
A driving force for polypeptide and protein collapse†
Antonello
Merlino
 a,
Nicola
Pontillo
a and
Giuseppe
Graziano
*b
a,
Nicola
Pontillo
a and
Giuseppe
Graziano
*b
aDipartimento di Scienze Chimiche, Università degli Studi di Napoli Federico II, Complesso Universitario di Monte Sant'Angelo, Via Cintia, 80126 Napoli, Italy
bDipartimento di Scienze e Tecnologie, Università del Sannio, Via Port’Arsa 11, 82100 Benevento, Italy. E-mail: graziano@unisannio.it; Fax: 39 0824 23013; Tel: +39 0824 305133
First published on 2nd December 2016
Abstract
Experimental measurements and computational results have shown that polypeptide chains, made up of 15–25 glycine residues, collapse to compact structures in water at room temperature. This contrasts with the classic idea that the burial of nonpolar side chains, i.e., the hydrophobic effect, is the driving force of collapse and folding of polypeptides and proteins. It is thus necessary to find a different driving force for polyglycine collapse. The present study aims at showing that the hydrophobic effect has to be re-defined in terms of decrease in solvent-excluded volume associated with chain collapse so that it is characterized by a gain in translational entropy of water molecules. This indicates that the presence of nonpolar side chains is not so important for polypeptide and protein collapse, even though it may be fundamental for the attainment of a unique folded structure.
Introduction
The molecular interactions driving the folding of globular proteins are still a matter of controversy and debate, notwithstanding the large number of performed investigations, because inconsistencies emerge frequently.1–4 In particular, recently, both computational and experimental data5–8 suggest that the hydrophobic effect, as originally pictured by Kauzmann,9 does not play a role in the collapse of polypeptide chains (note that collapse does not correspond to folding because collapsed conformations are a large number of compact structures characterized by different shapes, whereas the folded conformation consists of a set of structures that retain an overall single conformation). We would like to show that these “new” findings are useful to clarify that the hydrophobic effect, when correctly re-defined, is the driving force of polypeptide collapse.Pettitt and co-workers5 performed a 350 ns long MD simulation, at 300 K and 1 atm, of a chain of 25 glycine residues, Gly25, in the TIP3P water model,10 and found that it collapses to globule-shaped structures with an average radius of gyration, Rg ≈ 7 Å. This simulation result is in line with an earlier computational study6 and, more importantly, with experimental data7 that, somewhat unexpectedly, recorded the collapse in water of a chain of 20 glycine residues by means of fluorescence correlation spectroscopy measurements. Neuweiler and co-workers7 exactly wrote: “Taken together, the results showed that generic backbone interactions are present in polypeptides, providing a driving force sufficient to cause collapse”. They performed also measurements on a 47-residue chain of repetitive units of glycine and serine with the Ser OH group free to form H-bonds or blocked by acylation. The chain collapse occurred independently of the Ser OH group situation,7 confirming that both side chains and H-bonds do not play a fundamental role.
These results seem strange and unexpected because it is usually assumed that the driving force of protein collapse (and folding) is the hydrophobic effect. The latter, following the original Kauzmann's treatment,9 is usually associated with the burial of nonpolar side chains to avoid the unfavorable contact with water molecules (that can re-gain entropy), and to form a nonpolar core (i.e., the backbone should play no role). Polyglycines do not possess nonpolar side chains, should be unable to build up a nonpolar core, and so the hydrophobic effect seems to play no role in their collapse.11 Actually, the hydrophobic effect has a geometric origin that has to be clarified and readily grasped and, in the case of globular proteins, mainly depends on their polymeric nature (i.e., it has little to do with the presence of nonpolar side chains).
A dismissed structural feature
It is usually stated that the protein interior is rich of nonpolar groups, whereas the protein surface is characterized by the presence of charged and polar groups to have favorable interactions with water molecules. This is not strictly correct. Thirty years ago Miller, Janin, Lesk and Chothia12 published a very interesting analysis of the features of the solvent accessible surface area13 of the native state of 46 monomeric proteins. It resulted that: “The average water-accessible surface is found to be 57% nonpolar, 24% polar and 19% charged, with 5% root-mean-square variations. The molecular surface buried inside the protein is 58% nonpolar, 39% polar and 4% charged”.We repeated the analysis using 178 ultra-high resolution structures (i.e., solved at a resolution higher than 0.99 Å, with an average resolution of 0.91 Å) of monomeric proteins deposited in the Protein Data Bank (see ESI† for further details) and found similar results. The average water accessible surface area is: (59 ± 4)% nonpolar, (23 ± 5)% polar and (19 ± 5)% charged.
