Intramolecular cation–π interactions in protonated phenylalanine derivatives†
Received
26th October 2016
, Accepted 1st December 2016
First published on 1st December 2016
Abstract
The structures and properties of a series of phenylalanine (Phe) derivatives have been investigated in a joint computational and experimental infrared multiple photon dissociation (IRMPD) study. IRMPD spectra in the 1000–2000 cm−1 region show that protonation is localized on the amine group in all cases. Intramolecular cation–π interactions between the ammonium group and the phenyl ring heavily influence molecular geometries and properties such as gas phase basicity and proton affinity. By varying substituents on the phenyl ring, one can sensitively tune the cation–π interaction and, therefore, the molecular structure and properties. Variations in molecular structures and properties as a function of phenyl ring substitution are shown to correlate with substituent Hammett parameters.
Introduction
Non-covalent interactions are crucial to maintaining the three-dimensional structures of large molecules.1–8 Typically, individual non-covalent interactions have energies ranging from 4–20 kcal mol−1.3,9 Thus molecules can break individual non-covalent bonds relatively easily, facilitating, e.g., dynamic biological processes. However, while non-covalent interactions are individually weak and exist only ephemerally in biological systems, the cumulative energies of multiple non-covalent interactions can be significant and, combined, numerous non-covalent bonds can significantly stabilize large molecular structures.1,3,4
The cation–π interaction, which is a type of non-covalent charge–quadrupole interaction, has been identified as an important influence on the secondary structures of proteins.3,4,9–11 This interaction typically involves common metal cations (e.g., Na+, K+) and amino acid residues that contain conjugated π-systems (e.g., phenylalanine, tryptophan).1,3–6,10 The stabilizing effect of this interaction, 20–70 kJ mol−1, is similar to anion–π and hydrogen bonding interactions.11,12 To model cation–π interactions, researchers have studied the interactions between cationic species and small π-conjugated molecules such as benzene in the gas phase.13–15 However, while cation–π interactions in condensed phase biological systems have been relatively well characterized,3–5,8,9,16 isolated biologically relevant cation–π systems are much less studied.10,11,14,15,17,18 In 2008, McMahon and co-workers measured the binding energies of cation–π systems containing aromatic amino acids and free organic or metal cations (e.g., Na+).11 This work demonstrated enhancements in binding energies for phenylalanine (Phe), tryptophan (Trp), and tyrosine (Tyr) due to cation–π interactions involving the aromatic moiety. In 2010, Dunbar et al. studied the isolated, cationized complexes of M·Phe (M = Li+–Cs+, Ag+, Ba2+), and found that in all cases there was a clear preference for cation–π interaction between the metal ion and the side-chain aromatic ring.18 This showed that intramolecular charge–quadrupole interactions can have a defining effect on the molecular geometry of isolated biologically-relevant systems.
Much of the interest in non-covalent interactions stems from possible applications in structural control.2,5,9,16 One could envision fine-tuning molecular geometry via cation–π interactions by varying the quadrupole moment of a conjugated π-system through substitution with electron donating or electron withdrawing groups. For example, the quadrupole moment of benzene (ΘC6H6 = −33.3 × 10−40 C m2) can be inverted by complete fluorination to produce hexafluorobenzene (ΘC6F6 = 31.7 × 10−40 C m2); cations prefer to bind to facial sites of the former, and around the edge of the latter. However, the preferred binding site is not necessarily obvious when the ring is partially fluorinated or if other electron donating/withdrawing groups are present. An additional degree of complexity is introduced if the charge centre is tethered and constrained to occupy specific geometries. To begin addressing these issues, we have studied the structures and properties of a series of protonated Phe derivatives. The charge carrying proton in these systems is expected to be localized to the amino group of the amino acid moiety, and it is therefore constrained to the geometries that are imposed by the saturated carbon chain. The Phe derivatives vary based on aromatic substitution of the phenyl ring. This is expected to influence the local quadrupole moment of the ring moiety and, therefore, the possible intramolecular cation–π interactions. By exploring this series of molecules, we gain further insight into the subtle effect of the intramolecular cation–π interaction on molecular geometry.
