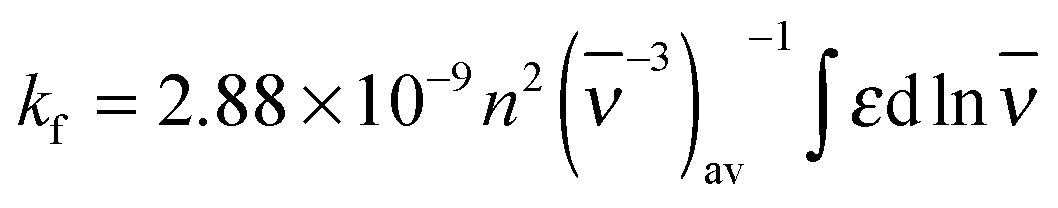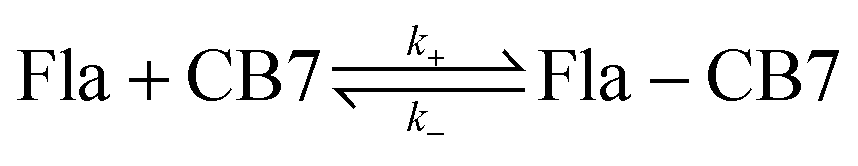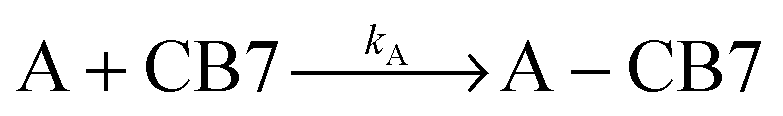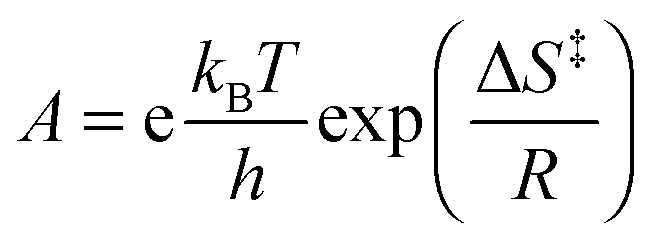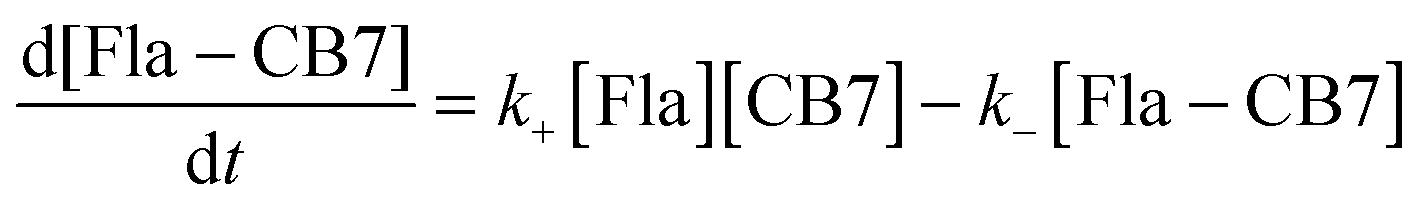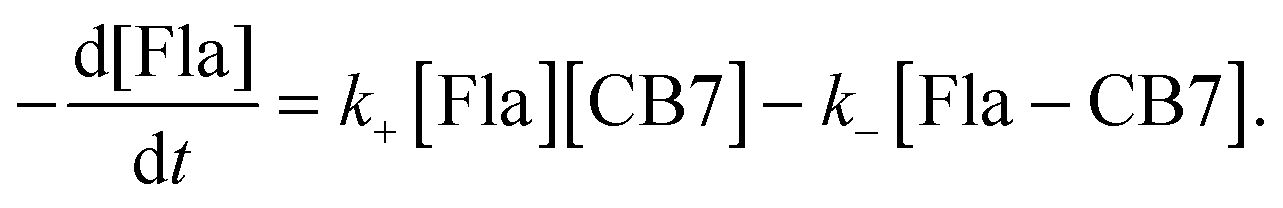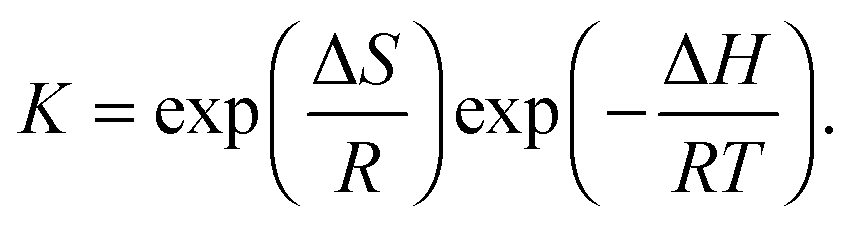Kinetics of the reversible inclusion of flavopereirine in cucurbit[7]uril†
Received
4th November 2016
, Accepted 24th November 2016
First published on 24th November 2016
Abstract
The temperature dependence of the kinetics of flavopereirine inclusion in cucurbit[7]uril (CB7) was studied by a stopped-flow method in water. The substantial blue-shift of the emission band and the ∼4-fold fluorescence quantum yield enhancement upon host–guest binding permitted the monitoring of the formation and dissociation of the flavopereirine–CB7 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex in real time. The competitive binding of the 1-adamantylammonium cation with extremely high affinity was exploited to selectively and very accurately determine the kinetic parameters of the exit of flavopereirine from the CB7 cavity. The rate constants of the ingression into and the egression from CB7 were found to be 9.0 × 107 M−1 s−1 and 1.6 s−1 at 298 K, respectively. Both processes had substantial activation enthalpy implying that a steric barrier had to be overcome in the course of the reversible encapsulation. The 31 ± 2 kJ mol−1 activation enthalpy of the entry into CB7 was comparable to the 37 ± 2 kJ mol−1 enthalpy change upon the dissociation of the complex.
:1 complex in real time. The competitive binding of the 1-adamantylammonium cation with extremely high affinity was exploited to selectively and very accurately determine the kinetic parameters of the exit of flavopereirine from the CB7 cavity. The rate constants of the ingression into and the egression from CB7 were found to be 9.0 × 107 M−1 s−1 and 1.6 s−1 at 298 K, respectively. Both processes had substantial activation enthalpy implying that a steric barrier had to be overcome in the course of the reversible encapsulation. The 31 ± 2 kJ mol−1 activation enthalpy of the entry into CB7 was comparable to the 37 ± 2 kJ mol−1 enthalpy change upon the dissociation of the complex.