These values, which also agree with other recent analyses,14 are important because they unequivocally demonstrate that it is not correct to state that nonpolar residues are buried in the protein core: 59% of the water accessible surface of the native state has a nonpolar character. In other words, the hydrophobic effect cannot be described as the tendency of nonpolar side chains to create a nonpolar core in the protein interior to avoid the “unfavorable” contact with water molecules.9 The rightness of this sentence is grounded on average structural features of globular proteins. In addition, one has to remind that the native structures of globular proteins do not resemble micelles; globular proteins are heteropolymers and the polymeric nature plays a major role in constraining the interior versus exterior distribution of both backbone and side chains.
In addition, the Miller–Janin–Lesk–Chothia analysis12 pointed out that a large fraction of peptide groups is buried and is involved in intramolecular H-bonds. The latter is a fundamental requirement to be satisfied because the same peptide groups form H-bonds with surrounding water molecules in unfolded conformations. Therefore, the formation of intramolecular H-bonds is not the driving force of protein collapse, even though it is fundamental for the stability of collapsed conformations and the attainment of a unique folded structure.15 In other words, the formation of H-bonds is the reason for the widespread occurrence of secondary structures, such as α-helices and β-sheets, that were identified, by Pauling and colleagues,16,17 for the ability to allow the formation of peptide–peptide H-bonds. If a polypeptide chain were not able to form a sufficient number of intramolecular H-bonds, it would be incapable to build up a stable and folded structure; i.e., it would be an intrinsically disordered protein.18
The hydrophobic effect
The expression hydrophobic effect is often used incorrectly in the scientific literature, especially in the biochemistry area. It is necessary to distinguish two different processes:19,20 (a) hydrophobic hydration refers to the thermodynamics characterizing the poor solubility of nonpolar species in water; i.e., in particular to the large negative entropy change causing this very low solubility around room temperature; (b) hydrophobic interaction refers to the attraction experienced by two or more nonpolar molecules in water, because the nonpolar cluster formation leads to an entropy gain for water molecules;20 a special case is the “intramolecular” hydrophobic interaction occurring in the case of globular protein collapse and folding.21 We try to provide a basic molecular interpretation.Hydrophobic hydration
In order to insert a solute molecule in a liquid, it is necessary to create a cavity suitable to host it. This is the basic consequence of the fact that each molecule possesses an intrinsic volume, and the void volume in a liquid is a very large fraction of the total volume, but it is partitioned in very small pieces which are not available to host another molecule,22,23 the solute molecule (i.e., the liquid state is a condensed phase of the matter). If cavity creation is performed at constant temperature, pressure and number of particles, the liquid volume increases by a quantity equal to the partial molar volume of the cavity itself. However, the shell region between the van der Waals surface of the cavity and its solvent accessible surface becomes excluded to the centers of solvent molecules (i.e., this constraint is a need to render empty the van der Waals volume of the cavity). This produces a solvent-excluded volume effect that causes a loss in