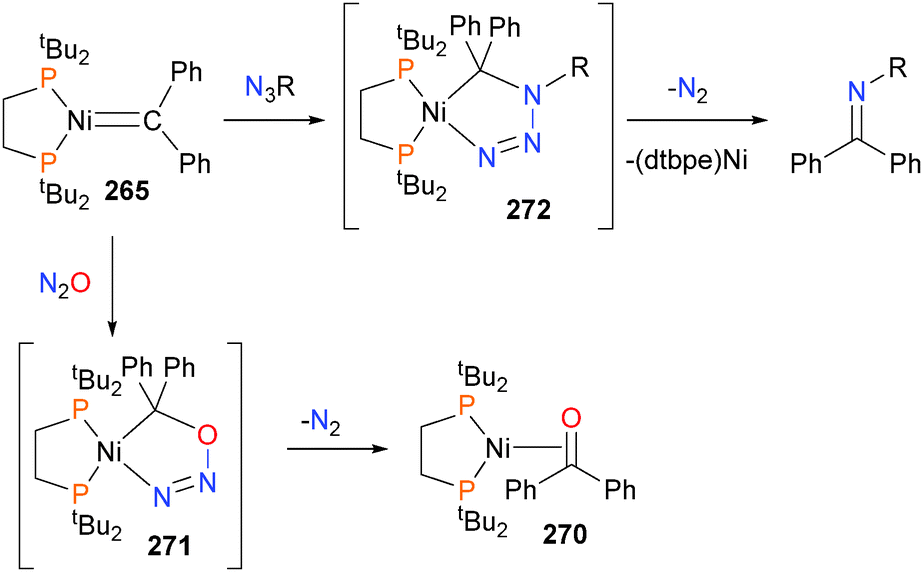 | ||
| Scheme 90 | ||
Dinuclear complex 273 has also been shown to bind to both CO2 and CS2 to give the η2 complexes 274 and 275, respectively.244 The CO2 adduct 274 was found to be thermally sensitive, decomposing via an O-atom transfer process to give oxidized phosphine ligand (dtbpeO), a dark precipitate presumed to be nickel(0) and the biscarbonyl adduct 124 (Scheme 91).
 | ||
| Scheme 91 | ||
In 2015, the Love group reported the first example of a well-defined 2-nickelaoxetane derived from the reaction of nickel(0) and an epoxide.245 It was found that reacting arene 273 with cyclic epoxides that featured a ketone group gave nickelaoxetane 276 in good yields. Most notably, in this case the stereochemical configuration of the epoxide was retained in the product metallacycle, as the cyclohexyl ring system prevents the C–C bond rotation previously observed by Hillhouse112 and Jamison.246 Performing the reaction at low temperatures allowed for the identification of an intermediate complex during this reaction, which was assigned as η2-ketone complex 277 (Scheme 92). DFT studies were performed, and it was found that a stepwise ring-opening/ring-closing mechanism that invoked the participation of two nickel(0) centres was significantly lower in energy than a concerted oxidative addition of the C–O bond of the epoxide. Reactivity studies on nickelaoxetane 276 found that in addition to the nickel chemistry already described here (protonolysis with acid, reduction with CO, oxidatively-induced reductive elimination etc.), deoxygenation of the nickelaoxetane ring was found to occur upon treatment with [Ph3C]BF4.
 | ||
| Scheme 92 | ||
The group of Jones has performed reactivity studies of a low-valent platinum complex 278 with esters,247 and found that C–O cleavage occurred to form acetate complexes of type 279. Subsequent disproportionation resulted in the formation of a bis(acetate) complex 280, as shown in Scheme 93.
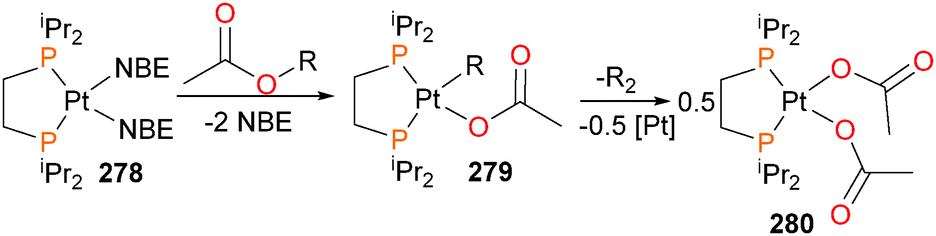 | ||
| Scheme 93 | ||
The Love group has also explored C–O and C–S oxidative addition chemistry with esters and thioesters (Scheme 94).248 In general, it was found that aryl esters underwent primarily Caryl–O oxidative addition to produce aryl–acetate nickel(II) complexes 281. In contrast, thioesters were found to undergo Cacyl–S bond cleavage, followed by rapid decarbonylation of the resulting acyl complex to form methyl–thiolate nickel(II) complexes 282.
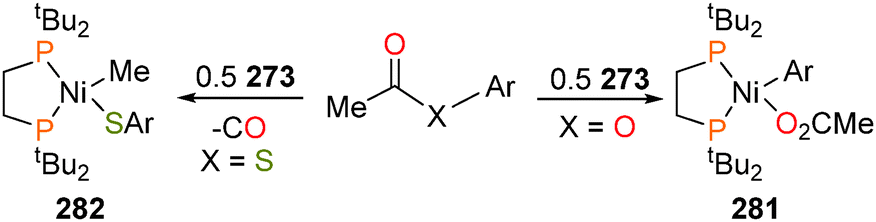 | ||
| Scheme 94 | ||
Muto et al. have found that arylpivalates can be coupled with azoles to generate biaryls using nickel catalysis.249 Mechanistic studies indicate that the combination of 283 and naphthylpivalate react when heated via the oxidative addition of the Caryl–O bond, generating the aryl–pivalate nickel(II) complex 284, which was fully characterized. Subsequent heating of 284 with the azole furnishes the cross-coupled product (Scheme 95).250,251 Nickel(0) complexes with bidentate phosphine ligands have also been demonstrated to be good catalysts for the oxidative coupling of ethylene and CO2 to form acrylate.252
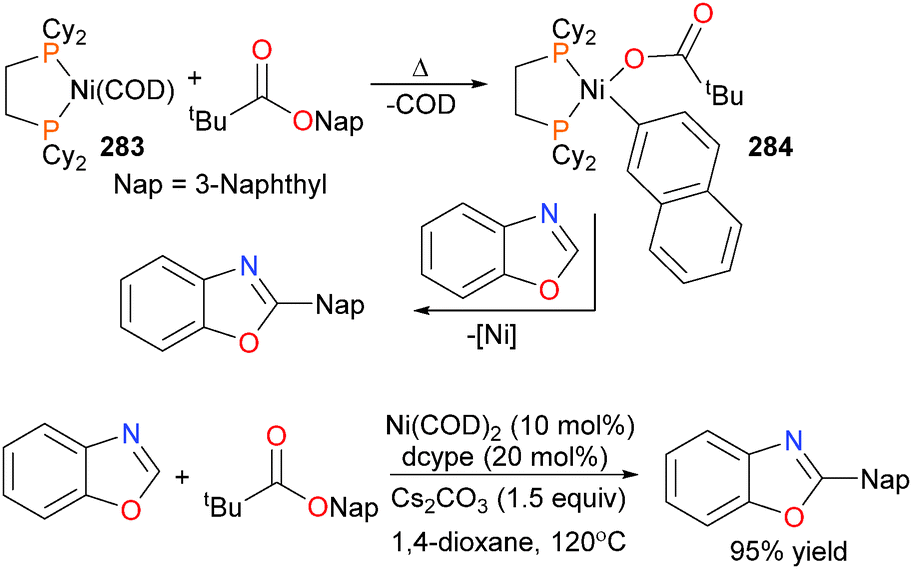 | ||
| Scheme 95 | ||
The Ogoshi group has done much work on the reactivity of nickel with aldehydes,253,254 ketones255,256 and esters.257 Recently, they have shown that cationic nickel enolate complex 285 can insert aldehydes into the Ni–C bond to generate the six-membered nickelacycles of type 286.258 Curiously, both 285 and 286 are competent catalysts for Tischenko-type coupling of aldehydes to form esters. Stoichiometric studies indicate that the first equivalent of aldehyde is not incorporated into the ester, as treating 286 with two equivalents of cyclohexylcarbaldehyde resulted in the quantitative formation of the ester shown in Scheme 96.
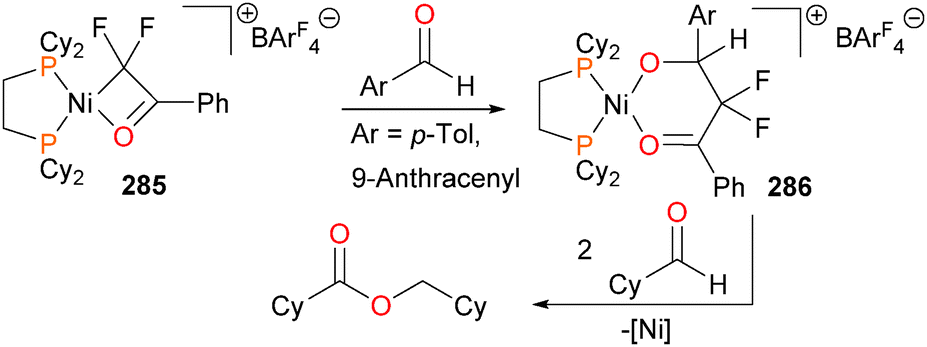 | ||
| Scheme 96 | ||
Nielsen and Doyle have recently reported catalytic cross-coupling of epoxides and boronic acids catalysed by nickel(0).259 From a catalytic reaction, the authors were able to isolate red crystals that were determined to be complex 287 by X-ray diffraction analysis (Scheme 97). The mechanism of formation of 287 is not clear, but the complex is catalytically competent, although an induction period indicates it is an off-cycle species rather than an intermediate.
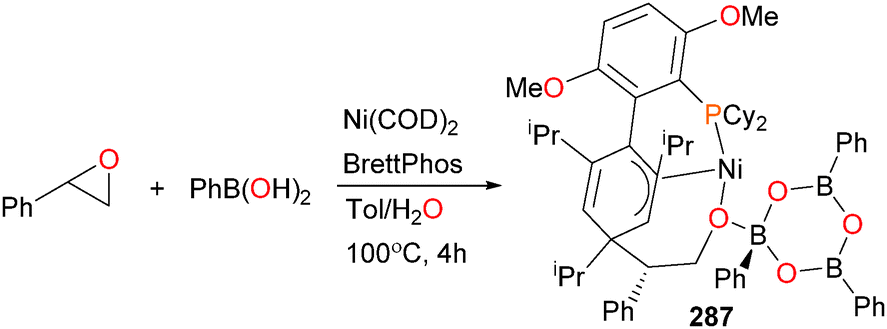 | ||
| Scheme 97 | ||
One of the earliest examples of C–O activation of aromatic methyl esters was reported by the Milstein group.224,225 Treating a pincer ligand precursor with palladium(II) complex 288 resulted in the release of methyl trifluoroacetate (Scheme 98), as detected by 1H NMR spectroscopy, and the formation of phenoxy complex 289via cleavage of the alkyl C–O bond. Complex 289 is produced in quantitative yield after heating for 3 hours at 80 °C in benzene. It is noteworthy that activation of the aromatic C–O bond is observed when the same aryl ether is treated with rhodium(I) as the metal. Related complexes of palladium260 and nickel225,261 have since been reported to undergo similar C–O activations.
