
Pathological levels of glucosylceramide change the biophysical properties of artificial and cell membranes†
Ana R. P.
Varela‡
abc,
Ana E.
Ventura‡
abc,
Ana C.
Carreira
ad,
Aleksander
Fedorov
b,
Anthony H.
Futerman
c,
Manuel
Prieto
b and
Liana C.
Silva
 *a
*a
aiMed.ULisboa – Research Institute for Medicines, Faculdade de Farmácia, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003 Lisboa, Portugal. E-mail: lianacsilva@ff.ulisboa.pt; Tel: +351 217 946 429
bCentro de Química-Física Molecular and Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
cDepartment of Biomolecular Sciences, Weizmann Institute of Science, Rehovot 76100, Israel
dCentro de Química e Bioquímica, DQB, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal
First published on 22nd November 2016
Abstract
Glucosylceramide (GlcCer) plays an active role in the regulation of various cellular events. Moreover, GlcCer is also a key modulator of membrane biophysical properties, which might be linked to the mechanism of its biological action. In order to understand the biophysical implications of GlcCer on membranes of living cells, we first studied the effect of GlcCer on artificial membranes containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), sphingomyelin (SM) and cholesterol (Chol). Using an array of biophysical methods, we demonstrate that at lower GlcCer/Chol ratios, GlcCer stabilizes SM/Chol-enriched liquid-ordered domains. However, upon decreasing the Chol content, GlcCer significantly increased membrane order through the formation of gel domains. Changes in pH disturbed the packing properties of GlcCer-containing membranes, leading to an increase in membrane fluidity and reduced membrane electronegativity. To address the biophysical impact of GlcCer in biological membranes, studies were performed in wild type and in fibroblasts treated with conduritol-B-epoxide (CBE), which causes intracellular GlcCer accumulation, and in fibroblasts from patients with type I Gaucher disease (GD). Decreased membrane fluidity was observed in cells containing higher levels of GlcCer, such as in CBE-treated and GD cells. Together, we demonstrate that elevated GlcCer levels change the biophysical properties of cellular membranes, which might compromise membrane-associated cellular events and be of relevance for understanding the pathology of diseases, such as GD, in which GlcCer accumulates at high levels.
Introduction
Glucosylceramide is a ubiquitous membrane lipid in mammalian cells and plays vital roles in cell maintenance and survival.1,2 It is one of the simplest glycosphingolipids (GSLs) and a precursor of complex GSLs.1 GlcCer has been implicated in lipid domain formation, and therefore it has also been suggested that GlcCer modulates a number of signalling events.3,4 Moreover, elevated levels of GlcCer are a hallmark of Gaucher disease (GD). The exact molecular mechanisms underlying the biological and pathological actions of GlcCer remain elusive. However, an increasing body of evidence supports the notion that GlcCer plays an important role in the modulation of membrane biophysical properties since it increases membrane order5,6 and promotes membrane structural alterations,6 which might trigger GlcCer-mediated actions.GlcCer is structurally similar to ceramide, with the only difference being a glucose moiety in the C1 position of the ceramide backbone. It is therefore not very surprising that GlcCer also displays a strong tendency to alter the biophysical and structural properties of fluid model membranes.6,7 By analogy with ceramide,8 GlcCer is able to form tightly packed gel domains in a fluid POPC bilayer,6 but displays a higher tendency to promote membrane shape changes.7 It is described that GlcCer has a low propensity to participate in the formation of sterol-enriched domains,9 but the miscibility of GlcCer in the liquid ordered (lo) phase is still higher than that of ceramide.10–12 Moreover, the addition of SM, or other glycosphingolipids (i.e. lactosylceramide (LacCer)), improves Chol/GlcCer miscibility13 probably due to packing defects created by the presence of different headgroups within the domains. In addition, GlcCer-induced biophysical changes are pH-dependent: a decreased ability to drive gel-fluid phase separation and increased tendency to induce membrane morphological alterations are observed upon acidification,7 suggesting that alterations in pH, such as those occurring along the endolysosomal pathway, might be sufficient to alter the membrane organization and the biophysical properties of membrane regions enriched in GlcCer.