Experimental
Experimental IRMPD spectra of a series of protonated Phe derivatives were recorded at the Centre Laser Infrarouge d'Orsay (CLIO) free electron laser (FEL) facility at the University of Paris XI.19,20 A detailed description of the experimental apparatus has been reported previously.12,21,22 Electrospray ionization (ESI) solutions of the Phe derivatives (100 μmol L−1) with 0.1% formic acid (ca. 5 μmol L−1) were prepared in methanol/water (50/50 vol%). The amino acids were used without further purification. The specific Phe derivatives chosen for study were: Phe, 3-fluorophenylalanine (3-F-Phe), 4-fluorophenylalanine (4-F-Phe), 2,5-difluorophenylalanine (2,5-F2-Phe), 3,5-difluorophenylalanine (3,5-F2-Phe), pentafluorophenylalanine (F5-Phe), 3-cyanophenylalanine (3-CN-Phe), 3-trifluoromethylphenylalanine (3-CF3-Phe), 3,4-dimethoxyphenylalanine (3,4-(OMe)2-Phe), and 4-nitrophenylalanine (4-NO2-Phe). Positive mode ESI was employed to produce the protonated Phe derivatives, which were studied in the gas phase. Nascent ions were transferred to a Bruker Esquire 3000+ ion trap mass spectrometer, where they were mass-selected and subsequently irradiated by the tunable output of the FEL over the 1000–2000 cm−1 range. Vibrational spectra were generated by recording the fragmentation efficiency of the protonated Phe derivatives as a function of FEL wavenumber.
Computational methods
To map the potential energy surfaces (PESs) of the various protonated and neutral Phe derivatives, a custom-written basin-hopping (BH) code that is interfaced with Gaussian 09 was employed.22–24 The molecules were modelled using the molecular mechanics Universal Force Field, which incorporated atomic partial charges calculated at the B3LYP/6-31+G(d,p) level of theory using the CHelpG partition scheme.25–28 For each random structural perturbation in the BH search, each of the dihedral angles in the given Phe derivative was given a random rotation of −5° ≤ ϕ ≤ 5°. For each cationic species, BH searches were carried out twice; once with protonation at the amine nitrogen atom, and once with protonation attached at carbonyl oxygen atom. In total, ca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures were sampled by the BH algorithm for each protonated structure. All of the isomers that were identified by the BH routine were pre-optimized with the semi-empirical PM6 method prior to treatment at the B3LYP/6-311++G(d,p) level of theory.29 Normal mode analyses were conducted to ensure that each isomer was a local minimum on the PES. This also served to calculate the harmonic vibrational spectrum for each structure, which was then used to assign the experimental IRMPD spectrum. DFT results and cluster XYZ coordinates are provided in the ESI.†
000 structures were sampled by the BH algorithm for each protonated structure. All of the isomers that were identified by the BH routine were pre-optimized with the semi-empirical PM6 method prior to treatment at the B3LYP/6-311++G(d,p) level of theory.29 Normal mode analyses were conducted to ensure that each isomer was a local minimum on the PES. This also served to calculate the harmonic vibrational spectrum for each structure, which was then used to assign the experimental IRMPD spectrum. DFT results and cluster XYZ coordinates are provided in the ESI.†
Results and discussion
IRMPD spectroscopy