Introduction
Cucurbit[n]urils (CBn), the family of macrocyclic molecular containers composed of n glycoluril units bound by a pair of methylene groups, have great potential in the delivery and controlled release of drugs.1–6 These water-soluble cavitands are nontoxic7,8 and can encapsulate molecules of pharmacological interest improving their solubility and bioavailability. They can serve as nanocapsules to promote the transportation of drugs into cells. Partial embedment in CBn may provide a formulation method for the clinical application of platinum-based anti-cancer agents.9,10 Inclusion in cucurbit[7]uril (CB7) significantly enhanced the thermal stability of ranitidine, a drug used for the treatment of excess stomach acid,11 and resulted in strongly bound complexes possessing pH-responsive behaviour for local anaesthetics.12 The confinement of the proton-pump inhibitor drugs lansoprazole and omeprazole in CB7 catalysed their transition into their physiologically active cyclic sulfonamide form and impeded their decomposition and dimerization.13
Due to the pharmaceutical importance of natural alkaloids, their self-assembly with CBn has received particular attention. The effect of the inclusion complex formation on the solubility and anticancer activity of camptothecin was revealed.14 Host–guest association with CB7 brought about only a 0.7 pKa unit increase in the basicity of norharmane15 but substantially inhibited the nucleophilic addition of hydroxide anions to sanguinarine cations and hindered the photooxidation.16 Berberine binding in CB7 resulted in about 500-fold fluorescence quantum yield growth,17 which was exploited to monitor the passage of biologically important compounds into liposomes and through the bacterial transmembrane protein.18 The intense fluorescence of CB7 complexes was used in the quantitative analysis of alkaloids in biological fluids.19,20
Despite the pivotal importance of the dissociation rate of the active ingredients in the design of pharmaceutical formulations, knowledge of the kinetics of the reversible host–guest complex formation of CB7 is scarce,21–23 and only a single study has been published on the activation parameters of entry into and exit from the nonpolar CB7 cavity.24 Large activation enthalpy was found for berberine ingression implying that considerable steric distortion occurred when the guest passed through the tight carbonyl-laced opening of CB7. Both inclusion and dissociation were much more rapid for the less spacious 6-methoxy-1-methylquinolinium25 and (R)-(+)-2-naphthyl-1-ethylammonium26 cations indicating the rate determining role of the molecular dimensions. Kaifer and co-workers observed an enormous solvent effect on the kinetics of the pseudorotaxane-type complex formation of dumbbell-shaped guests whose ingression into CB7 cavity was significantly faster in water than in DMSO.27 The quick 1:1 binding of the flavylium cationic form of an anthocyanin in CB7 was exploited to stabilize the dye colour since decomposition via hydration could not compete with the formation of the inclusion complex in water.28 Bohne and Thomas demonstrated with systematic studies that the CB7 complex of the 2-aminoanthracenium cation can form not only by direct inclusion but also via the initial encapsulation of 2-aminoanthracene followed by protonation.29 The substantial pKa increase upon host–guest binding was found to arise from the slower deprotonation in the cavity of CB7 than in water.
The present work focuses on the kinetics of the reversible confinement of flavopereirine (3-ethyl-12H-indolo[2,3-a]-quinolizinium perchlorate) (Fla) in CB7. This biologically important alkaloid, originally isolated from the bark of an Amazonian tree family known as Pao pereira in Brasil,30 may possess antiplasmodial and cytotoxic activity.31,32 The fluorescence characteristics of Fla are solvent-sensitive and significantly change upon interaction with nucleotides, DNA and chondroitin-6-sulfate.33 Therefore, inclusion in CB7 is expected to modify the fluorescence behaviour allowing the monitoring of the dynamics of complex formation in real time. Because host–guest complexes are in dynamic equilibrium with their components, direct measurements of association or dissociation contain information on the rate constants of ingression (k+) and egression (k−) alike. To enhance the accuracy of the obtained kinetic parameters, we intend to apply a simple straightforward fluorescence method for the separate determination of k−. Our main objectives are to reveal how the variation of temperature influences the kinetics of the elementary reaction steps and obtain information of the properties of the transition states. The formulae of the employed compounds are displayed in Scheme 1.
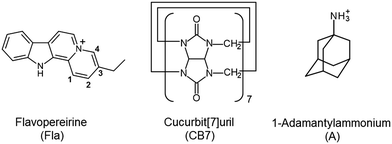 |
| | Scheme 1 Chemical structures of the compounds used. | |
Experimental
Flavopereirine perchlorate (Fla) (ChromaDex) and 1-adamantylamine (Aldrich) were used without further purification. The latter compound is fully protonated at neutral pH since its conjugated acid has a pKa value34 of 10.55. High-purity CB7 was kindly provided by Dr Anthony I. Day (University of New South Wales, Canberra, Australia). Experiments were carried out in double distilled water. Fla concentrations were determined spectrophotometrically using a molar absorption coefficient of 15070 M−1 cm−1 at 384 nm in water. The UV-visible absorption spectra were obtained on an Agilent Technologies Cary60 spectrophotometer. Corrected fluorescence spectra were recorded on a Jobin-Yvon Fluoromax-P photon counting spectrofluorometer. Stopped-flow measurements were performed using the same instrument using an Applied Photophysics RX2000 rapid mixing accessory and a pneumatic drive. The temperature of the samples was controlled using a Julabo F25-ED thermostat. Fluorescence decays were measured with the time-correlated single-photon counting technique using the previously described instrument.35 The results of spectrophotometric and fluorescence titrations as well as stopped-flow measurements were analysed using homemade programs written in MATLAB 7.9. Semiempirical calculations using the RM1 method were carried out using HyperChem 8.0 program.
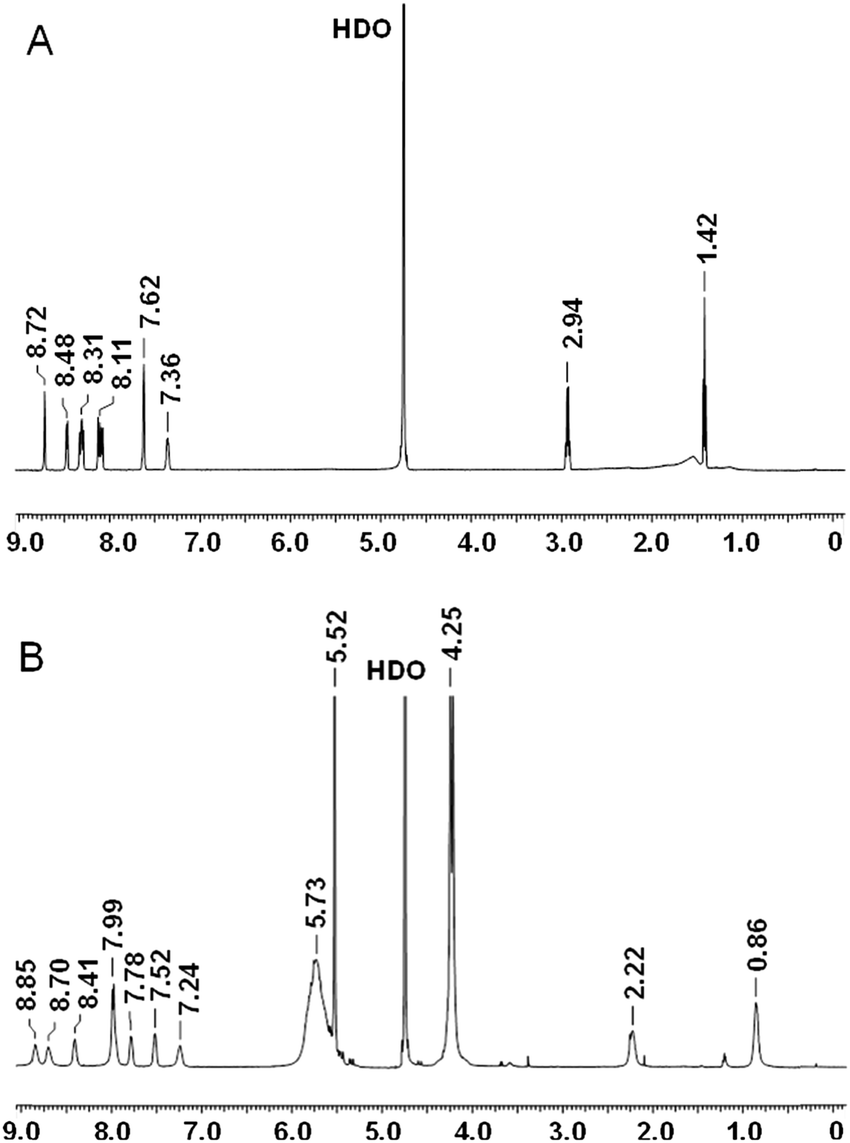
NMR spectra were acquired on a Varian 500 Inova spectrometer, running at 500 MHz for 1H. Chemical shifts (δ) are reported in ppm relative to residual solvent signals (CHCl3, 7.26 ppm for 1H NMR). The following abbreviations are used to indicate the multiplicity in 1H NMR spectra: s, singlet; d, doublet; t, triplet; m, multiplet; bs, broad signal. Assignments for Fla and the Fla–CB7 complex are as follows. Fla (D2O, ppm): 1.42 (t, 3H, CH3), 2.94 (q, 2H, CH2), 7.36 (m, 1H, arH), 7.62 (bs, 2H, arH), 8.08 (d, 1H, arH), 8.12 (d, 1H, arH), 8.29 (d, 1H, arH), 8.31 (d, 1H, arH), 8.48 (d, 1H, arH), 8.72 (s, 1H, arH, H-4). Fla–CB7 (D2O, ppm): 0.86 (bs, 3H, CH3), 2.22 (bs, 2H, CH2), 4.25 (d, 14H, CH2, CB7), 5.52 (s, 14H, CH, CB7), 5.73 (bs, 14H, CH2, CB7), 7.24 (bs, 1H, arH), 7.52 (bs, 1H, arH), 7.78 (bs, 1H, arH), 7.79 (bs, 3H, arH), 8.41 (bs, 1H, arH), 8.70 (bs, 1H, arH), 8.85 (bs, 1H, arH).