Considering the strong effects that GlcCer has on the biophysical properties of artificial membranes, it can be anticipated that pathological elevated levels of GlcCer, as observed in GD, might compromise the membrane structure and properties, which might be linked to pathology development. However, investigating the biophysical implications of altered GlcCer levels in this pathology is still an underexplored area. To address this issue, we first performed a series of studies in artificial membranes with higher lipid complexity. This strategy enables us to further understand the interplay of GlcCer with other lipids and better rationalize the effect of this lipid on the properties of complex biological membranes. Using multiple biophysical methodologies we showed that GlcCer not only associates with SM to drive gel-fluid phase separation, but it also interacts with both SM/Chol to increase the order of the lo phase, particularly when the Chol content of the mixtures is increased. In addition, elevated levels of GlcCer decreased the fluidity of membranes of living cells. Our results further support the hypothesis that local elevation in GlcCer levels in biological membranes might result in significant alterations in the membrane structure and packing properties, and might explain some of the mechanisms by which GlcCer accumulation triggers cell pathology in GD.
Experimental procedures
Materials and methods
POPC, C16-GlcCer (D-glucosyl-β-1,1′ N-palmitoyl-D-erythro-sphingosine), C16-SM (N-palmitoyl-D-erythro-sphingosylphosphorylcholine), Rho-DOPE (N-rhodamine-dipalmitoylphosphatidylethanolamine) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) (DOPE-biotin) were purchased from Avanti Polar Lipids (Alabaster, AL). t-PnA (trans-parinaric acid), Laurdan and NBD-DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4yl)) were purchased from Molecular Probes (Leiden, The Netherlands). Cholesterol and cell culture media were purchased from Sigma-Aldrich (Steinheim, Germany). Conduritol-B-epoxide (CBE) was purchased from Calbiochem (Darmstadt, Germany). All organic solvents were of UVASOL grade and were purchased from Merck (Darmstadt, Germany).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :106), Rho-DOPE and NBD-DPPE (at a probe/lipid ratio of 1:500 and 1:200, respectively), as previously described.3,4 The GUVs were then transferred to 8-well Ibidi® μ-slides that had been previously coated with avidin (at 0.1 mg ml−1) to improve GUV adhesion to the plate.4 MLV, LUV and GUV suspension medium was PBS buffer (10 mM sodium phosphate, 150 mM sodium chloride and 0.1 mM EDTA, pH 7.4) or citrate-phosphate buffer (100 mM citric acid and 200 mM sodium phosphate, pH 5.5).
:106), Rho-DOPE and NBD-DPPE (at a probe/lipid ratio of 1:500 and 1:200, respectively), as previously described.3,4 The GUVs were then transferred to 8-well Ibidi® μ-slides that had been previously coated with avidin (at 0.1 mg ml−1) to improve GUV adhesion to the plate.4 MLV, LUV and GUV suspension medium was PBS buffer (10 mM sodium phosphate, 150 mM sodium chloride and 0.1 mM EDTA, pH 7.4) or citrate-phosphate buffer (100 mM citric acid and 200 mM sodium phosphate, pH 5.5).
To chemically induce GD phenotype, human skin fibroblast cells were seeded in 6 or 12-well plates (Orange Scientific) and treated with none, 50 μM, 300 μM and 500 μM of CBE from 1 and up to 7 days. Every two days cell culture medium was replaced by fresh medium containing the adequate CBE concentration.
:21, vol:vol), with a protein inhibitor cocktail (200:1). Protein concentration was estimated using the BCA protein assay reagent (Pierce Chemical Co., Thermo Scientific, Rockford, IL, USA). Cell homogenates (20 μg of protein) were incubated at 37 °C with 10 μM C6-NBD-GlcCer in 50 μl of McIlvaine's buffer for 30 min. Reactions were terminated by adding chloroform/methanol (1:2 vol/vol). Lipids were extracted and separated by thin layer chromatography using chloroform:methanol:9.8 mM CaCl2 (60:35:8 vol/vol/vol) as the developing solvent, and quantified using a Typhoon 9410 variable mode imager and Image-QuantTL (GE Healthcare, Chalfont St Giles, UK).