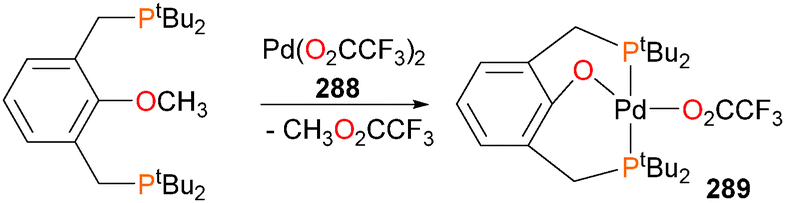 | ||
| Scheme 98 | ||
Mann and Hartwig authored one of the earliest reports of C–O reductive elimination from palladium(II) complexes in 1996.262 Shortly thereafter, Widenhoefer et al. published a more general study that demonstrated well-defined Caryl–O reductive elimination263 when they reported that a series of palladium(II) complexes of type 290 released aryl ethers upon gentle thermolysis (Scheme 99). Kinetic studies on the C–O reductive elimination of palladium(II) complexes of the type 290264 found that there is a rate enhancement for electron-withdrawing substituents on the palladium-bound aryl group, i.e. groups that can stabilize a buildup of negative charge during the transition state. The authors propose that this is likely indicative of a mechanism where the alkoxide ligand attacks the palladium-bound aryl group in an inner-sphere fashion to generate a zwitterionic Meisenheimer-type intermediate 291 before releasing free aryl ether and palladium(0) complex 292. Subsequent work found that use of a bulky, monodentate phosphine ligand allowed for the first catalytic formation of diaryl ethers,265 and it was found that steric bulk was the most important factor for C–O reductive eliminations in these systems.266 Related reductive eliminations of hydroxyl and aryl groups from palladium(II) have also been reported.267
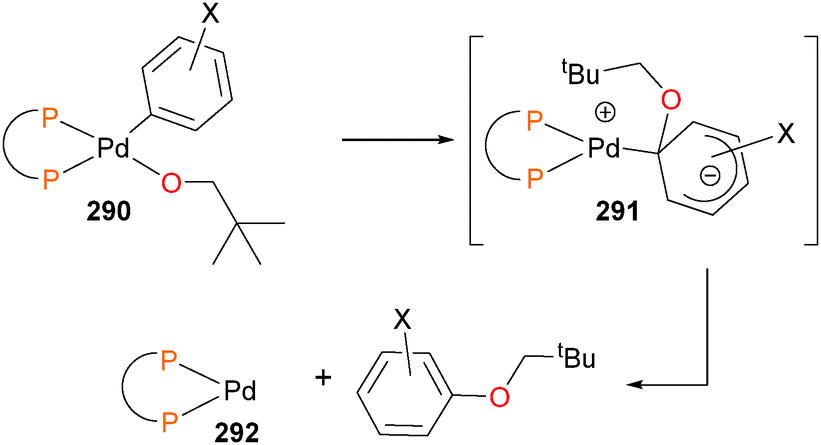 | ||
| Scheme 99 | ||
Williams and Goldberg have reported seminal work on the mechanism of C–O reductive elimination from platinum(IV) complexes 293.268–270 Dissociation of the alkoxide group from 293 generates a cationic five-coordinate intermediate 294, and the C–O bond forming step is likely the nucleophilic attack of alkoxide onto the electrophilic methyl group bound to platinum, resulting in reduction of platinum(IV) to platinum(II) and the formation of 295, as shown in Scheme 100. This type of nucleophilic attack was first demonstrated by Luinstra et al., who performed mechanistic studies on Shilov's methane oxidation system. It was found that platinum(IV) alkyls react with either water or chloride to form alcohols and alkyl chlorides, respectively. Deuterium labelling experiments found an inversion of sterochemical configuration at carbon, consistent with an SN2-type attack at the platinum-bound alkyl.271
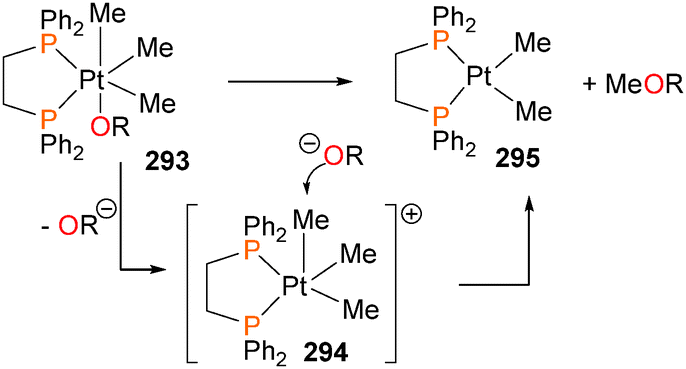 | ||
| Scheme 100 | ||
Sanford and co-workers have examined related chemistry with palladium,272,273 and found that for palladium complex 296, while C–O reductive elimination again proceeds through a five-coordinate intermediate (i.e.297), it is the palladium-bound carboxylate that couples with the phenylpyridyl ligand, resulting in direct reductive elimination at the metal centre, rather than nucleophilic attack of the alkoxide, that generates the C–O bond and palladium product 298 (Scheme 101). Direct C–O reductive elimination was demonstrated through several crossover experiments.274 It is worth noting that an earlier DFT study by a different group indicated that C–O reductive elimination occurred directly through the octahedral complex 296.275 Canty et al. have also explored C–O bond forming reactions with similar palladium complexes, but were unable to detect the palladium(IV) species responsible for C–O bond formation.276,277 The Sanford group has since expanded the scope this transformation to Csp3–O reductive elimination, and detailed mechanistic studies in this case point towards nucleophilic attack of alkoxide as the key C–O bond forming step.278
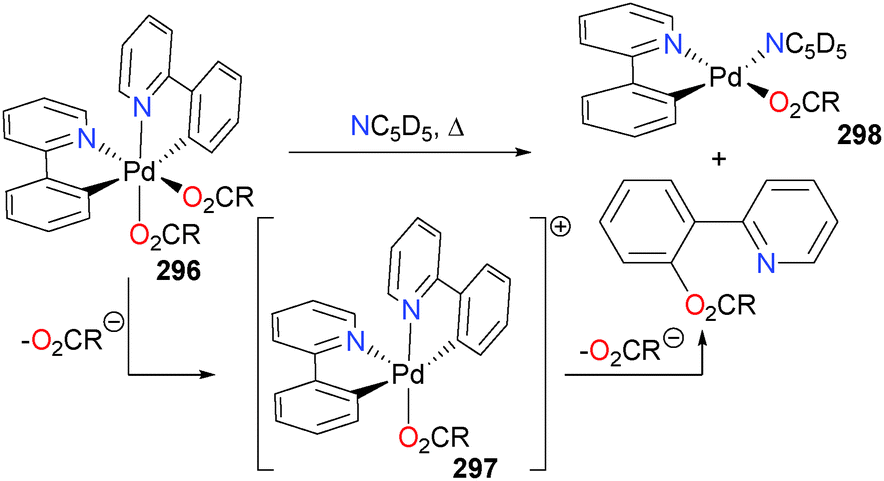 | ||
| Scheme 101 | ||
The first well-defined example of bimetallic carbon–heteroatom reductive elimination was reported by Powers and Ritter in a landmark study in 2009.279 The authors demonstrated that bimetallic palladium(III) complex 299, which is only stable at low temperatures but was nevertheless fully characterized, including by X-ray diffraction, produces aromatic acetate (see Scheme 102) in 64% yield via C–O reductive elimination.280–282 Labelling experiments demonstrate that both bridging and terminal acetates exchange at a rate much faster than C–O bond formation. Related bimetallic complexes of platinum(III) have been shown to be active in the oxidation of alkynes.283
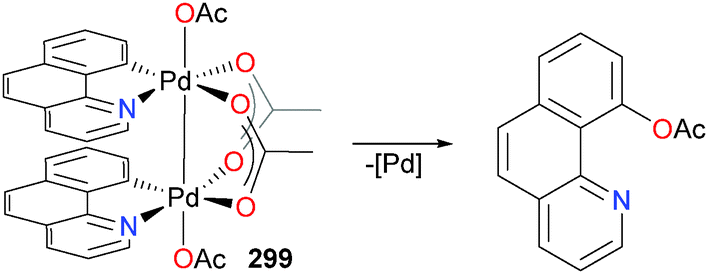 | ||
| Scheme 102 | ||
Recently, Camasso and Sanford284 have reported the synthesis and reactivity of a very rare example of a well-defined nickel(IV) complex 300 that can undergo carbon–heteroatom coupling reactions when treated with nucleophiles, as outlined in Scheme 103. Crucial to the reactivity observed is the ability of the tris(pyrazolyl)borate ligand to change binding modes from κ3 to κ2 upon reduction of the metal centre during the formation of product 301. Initial kinetic analyses found that the rate of the coupling reactions is first-order in both nickel species and NMe4X, which indicates an SN2-type mechanism of C–X bond formation. This proposal was also supported by a strong correlation between the initial rate of the coupling reaction and the Swain–Scott nucleophilicity parameters of the NMe4X reagent.
 | ||
| Scheme 103 | ||
Csp2–O bond formation has been shown to occur when cationic platinum alkyne complex 302 is treated with water to yield a 1:4 mixture of both the Markovnikov and anti-Markovnikov products, respectively (Scheme 104).285 Notably, the Markovnikov product 303 was much more susceptible to protonolysis than the acyl complex 304, reacting cleanly with HCl to produce ketones. Related water and/or alcohol additions to alkene complexes have also been reported.286
 | ||
| Scheme 104 | ||
The Sharp group has prepared platinum–oxo complex 305, and explored its reactivity with ethylene.287 Although the resulting reaction mixtures were complex,288 acetaldehyde was clearly identifiable as a major product. In an effort to isolate any potential intermediates in this oxidative transformation, ethylene was replaced with norbornene, and the resulting product characterized by NMR and X-ray diffraction as the binuclear platinaoxetane 306, formed in nearly quantitative yield. Furthermore, alkene insertion was found to be reversible,289 as treating 306 with norbornene derivatives, such as that shown in Scheme 105, gave the new platinaoxetane 307 in quantitative yield. Indeed, the reaction could also be pushed to completion in the opposite direction by adding a large excess of norbornene to 307 to reform 306. The kinetics of this exchange were monitored by NMR spectroscopy, and surprisingly, it was found that the reaction is zero-order in platinaoxetane 306. To test for the presence of a trace impurity that was catalyzing this exchange process, the rate of the reaction was examined as a function of initial concentration of 306, and a directly proportional increase in the rate of reaction relative to the initial concentration of 306 was observed. This confirmed the presence of some catalyst in the alkene exchange process. A screening of possible impurities was performed, and it was found that Lewis acids had a dramatic effect on the rate of the exchange reaction, with BF3OEt2 being the most efficient. The alkene exchange reaction occurred most rapidly when an excess of BF3 relative to platinum was used. Based on this, the authors propose that the actively exchanging species is a double Lewis acid adduct. The mono-BF3 adduct of 306 was later prepared and fully characterized.290 Further mechanistic studies also were consistent with protonations playing a key role in platinaoxetane formation.291
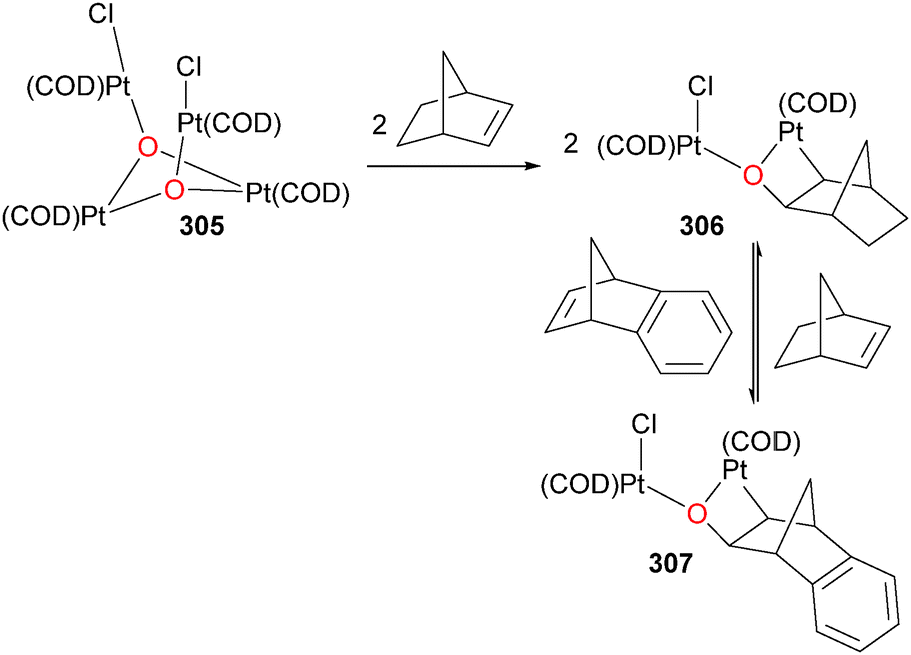 | ||
| Scheme 105 | ||
The reactivity of the related platinaoxetane 308 with small molecules was examined (Scheme 106),292,293 and it was found that complex 308 was amenable to insertion of a wide variety of electrophiles into the Pt–O bond to generate ring-expanded platinacycles. For instance, CO can insert into the Pt–O bond to form acyl–platinum complex 309. The electron-deficient alkyne DEAD also inserted cleanly to generate the platinadihydropyran 310, and similarly, tetracyanoethylene also inserted into the same bond. Reacting 308 with 2 equivalents of tBuNC first results in displacement of the COD ligand, yielding 311, while a third equivalent of isocyanide then inserts into the Pt–O bond to give the five-membered platinacycle 312. Lastly, reacting 308 with Br2 results in formation of the norbornene oxide via C–O bond formation, with 313 as the organometallic product, formed in quantitative yield.
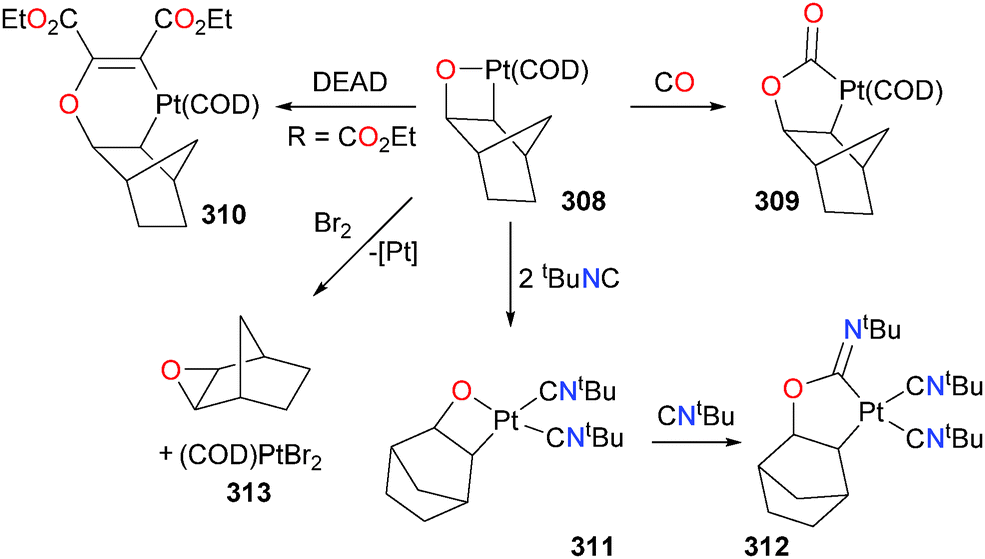 | ||
| Scheme 106 | ||
The Goldberg294 and Vedernikov295–297 groups, amongst others,298,299 have done significant work in the oxidation of group 10 metal hydrocarbyl complexes with oxidants such as dioxygen300–315 and H2O2.316–319 As these topics have recently been reviewed, they will not be covered in detail here.
Group 11 and 12 metals
The groups of Sadighi (see Scheme 107) and Marder have used NHC complexes of copper (i.e. complexes 314 and 315) for stoichiometric320–322 and catalytic functionalization of CO2,323 as well as the diborylation of aldehydes.324 Notably, insertion product 316, as well as the copper intermediates in the outlined catalytic cycles (complexes 317 and 318, respectively) have been characterized by X-ray crystallography. Related silver325 and gold clusters,326 also featuring NHC ligands, have also been used to fix CO2.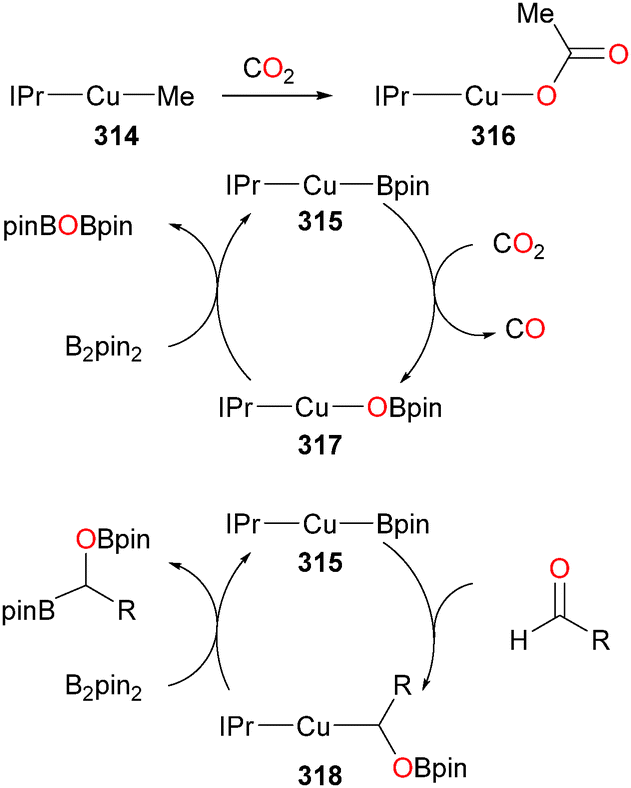 | ||
| Scheme 107 | ||
Binuclear gold–oxo complexes of the type 319 have been found to react with strained alkenes to generate oxygenated products.327 In the case of norbornene, auraoxetane 320 was isolated from the reaction mixture (Scheme 108). The organic products observed were a complex mixture of diols, aldehydes and an epoxide, all derived from norbornene.
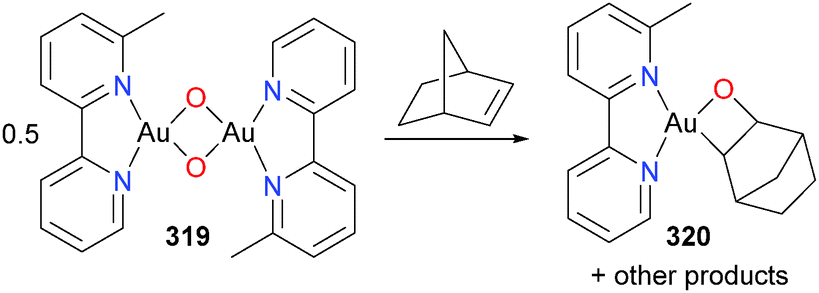 | ||
| Scheme 108 | ||
The Waterman group has reported that zinc(II) complex 319, when treated with 1 atm of CO2 over the course of 2 hours, yields 320, the formal insertion product of CO2 into the C–Si bond of the ligand backbone (Scheme 109).328 The product of insertion was characterized by X-ray diffraction studies, and preliminary kinetic data indicate that the reaction likely proceeds via stepwise insertion and rearrangement processes.