translational entropy of solvent molecules,20 as a consequence of the decrease in accessible configurational space (i.e., the entropy loss is not due to an increase of structural order; the iceberg model suggested by Frank and Evans24 long time ago is not correct25). The solvent-excluded volume effect is operative in all liquids, but its magnitude is markedly larger in water due to the small size of water molecules.20,26,27 This is particularly evident if one considers that the diameter of a water molecule is 2.8 Å,28 corresponding to the location of the first peak in the oxygen–oxygen radial distribution function of liquid water around room temperature.29 This diameter is the smallest among all the simple molecular liquids.20,30 The small size reflects also in the large number density characterizing liquid water:30 at 25 °C and 1 atm, 55.3 M for water, 24.5 M for methanol, 17.0 M for ethanol, 11.2 M for benzene, 10.3 M for carbon tetrachloride, and 9.2 M for cyclohexane. Therefore, the poor solubility of nonpolar species in water is due to the magnitude of the reversible work of cavity creation, ΔGc, which proves to be markedly larger than that in the other liquids20,26,30 (i.e., the van der Waals interactions between the nonpolar molecule and surrounding water molecules cannot overwhelm the large and positive ΔGc contribution). This is why the solubility of hydrocarbons in water scales with their water accessible surface area, WASA, with the latter quantity that is a measure of the solvent-excluded volume effect.31–33Hydrophobic interaction
Following the above reasoning, the nonpolar cluster formation is thermodynamically favored because it leads to a significant WASA decrease that corresponds to a decrease in solvent-excluded volume, and so to a significant gain in translational entropy of water molecules.20 The fundamental role of the translational entropy of water molecules emerges also in the theoretical approach developed by Kinoshita and co-workers.34–36 The same theoretical ideas should be used in the case of globular proteins, but it should be recognized that the latter can assume a very large number of different conformations in view of their polymeric nature. In the assumption that the van der Waals volume of the polypeptide chain does not change on passing from one conformation to the others, a fundamental distinguishing geometric property of such conformations is their WASA value. Note that the WASA of a given conformation has to correspond to the WASA of the cavity suitable to host it. The native state conformation should have the smallest WASA among all the possible conformations of a polypeptide chain.21 Therefore, the collapse of a globular protein can be described as a search over the conformational space of a given sequence to obtain the smallest WASA. The driving force for such a search is exactly the WASA decrease, that corresponds to a decrease in the solvent-excluded volume effect, and so to a significant gain in translational entropy of water molecules (note that the theoretical approach developed along these lines has proven to be able to rationalize the general occurrence of cold denaturation,21,37 the effect of stabilizing and destabilizing agents,38,39 the denaturation induced by high hydrostatic pressure,40 and to shed light on the extra-stability of thermophilic globular proteins,41 and on the coil-to-globule transition of PNIPAM42–44). An entropically driven helix formation model has also been proposed, grounded on the solvent-excluded volume effect.45 In other words, the collapse driving force is provided by the translational entropy gain of water molecules, it has a geometric origin, and is not related to the burial of nonpolar side chains. Their presence increases the magnitude of the effect, and provides important constraints to reduce the number of collapsed structures.46,47 This is why Gly25, totally lacking nonpolar side chains, populates globule-shaped structures in water around room temperature.A model for Gly25 collapse