Results
1. Influence of GlcCer on the biophysical properties of artificial membranes
Fig. 1 shows the variation of t-PnA fluorescence anisotropy (Fig. 1A), mean fluorescence lifetime (Fig. 1B) and the long lifetime component of the fluorescence intensity decay (Fig. 1C) in POPC/SM/Chol mixtures in the absence and the presence of C16-GlcCer. Increasing levels of C16-GlcCer in the mixtures lead to an increase in the fluorescence anisotropy and the mean fluorescence lifetime of t-PnA, irrespective of the composition of the mixtures, showing that C16-GlcCer increases membrane order. This effect is more pronounced for mixtures containing low Chol and low SM, i.e. for mixtures where the lo-phase fraction is up to 51% (Table S1, ESI†), and is much smaller for membranes containing a large fraction of the lo phase. This effect is similar to previous reports using ceramide,8 where a decreased ability to segregate into tightly packed gel domains alongside increased miscibility in the lo phase was observed upon increasing the lo fraction of the mixtures.5,9 This is further confirmed by the decrease in the long lifetime component of the t-PnA fluorescence intensity decay (Fig. 1C and Fig. S2C, E, ESI†). Nonetheless, even in mixtures containing a large lo fraction, the long lifetime component of t-PnA is very high and close to values typical of a gel phase (above 30 ns10). Moreover, four components were required to describe the fluorescence intensity decay of the probe, suggesting that the ability of GlcCer to segregate into gel domains is not completely abolished upon increasing the Chol and SM content of the quaternary mixtures at neutral pH.
 | ||
| Fig. 1 Fluorescence properties of t-PnA in POPC/C16-SM/Chol/C16-GlcCer at different pH values. t-PnA (A and D) fluorescence anisotropy, (B and E) mean fluorescence lifetime and (C and F) long lifetime component of the intensity decay in POPC/C16-SM/Chol mixtures containing 0 (solid black circles), 5 (open circles) and 10 (solid grey circles) mol% of C16-GlcCer. Measurements were performed at (A–C) pH 7.4 and (D–F) 5.5. Values are means ± SD of at least 3 independent experiments. | ||
Similar effects were observed when these mixtures were characterized at acidic pH (Fig. 1D–F). However, under acidic conditions, GlcCer is unable to induce gel-fluid phase separation in mixtures containing larger fractions of the lo phase, as shown by the decrease in the long lifetime component of t-PnA below values typical of the gel phase10 (Fig. 1F), and the disappearance of the fourth component of the analysis of the fluorescence intensity decay of the probe (Fig. S2D and F, ESI†).
The results further demonstrated that at pH 5.5, the overall order of the mixtures is lower compared to neutral pH. Decreased C16-GlcCer-enriched gel-domain formation has been previously observed in mixtures of POPC/C16-GlcCer1 and POPC/Chol/C16-GlcCer7 upon acidification.
Similar to the data obtained by fluorescence spectroscopy, analysis of the giant unilamellar vesicles (GUVs) by fluorescence microscopy (Fig. 2) showed that, regardless of the pH, C16-GlcCer drives an extensive gel-fluid phase separation in vesicles containing low Chol levels, as shown by the dark regions. Ternary mixtures containing larger Chol/SM levels display small areas labelled with Rho-DOPE (Fig. 2A) corresponding to the ld phase and an extensive area labelled with NBD-DPPE corresponding to the lo phase, in agreement with the lo phase fraction predicted for this mixture6 (Table S1, ESI†). The addition of C16-GlcCer to these mixtures does not result in detectable gel-fluid phase separation showing that C16-GlcCer becomes more miscible in the lo phase, similar to ceramide9 (Fig. 2B).
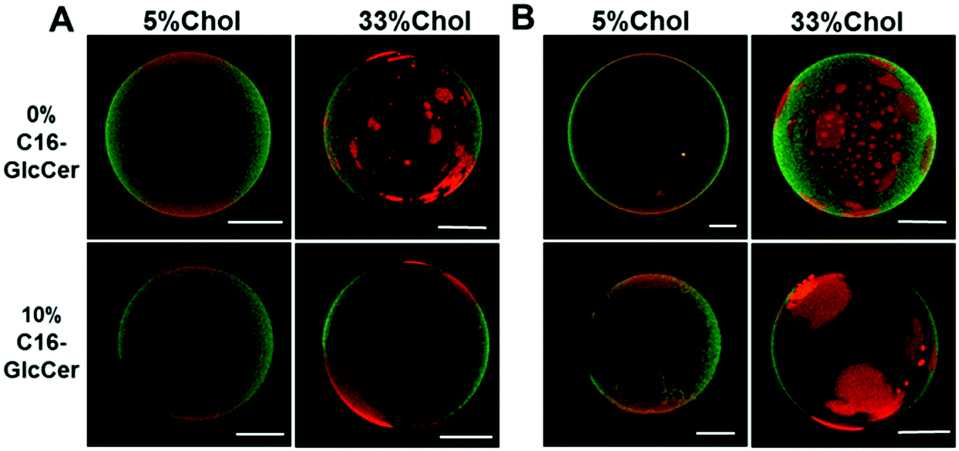 | ||
| Fig. 2 Confocal fluorescence microscopy of POPC/C16-SM/Chol and POPC/C16-SM/Chol/C16-GlcCer mixtures. 3D projection images from 0.4 μm confocal slices of POPC/C16-SM/Chol GUVs in the absence and the presence of 10 mol% of C16-GlcCer. Images correspond to the overlay of the NBD-DPPE (green) and Rho-DOPE (red) channels. Images were taken at (A) neutral (7.4) and (B) acidic (5.5) pH. Scale bar, 5 μm. | ||
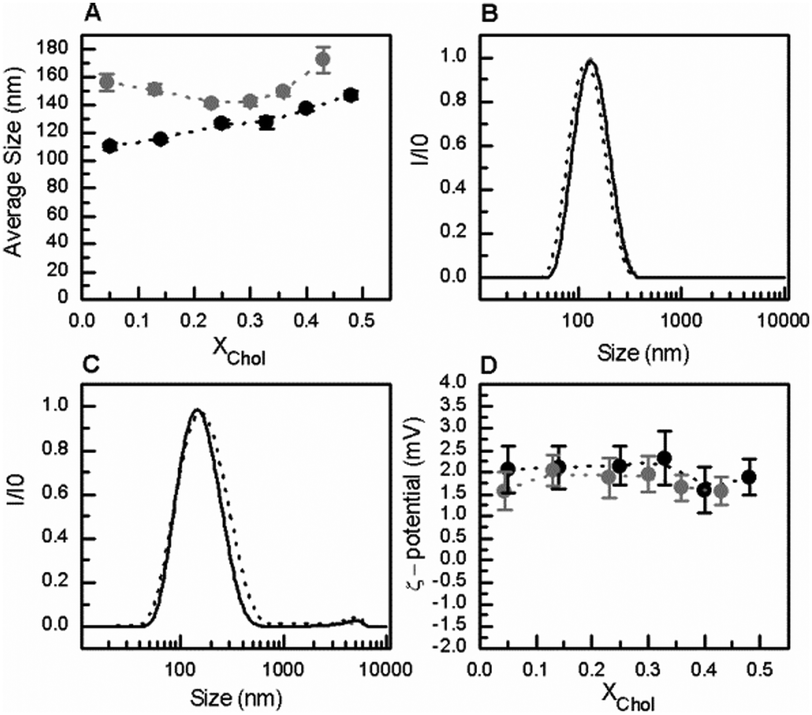
 | ||
| Fig. 3 Characterization of POPC/C16-SM/Chol/C16-GlcCer mixtures by electrophoretic and dynamic light scattering measurements, at neutral pH. (A) Average size and (D) ζ-potential of large unilamellar vesicles (LUVs) composed of POPC/C16-SM/Chol mixtures, containing 0 (black solid symbols), and 10 (grey solid circles) mol% of C16-GlcCer. (B–C) Normalized scattered light intensity of POPC/C16-SM/Chol/C16-GlcCer LUVs containing 26% (—), 51% (—), and 84% (---) of lo phase and (B) 0 and (C) 10 mol% of C16-GlcCer. Values are means ± SD of at least 3 independent experiments. | ||
Similar results were obtained at pH 5.5 (Fig. 4). The presence of GlcCer in the mixtures induced an increase in the overall size of the vesicles (Fig. 4A and C) but no significant changes were observed in the overall net surface charge of the vesicles (Fig. 4D). At acidic pH, the vesicles are slightly positively charged.
 | ||
| Fig. 4 Characterization of POPC/C16-SM/Chol/C16-GlcCer mixtures by electrophoretic and dynamic light scattering measurements at acidic pH (5.5). (A) Average size and (D) ζ-potential of POPC/C16-SM/Chol LUVs containing 0 (black solid circles) and 10 (grey solid circles) mol% of C16-GlcCer. (B and C) Normalized scattered light intensity of POPC/C16-SM/Chol/C16-GlcCer LUVs containing 26% (—), 51% (—), and 84% (---) of lo phase and (B) 0 and (C) 10 mol% of C16-GlcCer. Values are means ± SD of at least 3 independent experiments. | ||
2. Studies in living cells
To evaluate if GlcCer exerts similar effects on the biophysical properties of membranes from living cells, human skin fibroblasts were treated with CBE (conduritol-B-epoxide) for different time periods. CBE is an irreversible inhibitor of acid β-glucosidase12 (Fig. S3A, ESI†), and causes a time dependent accumulation of GlcCer (Fig. S3B, ESI†). To evaluate the effects of GlcCer on membrane order, cells were stained with the environment-sensitive probe Laurdan,13 and imaged by 2-photon microscopy (Fig. 5). Cells treated with CBE displayed higher Laurdan GP values, indicating an increased membrane order. This was observed both when the cell and the plasma membrane were analyzed (Fig. 5B and C, GP increased in, respectively, the bulk membranes and the PM from 0.41 and 0.50 in the control cells, to 0.49 and 0.58 in cells treated with 500 μM of CBE), suggesting that GlcCer accumulation likely impacts the organization and order of different cellular membranes. | ||
| Fig. 5 Effect on GlcCer accumulation on the membrane packing properties of human skin fibroblasts. (A) Laurdan imaging of control cells and cells treated with 50, 300 and 500 μM CBE for 7 days. Images in the upper panel show the overlay between emission at 400–460 nm (green channel) and 470–530 nm (red channel). Images in the lower panel show the GP images obtained by applying the GP function to the images from green and red channels pixel by pixel. Scale bars correspond to 15 μm. (B and C) Histograms of the GP values from (B) whole cells and (C) the plasma membranes (PM). Data from control and cells treated with 50, 300 and 500 μM CBE are shown in green, dark grey, light grey and black, respectively. The shift towards higher GP values in cells treated with higher concentrations of CBE indicates that these are more ordered, this being evidence that GlcCer accumulation increases membrane order. The differences in GP values were statistically significant (p < 0.001) when comparing CBE-treated cells both in relation to untreated cells and between the different CBE concentrations studied. | ||
A fibroblast cell line derived from patients with GD type I (N370S/N370S) was next compared to wild type human skin fibroblasts. Fig. 6 shows that t-PnA fluorescence anisotropy (Fig. 6A) and the mean fluorescence lifetime (Fig. 6B) are higher in mutant fibroblasts, suggesting an overall increase in membrane order compared to control fibroblasts. These results are compatible with an ordering effect promoted by GlcCer. This was further confirmed by the strong increase in the long lifetime component of the fluorescence intensity decay of t-PnA in GD fibroblasts to values close to those typical of the gel phase (Fig. 6C). Together these results show that the accumulation of GlcCer influences the packing properties of biological membranes.
 | ||
| Fig. 6 Fluorescence properties of t-PnA in wt and GD type I mutant fibroblasts. t-PnA (A) fluorescence anisotropy, (B) mean fluorescence lifetime and (C) long lifetime component of the intensity decay in the GD type I mutant (light grey) and wild type fibroblasts (dark grey). Values are means ± SD of at least 3 independent experiments. Statistically significant differences were obtained when comparing wt and mutant cells (p < 0.001 in A and C and p < 0.05 in B). | ||
Discussion
Interplay between GlcCer and lipid components in artificial membranes
The molecular mechanisms that connect the biophysical and biological properties of GlcCer were the main focus of the current study. Understanding the biophysical implications of a given lipid species in the complex setting of biological membranes requires the use of strategies that enable identifying particular biophysical features associated with that lipid. Accordingly, studies in artificial systems are mandatory to unravel the interplay of the lipid under study with membrane lipid components. It is already known that GlcCer segregates into domains with gel-phase properties, mainly due to its relatively small polar headgroup, high gel-to-fluid melting temperature and ability to function both as a donor and acceptor for H-bonding.1,7,14 In the present study we demonstrated that in artificial membranes with higher complexity GlcCer increases membrane order, irrespective of lipid composition. However, the types of phases induced by GlcCer are strongly dependent on the Chol and SM composition of the membrane. Accordingly, in mixtures containing up to 35 mol% of Chol, GlcCer induces a gel-fluid phase separation. Interestingly, the same amount of GlcCer (5 and 10 mol%) is not enough to induce gel-domain formation in binary POPC/GlcCer1 and ternary POPC/Chol/GlcCer7 mixtures. These results show that in the presence of SM, the miscibility of GlcCer in the fluid phase decreased, probably due to the association between SM and GlcCer, which contributes to gel-domain formation. This may result from the establishment of an efficient H-bond network between these two lipids. Similar interactions are known for artificial lipid mixtures and lipids extracted from red blood cells containing SM and ceramide in the absence and the presence of Chol,9,15–17 suggesting that this might be a common feature of different SLs.The phase separation of GlcCer and SM into gel domains is compromised by the presence of high Chol levels. Nonetheless, GlcCer is able to increase the order of the lo-phase. These observations are in agreement with previous studies showing that membranes containing high Chol fraction can only accommodate small proportions of GSLs in their sterol-enriched domains, particularly in the absence of other SLs.18–20
Our results further show that the organization and the physical properties of membranes enriched in GlcCer are pH-dependent, such that GlcCer has a decreased ability to induce gel domain formation under acidic conditions. The differences in the packing properties and membrane organization might arise from alterations in the H-bonding network between GlcCer molecules, neighboring lipids and water,18,21 and by the alteration of the orientation of the glucose moiety at the membrane interface.14,22 Moreover, an increase in the overall surface charge of the vesicles from electronegative to electropositive upon acidification indicates a shift from electron to proton association with the lipid groups at the membrane surface. Since no significant changes in the overall charge of the lipid constituents of the mixtures are expected,14,23–25 we suggest that alterations in the surface charge might also be driven by changes in the orientation of the lipid headgroups,26 alterations in H-bonding states and lipid organization, which would also contribute to a lower packing of the lipids and/or ability to form tightly packed gel domains.