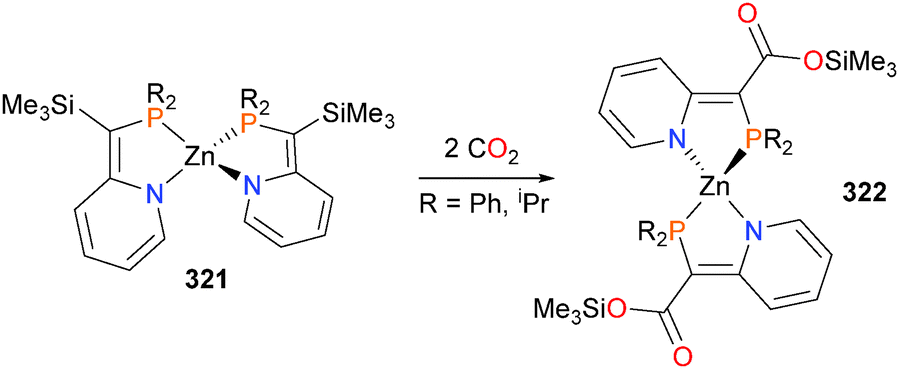 | ||
| Scheme 109 | ||
Summary
Group 8 alkoxo complexes can react with organic substrates via insertion–elimination mechanisms to form new products, as in the case of complex 160 and 167, or via nucleophilic attack of the donor atom, as seen for complex 164. Non-heme iron complexes such as 180 can undergo Csp2–F and Csp2–H hydroxylation, likely via radical pathways. Similarly, iron bis(imino)pyridine complexes can break Csp2–O bonds in esters via single electron processes.Low-valent iridium and rhodium complexes can ring-open epoxides via SN2-type mechanisms, and have been shown to undergo unexpected and non-trivial mechanisms of oxidation, as shown by Ritter et al.178 In addition, metal–ligand cooperativity can be exploited for the stereoselective alkynylation of phosphoramidates.228
The chemistry of nickel, platinum and palladium alkoxides is rich and varied. While nickel alkoxides can undergo oxidatively-induced reductive elimination to form Csp3–O bonds, heating them tends to favour β-hydride elimination followed by reductive elimination. Csp2–O oxidative addition from low-valent metals can be effected if the organic substrate is sufficiently reactive (such as esters) or if a directing group strategy is utilized. Copper has been successfully applied to the reduction of CO2 and aldehydes with B2pin2, while gold can be used to oxidize olefins to a variety of complexes, including epoxides, diols and the auraoxetane 320.
C–S bond cleavage and formation
Group 8 metals
Some of the earliest work on the activation of thiophene and other sulfur-containing organic molecules was done using group 8 metal carbonyl compounds (Scheme 110).329–335 For instance, refluxing benzothiophene with triiron dodecacarbonyl 323 yields the binuclear complex 324, resulting from oxidative addition of the C–S bond.336 | ||
| Scheme 110 | ||
Low-valent ruthenium complexes have also been demonstrated to cleave C–S bonds in thiophene derivatives. For instance, Borowski et al. have shown that reaction of 2-acetylthiophene with ruthenium bis(dihydrogen) complex 325 results in ring opening and hydrogenation of the thiophene moiety, forming 326 (Scheme 111).337 In addition, Komiya and co-workers have reported regioselective oxidative addition of substituted thiophenes when treated with ruthenium complex 327 and two equivalents of depe, yielding octahedral 328 complex as the major product.338
 | ||
| Scheme 111 | ||
Group 9 metals
Rheingold and co-workers have demonstrated preparation of three-legged piano stool complex 329via the oxidative addition of the C–S bond of alkynylsulfones with cobalt complex 330, as outlined in Scheme 112.339 Although not observed experimentally, the authors propose that the oxidative addition proceeds via the formation of an η2-alkyne complex. | ||
| Scheme 112 | ||
Jones et al. have described the reactivity of rhodium complex 331 with CS2.340 which gives a 4:1 mixture of η2-CS2 adduct 332 and dithioformate 334, which are separable by chromatography and were characterized by a variety of NMR techniques. Complex 333 can be treated with aniline for 15 days at 41 °C to yield a thioformamide (Scheme 113) upon workup by chromatopgraphy, likely produced from intermediate 334. Insertion of CS2 into related iridium hydrides has also been reported be Klein et al.,341 and the formation of thiocarbonyl compounds has also been observed elsewhere.342
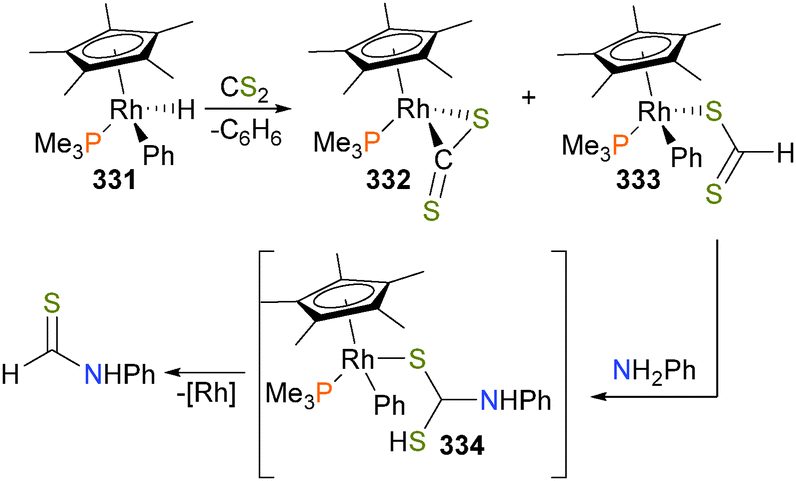 | ||
| Scheme 113 | ||
If a related dihydride rhodium(III) complex 335 is used as a starting material, sequential insertions occur to generate intermediate 336 followed by the bis(thiolate) complex 337.343 This is the first example of a reduction of CS2 to a methanedithiolate by a late transition metal complex. Immediately after the addition of CS2, the intermediate complex 336 can be observed by 1H NMR spectroscopy, but conversion to final product 337 is complete after minutes at room temperature (Scheme 114). The insertion of SO2 into group 9 alkyl and aryl complexes has also been reported.344
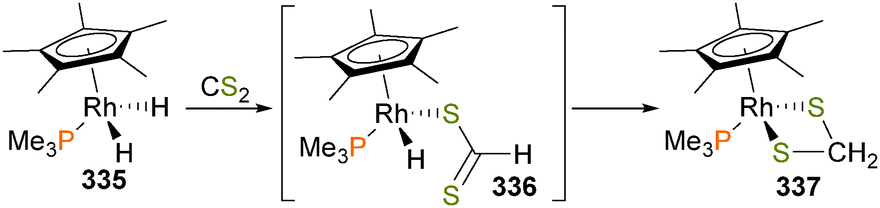 | ||
| Scheme 114 | ||
Heating complex 331 results in the elimination of benzene and the formation of a transient rhodium(I) species, which can undergo oxidative addition of the C–S bond of a variety of thiophenes (Scheme 115).345,346 Reductive elimination to regenerate the thiophene can be induced by heating complex 338 to 80 °C in the presence of PMe3. Based on mechanistic experiments, the authors propose that the process proceeds via coordination of the S-atom of thiophene to the metal centre prior to oxidative addition.347 Notably, a later DFT study found that S-atom coordination to Rh likely precedes an η2-C,S-bound intermediate which then undergoes subsequent C–S oxidative addition.348
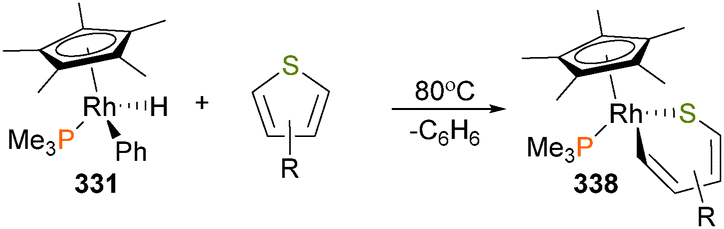 | ||
| Scheme 115 | ||
Interestingly, the use of an alternate rhodium precursor 339 resulted in the formation of the bimetallic complex 340, formed via C–C coupling after C–S oxidative addition (Scheme 116).349 The 1H NMR spectrum of complex 340 indicates that all eight of the methine protons are inequivalent and arranged in a linear array. This structure was confirmed by an X-ray diffraction study on a small crystal of 340.
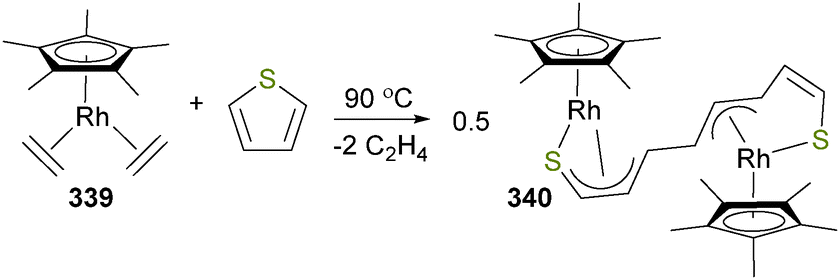 | ||
| Scheme 116 | ||
Regioselectivity of C–S oxidative addition in these Cp*rhodium complexes was found to be reversible350 and dictated primarily by steric factors.351 For instance, heating complex 331 with 2-methylbenzothiophene resulted in the formation of kinetic product 341, and then subsequent formation of the thermodynamic product 342 upon further heating, as shown in Scheme 117. Using a xylyl derivative of complex 331 as a starting material allowed the thermolysis to proceed under more mild conditions, and thus complexes 341 and 342 could be isolated independently and fully characterized. Kinetic experiments determined the conversion of 341 to 342 is first-order.
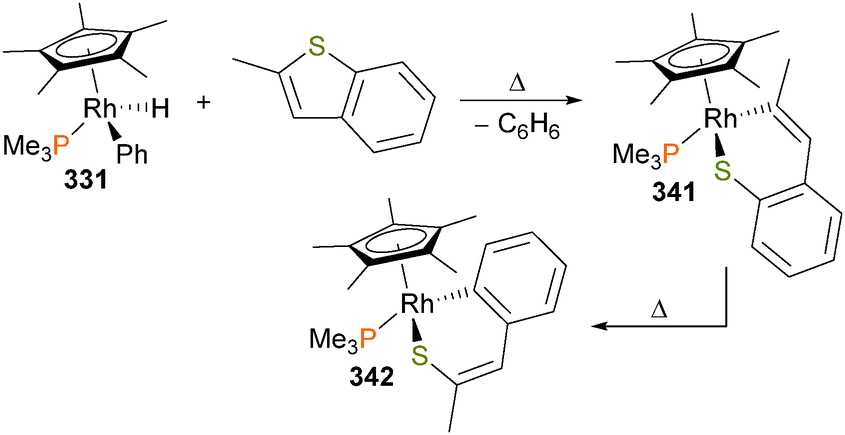 | ||
| Scheme 117 | ||
The cleavage of the C–S bond in thiophene can also be induced by treatment of the dicationic rhodium(III) complex 343 with two equivalents of base, which results in the cleavage of the C–S bond as well as the formation of a ketone group.352 This complex 344, was characterized by X-ray diffraction, and the reaction could be reversed by the addition of two equivalents of HOTf (Scheme 118). Thermolysis of complex 344 results in fragmentation of the remaining C–S bond and the release of tetramethylfuran as well as the tetrameric sulphide complex 345. This fragmentation process was later found to be unimolecular.353
 | ||
| Scheme 118 | ||
Trihydride complex 346 can also rupture the C–S bond of thiophene when refluxed in THF as solvent.354 The product 347 was assigned as a rhodium(III) metallacyclopropane based on the large coupling constants observed to 103Rh and 31P in the 13C NMR spectrum. Complex 347 could be protonated by using HBF4 or alkylated at the Rh–C bond using Ph3CPF6, forming complex 348 (Scheme 119). Treating 347 with MeI resulted instead in S-methylation.
 | ||
| Scheme 119 | ||
Binuclear rhodium hydride 349 reacts with the sterically protected 4,6-dimethyldibenzothiophene, which is one of the most difficult thiophene derivatives to activate.355 Thermolysis of dimer 349 in the presence of a large excess of 4,6-dimethyldibenzothiophene and 1 atm of H2 resulted in the formation of 350, isolated in 24% yield after chromatography on silica gel (Scheme 120).
 | ||
| Scheme 120 | ||
The bis(ethylene) complex 351 is the first reported example of a cobalt complex that can activate thiophene.356 Treatment of 351 with thiophene generates the bimetallic complex 352 (Scheme 121). Later studies found analogous reactivity with benzothiophene357 and dibenzothiophene.358 This binuclear motif of 352 has also been observed with rhodium.359,360
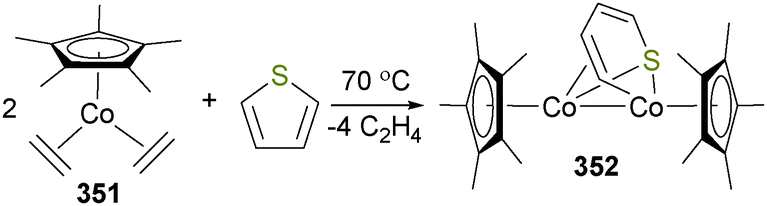 | ||
| Scheme 121 | ||
Iridium can also be used for the activation and desulfurization of thiophenes, as shown in Scheme 122.361–363 The reaction of iridium hydride complex 353 with thiophene in the presence of neohexene generates the binuclear bridging–sulfido complex 354, which can be isolated in 30% yield after purification by column chromatography.364 The 1,3-butadiene ligand can be displaced from the metal centres by the introduction of CO, or can be hydrogenated off with H2 to yield butane.
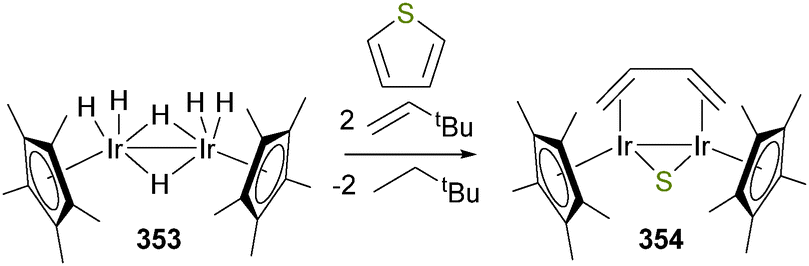 | ||
| Scheme 122 | ||
The activation of organic compounds bearing an SF5 group has recently been reported by the Braun group.365 Reacting rhodium dimer 355 with an equimolar amount of RSF5 (R = Ph, p-Tol, CF3) in the presence of an excess of HSiEt3 yields 356via S–F and C–S cleavage reactions. Mechanistic data indicates that the non-classical η2-silane complex 357 (Scheme 123) is an intermediate during this remarkable transformation.
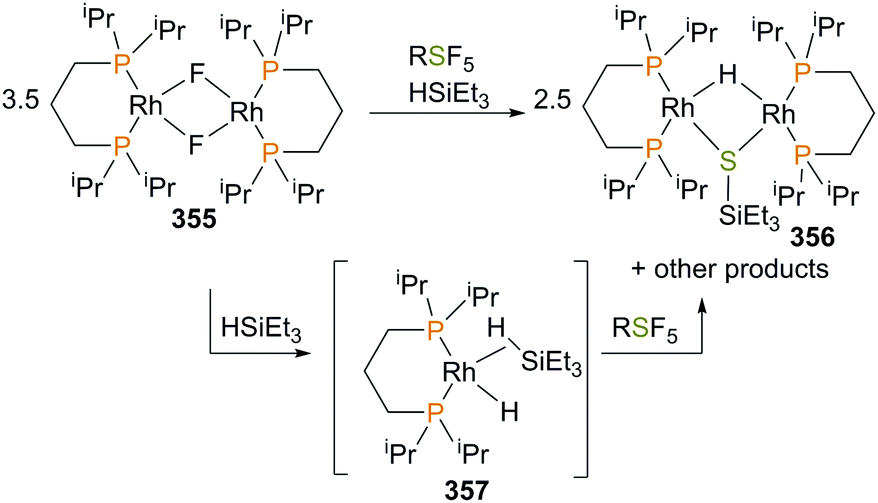 | ||
| Scheme 123 | ||
The Braun group has also reported that rhodium boryl complex 95 reacts rapidly with an equivalent of CS2 to form the thiocarbonyl complex 358, which is only stable in the presence of free PEt3. While a peak in the mass spectrum was observed for n = 2, a single, broad resonance was observed in the 31P NMR spectrum, along with no peak for free PEt3. Thus, the authors speculate that these data may be a sign of rapid exchange with free phosphine. Treating 358 with another equivalent of rhodium boryl 95 results in rapid precipitation of an orange product, which could be recrystallized and characterized as dinuclear complex 359, a remarkable bridging carbido complex of rhodium (Scheme 124).