IRMPD of the protonated Phe derivatives predominantly yields fragments associated with the loss of CO + H2O (m/z = 46 amu). Amongst the Phe derivatives studied here, the only exception to this trend is 3,4-(OMe)2-Phe·H+, which also exhibits product channels associated with loss of NH3 (m/z = 17 amu) and NH3 + CO + H2O (m/z = 63 amu). The IRMPD spectrum of Phe·H+ is plotted in Fig. 1A. The observed bandwidths of ca. 35 cm−1 are slightly broader than the typical CLIO line width of 25 cm−1,19 thus suggesting that there are at least two isomers contributing to the Phe·H+ spectrum. This is supported by our computational study, which identified three low energy N-protonated conformers of Phe·H+ at ΔG° = 0.0 kJ mol−1, 5.0 kJ mol−1, and 22.8 kJ mol−1, respectively. The XYZ atomic coordinates and calculated thermochemical data for all species are provided in the ESI† that accompanies this manuscript. Fig. 1B plots the calculated IR spectrum from the N-protonated global minimum conformer, while the calculated spectrum for the lowest O-protonated structure (isomer 4; 95.7 kJ mol−1) is plotted in Fig. 1C. Note that calculated spectra have been scaled by the commonly employed factor of 0.9679 to account for anharmonicity.30 Based on comparison between the measured and calculated spectra, we can confidently assign the observed vibrational bands to normal modes associated with the N-protonated global minimum conformer of Phe·H+. The peak at 1749 cm−1 is associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching motion, the peak at 1428 cm−1 is assigned to the NH3 umbrella motion, the band at 1141 cm−1 is attributed to the ∠COH bending mode, and the low-wavenumber shoulder at 1398 cm−1 is consistent with the combined ∠COH bend and NH3 wag/twist. Similar treatment for all the Phe derivatives studied enables the assignment of their measured IRMPD spectra, and a complete list of band assignments is provided for all species in the ESI.† The measured IRMPD spectra and calculated IR spectra for the global minima of the fluorinated Phe derivatives are provided in Fig. 2, and the spectra for the non-fluorinated derivatives are plotted in Fig. 3.
O stretching motion, the peak at 1428 cm−1 is assigned to the NH3 umbrella motion, the band at 1141 cm−1 is attributed to the ∠COH bending mode, and the low-wavenumber shoulder at 1398 cm−1 is consistent with the combined ∠COH bend and NH3 wag/twist. Similar treatment for all the Phe derivatives studied enables the assignment of their measured IRMPD spectra, and a complete list of band assignments is provided for all species in the ESI.† The measured IRMPD spectra and calculated IR spectra for the global minima of the fluorinated Phe derivatives are provided in Fig. 2, and the spectra for the non-fluorinated derivatives are plotted in Fig. 3.
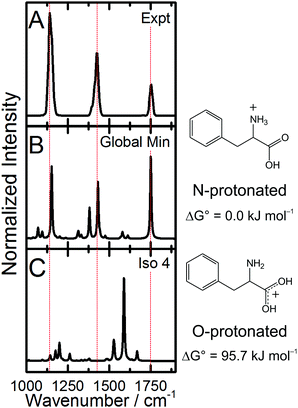 |
| | Fig. 1 (A) The measured IRMPD spectrum of Phe·H+, and the predicted spectra for the (B) global minimum and (C) lowest energy O-protonated structure as calculated at the B3LYP/6-311++G(d,p) level of theory. Calculated spectra have been scaled by 0.9679.30 | |
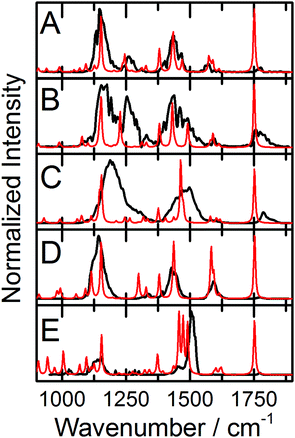 |
| | Fig. 2 The experimental IRMPD spectra (black traces) and calculated IR spectra (red traces) for the global minima of (A) 3-F-Phe·H+, (B) 4-F-Phe·H+, (C) 2,5-F2-Phe·H+, (D) 3,5-F2-Phe·H+, and © F5-Phe·H+. Calculations were conducted at the B3LYP/6-311++G(d,p) level of theory and calculated spectra are scaled by 0.9679.30 | |
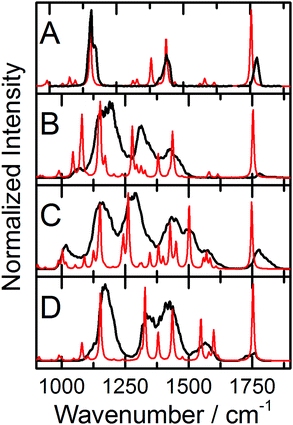 |
