
An atlas of endohedral Sc2S cluster fullerenes†
Li-Hua
Gan
*a,
Rui
Wu
a,
Jian-Lei
Tian
a and
Patrick W.
Fowler
*b
aSchool of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, China. E-mail: ganlh@swu.edu.cn
bDepartment of Chemistry, Sheffield University, Sheffield, S3 7HF, UK. E-mail: P.W.Fowler@sheffield.ac.uk
First published on 22nd November 2016
Abstract
Structural identification is a difficult task in the study of metallofullerenes, but understanding of the mechanism of formation of these structures is a pre-requisite for new high-yield synthetic methods. Here, systematic density functional theory calculations demonstrate that metal sulfide fullerenes Sc2S@Cn have similar cage geometries from C70 to C84 and form a close-knit family of structures related by Endo–Kroto insertion/extrusion of C2 units and Stone–Wales isomerization transformations. The stabilities predicted for favoured isomers by DFT calculations are in good agreement with available experimental observations, have implications for the formation of metallofullerenes, and will aid structural identification from within the combinatorially vast pool of conceivable isomers.
1. Introduction
Endohedral metallofullerenes (EMFs) are closed cage-shaped molecules with encaged metal atoms or clusters.1,2 Since their first synthesis in macroscopic amounts,3 EMFs have attracted extensive interest from chemists, physicists and materials scientists. To date, more than two hundred EMFs have been characterized, including mono-metallofullerenes, di-metallofullerenes, tri-metallofullerenes, and fullerenes incorporating clusters of various kinds.4 Amongst the EMFs, those in which the encapsuland is a metallic cluster have attracted attention because of their novel geometries and properties.5,6 However, as far as we are aware, the mechanism of formation of EMFs has not yet been clarified, even though so many EMFs have been reported. For small fullerenes, a recent theoretical study shows that C2 insertion (by the Endo–Kroto mechanism) can facilitate the formation of larger fullerenes without additional Stone–Wales (S–W) rotations.7 Where the cage is larger than C70, direct C2 insertion in classical isomers will form non-IPR (IPR = isolated pentagon rule8) isomers, which is not in agreement with experimental observations that all reported stable (neutral, bare) fullerenes are IPR-satisfying. Hence, C2 insertion alone cannot account for the formation of fullerenes. Some post-insertion step, for which S–W rotation is the best candidate, is a necessary stage in the formation of fullerenes.For endohedral metallic cluster fullerenes, formation processes may be similar to those for bare fullerenes, at least in terms of the cages involved, but they are likely to be affected by the presence of the additional participant. A pre-formed fullerene cage would almost certainly be broken by insertion of a metallic cluster into its interior, as the available windows, hexagonal, pentagonal or heptagonal faces, are small. It seems highly improbable that a metallic cluster will insert to form an EMF directly. Thus, reasonable formation paths would seem to require growth or shrinkage of a cage with a cluster inside, or curling up to form fullerene cages with simultaneous encapsulation of a metal atom/cluster, as is consistent with molecular dynamics simulations.9
Recent experiments show that non-classical fullerene cages with a heptagonal ring can also encapsulate metallic clusters,10 or can be captured as chlorofullerenes11 from the carbon-arc plasma in situ. Theoretical studies show that heptagonal rings may play an important role in the formation of bare fullerenes12,13 and trimetallic nitride template fullerenes.14 To predict the geometrical structures of some poorly characterized metallofullerenes and seek insight into their formation, it was decided to make a systematic study of classical and non-classical isomers Sc2S@Cn (n = 70–84). This was carried out with the help of an extended face-spiral algorithm for construction of cage candidates with small numbers of heptagonal faces. The results show that heptagon-including metallic sulfide fullerenes Sc2S@Cn are not competitive with classical Sc2S@Cn in terms of total energy. Interestingly, there are strong structural similarities amongst low-energy Sc2S@Cn isomers of equal and adjacent cage sizes. These similarities are given concrete form in terms of S–W isomerization and C2 insertion/extrusion transformations. Our results provide clues to finding new metallofullerenes from within the tens of thousands of conceivable structural isomers. They also give potentially useful information on the formation mechanisms of EMFs.