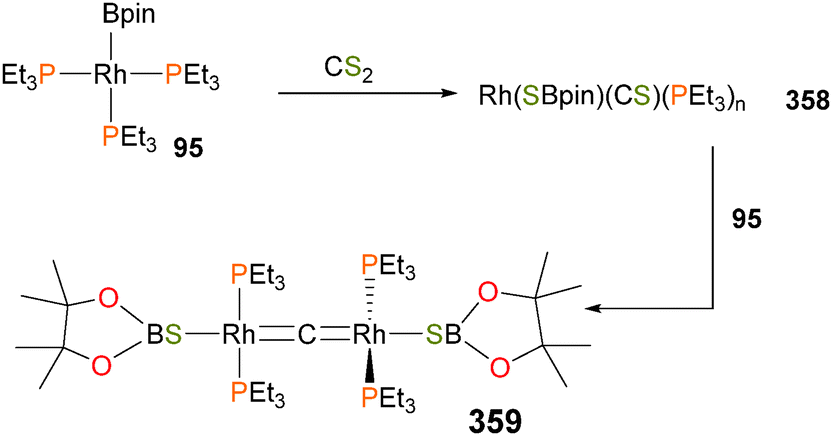 | ||
| Scheme 124 | ||
A report by Timpa et al. describes rhodium-catalyzed cross-coupling of aryl halides with thiols to generate aryl sulphides.366 The proposed mechanism of this transformation is described in Scheme 125. Reduction of precatalyst 360 with NaOtBu generates reactive intermediate 361, which undergoes oxidative addition of aryl halide to generate rhodium(III) complex 362. In the presence of thiol and base, transmetalation to form rhodium(III) thiolate 363 occurs, followed by C–S reductive elimination under thermolytic conditions to form rhodium(I) sulphide complex 364. Dissociation of sulphide from 364 regenerates the initial catalyst 361. Importantly, intermediates 363 and 364 were prepared independently and fully characterized, including X-ray diffraction studies.
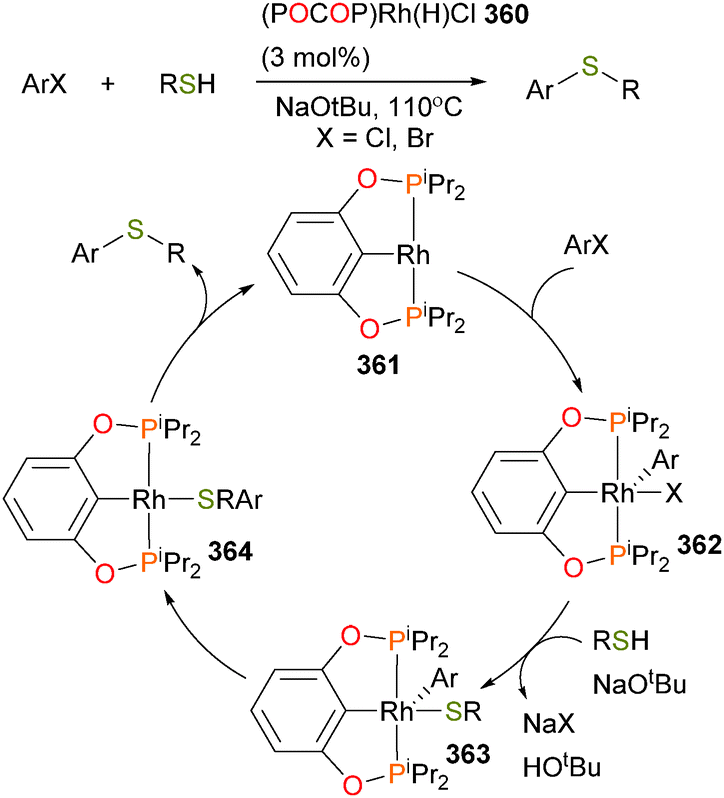 | ||
| Scheme 125 | ||
Group 10 metals
Yamamoto and co-workers described one of the earliest examples of reversible C–S oxidative addition and reductive elimination in 1986,367 when they reported that nickel(II) complexes 365 and 366 could interconvert via reversible oxidative addition and reductive elimination steps (Scheme 126). Variable temperature NMR studies revealed that 365 and 366 were in equilibrium. More recently, analogous reactivity using (dippe)palladium(0) has been reported by the Jones group.368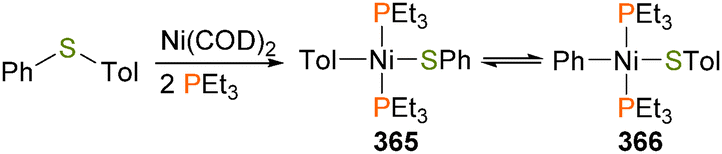 | ||
| Scheme 126 | ||
Campora et al. have reported that complex 254 reacts with one equivalent of CS2 to generate the dithialactone complex 367.369 Low-temperature NMR experiments revealed that the CS2 group inserts first into the Ni–Caryl bond prior to C–S reductive elimination. In addition, treating 367 with an additional two equivalents of CS2 yields Iber's salt 368 and releases the dithiolactone product (see Scheme 127).
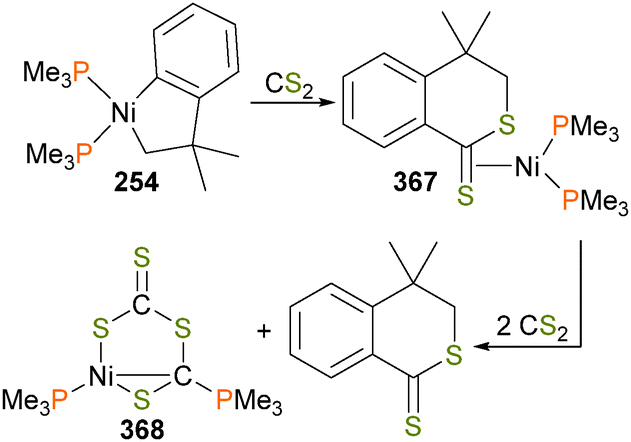 | ||
| Scheme 127 | ||
Vicic and Jones have shown that nickel(0) synthon 369 can reversibly activate the C–S bond of dibenzothiophene (forming complex 370), benzothiophene and thiophene, as shown in Scheme 128.370–372 On standing in THF for 5 days, complex 370 decomposes into a mixture of products, the bulk of which are equimolar amounts of complexes 371 and 372, derived from further C–S bond cleavage. Related reactivity has also been reported with platinum.373–378
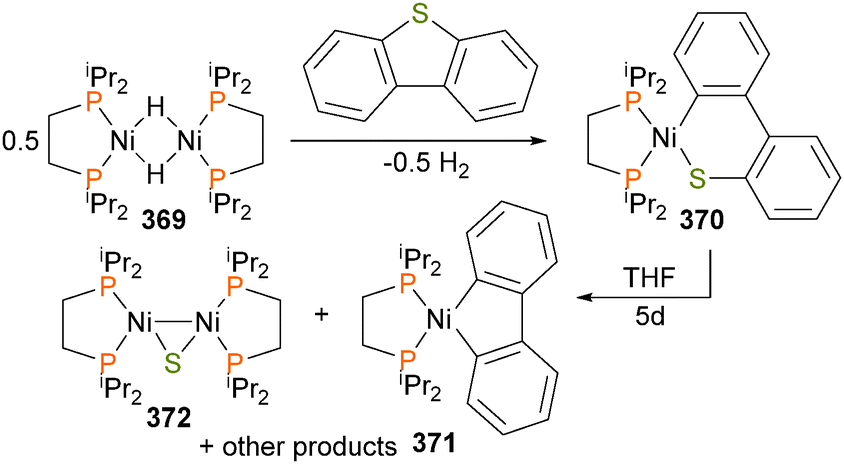 | ||
| Scheme 128 | ||
Curiously, when methyl-substituted dibenzothiophene complex 373 is dissolved in THF, a second C–S bond cleavage takes place to form the unexpected trinuclear379 complex 374 (Scheme 129). The location of the methyl group of the biphenyl moiety of 374 indicates that a C–H activation and migration event has also occurred.371 Other examples of hydrodesulfurization with group 10 complexes have been reported.380
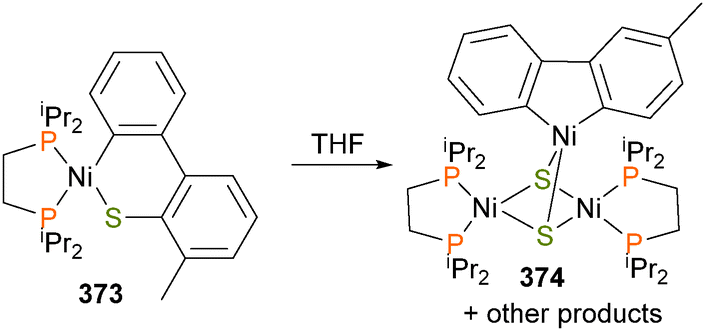 | ||
| Scheme 129 | ||
Vicic and Jones also reported one of the first examples of a transiently generated terminal sulfide of nickel.381 Mild heating of complex 375 resulted in the release of benzene and the quantitative formation of bridging sulphide dimer 376, likely formed via sulphide 317 (Scheme 130). If the same reaction took place in the presence of a nitrone, new product 378 was observed, characterized by NMR spectroscopy and X-ray diffraction studies.
 | ||
| Scheme 130 | ||
In 1994, Matsunaga et al. found that reacting the nickel(II) complex 379 with ethylene sulfide yielded the thianickelacyclobutane complex 380 after 7 hours at room temperature in THF (Scheme 131).382 The authors proposed that complex 379 first reductively eliminates butane to yield a reactive (bpy)nickel(0) complex, which can subsequently oxidatively add to the C–S bond of the thiirane to give the observed product. Consistent with this hypothesis, they found that using 118 as an alternate starting material allowed for the reaction to proceed under much milder conditions (30 minutes at 0 °C) with comparable yields. Expanding the substrate scope, the authors found that reaction of propylene sulfide with complex 118 resulted in a 6:1 mixture of isomers 381 and 382, indicating that steric interactions contribute appreciably to the site of oxidative addition.
 | ||
| Scheme 131 | ||
Han et al. have reported that elemental sulfur, S8, can oxidize nickel alkyl complexes bearing either bipyridine (107) or phosphines (254) as ancillary ligands (Scheme 132). The phosphine complex 383 can be converted to the bipyridine complex 384 by a simple ligand exchange reaction. Protonolysis of the Ni–C and Ni–S bonds with HCl yields aromatic thiol in 88% yield. Heating samples of 383 for 24 hours at 100 °C results in the formation of thioether via C–S reductive elimination. Initial mechanistic studies show that the rate of formation of thioether is faster in the presence of added phosphines. The authors propose that formation of an intermediate species, complex 385, which is detectable by NMR spectroscopy, thus directly precedes the C–S reductive elimination.383 Oxidation of platinum alkyls with S8 has also been reported.384,385
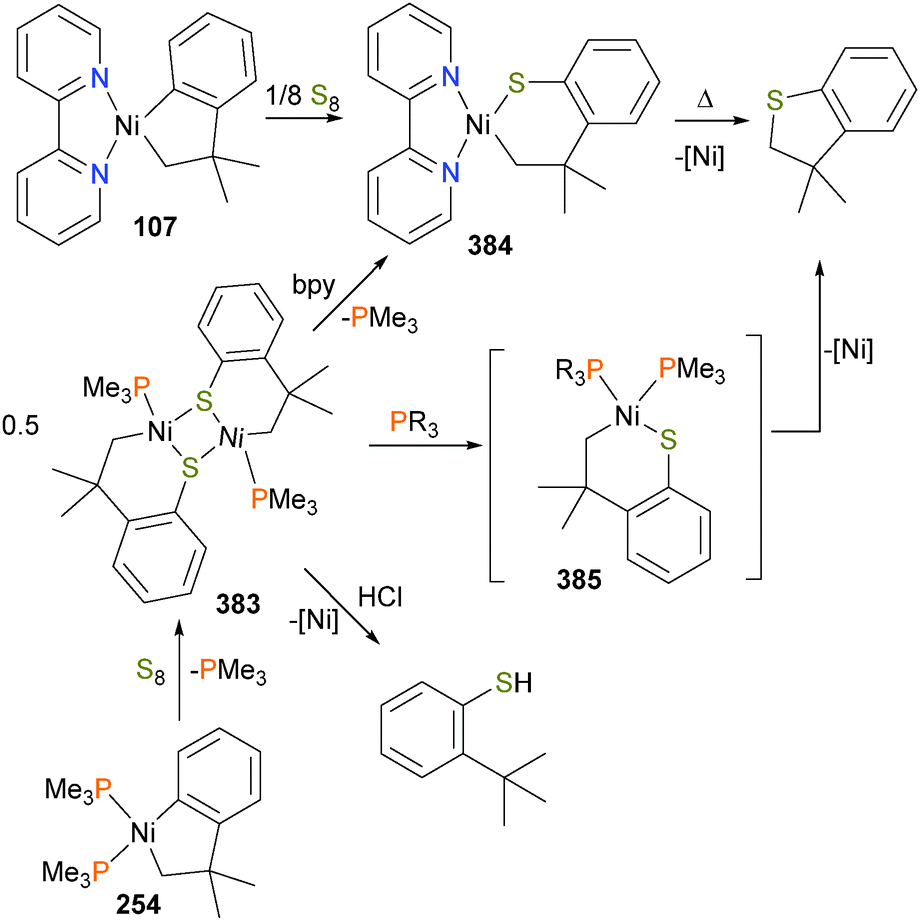 | ||
| Scheme 132 | ||
The Hartwig group has found that palladium(II) thiolate complexes, such as 386, undergo quite general C–S reductive elimination when heated (Scheme 133).386–388 Yields of the sulphide product were found to be highest when the reaction was performed in the presence of a suitable reagent (such as diphenylacetylene) to trap the palladium(0) product as 387, formed after reductive elimination. Detailed kinetic studies were performed, and the authors propose that reductive elimination occurs from a four-coordinate palladium(II) complex, and that coordination of a covalently-bound π-system (either from the sulphide or hydrocarbyl ligand) can serve to stabilize the transition state for reductive elimination.
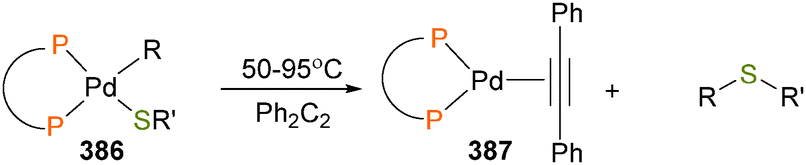 | ||
| Scheme 133 | ||
Inspired by Sanford (vide supra), Zhao and Dong have oxidized palladium(II) complex 388 with sulfonyl chlorides.389 Thermolyzing palladium(IV) complexes 389 in the presence of silver salts results in predominantly C–S reductive elimination to form aromatic sulfonyl products (Scheme 134), as well as the products of C–C and C–Cl reductive eliminations in minor amounts. Crossover experiments indicate that these reductive eliminations occur directly from palladium(IV).