| | Fig. 3 The experimental IRMPD spectra (black traces) and calculated IR spectra (red traces) for the global minima of (A) 3-CN-Phe·H+, (B) 3-CF3-Phe·H+, (C) 3,4-(Ome)2-Phe·H+, and (D) 4-NO2-Phe·H+. Calculations were conducted at the B3LYP/6-311++G(d,p) level of theory and calculated spectra are scaled by 0.9679.30 | |
Observation of the NH3 umbrella motion and the combined ∠COH bend and NH3 wag/twist provide direct evidence that protonation is localized on the amine group for all the Phe derivatives studied. This conclusion could be confirmed by the observation of an intense band predicted to be in the 3100–3175 cm−1 region (varying depending on the Phe derivative), which is associated with the H2N+–H⋯OC stretching mode of the shared proton. However, this band falls outside of the CLIO scan range and is likely to be significantly broadened owing to the anharmonic nature of the shared proton vibrational motion.31–34 It is also interesting to note the relatively low intensities of the CO stretching bands in the observed IRMPD spectra. This observation is surprising for two reasons; firstly, DFT calculations predict that the intensities of the CO stretching bands should be relatively intense when compared with the rest of the spectrum, and secondly, the measured CO stretching bands in similar systems are relatively intense.12,18,21,22 However, others have observed unexpectedly weak CO stretching bands in systems such as phosphorylated amino acids.35 This behavior can likely be attributed to relatively inefficient coupling between the CO stretching normal mode in the Phe·H+ derivatives and the dissociative threshold leading to production of CO and H2O. However, to properly model/interpret the observed IRMPD intensities, one must account for the dynamic energy flow during the IRMPD process.36
Assessing the influence of cation–π interactions
Having assigned the IRMPD spectra to the calculated global minima for the various Phe·H+ derivatives, we can return to the computational results for further interpretation. We are interested in assessing how charge–quadrupole interactions might affect the structures and properties of the protonated Phe derivatives. We must therefore define the molecular coordinates that are associated with the proposed cation–π interactions. In the Na+·benzene model system, the positively charged Na centre is located directly above/below the ring centre, while in the Na+·hexafluorobenzene system the charge centre lies in the C6F6 molecular plane.13,15,17 Thus, the cation–π interaction is defined by the distance from the ring centre to the Na+, and the angle formed between the surface normal passing through the ring centre and the vector that connects the ring centre and the Na+ cation. Owing to the fact that the charge centre (NH3+) in the Phe·H+ derivatives is constrained to a position defined by the carbon tether, we define two angles to describe the charge–quadrupole interaction; the angle formed between the surface normal passing through the ring centre (hereby defined as the z-axis) and the vector that connects the ring centre and the N atom as projected into the plane containing the H2C–phenyl bond (β) and z-axis, and as projected into the plane perpendicular to the H2C–phenyl bond and containing the z-axis (θ). These angles are shown in Fig. 4. We also calculated the distance from the ring centre to the N atom (dx-N), and from the ring centre to the nearest ammonium H atom (dx-H) as a means of estimating the charge–quadrupole separation. These coordinates are given in Table 1 for all the Phe·H+ species.