2. Computational details
The isomers to be considered are generated by an extension of the face-spiral algorithm to allow up to one heptagonal and 13 pentagonal faces.15 This approach is known to be complete in the size range. The nomenclature and labelling of isomers follow the face-spiral algorithm. Briefly, classical isomers are labelled by their positions in the sequence of canonical spirals, and a superscript (1h) is used to indicate a non-classical isomer with one heptagon, with a number indicating its position in the spiral order for these cages. Topological coordinates15 are used to provide initial cage structures, which are then optimized for charges 0, −2, −4 and −6, first at the semi-empirical PM3 level and then, for a selection of the best cages at each charge, at the B3LYP/3-21G level. Based on the energy ranking for the optimized cages with charge −4, and some other favoured isomers with different charges, the favoured cages are used as parents to construct Sc2S-based EMFs. The Sc2S moiety is placed close to the geometric centre of the cage, and irrespective of the initial shape and atom ordering (Sc–S–Sc or S–Sc–Sc) optimizes to a structure where S lies between the two Sc centres; this is a simple consequence of the fact that the moiety donates electrons to the cage, generating electrostatic repulsion between the newly formed ionic Sc centres. The numbers of isomers considered by nuclearity are shown in S1 (ESI†). Geometrical optimizations are performed at B3LYP/3-21G and then B3LYP/6-31G* levels on Sc2S@Cn with n from 70 to 84. All calculations are performed with Gaussian 09 software.16 The results for the favoured isomers are shown in Fig. 1 and Table 1, and the optimized coordinates calculated for the isomers of lowest energy are provided in S2 (ESI†). In the optimized structures for the favoured isomers, the Sc–S–Sc angle varies between 140° and 180° in most cases, with Sc2S@C76:19151 having a much smaller angle of 108°. The linear Sc–S–Sc moieties are found in Sc2S@C74:13333 and Sc2S@C84:51591 (see coordinates in S2, ESI†).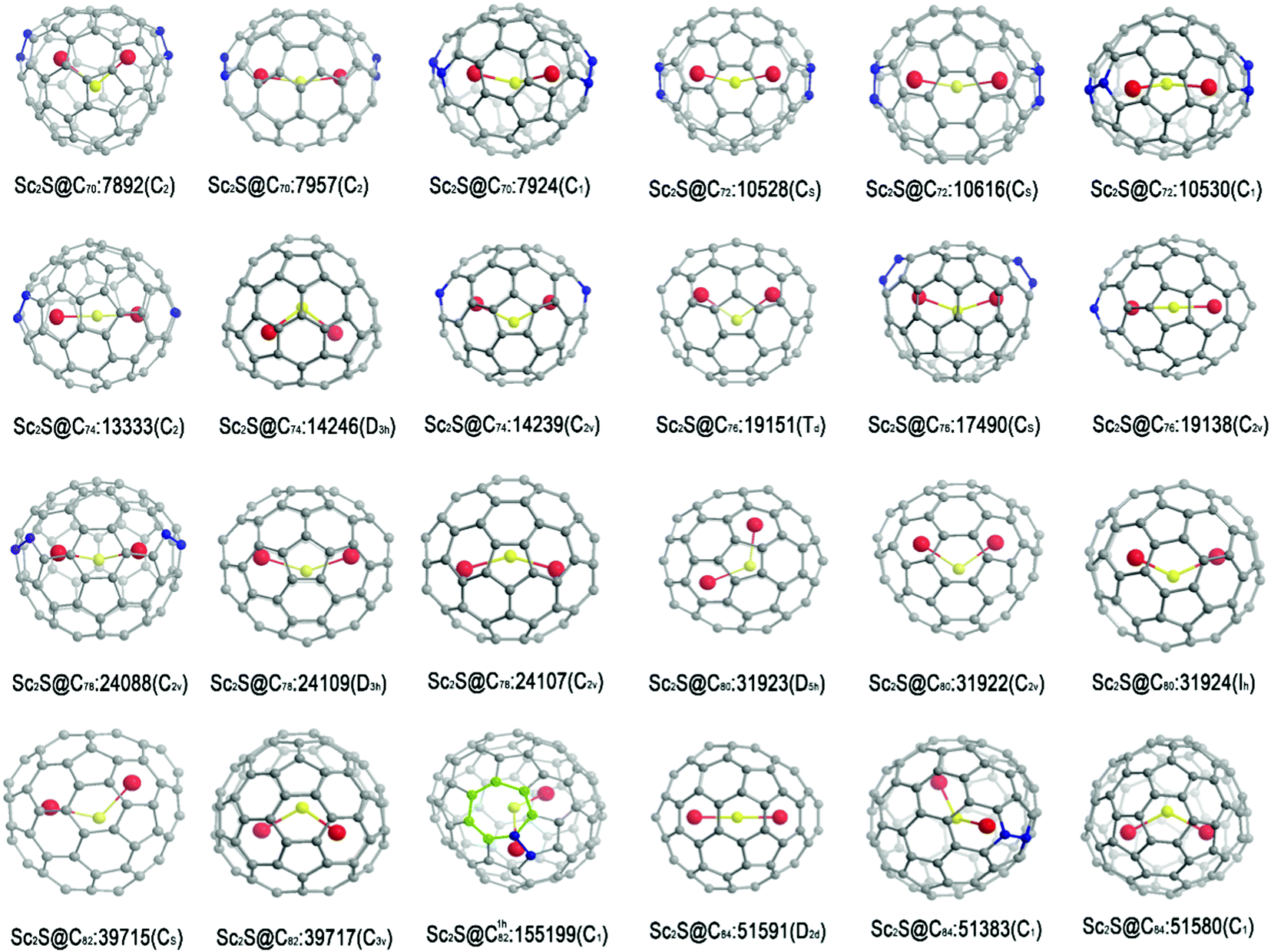 | ||
| Fig. 1 Optimized structures of the three isomers of lowest energy for each Sc2S@Cn with n from 70 to 84 as predicted at the B3LYP/6-31G* level. | ||
| Cage | N 55 | ΔE | Gap | Sc–Sc | Cage | N 55 | ΔE | Gap | Sc–Sc | Cage | N 55 | ΔE | Gap | Sc–Sc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C70:7892-C2 | 2 | 0.0 | 1.84 | 3.597 | C76:19151-Td | 0 | 0.0 | 1.45 | 3.801 | C82:39715-Cs | 0 | 0.0 | 1.72 | 4.073 |
| C70:7957-C2 | 2 | 20.8 | 1.54 | 4.584 | C76:17490-Cs | 2 | 11.3 | 1.32 | 4.546 | C82:39717-C3v | 0 | 0.3 | 2.08 | 4.008 |
| C70:7924-C1 | 2 | 20.9 | 1.35 | 4.241 | C76:19138-C1 | 1 | 12.0 | 0.76 | 4.599 | C1h82:155199(C1) | 1 | 12.8 | 1.81 | 4.129 |
| C72:10528-Cs | 2 | 0.0 | 1.85 | 4.225 | C78:24088-C2v | 2 | 0.0 | 1.76 | 4.207 | C82:39718-C2v | 0 | 14.4 | 1.20 | 4.363 |
| C72:10616-Cs | 2 | 13.9 | 1.93 | 4.425 | C78:24109-D3h | 0 | 1.7 | 0.97 | 4.485 | C84:51591-D2d | 0 | 0.0 | 1.68 | 4.621 |
| C72:10530-C1 | 2 | 19.1 | 1.66 | 3.979 | C78:24107-C2v | 0 | 3.7 | 1.53 | 4.168 | C84:51383-C1 | 1 | 10.4 | 1.90 | 4.447 |
| C74:13333-C2 | 2 | 0.0 | 1.68 | 4.651 | C80:31923-D5h | 0 | 0.0 | 1.12 | 4.108 | C84:51580-C1 | 0 | 12.3 | 1.73 | 4.163 |
| C74:14246-D3h | 0 | 11.8 | 1.14 | 3.815 | C80:31922-C2v | 0 | 9.0 | 1.67 | 3.840 | |||||
| C74:14239-C2v | 2 | 11.9 | 1.21 | 4.157 | C80:31924-Ih | 0 | 9.2 | 0.84 | 4.196 |
3. Results and discussion
Fig. 1 shows the structures for the three most favoured isomers for each of the eight formulas Sc2S@Cn with n = 70–84.It is shown in Table 1 that the isomer of lowest energy isomer for Sc2S@C70 has a large HOMO–LUMO gap (1.84 eV) and parent cage, the non-IPR C70:7892, with two pentagon adjacencies. The stability of this isomer is in good agreement with experimental results.17 In fact, this cage has recently been shown to be the parent of a geometrically and electronically similar cluster Sc2O.18 The two isomers next in energy order are also classical, but lie more than 20 kcal mol−1 higher.
It is shown in Table 1 that Sc2S@C72:10528 is the first favoured isomer in energy terms and has a large gap of 1.85 eV. The next most favoured isomer is higher in energy by 13.9 kcal mol−1 than the first, which corresponds to an equilibrium fractional population of only 3% at 2000 K. This isomer and others of higher energy are unlikely to be isolated in significant yield. The isomer predicted to be most favoured corresponds to experimental observations19a and recent theoretical results.19b The same parent cage is shared by the electronically similar cluster Sc2C2 in the metallic carbide fullerene Sc2C2@C72,20 and the electronically and geometrically similar cluster Sc2O in the metallic oxide fullerene Sc2O@C72.21
We have also studied Sc2S@C74.22 The results show the most favoured isomer to be IPR-violating and Sc2S cluster to be linear inside cage C74:13333. The next two isomers in order of energy lie 11.8 and 11.9 kcal mol−1 higher in energy than the first. The cage of the second favoured isomer is the only IPR isomer of C74, which itself is open-shell and hence reactive as a neutral cage, but is apparently passivated in the form of Sc2S@C74.
A molecule of stoichiometry Sc2S@C76 has been detected by mass spectrometry, but without further experimental characterization.19 Our calculations indicate that the isomer of lowest energy is Sc2S@C76:19151, followed by Sc2S@C76:17490, in agreement with recent theoretical results.23 The third isomer in order of energy is Sc2S@C76:19138. XRD characterization has shown that the C76:19151 cage also encapsulates the Sc2O cluster to form Sc2O@C76.24
For Sc2S@C78, the isomer predicted to have lowest energy has cage C78:24088, a non-IPR fullerene with two pentagon pairs, instead of the well-known IPR cage, C78-24109, which encapsulates two La atoms in the form of La2@C7825 in relatively high yield. Cage C78:24088 would be only ninth best in terms of the energy of the tetra-anion C784−, reminding us that a simple electron-transfer model does not account for the stability of Sc2S@Cn in every case.
The cage of the most favoured Sc2S@C80 is IPR-satisfying C80:31923, which gives the C80 tetra-anion of second lowest energy (see S3, ESI†). The tetra-anion with lowest calculated energy is based on Ih-symmetrical C80:31924, but the triply degenerate LUMO of this cage would tend to indicate acceptance six additional electrons, rather than four that Sc2S can offer, and it turns out that the isomer of Sc2S@C80 based on the Ih cage has only the third best predicted energy. The cage of the second favoured Sc2S@C80 is IPR-satisfying C80-31922, which has recently been shown to be the parent of a geometrically and electronically similar cluster Sc2O@C80.26 This case demonstrates some of the complexities of the interaction between cage and encapsuland, where predictions of stability based on electronic factors may be in conflict with simple steric matching of size and shape.
In terms of calculated relative energies of the tetra-anion (see S3, ESI†), C82:39717 should be the best cage for encapsulating a Sc2S cluster and, in fact, this metallic sulfide fullerene was the first to be reported for any nuclearity.27 Our calculations predict that Sc2S@C82:39717 and the isomer of lowest calculated energy, Sc2S@C82:39715, are essentially iso-energetic. Although evidently smaller than that of Sc2S@C82:39717, the HOMO–LUMO gap of Sc2S@C82:39715 (1.71 eV) is still relatively large, and this isomer has recently been reported in experiment.28 Interestingly, non-classical C1h82:155199, the cage of the third best isomer of Sc2S@C82 in our calculations, could serves as a structural bridge between the cages of two experimental isomers of Sc2S@C82, as shown in Fig. 2.
 | ||
| Fig. 2 The structural web (atlas) of Sc2S@Cn isomers. Arrows in red and purple indicate that a Stone–Wales isomerization can interconvert two isomers; colored edges indicate the atoms undergoing a Stone–Wales rotation. | ||
For Sc2S@C84, the calculations show that the favoured isomer is IPR-Sc2S@C84:51591, sharing a parent cage with Sc2C2@C84;29 the second lowest isomer is non-IPR Sc2S@C84:51383, with predicted high kinetic stability. Our recently reported calculations show that Sc2S@C84:51575 is favoured at high temperature (∼2800 K); this last isomer can transform to the most favourable isomer Sc2S@C84:51591 via S–W rotation.30
Geometry optimizations on Sc2S@Cn with non-classical candidate cages show none that are competitive with classical isomers. The best of the non-classical Sc2S@Cn isomers with n = 70 to 82 lie respectively 28.5, 32.5, 34.9, 27.8, 34.3, 17.9 and 28.2 kcal mol−1 higher than their classical competitors. Thus, isolation of non-classical isomers of these compounds is unlikely.
Cluster effect
I h-C80 is the commonest parent cage for encapsulation of TNT clusters, as demonstrated by numerous experimental observations.4–6 However, it is not favoured for encaging Sc2S. There are other indications of differences in stability for Sc2S and TNT clusters. For example, as mentioned above, C82:39718, the cage of the most favoured isomer of Sc3N@C8231 is a structural bridge between the cages of two favoured candidates, i.e., C82:39717 and C82:39715 for encaging TNT clusters. However, when the encaged cluster is Sc2S, the bridging isomer is the non-classical isomer C1h82:155199. Significantly, C2 addition to C82:39715, the favoured cage for encaging Sc2S, can form C84:51383, the parent cage of the second Sc2S@C84; however, C2 addition to C82:39718, the parent cage of the favoured Sc3N@C82 can form C84:51365, the parent cage of EMFs M3N@C84 (M = Tb, Tm and Gd).32,33 These results suggest that the encaged cluster mediates, or even controls, the formation mechanism; this important role could arise from differences in electron-donating capacity (six electrons for TNT cluster and four for the Sc2S or Sc2O cluster) and/or different degrees of geometrical match to the cages.Our recent calculations have shown that the lower symmetry and local deformations associated with introduction of a heptagonal ring favour encapsulation of mixed (and intrinsically less symmetrical) metal nitride clusters.14 In the present case, however, no non-classical Sc2S-based EMF is predicted to be competitive with those based on classical cages. Non-classical cages that include one heptagonal face will also tend to have more pentagon adjacencies, almost universally34 destabilizing in the neutral. As there are two metallic atoms rather than three, it may be easier for them to find suitable internal positions, whether the cage is symmetrical or not, especially as Sc2S has a low bending force constant35,36 and hence is intrinsically more flexible than an M3N cluster. This may account for the lesser role of non-classical cages in encapsulation of the Sc2S cluster.