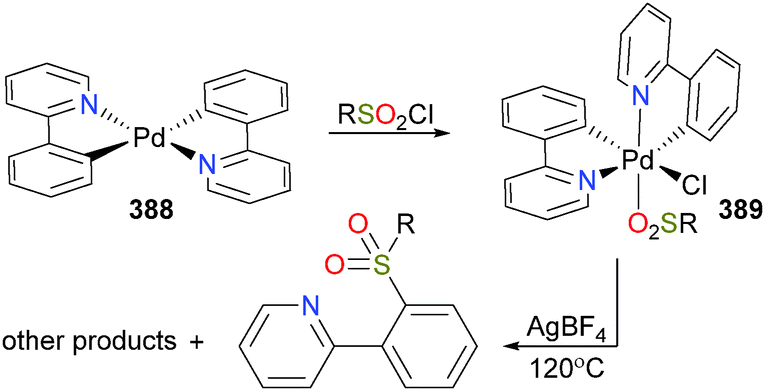 | ||
| Scheme 134 | ||
Kundu et al. have demonstrated that thioesters can also be activated with platinum(0) complexes,390 although high temperatures are required. Heating complex 278 with a 5-fold excess of thioester results in Cacyl–S bond cleavage. Subsequent heating of the platinum(II) complex 390 results in decarbonylation of the acyl ligand to generate a methyl thiolate complex 391, which can undergo further reaction with thioester to generate a bis(thiolate) complex 392 with concomitant release of acetone, as shown in Scheme 135. The same group has also examined similar reactivity of 278 with thioethers.391 Others have demonstrated related C–S oxidative addition reactions of sulfones with palladium392 and nickel.393
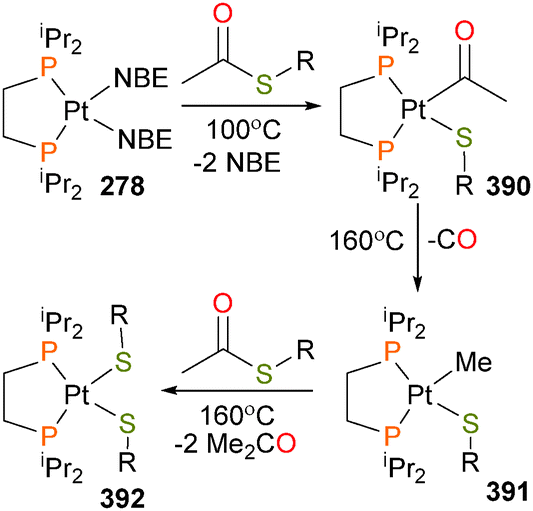 | ||
| Scheme 135 | ||
The insertion of SO2 into M–C bonds has been reviewed.394 An unusual example reported by Gates et al. results in the formation of a binuclear palladium complex 393, formed via insertion of SO2 into the Pd–Me bond of cationic 394 (Scheme 136).395
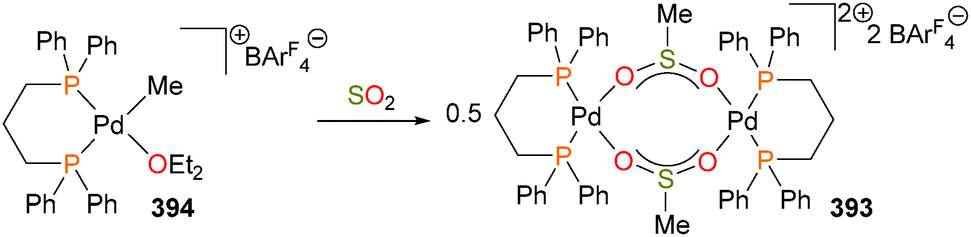 | ||
| Scheme 136 | ||
Group 11 and 12 metals
Well-defined examples of C–S bond formation with group 11 and 12 metals are rare, although there has been recent progress in the field of catalysis.396,397 Gold complex 395 has recently been used for the hydrothiolation of terminal alkynes with arylthiols, as outlined in Scheme 137. Intermediate organometallic complexes 396 and 397 have been isolated and fully characterized.398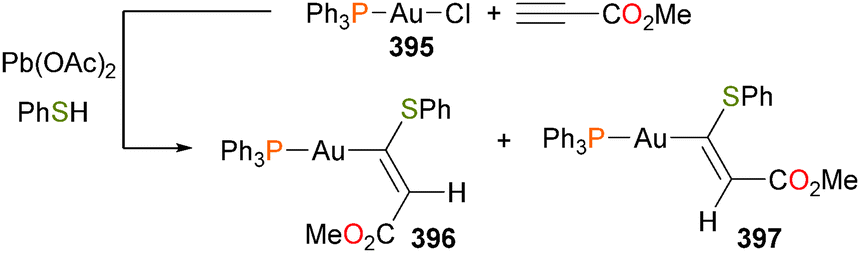 | ||
| Scheme 137 | ||
Miyamoto and co-workers have reported that Z-enethiolate complexes of gold can be used as cross-coupling reagents with aryl and alkyl halides (Scheme 138).399 TEMPO was found to be a valuable additive in these transformations, typically providing a boost in yield of the thioether product. These cross-couplings were shown to be stereoselective, as the Z-geometry of the thiolate is retained in the product.
 | ||
| Scheme 138 | ||
Summary
Hydrodesulfurization is an industrially important process, and many late transition metals have been shown to be capable of cleaving Csp2–S bonds under a range of conditions. Iron, rhodium, iridium and nickel are all capable of oxidative adding the Csp2–S bond of thiophene and thiophene derivatives, and in some cases subsequent C–S cleavage events also take place.CS2 is also prone to a myriad of reaction pathways. Rhodium hydrides such as 335 can insert the Csp2–S bond to make thiolate complexes, and more recently, rhodium boryl complex 95 has been shown to facilitate either 1 or 2 CS bond cleavage events, generating a RhCRh moiety in the case of the latter.
In terms of Csp2–S bond formation, Dong and co-workers have demonstrated reductive elimination upon heating complex 389, and the Ozerov group has extended this type of reactivity to the catalytic coupling of aryl halides and thiols.
Conclusions
The chemistry of late transition metals with carbon–heteroatom bonds is rich and varied, as demonstrated by the range of reactivity that has been described in the literature to date. We hope that the summary of reactivity that we have presented here will inspire synthetic chemists towards the development of new protocols that harness the fundamental chemistry of late transition metals.Abbreviations
| Ac | Acetyl, COMe |
| Ad | Adamantyl, C10H15 |
| Ar | Aryl |
| BArF4 | Tetrakis(3,5-bis(trifluoromethyl)phenyl)borate |
| Bn | Benzyl, CH2C6H5 |
| Boc | tert-Butyloxycarbonyl |
| bpy | 2,2′-Bipyridyl |
| BrettPhos | 2-(Dicyclohexylphosphino)-3,6-dimethoxy-2′,4′,6′-triisopropyl-1,1′-biphenyl |
| COD | 1,5-Cyclooctadiene |
| COT | 1,3,5,7-Cyclooctatetraene |
| Cp | Cyclopentadienyl, C5H5 |
| Cp* | Pentamethylcyclopentadienyl, C5Me5 |
| Cy | Cyclohexyl, C6H11 |
| DCM | Dichloromethane |
| depe | Bis(1,2-diethylphosphino)ethane |
| DFT | Density functional theory |
| Dipp | 2,6-Di-iso-propylphenyl |
| dmpe | Bis(1,2-dimethylphosphino)ethane |
| DPPBz | 1,2-Bis(diphenylphosphino)benzene |
| dppe | Bis(1,2-diphenylphosphino)ethane |
| DPPF | 1,1′-Bis(diphenylphosphino)ferrocene |
| dtbpe | Bis(1,2-di-tert-butylphosphine)ethane |
| Et | Ethyl, C2H5 |
| Et2O | Diethyl ether |
| Fc | Ferrocenium, Cp2Fe+ |
| HMDS | Hexamethyldizilazide, N(SiMe3)2 |
| iPr | Iso-propyl, C3H7 |
| Me | Methyl, CH3 |
| Mes | 2,4,6-Trimethylphenyl |
| NBE | Norbornene |
| NHC | N-Heterocyclic carbene |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser effect |
| Oct | Octyl |
| Ph | Phenyl, C6H5 |
| PhINTs | N-(p-Toluenesulfonyl)iminophenyliodinane |
| Pin | Pinacolato, OCMe2CMe2CO |
| p-Tol | para-Tolyl |
| Py | Pyridine, C5H4N |
| rt | Room temperature |
| t Bu | tert-Butyl |
| terpy | Terpyridine |
| Tf | Triflyl, SO2CF3 |
| TFA | Trifluoroacetic acid |
| TFE | 2,2,2-Trifluoroethanol |
| THF | Tetrahydrofuran |
| TMEDA | N,N,N′,N′-Tetramethylethylene-1,2-diamine |
| TMS | Trimethylsilyl, SiMe3 |
| Tol | Toluene, C7H8 |
| Ts | Tosyl, SO2-p-MeC6H4 |
| Xantphos | 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene |
| Xyl | 2,6-Dimethylphenyl |
Acknowledgements
We thank the University of British Columbia (Laird and 4YF to A. N. D.), NSERC (Discovery grant, Research Tools and Instrumentation grants; CGS-D3 and MSFSS to A. N. D.) and the Canada Foundation for Innovation for supporting our research. A. N. D. is grateful to the Izaak Walton Killam Foundation for a doctoral scholarship.Notes and references
- C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 Search PubMed.
- J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852–860 Search PubMed.
- J. F. Hartwig, Inorg. Chem., 2007, 46, 1936–1947 Search PubMed.
- P. S. Hanley and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8510–8525 Search PubMed.
- J. F. Hartwig, Nature, 2008, 455, 314–322 Search PubMed.
- V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 890–906, DOI:10.1021/acscatal.5b02021.
- J.-L. Renaud and S. Gaillard, Top. Organomet. Chem., 2015, 50, 83–144 Search PubMed.
- A. Corma, A. Leyva-Perez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 Search PubMed.
- R. Castarlenas, A. Di Giuseppe, J. J. Perez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211–222 Search PubMed.
- L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 Search PubMed.
- K. Ouyang, W. Hao, W. X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 Search PubMed.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 Search PubMed.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A. M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 Search PubMed.
- J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 Search PubMed.
- S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 Search PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1991, 113, 3404–3418 Search PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1991, 113, 6499–6508 Search PubMed.
- R. I. Michelman, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1991, 113, 5100–5102 Search PubMed.
- R. I. Michelman, R. G. Bergman and R. A. Andersen, Organometallics, 1993, 12, 2741–2751 Search PubMed.
- A. W. Holland and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 14684–14695 Search PubMed.
- D. J. Fox and R. G. Bergman, J. Am. Chem. Soc., 2003, 125, 8984–8985 Search PubMed.
- D. J. Fox and R. G. Bergman, Organometallics, 2004, 23, 1656–1670 Search PubMed.
- J. Zhang, T. B. Gunnoe and J. L. Petersen, Inorg. Chem., 2005, 44, 2895–2907 Search PubMed.
- D. Adhikari, F. Basuli, H. Fan, J. C. Huffman, M. Pink and D. J. Mindiola, Inorg. Chem., 2008, 4439–4441 Search PubMed.
- N. P. Mankad, P. Mueller and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 4083–4085 Search PubMed.
- E. R. King and T. A. Betley, Inorg. Chem., 2009, 48, 2361–2363 Search PubMed.
- J. T. Groves, J. Chem. Educ., 1985, 62, 928–931 Search PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 Search PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 Search PubMed.
- D. L. Broere, B. de Bruin, J. N. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574–11577 Search PubMed.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526 Search PubMed.
- T. Koreeda, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 7238–7239 Search PubMed.
- M. D. Seidler and R. G. Bergman, Organometallics, 1983, 2, 1897–1899 Search PubMed.
- M. D. Seidler and R. G. Bergman, J. Am. Chem. Soc., 1984, 106, 6110–6111 Search PubMed.
- M. R. Crimmin, R. G. Bergman and F. D. Toste, Angew. Chem., Int. Ed., 2011, 50, 4484–4487 Search PubMed.
- C. Zhao, M. R. Crimmin, F. D. Toste and R. G. Bergman, Acc. Chem. Res., 2014, 47, 517–529 Search PubMed.
- S. Takemoto, M. Kobayashi and H. Matsuzaka, J. Am. Chem. Soc., 2004, 126, 10802–10803 Search PubMed.
- S. Takemoto, S. Oshio, T. Kobayashi, H. Matsuzaka, M. Hoshi, H. Okimura, M. Yamshita, H. Miyasaka, T. Ishii and M. Yamashita, Organometallics, 2004, 23, 3587–3589 Search PubMed.
- W.-L. Man, W. W. Y. Lam and T.-C. Lau, Acc. Chem. Res., 2014, 47, 427–439 Search PubMed.
- W. L. Man, J. Xie, Y. Pan, W. W. Lam, H. K. Kwong, K. W. Ip, S. M. Yiu, K. C. Lau and T. C. Lau, J. Am. Chem. Soc., 2013, 135, 5533–5536 Search PubMed.
- W.-L. Man, W. W. Y. Lam, S.-M. Yiu, T.-C. Lau and S.-M. Peng, J. Am. Chem. Soc., 2004, 126, 15336–15337 Search PubMed.
- W. L. Man, J. Xie, P. K. Lo, W. W. Lam, S. M. Yiu, K. C. Lau and T. C. Lau, Angew. Chem., Int. Ed., 2014, 53, 8463–8466 Search PubMed.
- W. L. Man, W. W. Lam, H. K. Kwong, S. M. Yiu and T. C. Lau, Angew. Chem., Int. Ed., 2012, 51, 9101–9104 Search PubMed.
- H.-K. Kwong, W.-L. Man, J. Xiang, W.-T. Wong and T.-C. Lau, Inorg. Chem., 2009, 48, 3080–3086 Search PubMed.
- E. Kwan Huang, W.-M. Cheung, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Organometallics, 2013, 32, 733–736 Search PubMed.
- S. N. Brown, J. Am. Chem. Soc., 1999, 121, 9752–9753 Search PubMed.
- A. G. Maestri, S. D. Taylor, S. M. Schuck and S. N. Brown, Organometallics, 2004, 23, 1932–1946 Search PubMed.
- A. G. Maestri, K. S. Cherry, J. J. Toboni and S. N. Brown, J. Am. Chem. Soc., 2001, 123, 7459–7460 Search PubMed.
- C.-F. Leung, T.-W. Wong, T.-C. Lau and W.-T. Wong, Eur. J. Inorg. Chem., 2005, 773–778 Search PubMed.
- T. J. Crevier, B. K. Bennett, J. D. Soper, J. A. Bowman, A. Dehestani, D. A. Hrovat, S. Lovell, W. Kaminsky and J. M. Mayer, J. Am. Chem. Soc., 2001, 123, 1059–1071 Search PubMed.
- J. D. Soper, E. Saganic, D. Weinberg, D. A. Hrovat, J. B. Benedict, W. Kaminsky and J. M. Mayer, Inorg. Chem., 2004, 43, 5804–5815 Search PubMed.
- S. B. Munoz, 3rd, W. T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2015, 54, 10600–10603 Search PubMed.
- W. T. Lee, R. A. Juarez, J. J. Scepaniak, S. B. Munoz, 3rd, D. A. Dickie, H. Wang and J. M. Smith, Inorg. Chem., 2014, 53, 8425–8430 Search PubMed.