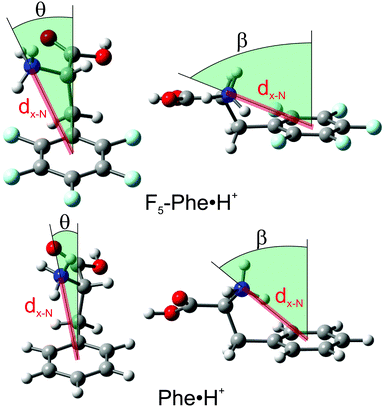 |
| | Fig. 4 The coordinates used to define the charge–quadrupole interaction in the Phe·H+ derivatives. The global minimum structures of Phe·H+ and F5-Phe·H+ are provided for comparison purposes. | |
Table 1 The calculated coordinates that define the charge–quadrupole in the Phe·H+ derivatives studied. Calculations were conducted at the B3LYP/6-311++G(d,p) level of theory
| Derivative |
d
x-N/Å |
d
x-H/Å |
θ
|
β
|
| 3,4(MeO)2-Phe |
3.718 |
2.874 |
16.258 |
47.532 |
| Phe |
3.770 |
2.955 |
14.554 |
48.075 |
| 3-F-Phe |
3.781 |
2.980 |
14.538 |
48.272 |
| 4-F-Phe |
3.798 |
2.990 |
13.098 |
48.483 |
| 3,5-F2-Phe |
3.805 |
3.020 |
16.093 |
48.744 |
| 3-CF3-Phe |
3.826 |
3.040 |
17.619 |
48.948 |
| 4-NO2-Phe |
3.837 |
3.063 |
13.973 |
48.789 |
| 3-CN-Phe |
3.850 |
3.071 |
16.695 |
49.488 |
| 2,5-F2-Phe |
4.215 |
3.698 |
43.012 |
62.560 |
| F5-Phe |
4.228 |
3.748 |
41.314 |
62.435 |
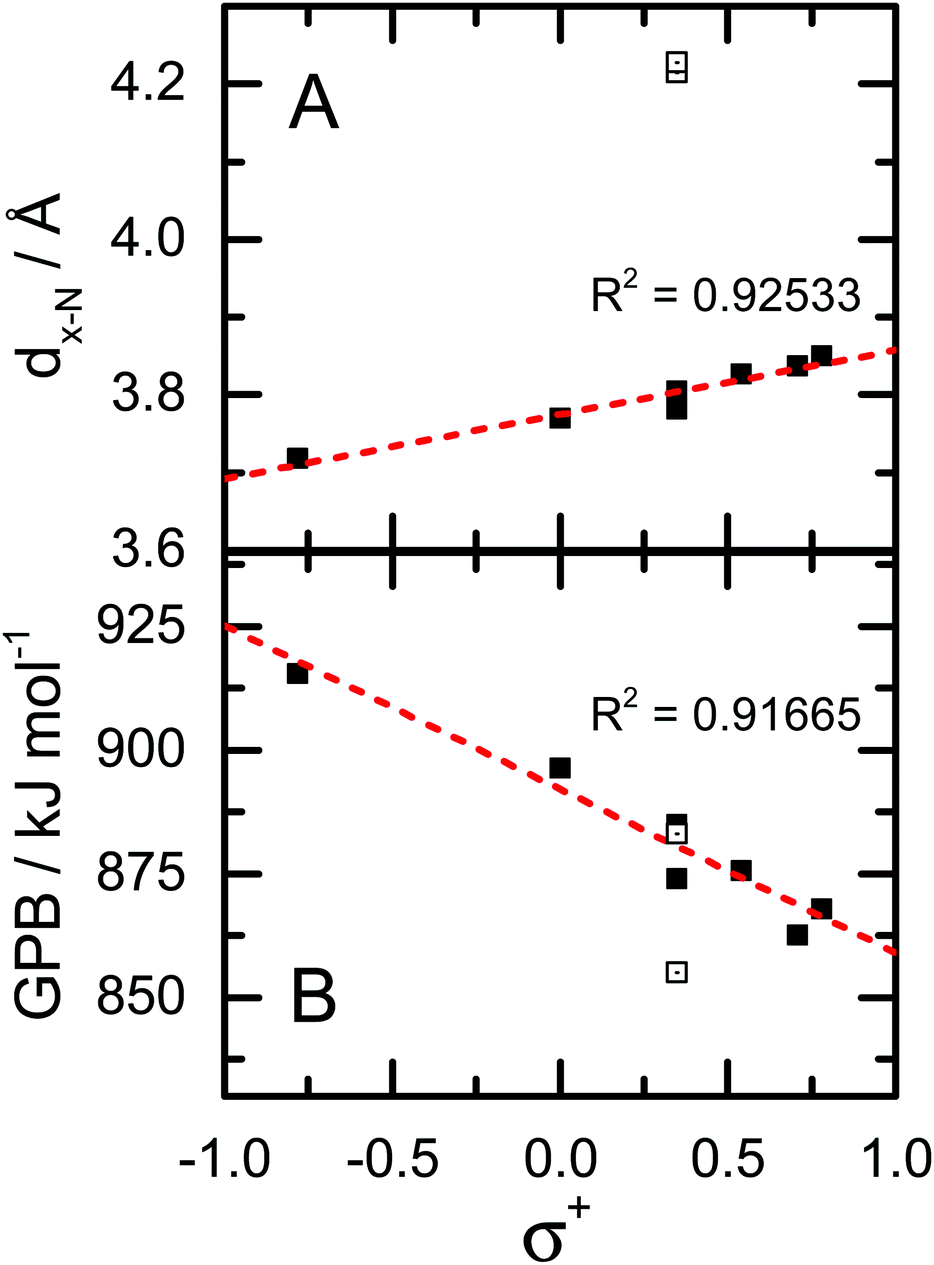
In examining the coordinates given in Table 1, one can identify a qualitative trend whereby the Phe·H+ derivatives which contain electron withdrawing groups exhibit larger β and θ angles, and longer dx-N and dx-H distances. In other words, when Phe is derivatized to include functional groups which act to remove electron charge density from the phenyl ring π-system, the most stable position for the charge centre migrates from a facial site towards an edge site. This, of course, is perfectly in line with expectations based on model charge–quadrupole interactions (e.g., Na+·benzene and Na+·hexafluorobenzene). To be quantitative, we explored the correlations between the calculated coordinates and Hammett (σ+) parameters for the various substituents.37,38 An example of this is shown in Fig. 5A, which plots the calculated dx-N values as a function of σ+. Note that, because our goal was to assess donation/withdrawal of electron density from the ring system rather than the specific stereoelectronic effects of substitution, we chose the largest value of σ+ from the para- and meta-substitution series. It is clear from Fig. 5A that the σ+ parameters, which provide a metric of quadrupole orientation (i.e., normal versus inverted), are strongly correlated with the calculated molecular geometries. In other words, the cation–π interaction has a significant impact on the geometries of the protonated Phe derivatives. However, there are two obvious outliers to the observed dx-N/σ+ correlation, which correspond to 2,5-F2-Phe·H+ and F5-Phe·H+. Since both species have F atoms in the 2-position of the phenyl ring, they can form intramolecular H–F hydrogen bonds. Indeed, the calculated H2N+–H⋯F internuclear distances are 1.81 Å and 1.91 Å for 2,5F2-Phe·H+ and F5-Phe·H+, respectively. This is substantially shorter than the sum of the van der Waals radii for H (1.20 Å) and F (1.47 Å), and is therefore indicative of intramolecular hydrogen bond (IMHB) formation. Being a much stronger interaction than the charge–quadrupole interaction, the IMHB interaction further drives the NH3+/ring system into an edgewise orientation.