Structural interdependence
The calculations here give a set of favoured structures for Sc2S-based EMFs with eight different cage sizes. As noted above, all predictions for isomers of lowest energy are in agreement with available experimental and theoretical data. Interestingly, there is an evident structural dependence among the parent cages of the low-energy isomers Sc2S@Cn. As shown in Fig. 2, a C2 unit added to C70:7892 can form C72:10528, and a further C2 addition forms C74:13333. The corresponding Sc2S-based EMFs are either those experimentally reported or theoretically predicted to be of lowest energy. A further addition leads to C76:17490, the parent cage of the second favoured isomer of Sc2S@C76. Further C2 addition to C76:17490 can form C78:22010, the parent cage of many TNT fullerenes;37,38 another C2 addition can form C1h80:112912, one Stone–Wales step away from the well-known Ih-symmetric C80:31924, the favoured cage for hexavalent (M3N) clusters. Carrying on the process of C2 addition can lead to the parent cage (C82:39717) of the first experimentally reported isomer of Sc2S@C82, and the parent cage of the recently reported, and essentially iso-energetic isomer, Sc2@C82:39715 with consecutive S–W rotations. C2 addition on C82: 39718 can form C84:51365, the parent of M3N@C84 (M = Tb, Tm and Gd).32,33In another part of the map, the unique IPR isomer of C70, i.e., C70:8149, has a tetra-anion energy that makes it the favoured candidate for encaging a cluster that would donate four electrons. C2 addition to C70:8149 can lead to C72:11188, fifth best in terms of its tetra-anion energy. In turn, C72:11188 can isomerize into a non-classical isomer C1h72:29907 with two pairs of fused pentagons, which can form C74:14246 via directly C2 addition. C74:14246 can form C76:19138 and C78:24088 via successive C2 addition. Interestingly, C2 addition on the parent cage of most favoured Sc2S@C80:31923 can lead to C82:39663, which has recently been proved to be the parent of Gd3N@C82,39,40 this cage is an S–W rotation away from the parent cage of the most favoured isomer Sc2S@C82:39715.
Exploration of the details encoded in the figure show that the vertical and diagonal connections in the map provide a route for expansion/shrinkage of all eight parent cages of low-energy isomers from Sc2S@C70 to Sc2S@C84.
C70:7892, the parent of the most favoured isomer of Sc2S@C70 can transform into C70:8111 via a C2 extrusion (C68:6094) and C2 insertion, and then isomerize into C70:8149; C68:6094 has been captured as C68Cl8 by in situ chlorination in the gas phase during radio-frequency synthesis41 and theoretically predicted to be the parent of Sc2O2.42 C72:10528, the parent of the most favoured isomer of Sc2S@C72, has a transformation to C72:11188, one of the molecules of lowest energy for this stoichiometry. The isomer of lowest energy Sc2S@C74:13333, can transform into the second-best Sc2S@C74:14246 as shown in Fig. 2. C76:14790 can transform into C76:19138, the second best cage for both tetra-valent and di-valent clusters, according to the relative energies of the anions. In fact, C76:19138 has been shown to encage divalent Sm.43 C76:19142 can transform into C76:19151 via an S–W rotation (not shown), the parent cage of lowest energy for Sc2S@C76. C78:24099 can transform into C78:24109 via an S–W rotation, the isomer most favoured for encaging Sc2O.44 C78-22010, the most favoured cage for the large metallic cluster Gd3N37 can transform into C78-24088, the most favoured cage for Sc2S. Interestingly, C70:7892, C72:10528, C74:13333, C76:17490 and C78:22010 all have similar transformations (extrusion/insertion and then isomerization) to their low energy counterparts. The Ih-C80 cage can transfer into D5h-C80via three kinds of path; the first is by growth, shrinkage and isomerization, the second is by shrinkage, growth and isomerization; the third path (at least in in a formal mathematical sense) is by a 36° rotation around a C5 axis of one half of Ih-C80 against the other. C82:39717 can isomerize into C82:39715, the parent cage of a recently experimentally reported isomer Sc2S@C8228via a non-classical bridge cage, C1h82-155199. Successive S–W rotations can transfer C84:51546 into the most favoured neutral cage C84:51590, and the parent cage of lowest energy Sc2S@C84, C84:51591.
In summary, favoured cages can grow or shrink in the vertical direction on the map, or isomerize in the horizontal direction into other favoured cages. Horizontal connections in the map typically provide a possible route from one favoured isomer to another of Sc2S@Cn. These include the global rotation of one hemisphere against another, which would have a high energy barrier for a pre-formed cluster, but for a mechanism involving combination of fragments would be a low-energy process. The parent fullerene cages and the low-energy Sc2S-based EMFs form a web of complex genealogical relationships. Many of the cages in the near region of this map are favoured for encapsulation of other types of mono-metallic, di-metallic or metallic clusters, all demonstrating common stabilizing substructures and motifs.
Formation mechanisms
The calculations of optimal structures demonstrate that C2 insertion/extrusion is not only a topological requirement but also a possible bridge for growth of favoured Sc2S-based EMFs, connecting isomers of low energy. As the most common metallic atom(s) or clusters would have extremely high barriers to insertion through a pentagon or hexagon of the fullerene, it seems likely that the Sc2S cluster would be encaged early in the process inside a fullerene (or fullerene-like) cage that could continue to grow or shrink via C2 insertion or extrusion, eventually forming a stable EMF. D3h-C78, Ih-C80, D5h-C80, C2v-C82, C3v-C82 and D2d-C84 are all IPR-satisfying and comprise the set of parent cages of most EMFs, but they cannot be formed by direct C2 insertion into a lower IPR fullerene, and so at least one S–W isomerization step is necessary. This remains true in models of growth from smaller clusters, since even if an IPR-satisfying cage of Cn+2 can be formed directly via C2 insertion into the heptagon of a non-classical isomer, an S–W isomerization would be needed for initial transformation from a classical isomer.Molecular dynamics simulations have shown that hot giant fullerenes can lose or gain carbon in high temperature conditions.45 This existence of a structural web such as shown in Fig. 2 suggests that, irrespective of the formation mechanism of Sc2S@Cn, there are generally many Sc2S@Cn species with different sizes, since any as-formed Sc2S@Cn can produce other metallic sulfide fullerenes via top-down46 or bottom-up47 paths and S–W isomerization. Experiment shows that there are always many Sc2S@Cn species in the soot,17,19a,48 and multiple species with different metallic atoms or clusters encaged inside different fullerene cages are produced under the same reaction conditions. For example, a mixture of several Sc2O@Cn, multiple Sc2C2@Cn, and even Sc3N@C80 species are found in soot produced under conditions designed to yield Sc2O@Cn by introducing CO2 as the oxygen source during the arcing process;21 a mixture of Sc2S@Cn and Sc2C2@C80 isomers is found in soot in a conventional Krätschmer–Huffman reactor for producing metallic sulfide fullerenes under an atmosphere of SO2.19 The difficulty in isolating a particular EMF is mainly a result of the diversity of EMFs in reaction mixtures.
Recently, Wang et al. identified four key structural motifs which govern the relative energies of anions of classical fullerene cages and thus can be used to search for suitable cages in endohedral metallofullerenes.49 This is a significant finding, but more rules would be desirable, since these motifs are compatible with many structural isomers. Our study helps to refine the predictive picture by setting the mass of experimental results scattered in the literature into the context of a relatively small family of low-energy isomers.
A further remark can be made. We have considered fullerenes encapsulating metallic sulfide clusters from the point of view of their structural interdependence. However, our main findings are likely to be valid for Sc2C2 and Sc2O in fullerene cages. The carbide Sc2C2 has similar electronic properties to Sc2S, and Sc2O and Sc2S are similar in both electronic and geometrical requirements. Thus, the present calculations may have implications for a larger family of EMFs, beyond the compounds of Sc2S.
4. Conclusion
Systematic DFT calculations demonstrate that there is a close structural interdependence amongst the favoured isomers of Sc2S@Cn with size from C70 to C84, and that one-heptagon non-classical Sc2S@Cn are generally much less competitive relative to classical cages in terms of total energetics. This is quite unlike the situation that we found for tri-metallic nitride fullerenes. These results indicate that formation of Sc2S@Cn and other EMFs is guided by the encaged clusters, and suggests a similar formation route in that cages featuring as preferred have closely related structures.As a whole, the stability of EMFs is determined by two main factors: one is electron transfer and the other is effect of size. Calculations show that many of the lowest energy isomers of Sc2O@Cn share the same cages with Sc2S@Cn apart from the cases with n = 74, 78 and 84. Since both Sc2O and Sc2S tend to donate 4 electrons to a fullerene cage, they tend to select the same isomer as host cage and these results indicate that electron transfer interactions play a vital role. Of course, the cluster sizes are different, and rankings are evidently different when comparing structures involving a series of cages of size Cn (when the sizes and shapes of both cages and encaged clusters play an important role).
Acknowledgements
The authors thank Gunnar Brinkmann (Ghent University) for cross-checking completeness of the face-spiral construction for the non-classical cages discussed here. Li-Hua Gan thanks the National Natural Science Foundation of China (51272216, 51472208) for financial support.References
- H. Shinohara, Rep. Prog. Phys., 2000, 63, 843–892 CrossRef CAS.
- A. Rodríguez-Fortea, A. L. Balch and J. M. Poblet, Chem. Soc. Rev., 2011, 40, 3551–3563 RSC.