- J. J. Scepaniak, J. A. Young, R. P. Bontchev and J. M. Smith, Angew. Chem., Int. Ed., 2009, 48, 3158–3160 Search PubMed.
- J. M. Smith, D. E. Mayberry, C. G. Margarit, J. Sutter, H. Wang, K. Meyer and R. P. Bontchev, J. Am. Chem. Soc., 2012, 134, 6516–6519 Search PubMed.
- T. M. Powers, A. R. Fout, S. L. Zheng and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 3336–3338 Search PubMed.
- P. N. Becker, M. A. White and R. G. Bergman, J. Am. Chem. Soc., 1980, 102, 5676–5677 Search PubMed.
- W. P. Weiner, M. A. White and R. G. Bergman, J. Am. Chem. Soc., 1981, 103, 3612–3614 Search PubMed.
- W. P. Weiner and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3922–3929 Search PubMed.
- A. L. Casalnuovo, J. C. Calabrese and D. Milstein, J. Am. Chem. Soc., 1988, 110, 6738–6744 Search PubMed.
- D. P. Klein, J. C. Hayes and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 3704–3706 Search PubMed.
- W. D. McGhee, T. Foo, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8543–8545 Search PubMed.
- D. S. Glueck, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 2719–2721 Search PubMed.
- D. S. Glueck, J. Wu, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1991, 113, 2041–2054 Search PubMed.
- D. M. Tellers, J. C. M. Ritter and R. G. Bergman, Inorg. Chem., 1999, 38, 4810–4818 Search PubMed.
- J. R. Fulton, A. W. Holland, D. J. Fox and R. G. Bergman, Acc. Chem. Res., 2002, 35, 44–56 Search PubMed.
- D. Rais and R. G. Bergman, Chem. – Eur. J., 2004, 10, 3970–3978 Search PubMed.
- A. W. Holland and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 9010–9011 Search PubMed.
- P. Zhao and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 11618–11619 Search PubMed.
- P. Zhao, C. Krug and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 12066 Search PubMed.
- C. Tejel, M. A. Ciriano, S. Jimenez, V. Passarelli and J. A. Lopez, Inorg. Chem., 2008, 47, 10220–10222 Search PubMed.
- Y.-W. Ge and P. R. Sharp, Organometallics, 1988, 7, 2234–2236 Search PubMed.
- Y.-W. Ge and P. R. Sharp, Inorg. Chem., 1992, 31, 379–384 Search PubMed.
- M. Rahim, C. White, A. L. Rheingold and K. J. Ahmed, Organometallics, 1993, 12, 2401–2403 Search PubMed.
- S. Takemoto, T. Honma and H. Matsuzaka, Organometallics, 2011, 30, 1013–1020 Search PubMed.
- I. Mena, M. A. Casado, V. Polo, P. Garcia-Orduna, F. J. Lahoz and L. A. Oro, Angew. Chem., Int. Ed., 2014, 53, 9627–9631 Search PubMed.
- K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 Search PubMed.
- S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492–2502 Search PubMed.
- A. Dauth and J. A. Love, Angew. Chem., Int. Ed., 2012, 51, 3634–3637 CrossRef CAS PubMed.
- B. de Bruin, P. H. Budzelaar and A. W. Gal, Angew. Chem., Int. Ed., 2004, 43, 4142–4157 Search PubMed.
- A. Dauth, M. W. Drover, T. Hajiashrafi, A. A. Tehrani and J. A. Love, J. Organomet. Chem., 2015, 791, 192–197 Search PubMed.
- A. Dauth, C. Rigling, J. Tsoung and J. A. Love, Chem. – Eur. J., 2013, 19, 17180–17191 Search PubMed.
- M. W. Drover, D. W. Beh, P. Kennepohl and J. A. Love, Chem. – Eur. J., 2014, 20, 13345–13355 Search PubMed.
- S. I. Kallane, T. Braun, M. Teltewskoi, B. Braun, R. Herrmann and R. Laubenstein, Chem. Commun., 2015, 51, 14613–14616 Search PubMed.
- S. I. Kallane, T. Braun, B. Braun and S. Mebs, Dalton Trans., 2014, 43, 6786–6801 Search PubMed.
- M. T. Whited, L. Qiu, A. J. Kosanovich and D. E. Janzen, Inorg. Chem., 2015, 54, 3670–3679 Search PubMed.
- M. T. Whited, A. J. Kosanovich and D. E. Janzen, Organometallics, 2014, 33, 1416–1422 Search PubMed.
- O. V. Ozerov, C. Guo, V. A. Papkov and B. M. Foxman, J. Am. Chem. Soc., 2004, 126, 4792–4793 Search PubMed.
- W. Weng, C. Guo, C. Moura, L. Yang, B. M. Foxman and O. V. Ozerov, Organometallics, 2005, 24, 3487–3499 Search PubMed.
- P. Surawatanawong and O. V. Ozerov, Organometallics, 2011, 30, 2972–2979 Search PubMed.
- G. S. Lemen and J. P. Wolfe, Top. Organomet. Chem., 2012, 46, 1–53 Search PubMed.
- L. A. Villanueva, K. A. Abboud and J. M. Boncella, Organometallics, 1994, 13, 3921–3931 Search PubMed.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 4708–4709 Search PubMed.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 7217–7218 Search PubMed.
- M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 8232–8245 Search PubMed.
- M. Yamashita, J. V. C. Vicario and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 16347–16360 Search PubMed.
- Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7734–7735 Search PubMed.
- G. Mann, J. F. Hartwig, M. S. Driver and C. Fernandez-Rivas, J. Am. Chem. Soc., 1998, 120, 827–828 Search PubMed.
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2090–2093 Search PubMed.
- K.-I. Fujita, M. Yamashita, F. Puschmann, M. M. Alvarez-Falcon, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9044–9045 Search PubMed.
- A. L. Monteiro and W. M. Davis, J. Braz. Chem. Soc., 2004, 15, 83–95 Search PubMed.
- S. L. Marquard, D. C. Rosenfeld and J. F. Hartwig, Angew. Chem., Int. Ed. Engl., 2010, 49, 793–796 Search PubMed.
- P. S. Hanley, S. L. Marquard, T. R. Cundari and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 15281–15284 Search PubMed.
- K. Koo and G. L. Hillhouse, Organometallics, 1995, 14, 4421–4423 Search PubMed.
- K. Koo and G. L. Hillhouse, Organometallics, 1996, 15, 2669–2671 Search PubMed.
- P. T. Matsunaga, C. R. Hess and G. L. Hillhouse, J. Am. Chem. Soc., 1994, 116, 3665–3666 Search PubMed.
- D. D. VanderLende, K. A. Abboud and J. M. Boncella, Inorg. Chem., 1995, 34, 5319–5326 Search PubMed.
- J. M. Boncella and L. A. Villanueva, J. Organomet. Chem., 1994, 465, 297–304 Search PubMed.
- L. A. Villanueva, K. A. Abboud and J. M. Boncella, Organometallics, 1992, 11, 2963–2965 Search PubMed.
- P. L. Holland, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1996, 118, 1092–11004 Search PubMed.
- S. Nieto, P. Arnau, E. Serrano, R. Navarro, T. Soler, C. Cativiela and E. P. Urriolabeitia, Inorg. Chem., 2009, 48, 11963–11975 Search PubMed.
- B. L. Lin, C. R. Clough and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 2890–2891 Search PubMed.
- C.-Y. Huang and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 9541–9544 Search PubMed.
- K. L. Jensen, E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2014, 136, 11145–11152 Search PubMed.
- S. Zhang, J. Wei, M. Zhan, Q. Luo, C. Wang, W. X. Zhang and Z. Xi, J. Am. Chem. Soc., 2012, 134, 11964–11967 Search PubMed.
- J. E. Ney and J. P. Wolfe, J. Am. Chem. Soc., 2006, 128, 15415–15422 Search PubMed.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2001, 123, 4623–4624 Search PubMed.
- D. J. Mindiola and G. L. Hillhouse, Chem. Commun., 2002, 1840–1841 Search PubMed.
- R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350–13351 Search PubMed.
- D. J. Mindiola, R. Waterman, V. M. Iluc, T. R. Cundari and G. L. Hillhouse, Inorg. Chem., 2014, 53, 13227–13238 Search PubMed.
- C. A. Laskowski, A. J. M. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771–773 Search PubMed.
- C. A. Laskowski and G. L. Hillhouse, Organometallics, 2009, 28, 6114–6120 Search PubMed.
- N. D. Harrold and G. L. Hillhouse, Chem. Sci., 2013, 4, 4011 Search PubMed.
- N. P. Mankad, W. E. Antholine, R. K. Szilagyi and J. C. Peters, J. Am. Chem. Soc., 2009, 131, 3878–3880 Search PubMed.
- B. A. Jazdzewski, P. L. Holland, M. Pink, V. G. Young, D. J. E. Spencer and W. B. Tolman, Inorg. Chem., 2001, 40, 6097–6107 Search PubMed.
- S. B. Harkins, N. P. Mankad, A. J. M. Miller, R. K. Szilagyi and J. C. Peters, J. Am. Chem. Soc., 2008, 130, 3478–3485 Search PubMed.
- Y.-W. Ge, Y. Ye and P. R. Sharp, J. Am. Chem. Soc., 1994, 116, 8384–8385 Search PubMed.
- G. Bai and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 1856–1859 Search PubMed.
- A. R. Dick, M. S. Remy, J. W. Kampf and M. S. Sanford, Organometallics, 2007, 26, 1365–1370 Search PubMed.
- A. V. Pawlikowski, A. D. Getty and K. I. Goldberg, J. Am. Chem. Soc., 2007, 129, 10382–10393 Search PubMed.
- A. Iglesias and K. Muniz, Helv. Chim. Acta, 2012, 95, 2007–2025 Search PubMed.
- K. Muniz, C. H. Hovelmann and J. Streuff, J. Am. Chem. Soc., 2008, 130, 763–773 Search PubMed.
- M. Ohashi, I. Takeda, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 18018–18021 Search PubMed.
- M. Ohashi, Y. Hoshimoto and S. Ogoshi, Dalton Trans., 2015, 44, 12060–12073 Search PubMed.
- V. Vreeken, M. A. Siegler, B. de Bruin, J. N. Reek, M. Lutz and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 7055–7059 Search PubMed.
- Y. Xie, J. Hu, Y. Wang, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 20613–20616 Search PubMed.
- L. Fan, L. Yang, C. Guo, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 4778–4787 Search PubMed.
- O. V. Ozerov, C. Guo, L. Fan and B. M. Foxman, Organometallics, 2004, 23, 5573–5580 Search PubMed.
- Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732–3735 Search PubMed.
- K. T. Sylvester, K. Wu and A. G. Doyle, J. Am. Chem. Soc., 2012, 134, 16967–16970 Search PubMed.
- F. E. Michael and B. M. Cochran, J. Am. Chem. Soc., 2006, 128, 4246–4247 Search PubMed.
- B. M. Cochran and F. E. Michael, J. Am. Chem. Soc., 2008, 130, 2786–2792 Search PubMed.
- C. Hahn, P. Morvillo and A. Vitagliano, Eur. J. Inorg. Chem., 2001, 419–429 Search PubMed.
- C. Hahn, P. Morvillo, E. Herdtweck and A. Vitagliano, Organometallics, 2002, 21, 1807–1818 Search PubMed.
- C. F. Bender, T. J. Brown and R. A. Widenhoefer, Organometallics, 2016, 35, 113–125 Search PubMed.
- P. Cao, J. Cabrera, R. Padilla, D. Serra, F. Rominger and M. Limbach, Organometallics, 2012, 31, 921–929 Search PubMed.
- A. Béthegnies, J.-C. Daran and R. Poli, Organometallics, 2013, 32, 673–681 Search PubMed.
- C. M. Kelly, D. H. Kwon, M. J. Ferguson, S. M. Bischof, O. L. Sydora, D. H. Ess, M. Stradiotto and L. Turculet, Angew. Chem., Int. Ed., 2015, 54, 14498–14502 Search PubMed.
- G. R. Owen, R. Vilar, A. J. P. White and D. J. Williams, Organometallics, 2003, 22, 4511–4521 Search PubMed.
- C. Munro-Leighton, S. A. Delp, E. D. Blue and T. B. Gunnoe, Organometallics, 2007, 26, 1483–1493 Search PubMed.
- E. D. Blue, A. Davis, D. Conner, T. B. Gunnoe, P. D. Boyle and P. S. White, J. Am. Chem. Soc., 2003, 125, 9435–9441 Search PubMed.
- L. A. Goj, E. D. Blue, S. A. Delp, T. B. Gunnoe, T. R. Cundari, A. W. Pierpont, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2006, 45, 9032–9045 Search PubMed.
- C. Boone, I. Korobkov and G. I. Nikonov, ACS Catal., 2013, 3, 2336–2340 Search PubMed.
- M. W. Johnson, S. L. Shevick, F. D. Toste and R. G. Bergman, Chem. Sci., 2013, 4, 1023–1027 Search PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1990, 112, 3234–3236 Search PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, Organometallics, 1991, 10, 3344–3362 Search PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, Organometallics, 1991, 10, 3326–3344 Search PubMed.
- A. W. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072–5085 Search PubMed.
- M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343–11345 Search PubMed.
- S. R. Caskey, M. H. Stewart, J. E. Kivela, J. R. Sootsman, M. J. A. Johnson and J. W. Kampf, J. Am. Chem. Soc., 2005, 127, 16750–16751 Search PubMed.
- Y. Mutoh, N. Kozono, M. Araki, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2010, 29, 519–522 Search PubMed.
- M. H. Stewart, M. J. A. Johnson and J. W. Kampf, Organometallics, 2007, 26, 5102–5110 Search PubMed.
- S. Takemoto, J. Ohata, K. Umetani, M. Yamaguchi and H. Matsuzaka, J. Am. Chem. Soc., 2014, 136, 15889–15892 Search PubMed.
- S. Ueno, E. Mizushima, N. Chatani and F. Kakiuchi, J. Am. Chem. Soc., 2006, 128, 16516–16517 Search PubMed.
- R. J. Trovitch, E. Lobkovsky, M. W. Bouwkamp and P. J. Chirik, Organometallics, 2008, 27, 6264–6278 Search PubMed.
- S. Sahu, M. G. Quesne, C. G. Davies, M. Durr, I. Ivanovic-Burmazovic, M. A. Siegler, G. N. Jameson, S. P. de Visser and D. P. Goldberg, J. Am. Chem. Soc., 2014, 136, 13542–13545 Search PubMed.
- S. Sahu, B. Zhang, C. J. Pollock, M. Durr, C. G. Davies, A. M. Confer, I. Ivanovic-Burmazovic, M. A. Siegler, G. N. Jameson, C. Krebs and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 12791–12802 Search PubMed.
- G. de Ruiter, N. B. Thompson, M. K. Takase and T. Agapie, J. Am. Chem. Soc., 2016, 138, 1486–1489 Search PubMed.
- D. Milstein, J. Am. Chem. Soc., 1982, 104, 5227–5228 Search PubMed.
- D. Milstein and J. C. Calabrese, J. Am. Chem. Soc., 1982, 104, 3773–3774 Search PubMed.
- M. J. Calhorda, A. M. Galvao, C. Unaleroglu, A. A. Zlota, F. Frolow and D. M. Milstein, Organometallics, 1993, 12, 3316–3325 Search PubMed.
- Y. Z. Han, M. S. Sanford, M. D. England and J. T. Groves, Chem. Commun., 2006, 549–551 Search PubMed.
- R. G. Bergman, Polyhedron, 1995, 14, 3227–3237 Search PubMed.