The conformational search of a polypeptide chain can be modelled in a simple, yet physically correct, manner, by looking at the cavity shape. It is well known that, keeping fixed the volume, the sphere is the 3D object possessing the smallest surface area. Therefore, the native state can be represented by a sphere, whereas the denatured conformations can be represented by prolate spherocylinders, all possessing the same van der Waals volume of the sphere representing the native state (note that the choice of prolate spherocylinders should be considered qualitatively correct because it has been shown that the denatured state of several globular proteins has a shape resembling a prolate ellipsoid48). From the geometric point of view, a prolate spherocylinder has a WASA larger than that of the corresponding sphere and such a difference increases on increasing the cylindrical length of the spherocylinder. This implies that the reversible work of cavity creation, ΔGc, increases with WASA, even though the van der Waals volume is fixed, because the magnitude of the solvent-excluded volume effect increases on increasing the cylindrical length of the prolate spherocylinder.49,50 A set of cavities has been selected to model Gly25. Since, according to the MD results of Pettitt and co-workers,5 the Gly25 collapsed state has Rg ≈ 7 Å and WASA ≈ 800 Å2, the selected sphere has a 6.6 Å radius and WASA = 804 Å2; since the average Gly25 extended structure has WASA ≈ 1650 Å2, the selected longest spherocylinder has WASA = 1612 Å2. The trend of ΔGc values versus WASA for the set of cavities, whose geometric measures are listed in Table 1, is shown in Fig. 1: an almost linear relationship is evident. The present calculations have been performed by means of classic scaled particle theory,51 SPT, that provides analytical formulae for both spherical and prolate spherocylindrical cavities,21 using the experimental density of water at 25 °C and 1 atm.52 It is worth noting that qualitatively similar results (i.e., the ΔGc increase caused by a rise of cavity WASA, on keeping fixed the van der Waals volume of the cavity) have been obtained not only by means of classic SPT calculations,49,50 but also by means of MD simulations in detailed water models by Wallqvist and Berne,53 and by Chandler and co-workers.54| a (Å) | l (Å) | V vdW (Å3) | WASA (Å2) | ΔGc (kJ mol−1) | ΔGc/WASA (J mol−1 Å−2) |
|---|---|---|---|---|---|
| Note: it is worth noting that the numbers in the last column have the dimensions of a surface tension, but they are not close to the experimental liquid–vapor surface tension of water at 25 °C and 1 atm. This is expected because the process of cavity creation in a liquid does not correspond to the creation of a liquid–vapor interface.30 In addition, these numbers depend on the size and shape of the cavity, as already pointed out by means of careful MD simulations,61 and simple classic SPT calculations.50 This is a further manifestation of the solvent-excluded volume effect associated with cavity creation, that is intrinsically a multi-particle and non-additive effect. | |||||
| 2.5 | 58.00 | 1204 | 1612 | 415.4 | 257.7 |
| 3.0 | 38.59 | 1204 | 1310 | 347.1 | 265.0 |
| 3.5 | 26.63 | 1204 | 1122 | 302.3 | 269.4 |
| 4.0 | 18.62 | 1204 | 998 | 271.9 | 272.4 |
| 4.5 | 12.93 | 1204 | 917 | 251.1 | 273.8 |
| 5.0 | 8.67 | 1204 | 863 | 237.1 | 274.7 |
| 6.6 | — | 1204 | 804 | 221.1 | 275.0 |
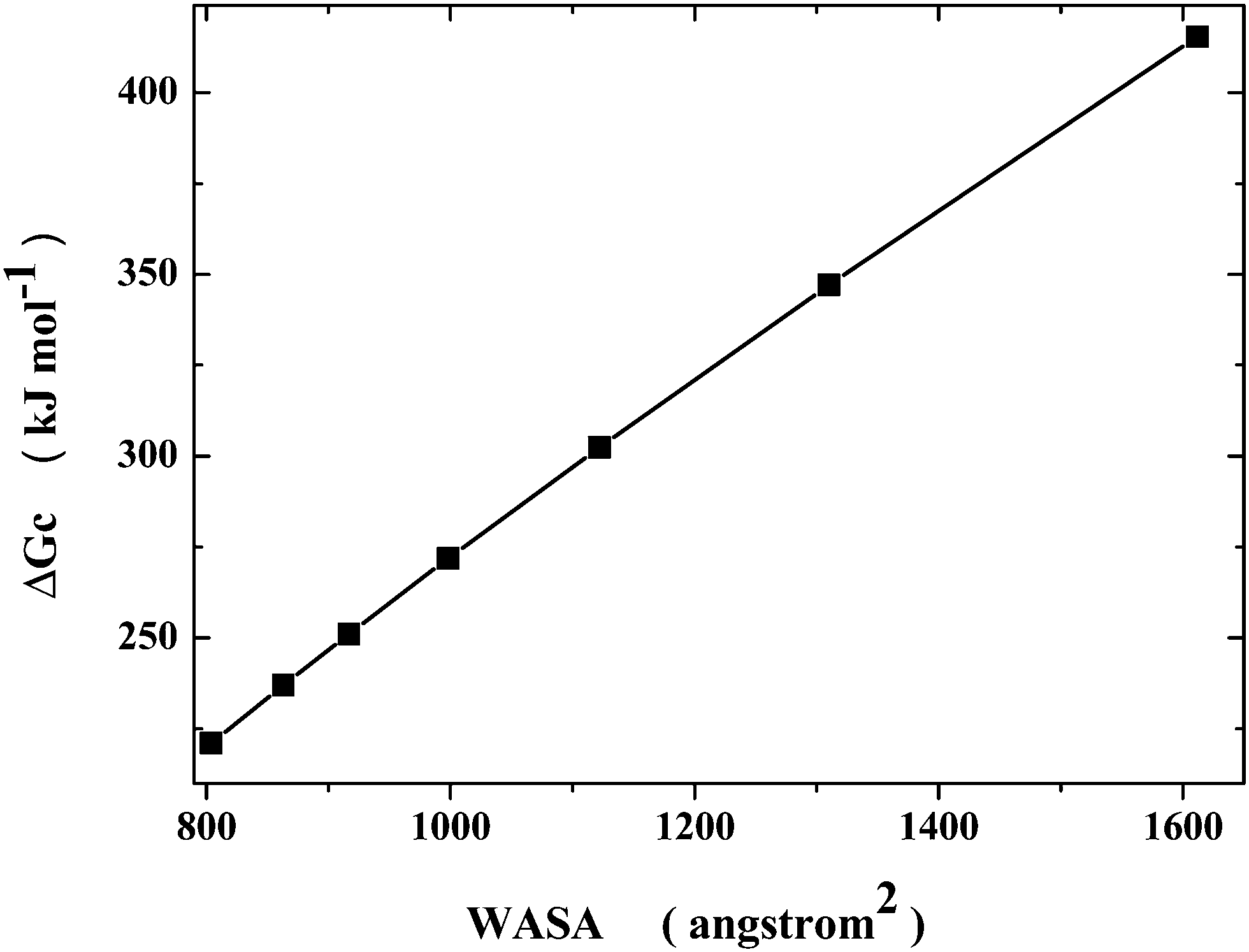 | ||
| Fig. 1 Dependence of ΔGc on WASA, according to classic SPT calculations, at 25 °C and 1 atm, for spherocylindrical cavities possessing the same van der Waals volume of a 6.6 Å radius sphere, using the experimental density of liquid water (see Table 1). | ||
On plotting the same ΔGc values as a function of the cylindrical length (see Fig. 2), one obtains a trend that resembles a side of the funnel that is considered to be the right description of the energy landscape for protein collapse and folding.55,56 The decrease in solvent-excluded volume drives the process; for Gly25, the Gibbs energy decreases by 194 kJ mol−1 on passing from the longest spherocylinder to the sphere, because water molecules gain translational entropy, and the chain becomes more and more compact. The geometric model is surely too crude to take into account all the details of protein collapse, but it should be able to clarify what is the actual driving force of the process.
 | ||
| Fig. 2 Dependence of ΔGc upon the cylindrical length for the whole series of spherocylindrical cavities listed in Table 1. | ||
The Gibbs energy gain given by ΔGc(sphere) − ΔGc(longest spherocylinder) = −194 kJ mol−1 is only one of the contributions governing the conformational stability of Gly25 (and globular proteins).21,37 The Gibbs energy change associated with collapse is given by:
| ΔGcollapse = ΔΔGc − T·ΔSconf + ΔEa | (1) |
| T·ΔSconf = −194 + 119 = −75 kJ mol−1 |
It is worth noting that a modified Gly20, where formation of backbone H-bonds was blocked by substituting the amide hydrogen atoms with methyl groups, NMe–Gly20, was much more soluble in water compared to Gly20 and showed no tendency to collapse.7 These experimental data are also confirmed by MD simulation results of Pettitt and co-workers.5 In the above Gibbs energy balance, the fundamental difference between Gly25 and NMe–Gly20 should be related to the magnitude of the ΔEa term. The intermolecular energetic interactions of the methylated amide groups of NMe–Gly20 with water molecules are surely stronger than the intramolecular energetic interactions among the same methylated amide groups. This means that the ΔEa term for NMe–Gly20 is positive and larger in magnitude than the estimate reported above, rendering ΔGcollapse positive and so stabilizing extended conformations. The devised theoretical approach is able to rationalize both the Gly25 collapse and the non-collapse of NMe–Gly20.
A careful computational analysis of Gly15 collapse has been performed by means of MD simulations by Asthagiri,60 leading to a rationalization different from the one presented above. He claims, in line with Pettitt and co-workers,5 that the intramolecular energetic attractions among peptide groups (not only H-bonds, but also dipole–dipole attractions) are the driving force for collapse because they provide a large and negative contribution to the Gibbs energy balance.60 Actually, the difference between the Asthagiri conclusion and the present one is only apparent. If all the energetic terms were grouped together, the Asthagiri calculations provide a scenario in which the ΔΔGc contribution drives chain collapse, in line with the present analysis, because the intramolecular energetic attractions are more than counterbalanced by the intermolecular energetic attractions between peptide groups and water molecules, that favor extended conformations. Therefore, the decrease in solvent-excluded volume, corresponding to a gain in translational entropy of water molecules, provides the driving force for polypeptide chain collapse.