Results
Effect of CB7 on the absorption and fluorescence properties
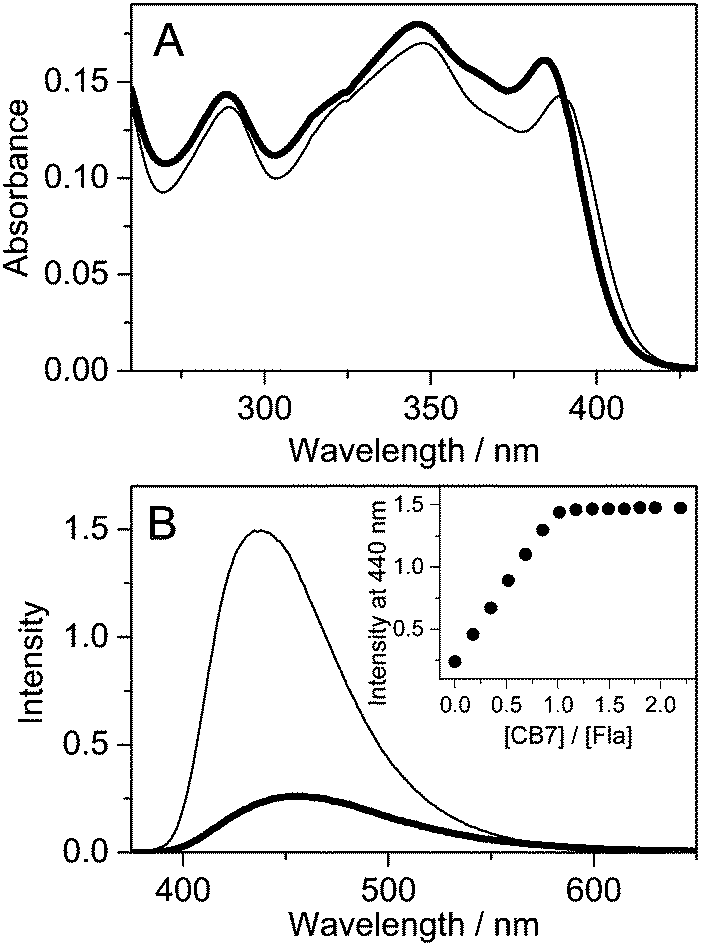
Fig. 1A presents the alteration of the absorption spectrum of 10.7 μM Fla upon addition of 26.1 μM CB7 in water. The lowest energy absorption maximum exhibits hypochromicity and about 4 nm red-shift due to inclusion complex formation. The confinement of Fla in the nonpolar cavity of CB7 leads to a substantial fluorescence intensity enhancement accompanied by a 21 nm hypsochromic displacement of the spectrum maximum (Fig. 1B). After an initial linear increase, the fluorescence intensity at 440 nm levels off at equimolar [CB7]/[Fla] concentration ratio, corroborating the 1:1 stoichiometry of binding.
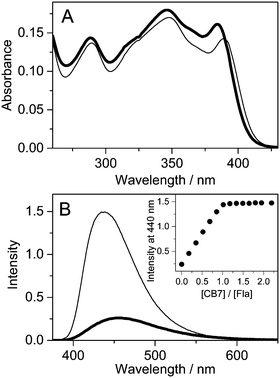 |
| | Fig. 1 Absorption (A) and fluorescence (B) spectra of 10.7 μM Fla in water (thick line) and in 26.1 μM CB7 aqueous solution (thin line) at 296 K (optical path 1 cm, excitation at 390 nm). Inset: Fluorescence intensity at 440 nm as a function of [CB7]/[Fla] concentration ratio. | |
The photophysical parameters of free and CB7-bound Fla are summarized in Table 1. Fig. S1 in the ESI† illustrates why the inclusion in the apolar CB7 cavity leads to a red-shift in the absorption spectra whereas a blue-shift in the fluorescence spectra. The energy of the lowest singlet-excited state (E(S1)) was calculated from the location of the intersection of the normalised absorption and fluorescence spectra plotted at the wavenumber scale. Remarkable increases in the quantum yield (Φf) and lifetime (τf) of fluorescence are observed upon encapsulation in CB7. The rate constants for radiative (kf) and nonradiative (knr) deactivations from the singlet-excited state are calculated using the expressions kf = Φf/τf and knr = (1 − Φf)/τf. The dominant change appears in the latter quantity, whereas kf grows only about 55% upon host–guest complexation. We have demonstrated that the photophysical characteristics of Fla are solvent dependent.36 The remarkable deuterium isotope effect on τf suggested that hydrogen bonding of the singlet-excited Fla offers an effective pathway for dissipation of excitation energy. The partial inclusion in the CB7 core provides a nonpolar, extremely nonpolarizable microenvironment for the guest37 and diminishes its interaction with water thereby decelerating radiationless deactivation from the singlet-excited state. The increase of kf in CB7 can be rationalized in terms of the Strickler–Berg equation38
| | 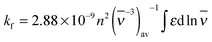 | (1) |
where
n denotes the refractive index of the microenvironment, (
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) −3
−3)
av−1 is an average over the fluorescence spectrum and the integral of molar absorption coefficients (
ε) extends over the first absorption band. This relationship predicts that
kf grows approximately as the cube of the emission energy. Hence, the blue-shift of the fluorescence spectrum due to the embedment in CB7 results in a larger
kf. On the basis of the comparison of the photophysical properties in various solvents
33 and CB7, we conclude that Fla senses somewhat less polar surroundings in CB7 than in acetonitrile.