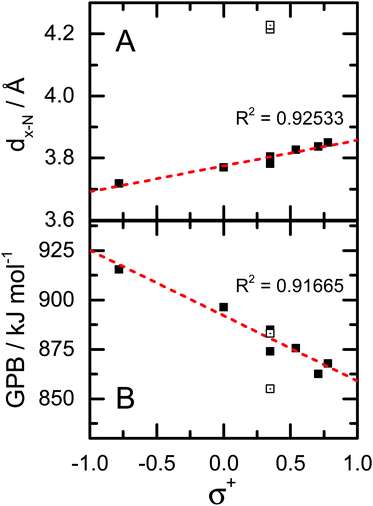 |
| | Fig. 5 (A) Correlation plot for dx-N and σ+. Outlying data points (open boxes) correspond to the 2,5-F2-Phe·H+ and F5-Phe·H+ derivatives. (B) Correlation plot for calculated gas phase basicity (GPB) and σ+. Data point for the 2,5-F2-Phe·H+ and F5-Phe·H+ derivatives are plotted as open squares. | |
Having modelled both the neutral and protonated Phe derivatives, we were also able to calculate gas phase proton affinities (Pas) and gas phase basicities (GPBs) per the equations:
| | 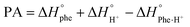 | (1) |
| | 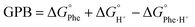 | (2) |
where the subscripts Phe and H
+ indicate the separate moieties, and Phe·H
+ indicates the protonated Phe derivative. The results of these calculations are tabulated in the ESI.
† The only experimentally determined value of which we are aware is the PA of Phe (922.9 kJ mol
−1),
39 which compares favourably with our calculated value of 926.0 kJ mol
−1. We find a strong negative correlation between the calculated GPBs and PAs of the Phe derivatives and the
σ+ parameters of the substituents (see
Fig. 5B); substitution with electron donating groups (EDGs) results in higher GPBs and PAs than does derivatization with electron withdrawing groups (EWGs). This can be rationalized by considering the geometry of the cation–π interaction. Electron density donation to the phenyl ring results in a facially oriented interaction and a shorter charge–quadrupole distance (
dx-N) than the edgewise interaction that is favoured by electron withdrawing groups. Since a monopole–quadrupole interaction has a 1/
r3 dependence, even modest differences in charge–quadrupole separation can result in substantial differences in interaction energies. Thus, protonated Phe derivatives which adopt a facially oriented cation–π geometry (those with EDGs) are stabilized relative to the edgewise interacting species (those with EWGs). Note that while the range of the calculated GPBs is large (
ca. 60 kJ mol
−1), it is consistent with the 20–70 kJ mol
−1 that one would expect for a cation–π interaction.
9,11,12
Conclusions
The potential energy surfaces of several Phe derivatives and their protonated analogues have been mapped using the basin hopping search algorithm. The IRMPD spectra of the protonated Phe derivatives were assigned by comparison with IR spectra calculated at the DFT level of theory. Protonation is found to be localized on the amino nitrogen atom in all cases. By comparing the geometries of the calculated global minima of the Phe·H+ derivatives, we find structural changes across the series that are consistent with an intramolecular ammonium group–π interaction. Phenyl moieties that contain electron withdrawing groups prefer to orient in an edgewise fashion to the charged ammonium centre, whereas phenyl rings which contain electron donating groups prefer to adopt a facial orientation. These geometric variations exhibit a strong positive correlation with the σ+ parameters of the substituents. Notable exceptions to this trend are the 2,5-F2-Phe·H+ and F5-Phe·H+ derivatives, whose geometries are more strongly influenced by the formation of H2N+–H⋯F intramolecular hydrogen bonds. We also find that calculated molecular properties (PA and GPB) correlate strongly with the intramolecular cation–π interactions. This suggests that, while much of the attention surrounding cation–π interactions have been directed towards supramolecular and condensed phase systems, intramolecular cation–π interactions could also have a significant impact on the chemical and physical properties of isolated molecules.
Acknowledgements
We gratefully acknowledge high performance computing support from the SHARCNET consortium of Compute Canada. We are also grateful to the Centre Laser Infrarouge d'Orsay (CLIO) team and technical support staff for the valuable assistance and hospitality. The authors would like to acknowledge financial support from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
Notes and references
- S. K. Burley and G. A. Petsko, FEBS Lett., 1986, 203, 139–143 CrossRef CAS PubMed.
- H. S. Choi, S. B. Suh, S. J. Cho and K. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12094–12099 CrossRef CAS.
- D. A. Dougherty, Science, 1996, 271, 163–168 CAS.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- C. Miller, Science, 1993, 261, 1692–1693 CAS.