- Y. Chai, T. Guo, C. Jin, R. E. Haufler, L. P. F. Chibante, J. Fure, L. Wang, J. M. Alford and R. E. Smalley, J. Phys. Chem., 1991, 95, 7564–7568 CrossRef CAS.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- J. Zhang, S. Stevenson and H. C. Dorn, Acc. Chem. Res., 2013, 46, 1548–1557 CrossRef CAS PubMed.
- T. Wang and C. Wang, Acc. Chem. Res., 2014, 47, 450–458 CrossRef CAS PubMed.
- J. S. Dang, W. W. Wang, J. J. Zheng, X. Zhao, E. Osawa and S. Nagase, J. Phys. Chem. C, 2012, 116, 16233–16239 CrossRef CAS.
- H. W. Kroto, Nature, 1987, 329, 529–531 CrossRef CAS.
- Q. Deng, T. Heine, S. Irle and A. A. Popov, Nanoscale, 2016, 8, 3796–3808 RSC.
- Y. Zhang, K. B. Ghiassi, Q. Deng, N. A. Samoylova, M. M. Olmstead, A. L. Balch and A. A. Popov, Angew. Chem., Int. Ed., 2015, 54, 495–499 CAS.
- Y. Z. Tan, R. T. Chen, Z. J. Liao, J. Li, F. Zhu, X. Lu, S. Y. Xie, J. Li, R. B. Huang and L. S. Zheng, Nat. Commun., 2011, 2, 1–6 Search PubMed.
- E. Hernandez, P. Ordejon and H. Terrones, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 193403 CrossRef.
- (a) W. W. Wang, J. S. Dang, J. J. Zheng and X. Zhao, J. Phys. Chem. C, 2012, 116, 17288–17293 CrossRef CAS; (b) W. W. Wang, J. S. Dang, J. J. Zheng, X. Zhao and S. Nagase, J. Phys. Chem. C, 2013, 117, 2349–2357 CrossRef CAS.
- L. H. Gan, D. Lei and P. W. Fowler, J. Comput. Chem., 2016, 37, 1907–1913 CrossRef CAS PubMed.
- P. W. Fowler and D. E. Manolopoulos, An Atlas of Fullerenes, Clarendon Press, Oxford, 1995 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox. Gaussian 09; Revision A. 02, Gaussian Inc., Pittsburgh, PA, 2009 Search PubMed.
- N. Chen, M. Mulet-Gas, Y. Y. Li, R. E. Stene, C. W. Atherton, A. Rodríguez-Fortea, J. M. Poblet and L. Echegoyen, Chem. Sci., 2013, 4, 180–186 RSC.
- L. Feng, M. Zhang, Y. Hao, Q. Tang, N. Chen, Z. Slanina and F. Uhlík, Dalton Trans., 2016, 45, 8142–8148 RSC.
- (a) N. Chen, C. M. Beavers, M. Mulet-Gas, A. Rodríguez-Fortea, E. J. Munoz, Y. Y. Li, M. M. Olmstead, A. L. Balch, J. M. Poblet and L. Echegoyen, J. Am. Chem. Soc., 2012, 134, 7851–7860 CrossRef CAS PubMed; (b) L.-J. Wang, R.-L. Zhong, S.-L. Sun, H.-L. Xu, X.-M. Pan and Z.-M. Su, Dalton Trans., 2014, 43, 9655 RSC.
- Y. Feng, T. Wang, J. Wu, L. Feng, J. Xiang, Y. Ma, Z. Zhang, L. Jiang, C. Shu and C. Wang, Nanoscale, 2013, 5, 6704–6707 RSC.
- M. Zhang, Y. Hao, X. Li, L. Feng, T. Yang, Y. Wan, N. Chen, Z. Slanina, F. Uhlík and H. Cong, J. Phys. Chem. C, 2014, 118, 28883–28889 CrossRef CAS.
- L. H. Gan, Q. Chang, C. Zhao and C. R. Wang, Chem. Phys. Lett., 2013, 570, 121–124 CrossRef CAS.
- P. Zhao, T. Yang, Y. J. Guo, J. S. Dang, X. Zhao and S. Nagase, J. Comput. Chem., 2014, 35, 1657–1663 CrossRef CAS PubMed.
- T. Yang, Y. Hao, L. Abella, Q. Tang, X. Li, Y. Wan, A. Rodríguez-Fortea, J. M. Poblet, L. Feng and N. Chen, Chem. – Eur. J., 2015, 21, 11110–11117 CrossRef CAS PubMed.
- B. Cao, T. Wakahara, T. Tsuchiya, M. Kondo, Y. Maeda, G. M. A. Rahman, T. Akasaka, K. Kobayashi, S. Nagase and K. Yamamoto, J. Am. Chem. Soc., 2004, 126, 9164–9165 CrossRef CAS PubMed.
- Q. Tang, L. Abella, Y. Hao, X. Li, Y. Wan, A. Rodríguez-Fortea, J. M. Poblet, L. Feng and N. Chen, Inorg. Chem., 2015, 54, 9845–9852 CrossRef CAS PubMed.
- L. Dunsch, S. Yang, L. Zhang, A. Svitova, S. Oswald and A. A. Popov, J. Am. Chem. Soc., 2010, 132, 5413–5421 CrossRef CAS PubMed.
- B. Q. Mercado, N. Chen, A. Rodríguez-Fortea, M. A. Mackey, S. Stevenson, L. Echegoyen, J. M. Poblet, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2011, 133, 6752–6760 CrossRef CAS PubMed.
- C. R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Angew. Chem., Int. Ed., 2001, 40, 397–399 CrossRef CAS PubMed.
- C. Zhao, D. Lei, L. H. Gan, Z. X. Zhang and C. R. Wang, ChemPhysChem, 2014, 15, 2780–2784 CrossRef CAS PubMed.
- T. Wei, S. Wang, F. Liu, Y. Tan, X. Zhu, S. Xie and S. Yang, J. Am. Chem. Soc., 2015, 137, 3119–3123 CrossRef CAS PubMed.
- C. M. Beavers, T. Zuo, J. C. Duchamp, K. Harich, H. C. Dorn, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2006, 128, 11352–11353 CrossRef CAS PubMed.
- T. Zuo, K. Walker, M. M. Olmstead, F. Melin, B. C. Holloway, L. Echegoyen, H. C. Dorn, M. N. Chaur, C. J. Chancellor, C. M. Beavers, A. L. Balch and A. J. Athans, Chem. Commun., 2008, 1067–1069 RSC.
- A. S. Matias, R. W. A. Havenith, M. Alcami and A. Ceulemans, Phys. Chem. Chem. Phys., 2016, 18, 11653–11660 RSC.
- Q. Deng and A. A. Popov, J. Am. Chem. Soc., 2014, 136, 4257–4264 CrossRef CAS PubMed.
- Q. Tang, L. Abella, Y. Hao, X. Li, Y. Wan, A. Rodríguez-Fortea, J. M. Poblet, L. Feng and N. Chen, Inorg. Chem., 2016, 55, 1926–1933 CrossRef CAS PubMed.
- C. M. Beavers, M. N. Chaur, M. M. Oimstead, L. Echegoyen and A. L. Balch, J. Am. Chem. Soc., 2009, 131, 11519–11524 CrossRef CAS PubMed.
- A. A. Popov, M. Krause, S. Yang, J. Wong and L. Dunsch, J. Phys. Chem. B, 2007, 111, 3363–3369 CrossRef CAS PubMed.
- M. Mulet-Gas, A. Rodríguez-Fortea, L. Echegoyen and J. M. Poblet, Inorg. Chem., 2013, 52, 1954–1959 CrossRef CAS PubMed.
- B. Q. Mercado, C. M. Beavers, M. M. Olmstead, M. N. Chaur, K. Walker, B. C. Holloway, L. Echegoyen and A. L. Balch, J. Am. Chem. Soc., 2008, 130, 7854–7855 CrossRef CAS PubMed.
- K. Yu. Amsharov, K. Ziegler, A. Mueller and M. Jansen, Chem. – Eur. J., 2012, 18, 9289–9293 CrossRef PubMed.
- L.-H. Gan, D. Lei and C. Zhao, RSC Adv., 2015, 5, 30409–30415 RSC.
- Y. Hao, L. Feng, W. Xu, Z. Gu, Z. Hu, Z. Shi, Z. Slanina and F. Uhlík, Inorg. Chem., 2015, 54, 4243–4248 CrossRef CAS PubMed.
- P. Zhao, M. Y. Li, Y. J. Guo, R. S. Zhao and X. Zhao, Inorg. Chem., 2016, 55, 2220–2226 CrossRef CAS PubMed.
- B. Saha, S. Irle and K. Morokuma, J. Phys. Chem. C, 2011, 115, 22707–22716 CrossRef CAS.
- J. Zhang, F. L. Bowles, D. W. Bearden, W. K. Ray, T. Fuhrer, Y. Ye, C. Dixon, K. Harich, R. F. Helm, M. M. Olmstead, A. L. Balsh and H. C. Dorn, Nat. Chem., 2013, 5, 880–885 CrossRef CAS PubMed.
- P. W. Dunk, M. Mulet-Gas, Y. Nakanishi, N. K. Kaiser, A. Rodríguez-Fortea, H. Shinohara, J. M. Poblet, A. G. Marshall and H. W. Kroto, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- N. Chen, M. N. Chaur, C. Moore, J. R. Pinzón, R. Valencia, A. Rodríguez-Fortea, J. M. Poblet and L. Echegoyen, Chem. Commun., 2010, 46, 4818–4820 RSC.
- Y. Wang, S. Díaz-Tendero, F. Martín and M. Alcamí, J. Am. Chem. Soc., 2016, 138, 1551–1560 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp07370k |
| This journal is © the Owner Societies 2017 |