Table 1 Variation of absorption (λa) and fluorescence (λf) maxima, energy of the lowest singlet-excited state (E(S1)), fluorescence quantum yield (Φf), fluorescence lifetime (τf), rate constant of fluorescence (kf) and nonradiative deactivation (knr) for 10.7 μM flavopereirine in water and 26.1 μM CB7 aqueous solution
| Solvent |
λ
a/nm |
λ
f/nm |
E(S1)/kJ mol−1 |
Φ
f
|
τ
f/ns |
k
f/107 s−1 |
k
nr/107 s−1 |
| H2O |
385 |
459 |
291.9 |
0.11 |
5.0 |
2.2 |
17.8 |
| 26.1 μM CB7 H2O |
389 |
438 |
294.7 |
0.43 |
12.8 |
3.4 |
4.5 |
Structure of the inclusion complex
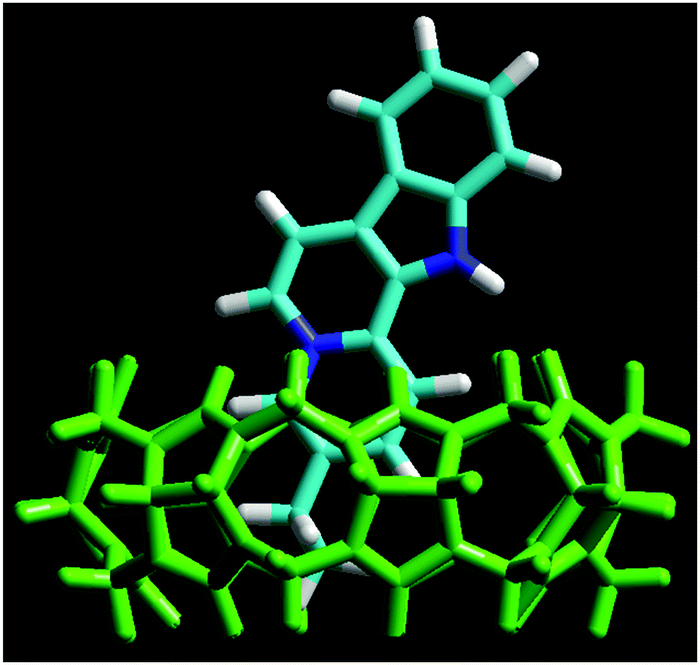
The energy-minimized structure of the Fla–CB7 complex was calculated by the RM1 semiempirical method using the HyperChem 8.0 program. The results (Fig. 2) show that the ethyl substituent of Fla is fully confined in the hydrophobic core of CB7, whereas the quinolizinium moiety is only partially encapsulated and its nitrogen is located in the vicinity of the carbonyl-rimmed portal of the host. The indole segment of Fla is not embedded. It protrudes into the aqueous phase. The charge of Fla is delocalized and mainly appears as low electron density on the peripheral hydrogens of the ring system. The carbon atom fusing the aromatic rings of quinolizinium also has a partial positive charge, which is located in the neighbourhood of the oxygen atoms at the entrance of CB7 cavity.
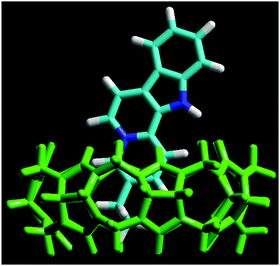 |
| | Fig. 2 Energy-minimized structure of the Fla–CB7 inclusion complex calculated by the RM1 method. Color codes: CB7, green; nitrogen, blue; carbon, light blue; and hydrogen, white. | |
1H NMR spectra of Fla and Fla–CB7 recorded in D2O (Fig. 3) corroborated the calculated structure of the inclusion complex. The addition of 1 equivalent of CB7 yielded remarkable spectral changes in the Fla spectrum (Fig. 3B). The signals of the ethyl group broadened and exhibited 0.6–0.7 ppm upfield shift indicating, in accordance with the results of calculations, that the ethyl group is fully embedded in the interior of CB7. Both downfield and upfield shifts of the resonances of the aromatic protons were found. Because of the poor solubility of both host and guest molecules in D2O, no exact assignments of the aromatic protons were made but probably the singlet peak of the methine proton (H-4, δ 8.72 ppm) adjacent to the nitrogen atom of the quinolizinium moiety as well as the H-1 and H-2 proton signals of this heterocycle shifted upfield due to the encapsulation in the hydrophobic environment.
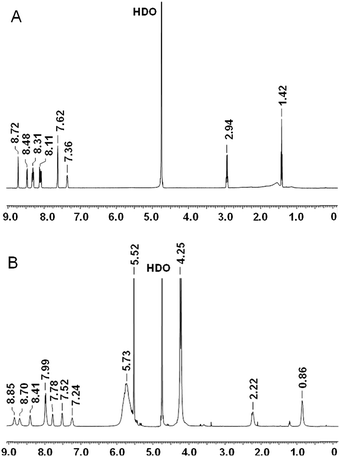 |
| | Fig. 3
1H NMR spectra of Fla (A) and the Fla–CB7 complex (B) recorded in D2O. | |
Highly accurate method for the selective determination of the egression rate constant
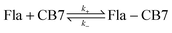
When the fluorescence intensity change due to a simple host–guest association is monitored in stopped-flow measurements, the kinetic traces have three unknown parameters: the rate constants of the formation and disassembly of the 1:1 complex and the ratio of the fluorescence efficiency for the free and bound guests at the monitoring wavelength. These variables can be calculated by least-squares fit of the experimental data but the results usually show considerable uncertainties. To enhance the accuracy of the derived values, we designed experiments whose results can be analysed by two-parameter fitting procedures.
The dissociation rate of the Fla–CB7 inclusion complex can be selectively measured by the addition of a high affinity binder (A), which rapidly occupies the cavity of practically all free CB7 thereby blocking the reverse formation of Fla–CB7 and promoting its dissociation.
| |  | (2) |
| | 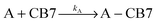 | (3) |
If A–CB7 has a very large stability constant, its dissociation is negligible on the timescale of the measurement. The concentration of A should be chosen to ensure that
k+[Fla] ≪
kA[A]. Moreover, A and A–CB7 should not absorb at the excitation wavelength used in the kinetic studies. 1-Adamantylammonium cation meets all of these requirements. Due to its ideal size complementarity and rigidity, it has an extremely large
39 (
KA = 1.7 × 10
14 M
−1) association constant with CB7. Assuming diffusion controlled complexation, whose rate constant is
kdiff = 6.5 × 10
9 M
−1 s
−1 in water,
40k−A =
kdiff/
K = 3.8 × 10
−5 s
−1 is estimated as an upper limit for the rate constant of A egression from CB7 cavity. This corresponds to
t1/2 = (ln
2)/
k−A > 5 h half-life for the first-order dissociation of the A–CB7 complex. Therefore, the rate of A exit from CB7 can be neglected on the time-scale of the stopped-flow experiments. The estimated
k−A value is close to the rate constant of
N-alkyl-1-adamantylammonium egression from CB7 ((2.4 ± 0.1) × 10
−5 s
−1) obtained by Isaacs' group using NMR spectroscopy
23 suggesting that A ingression into CB7 is indeed close to diffusion controlled.