- L. J. Newman and R. G. Bergman, J. Am. Chem. Soc., 1985, 107, 5314–5315 Search PubMed.
- D. S. Glueck, L. J. Newman Winslow and R. G. Bergman, Organometallics, 1991, 10, 1462–1479 Search PubMed.
- H. F. Luecke and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 11008–11009 Search PubMed.
- K. A. Woerpel and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 7888–7889 Search PubMed.
- J. C. M. Ritter and R. G. Bergman, J. Am. Chem. Soc., 1997, 119, 2580–2581 Search PubMed.
- J. C. M. Ritter and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 6826–6827 Search PubMed.
- D. P. Klein and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 3079–3080 Search PubMed.
- M. T. Whited and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 5874–5875 Search PubMed.
- M. T. Whited and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 16476–16477 Search PubMed.
- M. T. Whited and R. H. Grubbs, Organometallics, 2009, 28, 161–166 Search PubMed.
- M. T. Whited and R. H. Grubbs, Acc. Chem. Res., 2009, 42, 1607–1616 Search PubMed.
- M. T. Whited, Y. Zhu, S. D. Timpa, C.-H. Chen, B. M. Foxman, O. V. Ozerov and R. H. Grubbs, Organometallics, 2009, 28, 4560–4570 Search PubMed.
- A. W. Holland, D. S. Glueck and R. G. Bergman, Organometallics, 2001, 20, 2250–2261 Search PubMed.
- A. W. Holland and R. G. Bergman, Inorg. Chim. Acta, 2002, 341, 99–106 Search PubMed.
- B. E. Hauger, J. C. Huffman and K. G. Caulton, Organometallics, 1996, 15, 1856–1864 Search PubMed.
- D. G. H. Hetterscheid, J. M. M. Smits and B. de Bruin, Organometallics, 2004, 23, 4236–4246 Search PubMed.
- W. I. Dzik, J. M. M. Smits, J. N. Reek and B. de Bruin, Organometallics, 2009, 28, 1631–1643 Search PubMed.
- T. Ghatak, M. Sarkar, S. Dinda, I. Dutta, S. M. Rahaman and J. K. Bera, J. Am. Chem. Soc., 2015, 137, 6168–6171 Search PubMed.
- K. J. H. Young, O. A. Mironov and R. A. Periana, Organometallics, 2007, 26, 2137–2140 Search PubMed.
- M. P. del Rio, M. A. Ciriano and C. Tejel, Angew. Chem., Int. Ed., 2008, 47, 2502–2505 Search PubMed.
- C. Tejel, M. A. Ciriano, E. Sola, M. P. del Rio, G. Rios-Moreno, F. J. Lahoz and L. A. Oro, Angew. Chem., Int. Ed., 2005, 44, 3267–3271 Search PubMed.
- O. Blum and D. Milstein, J. Am. Chem. Soc., 1995, 117, 4582–4594 Search PubMed.
- T. C. Flood, M. Iimura, J. M. Perotti, A. L. Rheingold and T. E. Concolino, Chem. Commun., 2000, 1681–1682 Search PubMed.
- V. W. Day, W. G. Klemperer, S. P. Lockledge and D. J. Main, J. Am. Chem. Soc., 1990, 112, 2031–2033 Search PubMed.
- V. W. Day, T. A. Eberspacher, W. G. Klemperer and B. Zhong, J. Am. Chem. Soc., 1994, 116, 3119–3120 Search PubMed.
- B. de Bruin, M. J. Boerakker, J. J. J. M. Donners, B. E. C. Christiaans, P. P. J. Schlebos, R. de Gelder, J. M. M. Smits, A. L. Spek and A. W. Gal, Angew. Chem., Int. Ed. Engl., 1997, 36, 2064–2067 Search PubMed.
- M. Krom, R. G. E. Coumans, J. M. M. Smits and A. W. Gal, Angew. Chem., Int. Ed., 2001, 40, 2106–2108 Search PubMed.
- A. Dauth and J. A. Love, Angew. Chem., Int. Ed., 2010, 49, 9219–9224 Search PubMed.
- A. N. Desnoyer, A. Dauth, B. O. Patrick and J. A. Love, Dalton Trans., 2014, 43, 30–33 Search PubMed.
- B. de Bruin, M. J. Boerakker, J. A. W. Verhagen, R. de Gelder, J. M. M. Smits and A. W. Gal, Chem. – Eur. J., 2000, 6, 298–312 Search PubMed.
- Y. Yamamoto, X.-H. Han and J.-F. Ma, Angew. Chem., Int. Ed., 2000, 39, 1965–1968 Search PubMed.
- Y. Yamamoto, K. Sugawara and X.-H. Han, J. Chem. Soc., Dalton Trans., 2002, 195–211 Search PubMed.
- A. N. Desnoyer, S. Behyan, B. O. Patrick, A. Dauth, J. A. Love and P. Kennepohl, Inorg. Chem., 2016, 55, 13–15 Search PubMed.
- C. Tejel, M. A. Ciriano and V. Passarelli, Chem. – Eur. J., 2011, 17, 91–95 Search PubMed.
- M. R. Castillo, M. Martin, J. M. Fraile, J. A. Mayoral and E. Sola, Angew. Chem., Int. Ed., 2011, 50, 3240–3243 Search PubMed.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 Search PubMed.
- A. Bittner, T. Braun, R. Herrmann and S. Mebs, Chem. – Eur. J., 2015, 21, 12299–12302 Search PubMed.
- M. Ahijado, T. Braun, D. Noveski, N. Kocher, B. Neumann, D. Stalke and H. G. Stammler, Angew. Chem., Int. Ed., 2005, 44, 6947–6951 Search PubMed.
- C. Tejel, M. A. Ciriano, M. P. del Rio, F. J. van den Bruele, D. G. H. Hetterscheid, N. T. I. Spithas and B. de Bruin, J. Am. Chem. Soc., 2008, 130, 5844–5845 Search PubMed.
- P. Zhao, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9642–9643 Search PubMed.
- C. Krug and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 1674–1679 Search PubMed.
- C. Krug and J. F. Hartwig, Organometallics, 2004, 23, 4594–4607 Search PubMed.
- P. Zhao, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 3124–3125 Search PubMed.
- M. W. Drover, L. L. Schafer and J. A. Love, Dalton Trans., 2015, 44, 19487–19493 Search PubMed.
- J. Choi, Y. Choliy, X. Zhang, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2009, 131, 15627–15629 Search PubMed.
- S. Kundu, J. Choi, D. Y. Wang, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2013, 135, 5127–5143 Search PubMed.
- L. L. Santos, K. Mereiter and M. Paneque, Organometallics, 2013, 32, 565–569 Search PubMed.
- P. Lara, M. Paneque, M. L. Poveda, V. Salazar, L. L. Santos and E. Carmona, J. Am. Chem. Soc., 2006, 128, 3512–3513 Search PubMed.
- S. Conejero, M. Paneque, M. L. Poveda, L. L. Santos and E. Carmona, Acc. Chem. Res., 2010, 43, 572–580 Search PubMed.
- Y. Zhu, D. A. Smith, D. E. Herbert, S. Gatard and O. V. Ozerov, Chem. Commun., 2012, 48, 218–220 Search PubMed.
- M. E. van der Boom, S.-Y. Liou, Y. Ben-David, A. Vigalok and D. Milstein, Angew. Chem., Int. Ed. Engl., 1997, 36, 625–626 Search PubMed.
- M. E. Van der Boom, S.-Y. Liou, Y. Ben-David, L. J. Shimon and D. Milstein, J. Am. Chem. Soc., 1998, 120, 6531–6541 Search PubMed.
- L. E. Doyle, W. E. Piers and J. Borau-Garcia, J. Am. Chem. Soc., 2015, 137, 2187–2190 Search PubMed.
- L. E. Doyle, W. E. Piers, J. Borau-Garcia, M. J. Sgro and D. M. Spasyuk, Chem. Sci., 2016, 7, 921–931 Search PubMed.
- M. W. Drover, J. A. Love and L. L. Schafer, J. Am. Chem. Soc., 2016, 138, 8396–8399 Search PubMed.
- H. E. Bryndza, J. C. Calabrese and S. S. Wreford, Organometallics, 1984, 3, 1603–1604 Search PubMed.
- H. E. Bryndza, Organometallics, 1985, 4, 406–408 Search PubMed.
- T. L. Lohr, W. E. Piers and M. Parvez, Dalton Trans., 2013, 42, 14742–14748 Search PubMed.
- E. Carmona, E. Gutierrez-Puebla, J. M. Marin, A. Monge, M. Paneque, M. L. Poveda and C. Ruiz, J. Am. Chem. Soc., 1989, 111, 2883–2891 Search PubMed.
- P. T. Matsunaga and G. L. Hillhouse, J. Am. Chem. Soc., 1993, 115, 2075–2077 Search PubMed.
- P. T. Matsunaga, J. C. Mavropoulos and G. L. Hillhouse, Polyhedron, 1995, 14, 175–185 Search PubMed.
- T. Yamamoto, J. Ishizu, T. Kohara, S. Komiya and A. Yamamoto, J. Am. Chem. Soc., 1980, 102, 3758–3764 Search PubMed.
- Y.-J. Kim, K. Osakada, K. Sugita, T. Yamamoto and A. Yamamoto, Organometallics, 1988, 7, 2182–2188 Search PubMed.
- W. Zhou, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2015, 137, 7604–7607 Search PubMed.
- K. Koo and G. L. Hillhouse, Organometallics, 1998, 17, 2924–2925 Search PubMed.
- K. Koo, G. L. Hillhouse and A. L. Rheingold, Organometallics, 1995, 14, 456–460 Search PubMed.
- R. Han and G. L. Hillhouse, J. Am. Chem. Soc., 1997, 119, 8135–8136 Search PubMed.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 9976–9977 Search PubMed.
- I. Matas, J. Cámpora, P. Palma and E. Álvarez, Organometallics, 2009, 28, 6515–6523 Search PubMed.
- N. D. Harrold, R. Waterman, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2009, 131, 12872–12873 Search PubMed.
- J. S. Anderson, V. M. Iluc and G. L. Hillhouse, Inorg. Chem., 2010, 49, 10203–10207 Search PubMed.
- A. N. Desnoyer, E. G. Bowes, B. O. Patrick and J. A. Love, J. Am. Chem. Soc., 2015, 137, 12748–12751 Search PubMed.
- M. G. Beaver and T. F. Jamison, Org. Lett., 2011, 13, 4140–4143 Search PubMed.
- K. A. Manbeck, S. Kundu, A. P. Walsh, W. W. Brennessel and W. D. Jones, Organometallics, 2012, 31, 5018–5024 Search PubMed.
- A. N. Desnoyer, F. W. Friese, W. Chiu, M. W. Drover, B. O. Patrick and J. A. Love, Chem. – Eur. J., 2016, 22, 4070–4077 Search PubMed.
- K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 169–172 Search PubMed.
- K. Muto, J. Yamaguchi, A. Lei and K. Itami, J. Am. Chem. Soc., 2013, 135, 16384–16387 Search PubMed.
- H. Xu, K. Muto, J. Yamaguchi, C. Zhao, K. Itami and D. G. Musaev, J. Am. Chem. Soc., 2014, 136, 14834–14844 Search PubMed.
- M. L. Lejkowski, R. Lindner, T. Kageyama, G. E. Bodizs, P. N. Plessow, I. B. Muller, A. Schafer, F. Rominger, P. Hofmann, C. Futter, S. A. Schunk and M. Limbach, Chem. – Eur. J., 2012, 18, 14017–14025 Search PubMed.
- Y. Hoshimoto, M. Ohashi and S. Ogoshi, Acc. Chem. Res., 2015, 48, 1746–1755 Search PubMed.
- S. Ogoshi, K.-I. Tonomori, M.-A. Oka and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 7077–7086 Search PubMed.
- S. Ogoshi, M. Nagata and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 5350–5351 Search PubMed.
- T. Tamaki, M. Nagata, M. Ohashi and S. Ogoshi, Chem. – Eur. J., 2009, 15, 10083–10091 Search PubMed.
- M. Ohashi, T. Taniguchi and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 14900–14903 Search PubMed.
- R. Doi, K. Kikushima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 3276–3282 Search PubMed.
- D. K. Nielsen and A. G. Doyle, Angew. Chem., Int. Ed., 2011, 50, 6056–6059 Search PubMed.
- H. Weissman, L. J. W. Shimon and D. Milstein, Organometallics, 2004, 23, 3931–3940 Search PubMed.
- M. E. van der Boom, S.-Y. Liou, L. J. W. Shimon, Y. Ben-David and D. Milstein, Inorg. Chim. Acta, 2004, 357, 4015–4023 Search PubMed.
- G. Mann and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 13109–13110 Search PubMed.
- R. A. Widenhoefer, H. A. Zhong and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 6787–6795 Search PubMed.
- R. A. Widenhoefer and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6504–6511 Search PubMed.
- G. Mann, C. D. Incarvito, A. L. Rheingold and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 3224–3225 Search PubMed.
- G. Mann, Q. Shelby, A. H. Roy and J. F. Hartwig, Organometallics, 2003, 22, 2775–2789 Search PubMed.
- Y. Hayashi, S. Wada, M. Yamashita and K. Nozaki, Organometallics, 2012, 1073–1081 Search PubMed.
- B. S. Williams, A. W. Holland and K. I. Goldberg, J. Am. Chem. Soc., 1999, 121, 252–253 Search PubMed.
- B. S. Williams and K. I. Goldberg, J. Am. Chem. Soc., 2001, 123, 2576–2587 CrossRef CAS PubMed.
- N. A. Smythe, K. A. Grice, B. S. Williams and K. I. Goldberg, Organometallics, 2009, 28, 277–288 Search PubMed.
- G. A. Luinstra, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1993, 115, 3004–3005 Search PubMed.
- A. R. Dick, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790–12791 Search PubMed.
- J. B. Gary and M. S. Sanford, Organometallics, 2011, 30, 6143–6149 Search PubMed.
- J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974–10983 Search PubMed.
- Y. Fu, Z. Li, S. Liang, Q.-X. Guo and L. Liu, Organometallics, 2008, 27, 3736–3742 Search PubMed.
- A. J. Canty, M. C. Denney, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 1122–1131 Search PubMed.
- A. J. Canty, M. C. Denney, G. van Koten, B. W. Skelton and A. H. White, Organometallics, 2004, 23, 5432–5439 Search PubMed.
- N. M. Camasso, M. H. Perez-Temprano and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 12771–12775 Search PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 Search PubMed.
- D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050–17051 Search PubMed.
- D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14530–14536 Search PubMed.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2011, 45, 840–850 Search PubMed.
- M. Ochiai, Y.-S. Lin, J. Yamada, H. Misawa, S. Arai and K. Matsumoto, J. Am. Chem. Soc., 2004, 126, 2536–2545 Search PubMed.
- N. M. Camasso and M. S. Sanford, Science, 2015, 347, 1218–1220 Search PubMed.
- S. Carlisle, A. Matta, H. Valles, J. B. Bracken, M. Miranda, J. Yoo and C. Hahn, Organometallics, 2011, 30, 6446–6457 Search PubMed.
- C. Hahn, Organometallics, 2010, 29, 1331–1338 Search PubMed.
- E. Szuromi, H. Shan and P. R. Sharp, J. Am. Chem. Soc., 2003, 125, 10522–10523 Search PubMed.
- N. M. Weliange and P. R. Sharp, Organometallics, 2012, 31, 6823–6833 Search PubMed.
- E. Szuromi, J. Wu and P. R. Sharp, J. Am. Chem. Soc., 2006, 128, 12088–12089 Search PubMed.
- J. Wu and P. R. Sharp, Organometallics, 2008, 27, 1234–1241 CrossRef CAS.