- J. B. O. Mitchell, C. L. Nandi, S. Ali, J. K. McDonald, J. M. Thornton, S. L. Price and J. Singh, Nature, 1993, 366, 413 CrossRef.
- J. Singh and J. M. Thornton, J. Mol. Biol., 1990, 211, 595–615 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS.
- F. M. Siu, N. L. Ma and C. W. Tsang, J. Am. Chem. Soc., 2001, 123, 3397–3398 CrossRef CAS PubMed.
- R. H. Wu and T. B. McMahon, J. Am. Chem. Soc., 2008, 130, 12554–12555 CrossRef CAS PubMed.
- M. Burt, K. Wilson, R. Marta, M. Hasan, W. S. Hopkins and T. McMahon, Phys. Chem. Chem. Phys., 2014, 16, 24223–24234 RSC.
- B. C. Guo, J. W. Purnell and A. W. Castleman, Chem. Phys. Lett., 1990, 168, 155–160 CrossRef CAS.
- J. Sunner, K. Nishizawa and P. Kebarle, J. Phys. Chem., 1981, 85, 1814–1820 CrossRef CAS.
- J. C. Amicangelo and P. B. Armentrout, J. Phys. Chem. A, 2000, 104, 11420–11432 CrossRef CAS.
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- P. B. Armentrout and M. T. Rodgers, J. Phys. Chem. A, 2000, 104, 2238–2247 CrossRef CAS.
- R. C. Dunbar, J. D. Steill and J. Oomens, Phys. Chem. Chem. Phys., 2010, 12, 13383–13393 RSC.
- L. Mac Aleese, A. Simon, T. B. McMahon, J. M. Ortega, D. Scuderi, J. Lemaire and P. Maitre, Int. J. Mass Spectrom., 2006, 249, 14–20 CrossRef.
- J. M. Ortega, F. Glotin and R. Prazeres, Infrared Phys. Technol., 2006, 49, 133–138 CAS.
- W. S. Hopkins, R. A. Marta and T. B. McMahon, J. Phys. Chem. A, 2013, 117, 10714–10718 CrossRef CAS PubMed.
- W. S. Hopkins, R. A. Marta, V. Steinmetz and T. B. McMahon, Phys. Chem. Chem. Phys., 2015, 17, 28548–28555 RSC.
- M. J. Lecours, W. C. T. Chow and W. S. Hopkins, J. Phys. Chem. A, 2014, 118, 4278–4287 CrossRef CAS PubMed.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Comput. Chem., 1993, 14, 1504–1518 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- C. Ieritano, P. J. J. Carr, M. Hasan, M. Burt, R. A. Marta, V. Steinmetz, E. Fillion, T. B. McMahon and W. S. Hopkins, Phys. Chem. Chem. Phys., 2016, 18, 4704–4710 RSC.
- A. B. McCoy, J. L. Fry, J. S. Francisco, A. K. Mollner and M. Okumura, J. Chem. Phys., 2005, 122, 104311 CrossRef PubMed.
- J. Oomens, J. D. Steill and B. Redlich, J. Am. Chem. Soc., 2009, 131, 4310–4319 CrossRef CAS PubMed.
- J. D. Steill, J. Szczepanski, J. Oomens, J. R. Eyler and A. Brajter-Toth, Anal. Bioanal. Chem., 2011, 399, 2463–2473 CrossRef CAS PubMed.
- C. F. Correia, P. O. Balaj, D. Scuderi, P. Maitre and G. Ohanessian, J. Am. Chem. Soc., 2008, 130, 3359–3370 CrossRef CAS PubMed.
- P. Parneix, M. Basire and F. Calvo, J. Phys. Chem. A, 2013, 117, 3954–3959 CrossRef CAS PubMed.
- H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1958, 80, 4979–4987 CrossRef CAS.
- L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Phe and Phe·H+ structures, and thermochemical data for all derivatives. See DOI: 10.1039/c6cp07337a |
|
| This journal is © the Owner Societies 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a