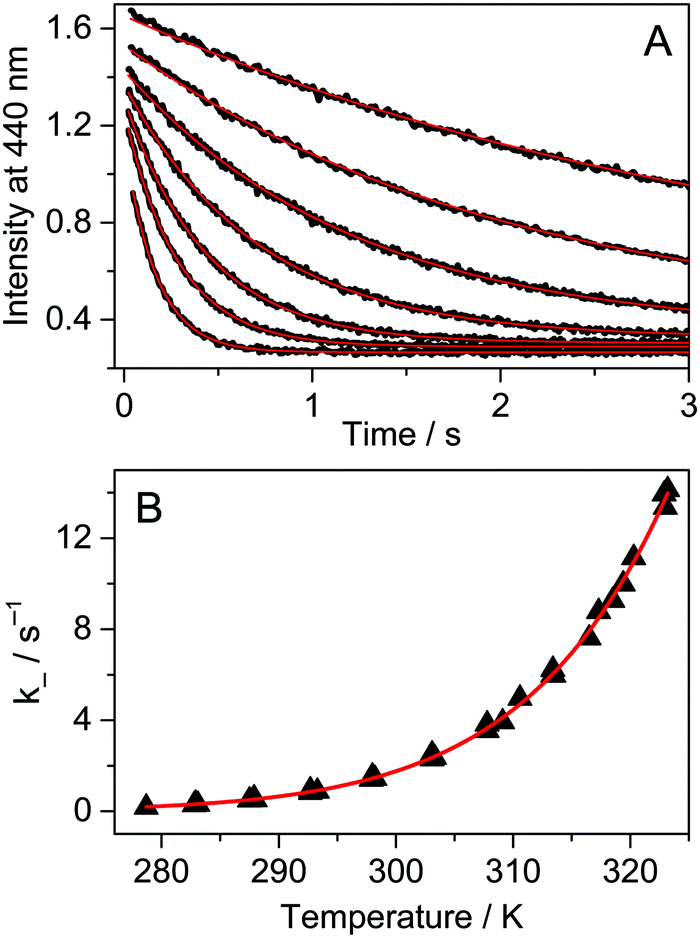
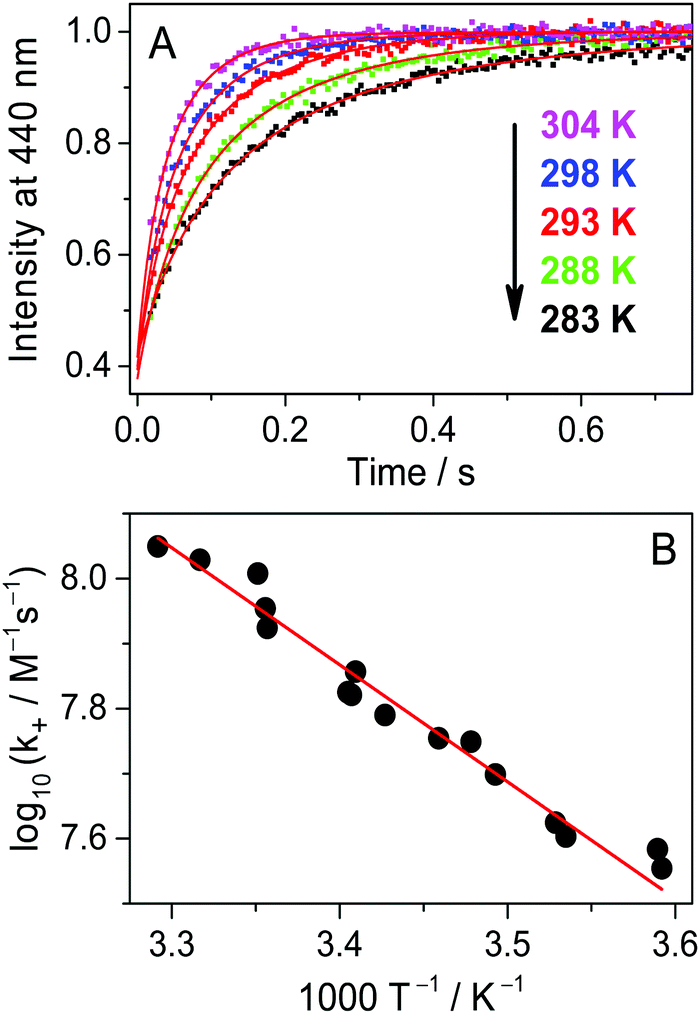
Fig. 4A presents the fluorescence intensity change at 440 nm after rapid addition of 3.0 μM A solution to Fla and the CB7 mixture at various temperatures. The initial concentrations of the latter two compounds were kept constant (0.15 μM). The dissociation of the Fla–CB7 complex is quickly followed by the entry of A into the CB7 cavity leading to very stable A–CB7. Consequently, A gradually replaces Fla in CB7 and the stopped-flow signal reflects the diminution of the Fla–CB7 concentration. The fluorescence intensity reaches the level observed for Fla aqueous solution indicating the total removal of Fla from CB7 into water. As Fla egression is a first-order process, its rate constant can be obtained by the nonlinear least-squares fit of an exponential function to the stopped-flow signal.
| | | I = I0exp(−k−t) + I∞ | (4) |
The method permits straightforward and highly accurate determination of
k− values. For Fla exit from CB7 at 298 K,
k− = 1.6 ± 0.2 s
−1 is calculated.
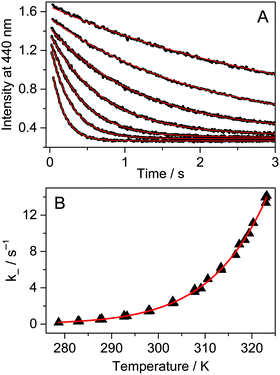 |
| | Fig. 4 (A) Stopped-flow signals after the addition of 3.0 μM 1-adamantylamine in water to 0.15 μM Fla and 0.15 μM CB7 aqueous solution at 283, 288, 293, 298, 303, 308 and 314 K (from top to down). Excitation and detection were performed at 350 and 440 nm, respectively. (B) Temperature dependence of the rate constant of Fla–CB7 complex dissociation in water. The red lines represent the results of the nonlinear least-squares analysis. | |
To obtain information on the activation parameters, the experiment was repeated at various temperatures (Fig. 4A). The fluorescence intensities immediately after rapid mixing lessen with the rise of temperature because of two reasons: (i) the fraction of encapsulated Fla becomes smaller due to the diminution of the binding constant and (ii) the fluorescence quantum yield slightly decreases at elevated temperature. The combined effect of these changes causes the diminution of the I0 and I∞ parameters in eqn (4) with increasing temperature. The initial segment of the stopped-flow signals gradually steepens with rising temperature indicating the acceleration of the exit of Fla from CB7. The analysis of the kinetic results using eqn (4) led to the rate constants presented in Fig. 4B. The temperature dependence of k− values was fitted with the Arrhenius equation k− = Aexp(−Ea/RT), where R denotes the gas constant. The calculated activation energies (Ea) and A factors are included in Table 2. On the basis of the transition-state theory, the meaning of these parameters can be interpreted by the following relationships:
| | 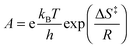 | (6) |
where
kB,
h, Δ
S‡, and Δ
H‡ denote the Boltzmann and Planck constants, standard entropy and enthalpy of activation, respectively. The Δ
S‡ and Δ
H‡ quantities derived according to
eqn (5) and (6) are also listed in
Table 2.
Table 2 Comparison of the kinetic and thermodynamic parameters for the reversible binding of Fla and berberine in CB7
| |
Flavopereirine |
Berberinea |
| Ingression |
Egression |
Ingression |
Egression |
|
Ref. 24.