- N. M. Weliange, E. Szuromi and P. R. Sharp, J. Am. Chem. Soc., 2009, 131, 8736–8737 Search PubMed.
- J. Wu and P. R. Sharp, Organometallics, 2008, 27, 4810–4816 Search PubMed.
- J. Wu and P. R. Sharp, Organometallics, 2009, 28, 6935–6943 Search PubMed.
- L. Boisvert and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 899–910 Search PubMed.
- A. N. Vedernikov, Acc. Chem. Res., 2012, 45, 803–813 Search PubMed.
- A. N. Vedernikov, Top. Organomet. Chem., 2010, 31, 101–121 Search PubMed.
- A. N. Vedernikov, Chem. Commun., 2009, 4781–4790 Search PubMed.
- A. R. Petersen, R. A. Taylor, I. Vicente-Hernandez, P. R. Mallender, H. Olley, A. J. White and G. J. Britovsek, J. Am. Chem. Soc., 2014, 136, 14089–14099 Search PubMed.
- A. T. Higgs, P. J. Zinn and M. S. Sanford, Organometallics, 2010, 29, 5446–5449 Search PubMed.
- M. L. Scheuermann and K. I. Goldberg, Chem. – Eur. J., 2014, 20, 14556–14568 Search PubMed.
- J. D. Prantner, W. Kaminsky and K. I. Goldberg, Organometallics, 2014, 33, 3227–3230 Search PubMed.
- M. L. Scheuermann, D. W. Boyce, K. A. Grice, W. Kaminsky, S. Stoll, W. B. Tolman, O. Swang and K. I. Goldberg, Angew. Chem., Int. Ed., 2014, 53, 6492–6495 Search PubMed.
- M. L. Scheuermann, U. Fekl, W. Kaminsky and K. I. Goldberg, Organometallics, 2010, 29, 4749–4751 Search PubMed.
- K. A. Grice and K. I. Goldberg, Organometallics, 2009, 28, 953–955 Search PubMed.
- M. L. Scheuermann, A. T. Luedtke, S. K. Hanson, U. Fekl, W. Kaminsky and K. I. Goldberg, Organometallics, 2013, 32, 4752–4758 Search PubMed.
- L. Boisvert, M. C. Denney, S. K. Hanson and K. I. Goldberg, J. Am. Chem. Soc., 2009, 131, 15802–15814 Search PubMed.
- V. V. Rostovtsev, J. A. Labinger, J. E. Bercaw, T. L. Lasseter and K. I. Goldberg, Organometallics, 1998, 17, 4530–4531 Search PubMed.
- A. V. Sberegaeva, P. Y. Zavalij and A. N. Vedernikov, J. Am. Chem. Soc., 2016, 138, 1446–1455 Search PubMed.
- W. G. Liu, A. V. Sberegaeva, R. J. Nielsen, W. A. Goddard, 3rd and A. N. Vedernikov, J. Am. Chem. Soc., 2014, 136, 2335–2341 Search PubMed.
- A. V. Sberegaeva, W. G. Liu, R. J. Nielsen, W. A. Goddard, 3rd and A. N. Vedernikov, J. Am. Chem. Soc., 2014, 136, 4761–4768 Search PubMed.
- J. R. Khusnutdinova, A. S. Maiorana, P. Y. Zavalij and A. N. Vedernikov, Inorg. Chim. Acta, 2011, 369, 274–283 Search PubMed.
- J. Zhang, E. Khaskin, N. P. Anderson, P. Y. Zavalij and A. N. Vedernikov, Chem. Commun., 2008, 3625–3627 Search PubMed.
- J. R. Khusnutdinova, L. L. Newman, P. Y. Zavalij, Y.-F. Lam and A. N. Vedernikov, J. Am. Chem. Soc., 2008, 130, 2174 Search PubMed.
- J. R. Khusnutdinova, P. Y. Zavalij and A. N. Vedernikov, Organometallics, 2007, 26, 2402–2413 Search PubMed.
- A. N. Vedernikov, S. A. Binfield, P. Y. Zavalij and J. R. Khusnutdinova, J. Am. Chem. Soc., 2006, 128, 82–83 Search PubMed.
- W. N. Oloo, P. Y. Zavalij and A. N. Vedernikov, Organometallics, 2013, 32, 5601–5614 Search PubMed.
- S. Pal and A. N. Vedernikov, Dalton Trans., 2012, 41, 8116–8122 RSC.
- J. R. Khusnutdinova, L. L. Newman and A. N. Vedernikov, J. Organomet. Chem., 2011, 696, 3998–4006 Search PubMed.
- W. Oloo, P. Y. Zavalij, J. Zhang, E. Khaskin and A. N. Vedernikov, J. Am. Chem. Soc., 2010, 132, 14400–14402 Search PubMed.
- N. P. Mankad, T. G. Gray, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 1191–1193 Search PubMed.
- C. M. Wyss, B. K. Tate, J. Bacsa, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2013, 52, 12920–12923 Search PubMed.
- C. Kleeberg, M. S. Cheung, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 19060–19063 CrossRef CAS PubMed.
- D. S. Laitar, P. Muller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 Search PubMed.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 1289, 11036–11037 Search PubMed.
- B. K. Tate, C. M. Wyss, J. Bacsa, K. Kluge, L. Gelbaum and J. P. Sadighi, Chem. Sci., 2013, 4, 3068 Search PubMed.
- T. J. Robilotto, J. Bacsa, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2012, 51, 12077–12080 Search PubMed.
- M. A. Cinellu, G. Minghetti, F. Cocco, S. Stoccoro, A. Zucca and M. Manassero, Angew. Chem., Int. Ed., 2005, 44, 6892–6895 Search PubMed.
- G. I. McGrew, P. A. Khatri, W. E. Geiger, R. A. Kemp and R. Waterman, Chem. Commun., 2015, 51, 15804–15807 Search PubMed.
- R. B. King and F. G. A. Stone, J. Am. Chem. Soc., 1960, 82, 4557–4562 Search PubMed.
- H. D. Kaesz, R. B. King, T. A. Manuel, A. D. Nichols and F. G. A. Stone, J. Am. Chem. Soc., 1960, 82, 4749–4750 CrossRef CAS.
- P. Hubener and E. Weiss, J. Organomet. Chem., 1977, 129, 105–115 Search PubMed.
- A. E. Ogilvy, M. Draganjac, T. B. Rauchfuss and S. R. Wilson, Organometallics, 1988, 7, 1171–1177 Search PubMed.
- A. J. Arce, Y. De Sanctis, A. Karam and A. J. Deeming, Angew. Chem., Int. Ed. Engl., 1994, 33, 1381–1383 CrossRef.
- A. J. Arce, A. Karam, Y. De Sanctis, M. V. Capparelli and A. J. Deeming, Inorg. Chim. Acta, 1999, 285, 277–282 CrossRef CAS.
- P. Mathur, S. Boodida, R. S. Ji and S. M. Mobin, J. Organomet. Chem., 2009, 694, 3043–3045 Search PubMed.
- R. B. King, P. M. Treichel and F. G. A. Stone, J. Am. Chem. Soc., 1961, 83, 3600–3604 Search PubMed.
- A. F. Borowski, S. Sabo-Etienne, B. Donnadieu and B. Chaudret, Organometallics, 2003, 22, 4803–4809 CrossRef CAS.
- J. G. Planas, M. Hirano and S. Komiya, Chem. Commun., 1999, 1793–1794 Search PubMed.
- J. M. O'Connor, K. D. Bunker, A. L. Rheingold and L. Zakharoc, J. Am. Chem. Soc., 2005, 127, 4180–4181 Search PubMed.
- W. D. Jones, V. L. Chandler and F. J. Feher, Organometallics, 1990, 9, 164–174 Search PubMed.
- D. P. Klein, G. M. Kloster and R. G. Bergman, J. Am. Chem. Soc., 1990, 112, 2022–2024 CrossRef CAS.
- A. B. Rivas, J. M. Gascon, F. J. Lahoz, A. I. Balana, A. J. Pardey, L. A. Oro and J. J. Perez-Torrente, Inorg. Chem., 2008, 47, 6090–6104 Search PubMed.
- W. D. Jones and A. D. Selmeczy, Organometallics, 1992, 11, 889–893 Search PubMed.
- L. Lefort, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 1420–1425 Search PubMed.
- W. D. Jones and L. Dong, J. Am. Chem. Soc., 1991, 113, 559–564 Search PubMed.
- W. D. Jones, D. A. Vicic, R. M. Chin, J. H. Roache and A. W. Myers, Polyhedron, 1997, 16, 3115–3128 Search PubMed.
- L. Dong, S. B. Duckett, K. F. Ohman and W. D. Jones, J. Am. Chem. Soc., 1992, 114, 151 Search PubMed.
- T. A. Atesin and W. D. Jones, Organometallics, 2008, 27, 3666–3670 Search PubMed.
- R. M. Chin and W. D. Jones, Angew. Chem., Int. Ed. Engl., 1992, 31, 357–358 Search PubMed.
- A. W. Myers, W. D. Jones and S. M. McClements, J. Am. Chem. Soc., 1995, 117, 11704–11709 Search PubMed.
- A. W. Myers and W. D. Jones, Organometallics, 1996, 15, 2905–2917 Search PubMed.
- A. E. Skaugset, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 1992, 114, 8521–8526 Search PubMed.
- Q. Feng, H. Krautscheid, T. B. Rauchfuss, A. E. Skaugset and A. Venturelli, Organometallics, 1995, 14, 297–304 Search PubMed.
- C. Bianchini, P. Frediani, V. Herrera, M. V. Jimenez, A. Meli, L. Rincon, R. Sanchez-Delgado and F. Vizza, J. Am. Chem. Soc., 1995, 117, 4333–4346 Search PubMed.
- S. S. Oster, M. R. Grochowski, R. J. Lachicotte, W. W. Brennessel and W. D. Jones, Organometallics, 2010, 29, 4923–4931 Search PubMed.
- W. D. Jones and R. M. Chin, Organometallics, 1992, 11, 2698–2700 Search PubMed.
- N. H. Chan, J. H. Roache and W. D. Jones, Inorg. Chim. Acta, 2015, 437, 36–40 Search PubMed.
- W. D. Jones and R. M. Chin, J. Organomet. Chem., 1994, 472, 311–316 Search PubMed.
- S. T. H. Willems, P. H. Budzelaar, N. N. P. Moonen, R. de Gelder, J. M. M. Smits and A. W. Gal, Chem. – Eur. J., 2002, 8, 1310–1320 Search PubMed.
- S. Luo, A. E. Skaugset, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 1992, 114, 1732–1735 Search PubMed.
- C. Bianchini, A. Meli, M. Peruzzini, F. Vizza, S. Moneti, V. Herrera and R. Sanchez-Delgado, J. Am. Chem. Soc., 1994, 116, 4370–4381 Search PubMed.
- H. E. Selnau and J. S. Merola, Organometallics, 1993, 12, 1583–1591 Search PubMed.
- M. R. Grochowski, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 2661–2667 Search PubMed.
- W. D. Jones and R. M. Chin, J. Am. Chem. Soc., 1994, 116, 198–203 Search PubMed.
- L. Zamostna, T. Braun and B. Braun, Angew. Chem., Int. Ed., 2014, 53, 2745–2749 Search PubMed.
- S. D. Timpa, C. J. Pell and O. V. Ozerov, J. Am. Chem. Soc., 2014, 136, 14772–14779 Search PubMed.
- K. Osakada, M. Maeda, Y. Nakamura, T. Yamamoto and A. Yamamoto, J. Chem. Soc., Chem. Commun., 1986, 442–443 Search PubMed.
- L. Munjaja, W. W. Brennessel and W. D. Jones, Organometallics, 2015, 34, 4574 Search PubMed.
- J. Campora, E. Gutierrez, A. Monge, P. Palma, M. L. Poveda, C. Ruiz and E. Carmona, Organometallics, 1994, 13, 1728–1745 Search PubMed.
- D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1997, 119, 10855–10856 Search PubMed.
- D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 7606–7617 Search PubMed.
- J. Torres-Nieto, W. W. Brennessel, W. D. Jones and J. J. Garcia, J. Am. Chem. Soc., 2009, 131, 4120–4126 Search PubMed.
- D. A. Vicic and W. D. Jones, Organometallics, 1998, 17, 3411–3413 Search PubMed.
- L. Munjaja, W. W. Brennessel and W. D. Jones, Organometallics, 2015, 34, 1716–1724 Search PubMed.
- T. A. Atesin, A. C. Atesin, K. Skugrud and W. D. Jones, Inorg. Chem., 2008, 47, 4596–4604 Search PubMed.
- K. Yu, H. Li, E. J. Watson, K. L. Virkaitis, G. B. Carpenter and D. A. Sweigart, Organometallics, 2001, 20, 3550–3559 Search PubMed.
- A. Arevalo, S. Bernes, J. J. Garcia and P. M. Maitlis, Organometallics, 1999, 18, 1680–1685 Search PubMed.
- A. Oviedo, A. Arévalo, M. Flores-Alamo and J. J. García, Organometallics, 2012, 31, 4039–4045 Search PubMed.
- M. S. Palmer and S. Harris, Organometallics, 2000, 19, 2114–2124 Search PubMed.
- M. M. Shoshani and S. A. Johnson, Inorg. Chem., 2015, 54, 11977–11985 Search PubMed.
- D. A. Vicic and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 4070–4071 Search PubMed.
- P. T. Matsunaga and G. L. Hillhouse, Angew. Chem., Int. Ed. Engl., 1994, 33, 1748–1749 Search PubMed.
- R. Han and G. L. Hillhouse, J. Am. Chem. Soc., 1998, 120, 7657–7658 Search PubMed.
- M. S. Morton, R. J. Lachicotte, D. A. Vicic and W. D. Jones, Organometallics, 1999, 18, 227–234 Search PubMed.
- A. Sivaramakrishna, B. C. E. Makhubela, J. R. Moss and G. S. Smith, J. Organomet. Chem., 2010, 695, 1627–1633 Search PubMed.
- D. Baranano and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 2937–2938 Search PubMed.
- G. Mann, D. Baranano, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205–9219 Search PubMed.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 Search PubMed.
- X. Zhao and V. M. Dong, Angew. Chem., Int. Ed., 2011, 50, 932–934 Search PubMed.
- S. Kundu, W. W. Brennessel and W. D. Jones, Organometallics, 2011, 30, 5147–5154 Search PubMed.
- S. Kundu, B. E. R. Snyder, A. P. Walsh, W. W. Brennessel and W. D. Jones, Polyhedron, 2013, 58, 99–105 Search PubMed.
- E. Lee and D. V. Yandulov, J. Organomet. Chem., 2011, 696, 4095–4103 Search PubMed.
- T. Schaub, M. Backes, O. Plietzsch and U. Radius, Dalton Trans., 2009, 7071–7079 Search PubMed.
- G. J. Kubas, Acc. Chem. Res., 1994, 27, 183–190 Search PubMed.
- D. P. Gates, P. S. White and M. Brookhart, Chem. Commun., 2000, 47–48 Search PubMed.
- T. Tamai, K. Fujiwara, S. Higashimae, A. Nomoto and A. Ogawa, Org. Lett., 2016, 18, 2114–2117 Search PubMed.
- C. Munro-Leighton, S. A. Delp, N. M. Alsop, E. D. Blue and T. B. Gunnoe, Chem. Commun., 2008, 111–113, 10.1039/b715507g.
- S. S. Zalesskiy, V. N. Khrustalev, A. Y. Kostukovich and V. P. Ananikov, Organometallics, 2015, 34, 5214–5224 Search PubMed.
- K. Miyamoto, M. Hirobe, M. Uchiyama and M. Ochiai, Chem. Commun., 2015, 51, 7962–7965 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |