
Influence of π-conjugation structural changes on intramolecular charge transfer and photoinduced electron transfer in donor–π–acceptor dyads†
So-Yoen
Kim
a,
Yang-Jin
Cho
a,
Ah-Rang
Lee
b,
Ho-jin
Son
a,
Won-Sik
Han
*b,
Dae Won
Cho
*ac and
Sang Ook
Kang
*a
aDepartment of Advanced Materials Chemistry, Korea University (Sejong), Sejong, 30019, South Korea. E-mail: hjson@korea.ac.kr; sangok@korea.ac.kr
bDepartment of Chemistry, Seoul Women's University, Seoul, 01797, South Korea. E-mail: wshan@swu.ac.kr
cCenter for Photovoltaic Materials, Korea University (Sejong), Sejong, 30019, South Korea. E-mail: dwcho@korea.ac.kr
First published on 23rd November 2016
Abstract
The influence of π-conjugation structural changes on photoinduced electron transfer (PET) and intramolecular charge transfer (ICT) processes in π-conjugated donor (D)–acceptor (A) dyads (D–π–A) was investigated. Three types of D–π–A dyads were prepared through the modification of the structure of their π-conjugated linker, including D–π–A (1) and D–πtw–A (2) having a twisted π-conjugation, and D–π–Si–π–A (3) with a π-conjugation severed by a Si-atom. In these dyads, carbazole (Cz) and oxadiazole (Oz) moieties act as an electron donor and acceptor, respectively. The emission maxima of dyads 1 and 3 red-shifted with the increase in polarity, which could be attributed to the ICT process. The fluorescence lifetimes of dyads 1 and 3 were 2.64 and 4.29 ns in CH2Cl2, respectively. In contrast, dyad 2 showed dual emission at 350 and 470 nm in CH2Cl2. The emission of dyad 2 at 380 nm corresponded to the monomer fluorescence in the locally excited state. Moreover, the emission at 470 nm increased simultaneously with the diminishing of the fluorescence at 380 nm. This emission band can be assigned as the intramolecular exciplex emission, and showed a strong solvatochromic shift. The low emission quantum yield (<3%) of dyad 2 is due to the PET process. In dyad 2, the cationic and anionic radical species generated by the PET process were confirmed by femtosecond transient absorption (fs-TA) spectroscopy. Upon photoexcitation at 290 or 340 nm, the A or D moieties can be selectively excited. Upon excitation at 290 nm, the acceptor moiety can be excited to the 1A* state, thus the photoinduced hole transfer (PHT) takes place from 1A* to D through the HOMO levels within a few picoseconds. On the other hand, when the donor moiety is excited at 340 nm, the PET process occurs from 1D* to A. Based on the fs-TA studies, it was found that the dynamics and mechanisms for the electron (or charge) transfer were strongly affected by the variation of the π-conjugation of the linker. Herein, we can conclude that the PET and ICT processes are strongly influenced by the π-conjugation properties and their mechanisms are also affected by whether selective excitation of the donor or acceptor moiety occurs. Moreover, unit electron transfers (PET or PHT) were observed dominantly in the dyads having severed/twisted linkers in π-conjugation. However, dyad 1 possessing a well-conjugated linker showed a partial charge transfer character.
Introduction
Intramolecular charge transfer (ICT) is one of the numerous deactivation processes of excited molecules.1 The ICT process occurs in push–pull molecules having a donor and an acceptor moiety acting as an electron source and electron receiving species, respectively.2 Usually, the investigation of the ICT process is performed on aromatic molecules possessing donor and acceptor functional groups. Alternatively, in a dyad system the donor and acceptor moieties are connected by a linker.3–6 Therefore, the photophysical properties of dyads are affected by the nature of their linker. Two types of linkers have been introduced in dyads, i.e., π-conjugated and non-conjugated (or aliphatic) linkers. In the ICT process with a dyad having a π-conjugated linker, the overall charge distribution is shifted to the acceptor moiety through the π-conjugated linker in the excited state. The whole dyad molecule including the linker can be regarded as one single chromophore in the ICT process. Therefore, the ICT acronym can be used to refer to fully π-conjugated dyads.7,8 A dyad π-conjugated by donor and acceptor moieties induces a large dipole moment change in the excited state. Therefore, the ICT process has been proposed to be responsible for the strongly shifted Stokes emission, which is very sensitive to the solvent polarity.9On the other hand, photoinduced electron transfer (PET) implies an electron transfer process from the donor to acceptor in the excited state.10 Consequently, the PET process also demands the presence of donor and acceptor moieties or molecules analogous to the ones used in the ICT process. For this reason, PET and ICT processes are often considered virtually as the same process. In the intermolecular PET process, the donor and the acceptor can be fully separated as individual species in solution.11–14 When the donor and acceptor are connected through a non-conjugated (or aliphatic) linker as in dyads,15 a unit charge is separated, i.e., redox reactions take place in the excited state as a result of the PET process. For the intramolecular PET process on a dyad with an aliphatic linker, there has been considerable debate concerning the role of the bridging linker.16 A super-exchange mechanism for the PET process is generally accepted in dyad systems having a lack of π-conjugation between the donor and the acceptor.
Two hypotheses have been proposed to explain rapid PET processes over a long distance: (1) a through-bond interaction, and (2) a through-space interaction between the donor and the acceptor.17,18 Although many experimental results have been collected to prove the above hypotheses, the π-conjugation role of the linker and the electronic coupling between the donor and acceptor moieties should be examined in detail. Moreover, in spite of the great differences between PET and ICT processes mentioned above, these terms have been used interchangeably.7,8
In this work, in order to understand the crucial discrepancy between ICT and PET in dyad systems and the role of π-conjugated linkers, we prepared three different dyads: π-conjugated D–π–A (1) and D–π–Si–π–A (2) with π-conjugation severed by a Si atom, and D–πtw–π–A (3) having a twisted π-conjugation by steric hindrance, in which D and A are carbazole (Cz) and 2,5-diphenyl-1,3,4-oxadiazole (DPOz) moieties, respectively (Scheme 1). DPOz is also a typical chromophore containing a five-membered heterocycle.19
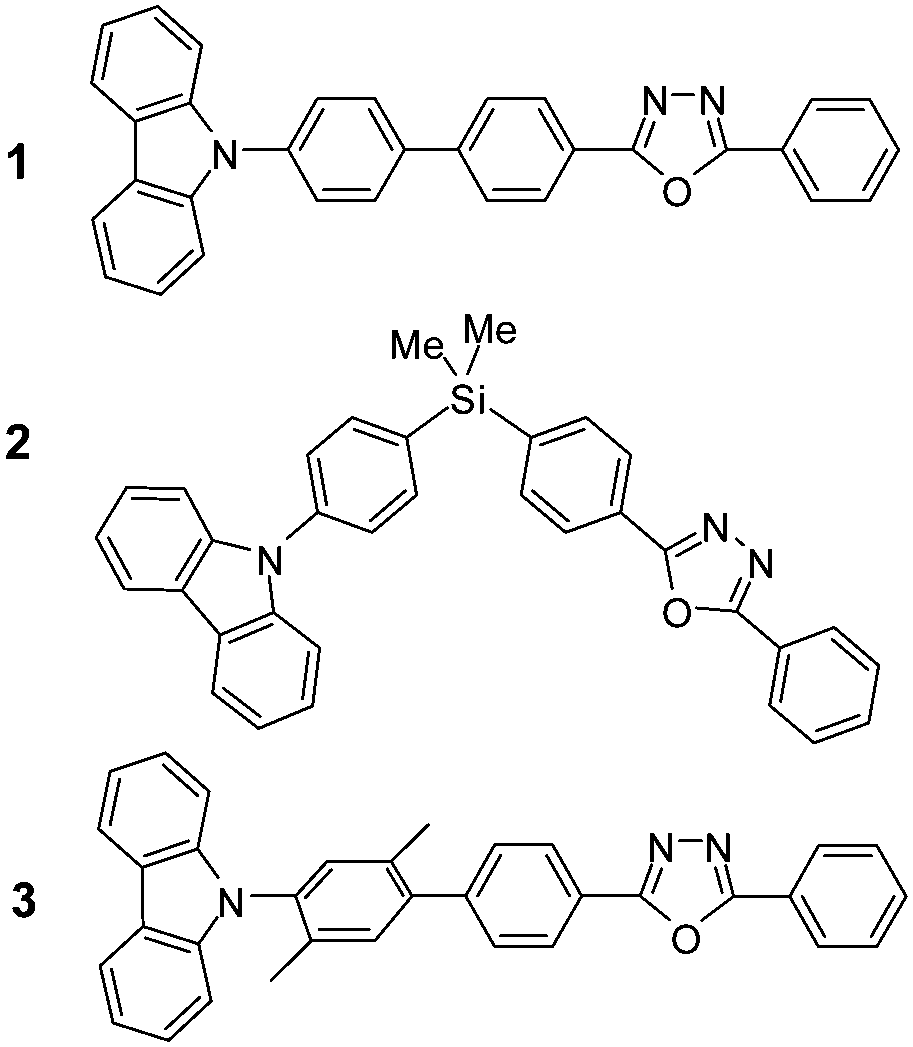 | ||
| Scheme 1 Molecular structures of dyads 1–3. | ||
The fluorescence of dyad 2 markedly decreased due to the PET and the photoinduced hole transfer (PHT) processes, which were confirmed by femtosecond transient absorption (fs-TA) spectroscopy. As a result, the cationic and anionic radicals generated by the PET (or PHT) process were detected in the fs-TA spectra. The π-conjugation of dyads 2 and 3 was interrupted, thus the donor and acceptor moieties acted as independent chromophores even in a single dyad system. For example, distinct excitation wavelength effects were observed in dyad 2; the PET process took place upon photoexcitation of the donor moiety, while the PHT process dominantly occurred by excitation of the acceptor moiety. These trends were also observed in dyads 1 and 3, but the contribution was minor and the dynamics were very short compared to those of dyad 2.
Results and discussion
Steady-state absorption and emission spectroscopic properties
Dyad 1 exhibited an intense absorption band at around 294 nm, as shown in Fig. 1a. The absorption maximum of phenyl Cz was overlapped with that of DPOz at around 285 nm. The extinction coefficient (ε) value of dyad 1 at this wavelength was ∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1, which is comparable with the summation of Cz and DPOz values. Indeed, the reported ε values for phenyl Cz and DPOz are 13400 M−1 cm−1 at 290 nm and >30000 M−1 cm−1 at 280 nm, respectively.20,21 An additional intense absorption band at around 340 nm was observed, resulting from the extension of the π-conjugation of Cz to the acceptor DPOz moiety through the biphenyl linker. The ε values for Cz, DPOz, and dyads are listed in Table 1.
000 M−1 cm−1, which is comparable with the summation of Cz and DPOz values. Indeed, the reported ε values for phenyl Cz and DPOz are 13400 M−1 cm−1 at 290 nm and >30000 M−1 cm−1 at 280 nm, respectively.20,21 An additional intense absorption band at around 340 nm was observed, resulting from the extension of the π-conjugation of Cz to the acceptor DPOz moiety through the biphenyl linker. The ε values for Cz, DPOz, and dyads are listed in Table 1.
 | ||
| Fig. 1 Absorption (a) and excitation (b) spectra of dyads 1–3 in CH2Cl2. Values in parentheses indicate the monitoring emission wavelengths. | ||
Intense absorption bands of dyads 2 and 3 were observed at around 295 nm, and can be attributed to the π,π* transition for either the Cz or the DPOz moiety (Fig. 1a). For dyad 2, the π-conjugation between Cz and Oz moieties is severed by a Si atom. The weak shoulder absorption band at around 340 nm can be assigned to the n,π*-transition of the Cz moiety, since no absorption band of the DPOz moiety was observed at around 340 nm.
For dyad 3, the π-conjugation is twisted due to the steric hindrance of the dimethyl phenyl group in the linker. Dyad 3 exhibited a weak absorption band at around 340 nm compared to that of dyad 1, but moderately intense compared to that of dyad 2. This result indicates that the π-conjugation of dyad 3 is partially hampered. The absorption spectra of dyads 1–3 were not affected by the solvent polarity, as listed in Table S1 and shown in Fig. S1 in the ESI.†
In contrast to the weak solvent polarity dependency observed in the absorption spectra, the fluorescence emissions of dyads 1–3 were markedly red-shifted with increasing solvent polarity, as shown in Fig. 2a and c. The large Stokes shift in polar solvents can be attributed to the large dipole moment change in the excited state. The excitation spectra of dyads 1–3 were similar to their absorption spectra shown in Fig. 1. However, the intensities at longer wavelengths over 300 nm in the excitation spectra were higher than those in the absorption spectra. Since the Cz moiety can only be observed with longer wavelength light, the emissive state is attributed to the absorption of the Cz moiety rather than that of the DPOz moiety. The reason why the absorption of DPOz contributes relatively less to the emissive state will be discussed in detail later.
 | ||
| Fig. 2 Emission spectra of dyads (a) 1, (b) 2, and (c) 3 measured in various solvents: (1) n-hexane, (2) chloroform, (3) THF, (4) CH2Cl2, and (5) CH3CN. λex = 290 nm. (d) Lippert–Mataga plots for dyads 1–3 measured in various solvents. | ||
Dyad 2 showed a dual emission at around 354 nm and at longer wavelengths in various solvents (except n-hexane) as shown in Fig. 2b. The emission at short wavelengths can be assigned as the fluorescence from the locally excited (LE) state. Regarding the emission spectral features and maximum wavelength, the overall emission behaviour from the LE state was closer to the fluorescence character of Cz22 rather than that of DPOz.23 The emission maximum from the LE state was slightly affected by the solvent polarity. In comparison, the longer wavelength emission was red-shifted with increasing solvent polarities. No concentration effect was observed on the ratio of intensities between the 510 and 370 nm emission bands for dyad 2 in CH2Cl2, as shown in Fig. S2a in the ESI.† The absence of a concentration effect can exclude an intermolecular interaction process as a cause of the long wavelength emission. Thus, the longer wavelength emission can originate from an intramolecular interaction, namely the exciplex formation, between the excited Cz and DPOz moieties. It should be pointed out that the exciplex emission showed a marked red-shift according to the increase of solvent polarity. This result implies that the exciplex possesses a charge transfer character.