Derived from the temperature dependence of the binding constant.
|
|
k at 298 K |
(9.0 ± 0.9) × 107 M−1 s−1 |
1.6 ± 0.2 s−1 |
(1.9 ± 0.1) × 107 M−1 s−1 |
0.81 ± 0.08 s−1 |
|
A
|
(6.1 ± 3.5) × 1013 M−1 s−1 |
(6.3 ± 2.6) × 1012 s−1 |
(2.0 ± 1.0) × 1013 M−1 s−1 |
(1.8 ± 1.0) × 1012 s−1 |
|
E
a
|
33 ± 2 kJ mol−1 |
72 ± 2 kJ mol−1 |
35 ± 2 kJ mol−1 |
71 ± 2 kJ mol−1 |
| ΔH‡ |
31 ± 2 kJ mol−1 |
69 ± 2 kJ mol−1 |
32 ± 2 kJ mol−1 |
69 ± 2 kJ mol−1 |
| ΔS‡ |
11 ± 7 J mol−1 K−1 |
−8 ± 3 J mol−1 K−1 |
2.6 ± 1.9 J mol−1 K−1 |
−19 ± 3 J mol−1 K−1 |
|
K at 298 K |
(6.1 ± 0.4) × 107 M−1 |
(2.4 ± 0.3) × 107 M−1 |
| ΔHb |
−37 ± 2 kJ mol−1 |
−37 ± 2 kJ mol−1 |
| ΔSb |
25 ± 7 J mol−1 K−1 |
17 ± 6 J mol−1 K−1 |
Kinetics of Fla ingression in CB7
The mixing of equimolar Fla and CB7 solutions results in fluorescence intensity enhancement because host–guest binding leads to an almost 4-fold increase in the fluorescence quantum yield of Fla (Table 1). To decelerate the inclusion complex formation, dilute (0.18 μM) reactant solutions were used. Fig. 5A presents the normalized fluorescence intensity versus time profiles at various temperatures. Above 304 K, the accuracy of measurements diminishes due to the fast increase of the stopped-flow signals. The experimental data can be described well by assuming simple 1:1 encapsulation equilibrium. In this case, the rates of Fla–CB7 formation and Fla concentration diminution are given as follows:| | 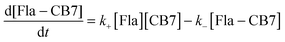 | (7) |
| | 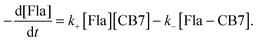 | (8) |
The rate constant of Fla confinement in CB7 (k+) was calculated by the nonlinear least-squares fit of the numerical solution of eqn (7) and (8) to the results of stopped-flow measurements. To diminish the uncertainty of k+, k− values determined by independent experiments (vide supra) were kept constant in the regression analysis. Further details of data analysis are provided in the ESI.† The logarithm of the calculated k+ values is plotted against reciprocal temperature in Fig. 5B and the kinetic parameters of host–guest binding are summarized in Table 2. Substantial activation energy is found for the inclusion in CB7, whereas the Arrhenius pre-exponential factor is more than one order of magnitude larger for the association than for the reverse process.
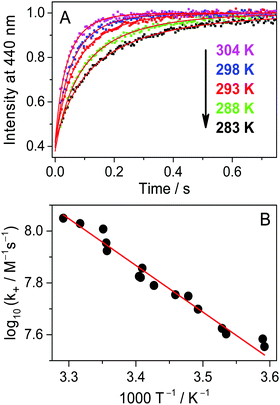 |
| | Fig. 5 (A) Alteration of stopped-flow results with temperature at 0.18 μM initial Fla and CB7 concentrations in water. Excitation and detection were carried out at 384 and 440 nm, respectively. (B) Arrhenius plot of the rate constant of Fla inclusion in CB7. | |
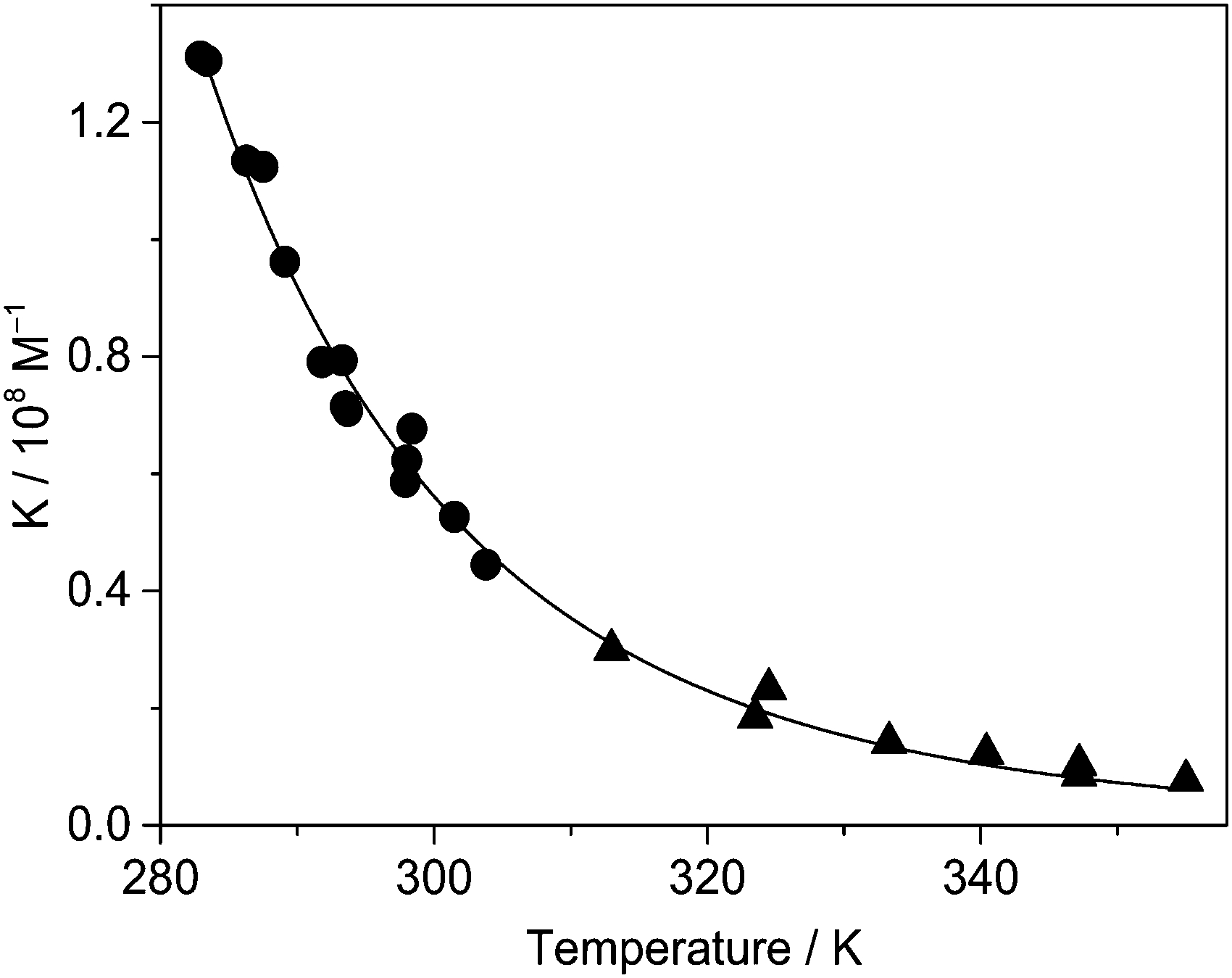
Temperature dependence of the binding constant
The considerable fluorescence intensity enhancement upon Fla confinement in CB7 (Fig. 1) was employed to measure the binding constant (K). As a representative example, Fig. S2 in the ESI† presents the result of fluorescence titration at three temperatures. The analysis of these data led to K = 7.7 × 106 M−1 for the association constant at 355 K. The significant growth of K with decreasing temperature raised the uncertainty of the determined values around room temperature because increasing segments of the titration curves consisted of two straight lines, which did not contain information about the binding affinity. Therefore, association constants were derived as a ratio of the rate constants of the forward and reverse reactions, K = k+/k− below 304 K. Above this temperature the accuracy of k+ is not high enough to calculate reliable K from kinetic measurements due to the accelerated complex formation and the limited time resolution of the stopped-flow technique, but fluorescence titrations provide accurate results. Fig. 6 demonstrates that the binding constants obtained by stopped-flow and direct fluorescence titration experiments complement each other well. The line represents the result of a nonlinear least-squares fit of the relationship| | 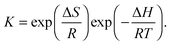 | (9) |
The calculated function matches well the experimental data, and the acquired enthalpy (ΔH) and entropy (ΔS) changes are incorporated in Table 2.