Conclusion
The present study has been devoted to clarify a puzzling experimental and computational datum,5,7 the collapse of Gly25 in water at room temperature. The latter corresponds to a decrease in solvent-excluded volume that is the right description of hydrophobic interactions, and leads to a significant gain in translational entropy of water molecules. This entropy gain is the driving force of polypeptide and protein collapse.Acknowledgements
We would like to thank Dr Luigi Vitagliano, Istituto di Biostrutture e Bioimmagini, CNR, for several stimulating discussions on protein collapse and folding over the years.References
- H. S. Chan and K. A. Dill, Phys. Today, 1993, 46, 24–32 CrossRef CAS.
- E. Shakhnovich, Chem. Rev., 2006, 106, 1559–1588 CrossRef CAS PubMed.
- K. A. Dill, S. B. Ozkan, M. S. Shell and T. R. Weikl, Annu. Rev. Biophys., 2008, 37, 289–316 CrossRef CAS PubMed.
- A. Ben-Naim, Myths and Verities in Protein Folding Theories, World Scientific, Singapore, 2015 Search PubMed.
- D. Karandur, R. C. Harris and B. M. Pettitt, Protein Sci., 2016, 25, 103–110 CrossRef CAS PubMed.
- H. T. Tran, A. Mao and R. V. Pappu, J. Am. Chem. Soc., 2008, 130, 7380–7392 CrossRef CAS PubMed.
- D. P. Teufel, C. M. Johnson, J. K. Lum and H. Neuweiler, J. Mol. Biol., 2011, 409, 250–262 CrossRef CAS PubMed.
- A. Moglich, K. Joder and T. Kiefhaber, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12394–12399 CrossRef PubMed.
- W. Kauzmann, Adv. Protein Chem., 1959, 14, 1–63 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. Gong, L. L. Porter and G. D. Rose, Protein Sci., 2011, 20, 417–427 CrossRef CAS PubMed.
- S. Miller, J. Janin, A. M. Lesk and C. Chothia, J. Mol. Biol., 1987, 196, 641–656 CrossRef CAS PubMed.
- B. Lee and F. M. Richards, J. Mol. Biol., 1971, 55, 379–400 CrossRef CAS PubMed.
- G. Graziano and A. Merlino, Biochim. Biophys. Acta, 2014, 1844, 850–858 CrossRef CAS PubMed.
- G. D. Rose, P. J. Fleming, J. R. Banavar and A. Maritan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16623–16633 CrossRef CAS PubMed.
- L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 251–256 CrossRef CAS.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 CrossRef CAS PubMed.
- W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed. Engl., 1993, 32, 1545–1579 CrossRef.
- G. Graziano, Pure Appl. Chem., 2016, 88, 177–188 CrossRef CAS.
- G. Graziano, Phys. Chem. Chem. Phys., 2010, 12, 14245–14252 RSC.
- A. Pohorille and L. R. Pratt, J. Am. Chem. Soc., 1990, 112, 5066–5074 CrossRef CAS PubMed.
- G. Graziano, Biophys. Chem., 2003, 104, 393–405 CrossRef CAS PubMed.
- H. S. Frank and M. W. Evans, J. Chem. Phys., 1945, 13, 507–532 CrossRef CAS.
- P. Buchanan, N. Aldiwan, A. K. Soper, J. L. Creek and C. A. Koh, Chem. Phys. Lett., 2005, 415, 89–93 CrossRef CAS.
- B. Lee, Biopolymers, 1985, 24, 813–823 CrossRef CAS PubMed.
- B. Madan and B. Lee, Biophys. Chem., 1994, 51, 279–289 CrossRef CAS PubMed.
- G. Graziano, Chem. Phys. Lett., 2004, 396, 226–231 CrossRef CAS.