Most chromophores show single emission bands, which are shifted according to the solvent polarity. However, many reports have explained that a dual emission can occur due to the presence of a significant activation energy barrier between two states, accompanied by structural changes between initial and final emissive states. For example, some ICT molecules show a dual emission from the LE state and a twisted intramolecular charge transfer (TICT) state,24,25 as there is an energy barrier for internal rotation. In addition, other examples of energy barriers can be easily found, such as the excited state intramolecular proton transfer (ESIPT)26,27 and the intramolecular exciplex (or excimer) formation28,29 reactions. Therefore, the observation of the dual emission in dyad 2, especially in CH2Cl2 and CH3CN, implies that a significant energy barrier exists between two emissive states. We preliminarily suggest that this activation energy barrier correlates with the structural change required to form the charge-transfer type exciplex.28,30
Solvent polarity dependency
As it was found that a larger dipole moment change caused a larger Stokes shift in the emission spectra, the solvent polarity effect was analysed in terms of the difference in the dipole moments between the ground and excited states using a Lippert–Mataga plot (Fig. 2c).9 The Stokes shift between the absorption and emission maxima, in wavenumbers (Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ), can be expressed using eqn (1):
), can be expressed using eqn (1): | (1) |
The Onsager radius was 10.76, 10.25, and 10.76 Å for dyad 1, 2, and 3, respectively, as determined by ab initio calculations (B3LYP method, 6-31G(d,p) basis). As shown in Fig. 2d, the Stokes shift for emissions changed linearly with Δf. Using the slope value in eqn (1), the dipole moment change (Δμ) was estimated to be 15.6, 19.3, and 17.2 D for dyad 1, 2, and 3, respectively, as listed in Table 1. Solely Cz and DPOz showed a weak solvent polarity effect. Indeed, the Δμ values are approximately 3.5 and 3.8 D for Cz26,27 and DPOz,23 respectively. Therefore, the large Δμ values in the dyads can be attributed to the donor–acceptor interaction in the dipolar structures. The large Δμ value of dyad 1 is acceptable, because of the well-conjugated π-electrons. In this case, the charge is able to transfer to the acceptor moiety quite easily. Although the electronic communication through the π-conjugation was expected to be poor, larger Δμ changes were observed in dyads 2 and 3. In order to explain these results, we performed time-resolved spectroscopic investigations as follows.
Time-resolved emission spectral properties
Fig. 3 shows the time-resolved fluorescence spectra of dyad 2 in CH2Cl2 obtained by femtosecond pulse excitation. At early delay times, dyad 2 only showed a time-resolved emission at around 380 nm, which corresponded to the monomer fluorescence in the LE state. Moreover, the emission at 470 nm increased simultaneously with the diminishing of the fluorescence at 380 nm. The emission lifetime monitored at short wavelengths (380 nm) was approximately 670 ps. On the other hand, the temporal profile of emission monitored at long wavelengths showed the rise component of approximately 670 ps, as shown in the inset of Fig. 3. The decay or rise times of monomer and exciplex emissions did not change upon varying the concentration up to 10 times, as shown in Fig. S2b in the ESI.† This is consistent with the trends observed for the steady-state emission. The absence of a concentration dependency implies that the exciplex emission is due to an intramolecular interaction. We suggest that this time constant of 670 ps corresponds to the exciplex formation. Especially, the geometrical changing dynamics can be proposed to form a stable intramolecular exciplex between donor and acceptor moieties. The emission lifetimes of the dyads are listed in Table 2.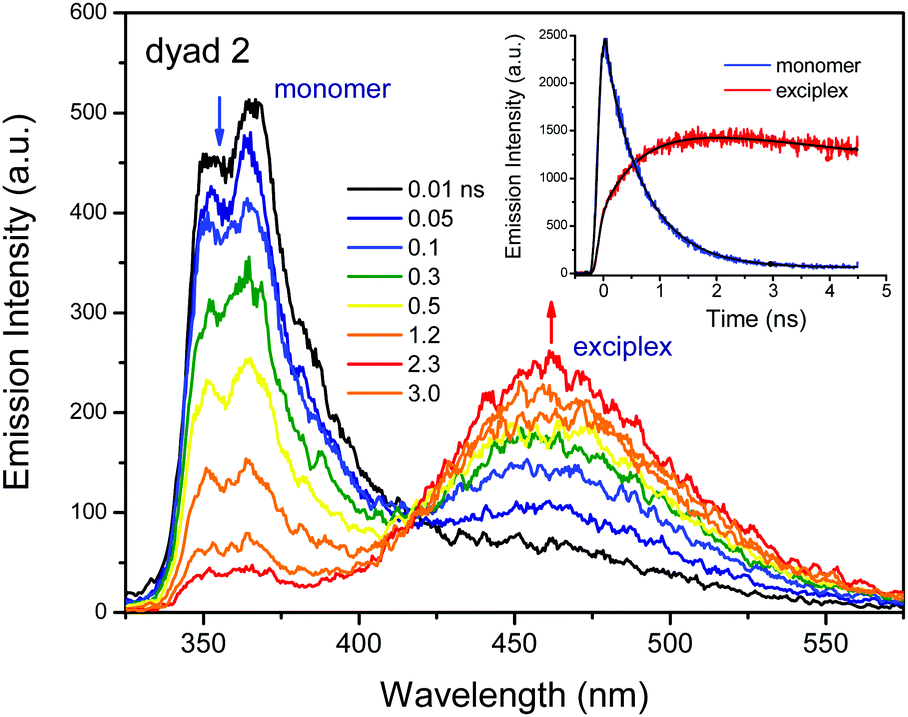 | ||
| Fig. 3 Time-resolved emission spectra of dyad 2 in CH2Cl2. Excitation wavelength is 290 nm. Inset figure indicates the decay profiles monitored at different wavelength ranges: 350–400 and 450–550 nm for monomer and exciplex, respectively. | ||
The decay time of the exciplex emission was 21.0 ns. This emission exhibited a strong solvatochromic effect, suggesting that the exciplex had the largest dipole moment change as explained above, as well as a charge transfer character. A charge transfer type exciplex can be formed by the association of different (donor and acceptor) moieties within one large molecule, in which a fraction of the electronic charge is transferred between the donor and the acceptor. The resulting electrostatic attraction provides a stabilizing force for the charge-transfer-type exciplex. We will discuss about this issue later using the transient absorption results.
Dual emissions are clearly observed in polar solvents, thus also the monomer and the exciplex emissions can be classified by measuring the time-resolved emission spectra in a polar solvent. For example, the exciplex emission was not observed in a nonpolar solvent such as n-hexane. Therefore, the exciplex formation may be limited due to the high energy barrier. The energy barrier in polar solvents was lower compared with that in nonpolar solvents. On the other hand, the charge separation in polar solvents may induce the structural change able to form the exciplex by electrostatic interactions between oppositely charged moieties.
It is well known that the fluorescence emission quantum yield (ϕem) of Cz is 0.3.22 However, the ϕem values of dyads 1 and 3 were 0.23 and 0.14, respectively, which are smaller than that of Cz, as listed in Table 2. Especially, the ϕem values of the monomer and the exciplex in dyad 2 were extremely lower than 0.02. This means that 97% of excited dyad 2 deactivates through a nonradiative process. The major deactivation process in dyads 1 and 3 is also a nonradiative process. In order to understand this nonradiative deactivation process, we carried out an fs-TA study as follows.
Transient absorption spectral properties
The fs-TA spectral properties of dyads 1–3 in CH2Cl2 were investigated upon photoexcitation at 290 and 340 nm, respectively.In dyad 2, the composing moieties act as independent molecules due to the interruption of the electronic communication through π-conjugation between the donor and the acceptor, thus they maintain their intrinsic spectroscopic properties. As mentioned above, the absorption band at 290 nm corresponds to the π,π* transition of both donor and acceptor moieties. However, the DPOz moiety absorbs a large portion of light at 290 nm, because the molar extinction coefficient of DPOz is higher compared to that of the Cz moiety at 290 nm. On the other hand, the light at 340 nm can be absorbed dominantly by the Cz moiety, because the absorption band at around 340 nm corresponds to the n,π* transition of the Cz moiety.
Upon photoexcitation at 290 nm, the intense TA band of dyad 2 was observed at around 550 nm, as shown in Fig. 4A(a and b). This strong TA band was assigned to the S1 → Sn transitions of the DPOz moiety. This TA band decayed very quickly with a time constant of 1.13 ± 0.02 ps. The TA band at 780 nm increased simultaneously with a rise time constant of 1.19 ± 0.06 ps (Fig. 4A(b and c)). This characteristic band at 780 nm can be assigned as the TA band of the Cz+˙ species. The TA spectra showed an isosbestic point at 615 nm (Fig. 4A(b)). The observation of the isosbestic point in the TA spectra proves that two excited species (1DPOz* and Cz+˙) quantitatively change at the equilibrium. It is noteworthy that the intensity of TA of 1DPOz* is very high. This means that the extinction coefficient value for the S1 → Sn transitions of the DPOz moiety may be very high compared to that of the Cz+˙ species equivalently generated.
 | ||
| Fig. 4 The fs-TA spectra of dyad 2 in CH2Cl2 were measured using different excitation wavelengths (column A; 290 nm, and column B; 340 nm) at early delay times (0–5 ps) after 100 fs pulse excitation. Chirping uncorrected contour images (a), chirping corrected transient absorption spectra (b) and decay profiles (c). The decay and rise profiles in (c) were monitored at selected wavelengths, respectively, as mentioned in the figures. | ||
Briefly, the 1DPOz* species is dominantly generated by excitation with a 290 nm pulse, then it is reduced and the counter Cz moiety is concurrently oxidised. The possible mechanism for the generation of the Cz+˙ species from 1DPOz* is a photoinduced hole transfer (PHT) process, as shown in Scheme 2a. When the acceptor (DPOz) is excited upon irradiation with a 290 nm pulse, a vacancy (hole) is formed in the highest occupied molecular orbital (HOMO) of the acceptor. This hole moves from the HOMO of the acceptor to the HOMO of the donor, so it is named hole-transfer. It should be noticed that the actual event in the PHT process is the electron transfer from the donor to the acceptor via the HOMO levels, as shown in Scheme 2a. Therefore, the PHT process has been named as such to distinguish it from the term of the PET process occurring in the excited state.
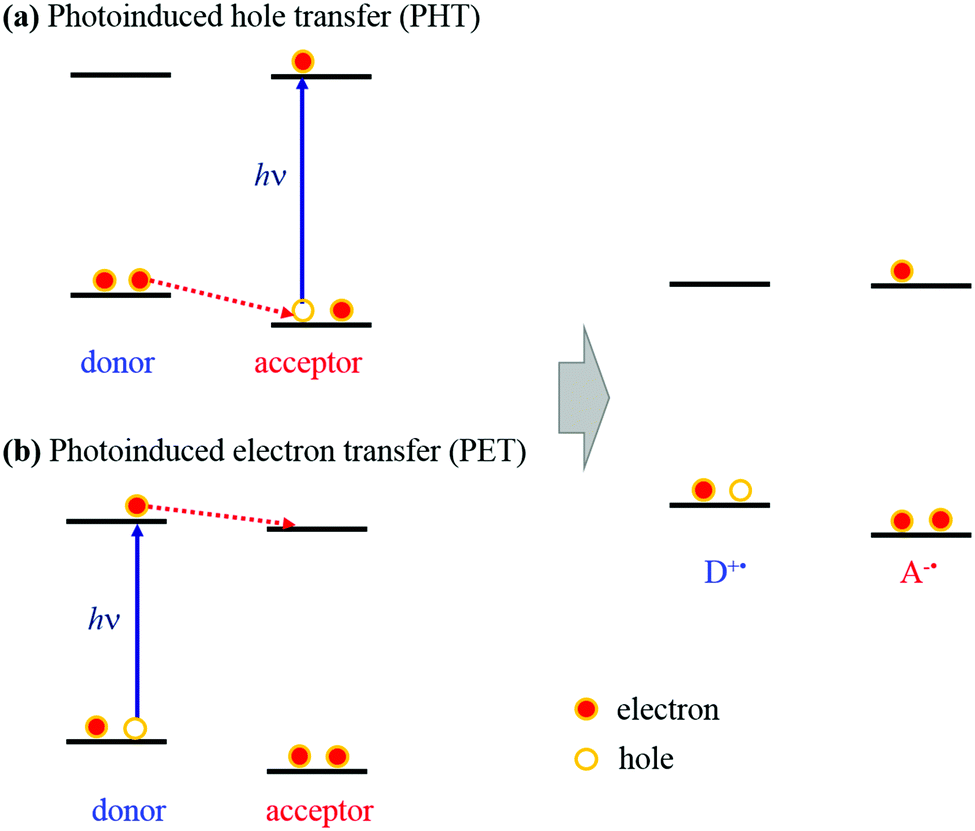 | ||
| Scheme 2 Schematic of the PHT (a) and PET (b) mechanisms in dyads. | ||
During the hole-transfer occurring via the HOMO levels, the ultimate transient species (cation and anion radical species) should be generated at the same time as displayed in eqn (2).
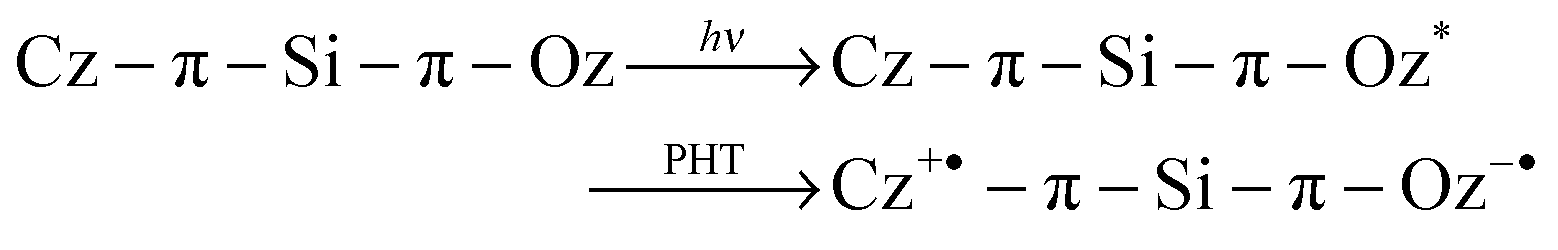 | (2) |
On the other hand, the Cz moiety can absorb the excitation light at 340 nm. As shown in Fig. 4B(b), at early delay times, the characteristic TA band was observed for the S1 → Sn transitions of 1Cz* at around 640 nm.8 Moreover, a weak TA band for Cz+˙ was observed at around 780 nm. The TA spectra of all species did not change in the early delay time ranges of ca. 5 ps, as shown in Fig. 4B(c).
As shown in the spectra in Fig. 5B (λex = 340 nm), measured in a long delay time range of 5 ns with a delay time interval of 25 ps, the 1Cz* band at 640 nm decayed with a time constant of 624 ± 70 ps. The TA band at 780 nm increased together with a rise time constant of 670 ± 15 ps, which accompanied the decay of the 1Cz* band (Fig. 5B(c)). These rise and decay dynamics in the TA spectra are identical to the decay time of fluorescence and the rise time of exciplex emission shown in Fig. 3. The fact that the decay time of the 1Cz* species in the TA spectra is almost identical to the fluorescence decay time monitored at short wavelengths further confirms that the fluorescence at 350 nm is due to the monomeric 1Cz* species. In the fluorescence spectra shown in Fig. 3, 1Cz* forms an exciplex with the DPOz moiety with 670 ps. On the other hand, in the TA spectra of Fig. 5B, 1Cz* changes to a Cz+˙ species with the same time scale of the fluorescence lifetime. This implies that exciplex formation and the PET process from 1Cz* to DPOz take place within the lifetime of the 1Cz* species.
 | ||
| Fig. 5 The TA spectra of dyad 2 in CH2Cl2 are measured using different excitation wavelengths (column A; 290 nm, and column B; 340 nm) at long delay times (0–5000 ps) after 100 fs pulse excitation. Contour images (a), transient absorption spectra (b) and decay profiles (c). The decay and rise profiles in (c) were monitored at selected wavelengths, respectively, as mentioned in figures. | ||
Radical species have a doublet multiplicity, thus the radiative transition from radical species is forbidden according to the selection rule in terms of spin multiplicity. Therefore, anion or cation radicals are classified as non-emissive species. Indeed, the PET process is a major deactivation process in dyad 2, thus the ϕem values of the monomer and exciplex are very small.
Although dyad 2 has a V-shaped structure, the initial orientation of the donor and acceptor moieties might not be favourable to form an intramolecular exciplex. Actually, based on X-ray crystallographic investigations,31 the phenyl Cz moiety possesses an orthogonal geometry to the DPOz moiety at the Si atom. Therefore, a structural change would be required to allow a face-to-face geometry, which could be favourable to form a charge-transfer type exciplex as demonstrated in the following eqn (3):
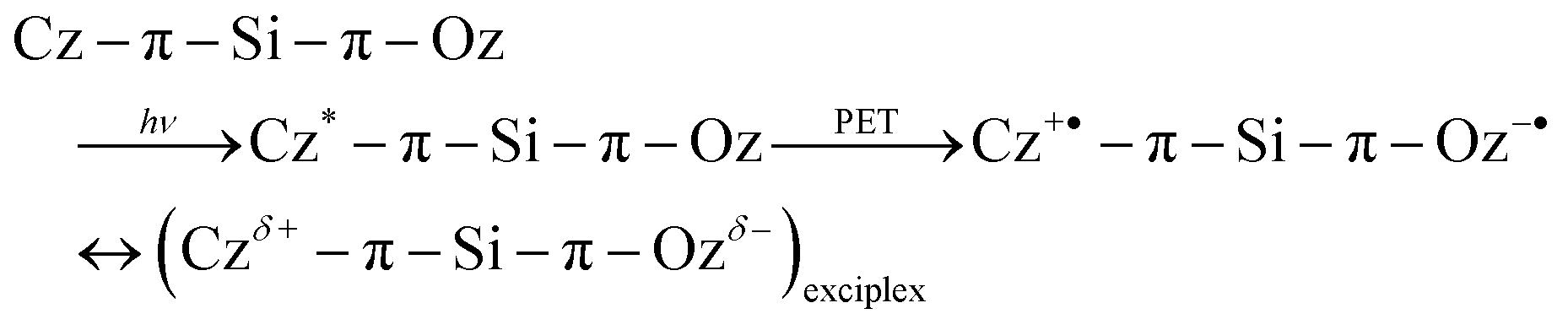 | (3) |
Fig. 5A shows the TA spectra measured at longer delay times upon excitation at 290 nm with a delay time interval of 25 ps. As mentioned above, 1DPOz* quickly vanishes to form a DPOz˙− species though the PHT process within a few picoseconds. Therefore, under the conditions of 25 ps interval, the TA band of 1Cz* is mainly observed at 615 nm, while the TA band of extant 1DPOz* is observed at 550 nm (Fig. 5A(b)). The TA intensity of extant 1DPOz* after the fast PHT process is comparable with that of 1Cz*; this may be due to the extremely high extinction coefficient of 1DPOz*.
The decay time of 1Cz* was 627 ± 14 ps, concurrently the TA bands for the Cz+˙ and DPOz−˙ species increased at around 780 and 465 nm, respectively, as shown in Fig. 5A(b). The isosbestic points were observed at around 680 and 480 nm, indicating that the PET process progressed quantitatively from 1Cz* to DPOz, generating the Cz+˙ and DPOz−˙ species.
When the DPOz moiety was excited at 290 nm, the PHT process was the major process. The extant 1Cz* trace progressed to the PET process or the exciplex formation, similar to the case of directly excited 1Cz* species at 340 nm. Both radical species were not expired until 5 ns in their temporal profiles, as shown in Fig. 5c. However, the recombination dynamics between oppositely charged radicals were not examined because of the time delay limitation of our instrument. The emission lifetime of the exciplex was 21 ns. It can be suggested that the decay dynamics of the exciplex is not correlated with the electron recombination process.
Dyad 3 has a twisted structure caused by the steric hindrance of the two methyl groups in the biphenyl linker (see Scheme S1 in the ESI†). The DPOz moiety absorbs light at 290 nm dominantly, so the intense TA band can be attributed to the 1DPOz* species at 550 nm at initial delay times, upon photoexcitation at 290 nm (Fig. 6A). The 1DPOz* band decayed with a time constant of 0.45 ± 0.01 ps, which is faster than that in dyad 2. This fast decay of 1DPOz* may be attributed to the direct connection of the π-conjugated moieties in spite of the twisted conformation.
 | ||
| Fig. 6 The fs-TA spectra of dyad 3 in CH2Cl2 were measured using different excitation wavelengths (column A; 290 nm, and column B; 340 nm) at early delay times (0–30 ps) after 100 fs pulse excitation. Chirping uncorrected contour images (a), chirping corrected transient absorption spectra (b) and decay profiles (c). The decay and rise profiles in (c) were monitored at selected wavelengths, respectively, as mentioned in figures. | ||
The formation of a Cz+˙ species was analysed by the rise of the profile at 780 nm, which was fitted as two rising components (0.43 ± 0.03 and 6.7 ± 0.3 ps), as shown in Fig. 6A(c). The decay time of the shorter one is identical to the short decay time of 1DPOz* monitored at 550 nm, as shown in Fig. 6A(c). These dynamics were assigned as for the PHT process. Isosbestic points were observed at around 680 and 500 nm. At initial delay times, the 1DPOz* band vanished very quickly through the PHT process with a decay time of ca. 0.45 ps, and consecutively the TA bands for the Cz+˙ and DPOz−˙ species increased with a decay time of ca. 6.7 ± 0.3 ps. Based on the above results, it can be concluded that the Cz+˙ species is generated by two different pathways, i.e., the PHT and PET processes. The longer dynamics of ca. 6.7 ps indicates a rotational motion of the linker. We suggested that the twisted linker was further rotated to form an orthogonal geometry. In this orthogonal geometry, the π-conjugation of the linker is interrupted, and a unit electron transfer is more favourable than a partial charge transfer. So the PET process is facilitated rather than the ICT process in orthogonal geometry. The PHT and PET processes are non-radiative decay processes, generating radical species. The reaction relation in the excited state is expressed by eqn (4):
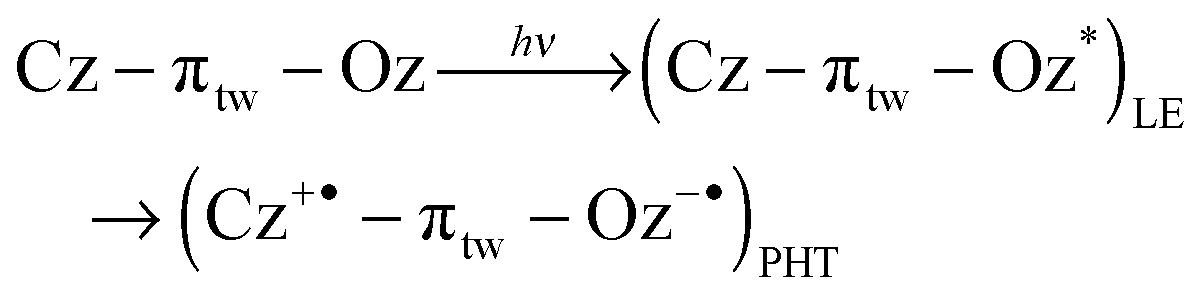 | (4) |
 | (5) |
As shown in Fig. S3 in the ESI,† the overall TA bands decayed exponentially. The decay times monitored at 500 and 780 nm were ca. 2.4 ± 0.5 and 2.7 ± 0.3 ns for the excitation at 290 and 340 nm, respectively, which can be assigned to the electron (charge) recombination times. On the other hand, it is well known that many organic chromophores having single bonds between aromatic rings change their structure toward a planar geometry in the excited state. In this geometry the ICT process is more favourable as the radiative decay process, which can be investigated using the emission dynamics. Actually, the emission lifetime (4.29 ns) of the ICT state is longer than the charge recombination time.
In dyad 1, the Cz and DPOz moieties are connected by a well π-conjugated linker, thus the π-electrons are delocalized within the whole dyad molecule in the ground state. When dyad 1 was excited at 290 nm, the TA spectra showed a broad band at around 570 nm at initial delay times, then this band slightly shifted to shorter wavelengths (ca. 560 nm) according to the delay time, and a shoulder band at 500 nm was observed, as shown in Fig. 7A(b). Both TA bands decayed with the same decay time. Therefore, it could be concluded that these bands originated from the same state, and could be attributed to the ICT state of dyad 1. The trace conformer having an orthogonal geometry against the Cz moiety deactivated through the PET rather than the ICT process. However, the contribution of the PET process was smaller than that of the ICT process (C1 ≫ C2 in relation (6), where C1 and C2 indicate the contribution coefficient of the ICT and PET processes), since the Cz+˙ band was very weak.
 | (6) |
Upon excitation at 340 nm, dyad 1 showed a quite broad TA band centred at 600 nm at early delay times. Then, this band shifted to short wavelengths (ca. 560 nm) according to the delay time, as shown in Fig. 7B(b). The TA spectral shape differed from that obtained by the excitation at 290 nm. Especially, the TA at longer wavelengths around 650 nm was more intense than that of the 290 nm excitation, probably contributing to the transition of the 1Cz* species.8
As shown in the decay profiles of Fig. 7c, the decay time of the 1Cz* species monitored at 640 nm was 1.2 ps. Moreover, the spectral shift occurred in this decay time range. Usually, the blue-shift of the TA band has been explained as the stabilization of the adiabatic potential energy surface. Excited dyad 1 is stabilized in the ICT state, since the negative partial charge localizes to the acceptor moiety as explained in eqn (6). However, in the case of the 340 nm excitation, the PET process was not the major deactivation process, because the TA intensity of Cz+˙ species at 780 nm was very weak.
 | ||
| Fig. 7 The fs-TA spectra of dyad 1 in CH2Cl2 were measured using different excitation wavelengths (column A; 290 nm, and column B; 340 nm) at early delay times (0–30 ps) after 100 fs pulse excitation. Chirping uncorrected contour images (a), chirping corrected transient absorption spectra (b) and decay profiles (c). The decay and rise profiles in (c) were monitored at selected wavelengths, respectively, as mentioned in figures. | ||
The TA spectra of dyad 1 at long delay times are shown in Fig. S4 in the ESI,† in which the decay time constant is ca. 2.9 ± 0.3 ns. This decay time is similar to that of the fluorescence lifetime. The TA spectral shape did not change. This indicates that the TA spectral behaviours at longer delay times originate from the same long-lived ICT state as the emissive state.
Conclusions
The PET and ICT processes are strongly influenced by the π-conjugation properties. In order to investigate the role of the π-conjugation in dyads, three types of D–π–A dyads were prepared through the modification of the π-conjugation of their linker, i.e., well conjugated, twisted, and severed by a Si-atom. The PET and ICT processes were strongly influenced by the π-conjugation properties. The unit electron transfers (PET or PHT) were dominantly observed in the dyads having severed/twisted linkers in the π-conjugation. However, the dyad possessing a well π-conjugated linker showed a partial charge transfer character. Therefore, the severance of π-conjugation between the donor and the acceptor causes a unit-electron transfer rather than a partial charge transfer. The electron transfer mechanisms are affected by whether selective excitation of the donor or acceptor moiety occurs. Actually, the PET process was observed upon excitation of the donor moiety, while the PHT process was prevalently observed upon excitation of the acceptor.Experimental
Picosecond time-resolved emission measurements
Time-resolved fluorescence spectra were measured using a streak scope (Hamamatsu Photonics, C10627) equipped with a polychromator (Princeton Instruments, Acton SpectroPro SP-2300). An ultrashort laser pulse was generated with a Ti:sapphire oscillator (Spectra Physics, MaiTai SP) pumped with a diode-pumped solid-state laser and the high power pulses are generated using a Ti:sapphire regenerative amplifier (Spectra Physics, Spitfire Ace). For excitation of the sample, the output of the Ti:sapphire regenerative amplifier was converted to 330 nm by using an optical parametric amplifier (Spectra Physics, TOPAS prime). The instrument response function was also determined by measuring the scattered laser light to analyse the temporal profile. This method provides a time resolution of approximately 15 ps after the deconvolution procedure. The temporal emission profiles were well fitted to an exponential function. The residuals for each system were less than 1.1.Femtosecond transient absorption measurements
The sub-picosecond time-resolved absorption spectra were collected using a pump–probe transient absorption spectroscopy system (Ultrafast Systems, Helios). The pump light was generated by using a regenerative amplified titanium sapphire laser system (Spectra Physics, Spitfire Ace, 1 kHz) pumped by a diode-pumped Q-switched laser (Spectra Physics, Empower). The seed pulse was generated using a titanium sapphire laser (Spectra Physics, MaiTai SP). The pulses (290 and 340 nm) generated from an optical parametric amplifier (Spectra Physics, TOPAS prime) were used as the excitation pulse. And a white light continuum pulse, which was generated by focusing the residual of the fundamental light to a thin Sapphire crystal after the controlled optical delay, was used as a probe beam and directed to the sample cell with 2.0 mm of the optical path and detected with a CCD detector installed in the absorption spectroscope. The pump pulse was chopped by the mechanical chopper synchronized to one-half of the laser repetition rate, resulting in a pair of the spectra with and without the pump, from which the absorption change induced by the pump pulse was estimated.General information
All the experimental procedures were carried out under a dry nitrogen or an argon atmosphere using standard Schlenk techniques. Tetrahydrofuran (THF) was distilled freshly over sodium benzophenone. The 1H and 13C NMR spectra were recorded on a Bruker Fourier 300 MHz spectrometer, which was operated at 300.1 and 75.4 MHz, respectively. 1H and 13C NMR chemical shifts were measured in CDCl3 and referenced to relative peaks of CHCl3 (7.26 ppm for 1H NMR) and CDCl3 (77.16 ppm for 13C NMR). The elemental analyses were performed using a Carlo Erba Instrument CHNS-O EA 1108 analyser. The HR-MS analysis was performed using a high sensitive LC/MS/MSn (n = 10) spectrometer (Thermo Fisher Scientific, LCQ Fleet Hyperbolic Ion Trap MS/MSn Spectrometer).1,4-Dibromobenzene, carbazole, benzhydrazide, 4-benzoyl chloride, sodium bicarbonate, copper sulfate, potassium carbonate, phosphorus oxychloride, bis(pinacolato)diboron, potassium acetate, 1,4-dibromo-2,5-dimethylbenzene, [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane, tetrakis(triphenylphosphine)palladium(0), n-BuLi (2.5 M solution in n-hexane), dimethyldichlorosilane, and sodium carbonate were purchased from Aldrich or TCI and used without further purification. The starting materials, 2-(4-bromophenyl)-5-phenyl-1,3,4-oxadiazole (DPOz-Br),32 9-(4-bromophenyl)carbazole (PCz-Br),33 9-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2′-yl)phenyl)-9H-carbazole (PCz-BE),34 were prepared according to the previously reported procedures.
Synthesis
Synthetic details for dyads 1 and 2 are reported elsewhere,31 and provided in the ESI.†:v = 18:6) was stirred at 70 °C for overnight under a nitrogen atmosphere. After cooling to room temperature, the mixture was poured into distilled water and extracted with dichloromethane. The combined organic layers were dried over MgSO4 and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography by using ethyl acetate/n-hexane (v:v = 1/5) to give a product. Yield: 42%; 1H NMR (300 MHz, CDCl3, δ): 8.28 (d, J = 6.6 Hz, 2H), 8.18–8.22 (m, 4H), 7.64 (d, J = 8.4 Hz, 2H), 7.55–7.60 (m, 3H), 7.44 (t, J = 7.5 and 6.9 Hz, 2H), 7.38 (s, 1H), 7.29–7.34 (m, 3H), 7.17 (d, J = 8.1 Hz, 2H), 2.35 (s, 3H), 2.02 (s, 3H); 13C NMR (75 MHz, CDCl3, δ): 164.7, 164.6, 144.9, 141.2, 141.04, 135.6, 134.7, 134.4, 132.7, 131.8, 131.0, 130.0, 129.2, 126.9, 125.9, 124.0, 123.1, 122.7, 120.4, 119.6, 109.9, 20.1, 17.2 HRMS: calculated for C34H25N3O: 491.1998, found: 491.1996. Elemental analysis: calculated for C34H25N3O: C, 83.07; H, 5.13; N, 8.55, found: C, 83.05; H, 5.12; N, 8.56.
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2014R1A6A1030732 and NRF-2014R1A1A-2004199). This research was supported by the International Science and Business Belt Program through the Ministry of Science, ICT and Future Planning (2015K000287), and Seoul Women's University (2015).Notes and references
- E. Lippert, W. Luder and H. Boos, in Advances in Molecular Spectroscopy, ed. A. Mangini, Pergamon, Oxford, 1962, pp. 443–454 Search PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4031 CrossRef PubMed.
- R. Ali, S. S. Razi, R. C. Gupta, S. K. Dwivedi and A. Misra, New J. Chem., 2016, 40, 162–170 RSC.
- A. Felouat, A. D'Aléo, A. Charaf-Eddin, D. Jacquemin, B. L. Guennic, E. Kim, K. J. Lee, J. H. Woo, J.-C. Ribierre, J. W. Wu and F. Fages, J. Phys. Chem. A, 2015, 119, 6283–6295 CrossRef CAS PubMed.
- Y.-J. Cho, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 9702–9708 RSC.
- S.-Y. Kim, Y.-J. Cho, G. F. Jin, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2015, 17, 15679–15682 RSC.
- V. Novakova, P. Hladík, T. Filandrová, I. Zajícová, V. Krepsová, M. Miletin, J. Lenčod and P. Zimcik, Phys. Chem. Chem. Phys., 2014, 16, 5440–5446 RSC.
- Y.-J. Cho, A.-R. Lee, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 22921–22928 RSC.
- J. R. Lakowicz, Principle of Fluorescence Spectroscopy, Springer, Singapore, 3rd edn, 2010 Search PubMed.
- J. S. Connolly and J. R. Bolton, in Photoinduced Electron transfer, ed. M. A. Fox and M. Chanon, Elsevier, New York, 1989, vol. 4, pp. 303–393 Search PubMed.
- K. Wynne, C. Galli and R. M. Hochstrasser, J. Chem. Phys., 1994, 100, 4797–4810 CrossRef CAS.
- T. Ganguly, D. K. Sharma, S. Gauthier, D. Gravel and D. Durocher, J. Phys. Chem., 1992, 96, 3757–3766 CrossRef CAS.
- K. B. Yoon, S. M. Hubig and J. K. Kochi, J. Phys. Chem., 1994, 98, 3865–3871 CrossRef CAS.
- D. W. Cho, M. Fujitsuka, U. C. Yoon and T. Majima, J. Photochem. Photobiol., A, 2007, 190, 101–109 CrossRef CAS.
- D. W. Cho, M. Fujitsuka, A. Sugimoto, U. C. Yoon, P. S. Mariano and T. Majima, J. Phys. Chem. B, 2006, 110, 11062–11068 CrossRef CAS PubMed.
- D. W. Cho, M. Fujitsuka, U. C. Yoon and T. Majima, Phys. Chem. Chem. Phys., 2008, 10, 4393–4399 RSC.
- J. R. Miller, Noun J. Chim., 1987, 11, 83–89 CAS.
- N.-C. C. Yang, S.-L. Zhang, M. J. Lang, S. Goodman, C. Durnell, G. R. Fleming, H. L. Carrell and R. M. Garavito, in Advances in Chemical Physics, Part 1: Electron Transfer-From Isolated Molecules to Biomolecules, ed. J. Jortner and M. Bixon, Series ed. I. Prigogine and S. A. Rice, John Wiley & Sons, Inc., 1999, vol. 106, pp. 645–666 Search PubMed.
- Phosphor Handbook, ed. S. Shionoya and W. M. Yen, CRC Press, New York, 1998 Search PubMed.
- A.-R. Hwang, W.-S. Han, K.-R. Wee, H. Y. Kim, D. W. Cho, B. K. Min, S. W. Nam, C. Pac and S. O. Kang, J. Phys. Chem. C, 2012, 116, 1973–1986 CrossRef CAS.
- R. E. Ballard, A. Titchard and R. Wyatt, Spectrochim. Acta, 1967, 23A, 1889–1897 CrossRef.
- Y.-J. Cho, K.-R. Wee, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2014, 16, 4510–4521 RSC.
- C. Rulliere and P. C. Roberge, Chem. Phys. Lett., 1983, 97, 247–252 CrossRef CAS.
- K. Rotkiewicz, K. H. Grellman and Z. R. Grabowski, Chem. Phys. Lett., 1973, 19, 315–318 CrossRef CAS.
- Y. H. Kim, D. W. Cho, M. Yoon and D. Kim, J. Phys. Chem., 1996, 100, 15670–15676 CrossRef CAS.
- J. R. Choi, S. C. Jeoung and D. W. Cho, Chem. Phys. Lett., 2004, 385, 384–388 CrossRef CAS.
- Y. H. Kim, S.-G. Roh, S.-D. Jung, M.-A. Chung, H. K. Kim and D. W. Cho, Photochem. Photobiol. Sci., 2010, 9, 722–729 Search PubMed.
- D. W. Cho, M. Fujitsuka, A. Sugimoto, U. C. Yoon, D. W. Cho and T. Majima, Phys. Chem. Chem. Phys., 2014, 16, 5779–5784 RSC.
- D. W. Cho, M. Fujitsuka, A. Sugimoto and T. Majima, J. Phys. Chem. A, 2008, 112, 7208–7213 CrossRef CAS PubMed.
- D. W. Cho and D. W. Cho, New J. Chem., 2014, 38, 2233–2236 RSC.
- A.-R. Lee, J. Lee, J. Lee and W.-S. Han, Org. Electron., 2016, 38, 222–229 CrossRef CAS.
- W. Kwon, B. Ahn, D. M. Kim, Y. G. Ko, S. G. Hahm, Y. Kim, H. Kim and M. Ree, J. Phys. Chem. C, 2011, 115, 19355–19363 CrossRef CAS.
- W. S. Han, H. J. Son, K. R. Wee, K. T. Min, S. Kwon, I. H. Suh, S. H. Choi, D. H. Jung and S. O. Kang, J. Phys. Chem. C, 2009, 113, 19686–19693 CrossRef CAS.
- J. Y. Jeon, T. J. Park, W. S. Jeon, J. J. Park, J. Jang, J. H. Kwon and J. Y. Lee, Chem. Lett., 2007, 36, 1156–1157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06566j |
| This journal is © the Owner Societies 2017 |