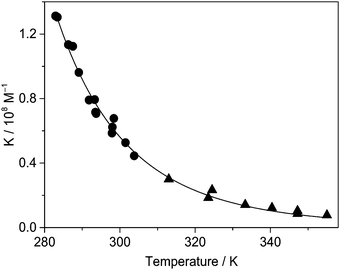 |
| | Fig. 6 Binding constant of Fla encapsulation in CB7 in water as a function of temperature derived from stopped-flow measurements (●) and fluorescence titrations (▲). | |
Discussion
NMR measurements in agreement with quantum chemical calculations showed the partial encapsulation of Fla in CB7. The predominant fraction of the substituted six-membered heterocyclic ring and its ethyl moiety were located inside the cavitand. Hydrophobic interactions and electrostatic attraction with the considerable negative charge density of the portal and the inner surface of the macrocycle41 promoted the embedment in the apolar core of the host. The considerable fluorescence intensity increase upon confinement and the high fluorescence quantum yield of the inclusion complex allowed the use of very low (<0.2 μM) Fla concentrations in our kinetic studies. This offered several advantages: (i) the bimolecular host–guest binding became slow even in neat water, (ii) the kinetic traces had more than 100 data points permitting accurate determination of the fitted parameters, (iii) addition of competing ions was not needed to monitor the association kinetics in the time range accessible to the stopped-flow method, which (iv) simplified the data analysis and allowed the determination of activation parameters.
All experimental results could be rationalized by a one step binding equilibrium. Intermediate or exclusion complex formation was not observed. Intermediary association complexes were found when cucurbit[6]uril (CB6) served as a host for 4-methylbenzylammonium,42 or cyclohexylmethylammonium43,44 as well as for 2-aminoanthracenium confinement in CB7.29 The hydrogens of the NH3+ substituent of these guests have considerable partial positive charge, which facilitates electrostatic interaction and hydrogen bonding with the electron-rich oxygens at the rim of CBn cavitands. These effects stabilize the exclusion complex in which the hydrophobic moiety remains in the aqueous phase. Nau and co-workers proposed the transformation of an ammonium type of exclusion complex into the thermodynamically more stable inclusion complex in a “flip-flop” manner.43,44 Such a mechanism is not possible for Fla binding because it cannot form hydrogen bonds with CB7 and its positive charge is delocalized.
The results summarized in Table 2 demonstrate that Fla has a high binding affinity to CB7. The inclusion is an exothermic enthalpy-controlled process (ΔH = −37 kJ mol−1). The entropic term −TΔS contributes ≈−7.5 kJ mol−1 to the driving force of complexation. The entropy gain arises from the expulsion of water molecules from the cavity of CB7 and from the solvate shell of Fla upon the encapsulation. The entropy loss caused by the host–guest association only partly compensates for these effects. The rate constant of ingression of Fla into CB7 is ∼70-fold lower (k+ = 9.0 × 107 M−1 s−1 at 298 K) than that of the diffusion-controlled process (kdiff = 6.5 × 109 M−1 s−1) in water.45 The substantial activation enthalpy (31 kJ mol−1) decelerates the binding process. In addition to the activation enthalpy, the binding enthalpy must be supplied to induce the dissociation of the Fla–CB7 complex. Because of the high activation barrier, the exit from CB7 is slow and has a rate constant of 1.6 s−1 at 298 K. Our results suggest that the entry into and exit from the relatively rigid CB7 host is sterically hindered.
Activation parameters of the confinement in CB7 were reported only for berberine.24 The activation enthalpies (ΔH‡) of the ingression and egression agree within the limit of experimental errors for Fla and berberine (Table 2). This may indicate that an oval distortion of the carbonyl-laced entrance of CB7 is needed for the penetration of these guests into the interior of the macrocycle as well as for the dissociation of the produced complex. Such a structural change was found to occur when bulky guests were reversibly accommodated in the smaller CB6 cavitand.43,44,46,47 The rate constant of the ingression into CB7 is almost 5-fold larger for Fla than for berberine. This difference is primarily attributed to the more favourable activation entropy of Fla–CB7 formation. The ingression of the ethyl moiety of Fla into CB7 probably leads to a more loosely bound transition state in which fewer degrees of freedom become limited than in the case of the entry of the dimethoxybenzene moiety of berberine. The positive ΔS‡ of the inclusion complex formation implies that the partial desolvation of the guest and the interior of CB7 more significantly overcompensate the entropy diminution due to the ordering of the reactants into the transition state. The negative ΔS‡ for the egression from CB7 indicates that the deliberation of the degrees of freedom in going to the transition state only partially balances the unfavourable entropy change caused by the coordination of water molecules. Because of the more loosely bound structure of the activated complex, the exit of Fla from CB7 has a less negative ΔS‡ than the corresponding process for berberine. Therefore, the dissociation rate of Fla–CB7 is a factor of two higher than that of berberine–CB7. The larger stability constant (K) of the former complex originates from the more substantial entropy gain upon Fla encapsulation. Fla fits less tightly in CB7 than in berberine. As a consequence, the entropy diminution brought about by the restricted movement of the guest in CB7 offsets the entropy gain due to the release of water molecules upon binding to a lesser extent when Fla is confined instead of berberine. The release of high-energy water molecules from the apolar core of cucurbiturils is the most important factor determining the stability of the inclusion complexes.48,49 No difference was found between the enthalpies of CB7 complex formation of Fla and berberine because the desolvation of these hydrophobic compounds has a similar and small effect on the exothermicity of the inclusion.
In the case of Fla at 298 K, the rate constant of ingression into CB7 (k+ = 9.0 × 107 M−1 s−1) falls between the values reported for (R)-(+)-2-naphthyl-1-ethylammonium26 (k+ = 6.3 × 108 M−1 s−1) and ethyl benzyl ketone50 (k+ = 4.6 × 106 M−1 s−1). Despite the 7-fold slower entry of Fla than (R)-(+)-2-naphthyl-1-ethylammonium, the binding constant of Fla (K = 6.1 × 107 M−1 at 298 K) is significantly higher than that of the latter compound26 (K = 1.1 × 107 M−1) indicating the lack of correlation between the kinetic parameters and the binding affinity. Because of weaker steric hindrance, the smaller size of (R)-(+)-2-naphthyl-1-ethylammonium permits more rapid egression from CB7 (k− = 55 s−1)26 which results in a lower stability of the inclusion complex compared with that of Fla–CB7.