- J. M. Sorenson, G. Hura, R. M. Glaser and T. Head-Gordon, J. Chem. Phys., 2000, 113, 9149–9161 CrossRef CAS.
- G. Graziano, J. Phys. Chem. B, 2006, 110, 11421–11426 CrossRef CAS PubMed.
- R. B. Hermann, J. Phys. Chem., 1972, 76, 2754–2759 CrossRef CAS.
- C. Chothia, Nature, 1974, 248, 338–339 CrossRef CAS.
- P. A. Karplus, Protein Sci., 1997, 6, 1302–1307 CrossRef CAS PubMed.
- Y. Harano and M. Kinoshita, Biophys. J., 2005, 89, 2701–2710 CrossRef CAS PubMed.
- T. Yoshidome and M. Kinoshita, Phys. Chem. Chem. Phys., 2012, 14, 14554–14566 RSC.
- H. Oshima and M. Kinoshita, J. Chem. Phys., 2015, 142, 145103 CrossRef PubMed.
- G. Graziano, Phys. Chem. Chem. Phys., 2014, 16, 21755–21767 RSC.
- G. Graziano, Phys. Chem. Chem. Phys., 2011, 13, 12008–12014 RSC.
- G. Graziano, Phys. Chem. Chem. Phys., 2011, 13, 17689–17695 RSC.
- G. Graziano, Biopolymers, 2015, 103, 711–718 CrossRef CAS PubMed.
- G. Graziano, Biopolymers, 2016, 104, 856–863 Search PubMed.
- G. Graziano, Phys. Chem. Chem. Phys., 2015, 17, 27750–27757 RSC.
- G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 14426–14433 RSC.
- G. Graziano, Phys. Chem. Chem. Phys., 2016, 18, 25601–25608 RSC.
- Y. Snir and R. D. Kamien, Science, 2005, 307, 1067 CrossRef CAS PubMed.
- M. J. Behe, E. E. Lattman and G. D. Rose, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 4195–4199 CrossRef CAS.
- A. Q. Zhou, C. S. O’Hern and L. Regan, Proteins, 2014, 82, 2574–2584 CrossRef CAS PubMed.
- H. T. Tran and R. V. Pappu, Biophys. J., 2006, 91, 1868–1886 CrossRef CAS PubMed.
- G. Graziano, J. Phys. Chem. B, 2009, 113, 11232–11239 CrossRef CAS PubMed.
- G. Graziano, J. Mol. Liq., 2015, 211, 1047–1051 CrossRef CAS.
- R. A. Pierotti, Chem. Rev., 1976, 76, 717–726 CrossRef CAS.
- G. S. Kell, J. Chem. Eng. Data, 1975, 20, 97–105 CrossRef CAS.
- A. Wallqvist and B. J. Berne, J. Phys. Chem., 1995, 99, 2885–2892 CrossRef CAS.
- A. J. Patel, P. Varilly and D. Chandler, J. Phys. Chem. B, 2010, 114, 1632–1637 CrossRef CAS PubMed.
- K. A. Dill and H. S. Chan, Nat. Struct. Biol., 1997, 4, 10–19 CrossRef CAS PubMed.
- J. N. Onuchic and P. G. Wolynes, Curr. Opin. Struct. Biol., 2004, 14, 70–75 CrossRef CAS PubMed.
- M. C. Baxa, E. J. Haddadian, J. M. Jumper, K. F. Freed and T. R. Sosnick, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15396–15401 CrossRef CAS PubMed.
- K. A. Sharp, E. O’Brien, V. Kasinath and A. J. Wand, Proteins, 2015, 83, 922–930 CrossRef CAS PubMed.
- F. Fogolari, A. Corazza, S. Fortuna, M. A. Soler, B. Van Schouwen, G. Brancolini, S. Corni, G. Melacini and G. Esposito, PLoS One, 2015, 10, e0132356 Search PubMed.
- D. Asthagiri, 2016, arXiv:1605.00350v2.
- R. C. Harris and B. M. Pettitt, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14681–14686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: List of the PDB codes of the 178 ultra-high resolution protein structures analyzed, and results of the calculations. See DOI: 10.1039/c6cp07397b |
| This journal is © the Owner Societies 2017 |