Conclusions
Fla produces a remarkably stable 1:1 inclusion complex with CB7 in water. Only the ethyl group and the substituted 6-membered nitrogen heterocyclic moiety of the alkaloid are embedded in the hydrophobic core of the host macrocycle. The large equilibrium constant for Fla–CB7 formation is attributed primarily to the considerable enthalpy gain but the entropy growth upon the release of water from the CB7 cavity and the solvate shell of the reactants also has a favourable contribution. The complexation is significantly slower than a diffusion-controlled process because Fla cannot pass freely through the portal of CB7. The rate constant of ingression is controlled by steric effects, which depend on the relative size of the guest and the openings of the host macrocycle. In addition to these factors, the enthalpy change of host–guest binding affects the rate of egression from CB7. The entry into this molecular container is expected to occur without formation of an intermediate if the guest molecule possesses delocalised positive charge and is unable to form a hydrogen bond with the carbonyl-laced opening of the host. The fluorescence method based on the competitive binding of the 1-adamantylammonium cation to CB7 can also be used for the simple, rapid, and highly accurate determination of the egression rate constant in the case of other guests.
Acknowledgements
This work was supported by the BIONANO_GINOP-2.3.2-15-2016-00017 project and by the Hungarian Scientific Research Fund (OTKA, Grant K104201). The authors are grateful for the support.
References
- D. H. Macartney, Isr. J. Chem., 2011, 51, 600–615 CrossRef CAS.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- S. Walker, R. Oun, F. J. McInnes and N. J. Wheate, Isr. J. Chem., 2011, 51, 616–624 CrossRef CAS.
- K. M. Park, K. Suh, H. Jung, D. W. Lee, Y. Ahn, J. Kim, K. Baek and K. Kim, Chem. Commun., 2009, 71–73 RSC.
- I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed.
-
D. H. Macartney, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2013 Search PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 CAS.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PLoS One, 2010, 5, e10514 Search PubMed.
- A. R. Kennedy, A. J. Florence, F. J. McInnes and N. J. Wheate, Dalton Trans., 2009, 7695–7700 RSC.
- N. J. Wheate, D. P. Buck, A. I. Day and J. G. Collins, Dalton Trans., 2006, 451–458 RSC.
- R. B. Wang and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1955–1960 CAS.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2010, 8, 247–252 CAS.
- N. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398–5401 CrossRef CAS PubMed.
- N. Dong, S. F. Xue, Q. J. Zhu, Z. Tao, Y. Zhao and L. X. Yang, Supramol. Chem., 2008, 20, 659–665 CrossRef CAS.
- R. Wang, I. W. Wyman, S. Wang and D. H. Macartney, J. Inclusion Phenom. Macrocyclic Chem., 2009, 64, 233–237 CrossRef CAS.
- Z. Miskolczy, M. Megyesi, G. Tárkányi, R. Mizsei and L. Biczók, Org. Biomol. Chem., 2011, 9, 1061–1070 CAS.
- M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410–3416 CAS.
- G. Ghale, A. G. Lanctôt, H. T. Kreissl, M. H. Jacob, H. Weingart, M. Winterhalter and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 2762–2765 CrossRef CAS PubMed.
- C. F. Li, L. M. Du and H. M. Zhang, Spectrochim. Acta, Part A, 2010, 75, 912–917 CrossRef.
- C. F. Li, L. M. Du, W. Y. Wu and A. Z. Sheng, Talanta, 2010, 80, 1939–1944 CrossRef CAS PubMed.
- C. Bohne, Chem. Soc. Rev., 2014, 43, 4037–4050 RSC.
- M. H. Tootoonchi, S. Yi and A. E. Kaifer, J. Am. Chem. Soc., 2013, 135, 10804–10809 CrossRef CAS PubMed.
- P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093–14102 CrossRef CAS PubMed.
- Z. Miskolczy and L. Biczók, J. Phys. Chem. B, 2014, 118, 2499–2505 CrossRef CAS PubMed.
- Z. Miskolczy, J. G. Harangozó, L. Biczók, V. Wintgens, C. Lorthioir and C. Amiel, Photochem. Photobiol. Sci., 2014, 13, 499–508 CAS.
- H. Tang, D. Fuentealba, Y. H. Ko, N. Selvapalam, K. Kim and C. Bohne, J. Am. Chem. Soc., 2011, 133, 20623–20633 CrossRef CAS PubMed.
- S. Senler, B. Cheng and A. E. Kaifer, Org. Lett., 2014, 16, 5834–5837 CrossRef CAS PubMed.
- B. Held, H. Tang, P. Natarajan, C. P. da Silva, V. de Oliveira Silva, C. Bohne and F. H. Quina, Photochem. Photobiol. Sci., 2016, 15, 752–757 CAS.
- S. S. Thomas and C. Bohne, Faraday Discuss., 2015, 185, 381–398 RSC.
- N. A. Hughes and H. Rapoport, J. Am. Chem. Soc., 1958, 80, 1604–1609 CrossRef CAS.
- J. C. P. Steele, N. C. Veitch, G. C. Kite, M. S. J. Simmonds and D. C. Warhurst, J. Nat. Prod., 2002, 65, 85–88 CrossRef CAS.
- M. Reina, W. Ruiz-Mesia, M. López-Rodríguez, L. Ruiz-Mesia, A. González-Coloma and R. Martínez-Díaz, J. Nat. Prod., 2012, 75, 928–934 CrossRef CAS PubMed.
- Z. Miskolczy, M. Megyesi, L. Biczók and H. Görner, Photochem. Photobiol. Sci., 2011, 10, 592–600 CAS.
- X. Yao, X. Wang, T. Jiang, X. Ma and H. Tian, Langmuir, 2015, 31, 13647–13654 CrossRef CAS PubMed.
- M. Megyesi and L. Biczók, J. Phys. Chem. B, 2010, 114, 2814–2819 CrossRef CAS PubMed.
- M. Megyesi, L. Biczók and H. Görner, Photochem. Photobiol. Sci., 2009, 8, 556–561 CAS.
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 4387–4390 CrossRef CAS.
- Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222–2233 CrossRef CAS PubMed.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570–3581 CrossRef CAS PubMed.
-
S. L. Murov, I. Carmichael and G. L. Hug, Handbook of photochemistry, Marcel Dekker, New York, 1993 Search PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- R. Hoffmann, W. Knoche, C. Fenn and H.-J. Buschmann, J. Chem. Soc., Faraday Trans., 1994, 90, 1507–1511 RSC.
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 3155–3160 CrossRef CAS.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef PubMed.
-
M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, FL, 3rd edn, 2006 Search PubMed.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o1060–o1062 CAS.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1321–o1322 CAS.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318–15323 CrossRef CAS PubMed.
- F. Biedermann, M. Vendruscolo, O. A. Scherman, A. De Simone and W. M. Nau, J. Am. Chem. Soc., 2013, 135, 14879–14888 CrossRef CAS PubMed.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem. – Eur. J., 2007, 13, 7223–7233 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp07553c |
|
| This journal is © the Owner Societies 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
István
Jablonkai
b
*a and
István
Jablonkai
b