Influence of GlcCer in cell membrane biophysical properties
Intracellular levels of GlcCer depend on the balance between its rate of synthesis and degradation. A deficiency in acid β-glucosidase promotes the accumulation of GlcCer, primarily in the lysosome, leading to cell and tissue dysfunction,27,28 as occurs in GD.29,30 Even though GlcCer has been extensively studied, particularly regarding its role in GD,29–33 little attention has been paid to its biophysical properties. GlcCer is thought to be associated with lipid domains, from where it might exert its biological action through alterations in membrane biophysical properties.8 In this and in our previous studies, GlcCer was indeed shown to change the biophysical properties of fluid model membranes containing a distinct lipid composition. In particular, an increase in the content of GlcCer results in membrane ordering, either due to the formation of a GlcCer-enriched gel phase or a GlcCer-enriched lo-phase, depending on the lipid composition of the artificial membranes. Now, using complementary techniques and different experimental strategies, we assessed the effect of GlcCer on membranes of living cells, through comparison of the biophysical properties of wild type human fibroblasts and cells with those of the chemically induced GD phenotype or carrying a mutation homozygote for the N370S. Enrichment in GlcCer levels leads to an increase in the overall order of the membranes, and particularly of the PM. These results are consistent with data obtained in model membranes that demonstrated an increase in the packing properties of the membranes upon increasing GlcCer content. Moreover, the very long lifetime component of t-PnA fluorescence intensity decay shows that regions that display properties resembling a gel-like phase are present in GD fibroblasts. The relatively high Laurdan GP values obtained in CBE-treated cells further support this conclusion. Whether these ordered regions are driven by the segregation of GlcCer into gel domains, or due to association between GlcCer and other membrane lipids prone to form a gel phase, such as ceramides, SM or even saturated phospholipids, is at present not known. These ordered regions might also result from the interaction between GlcCer and Chol that, by analogy with what has been previously suggested for ceramide-Chol interplay,9,34,35 might lead to the formation of domains with properties intermediate between the lo- and the gel-enriched phases, also reported as complex gel-like phases.17 The interactions that occur in cell membranes are complex, involving different lipids and proteins, which, in addition to GlcCer accumulation, will be important determinants in the development of GD pathophysiology. Nevertheless, the results obtained in the present study point towards a change in the biophysical properties of the membrane, which could clearly influence diverse cellular processes, including lipid and protein trafficking, sorting and recycling36–38 that can ultimately impair cell functions. Further investigation of the biophysical changes associated with accumulation of GlcCer in GD might therefore contribute to understanding of the molecular mechanisms underlying this complex disease.Conclusions
The present study demonstrated that in complex membranes, GlcCer increases membrane order, although this property is modulated by Chol. The ability of GlcCer to stabilize lo domains further supports the role of this lipid in the formation of membrane specialized domains, suggesting that GlcCer-mediated biological action might be linked to lipid domain formation and GlcCer-induced biophysical changes. This might have important consequences, particularly taking into account that an abnormal increase in GlcCer, such as observed in GD, drives significant alterations in the packing properties of biological membranes. The increased membrane rigidity observed in cells from GD patients might disturb several cellular processes, which could underlie some of the mechanisms involved in the pathology of GD.Acknowledgements
This work was supported by Fundação para a Ciência e Tecnologia (FCT), Portugal grants: PTDC/QUI-BIQ/111411/2009, PTDC/BBB-BQB/0506/2012, PTDC/BBB-BQB/3710/2014, RECI/CTM-POL/0342/2012, SFRH/BD/69982/2010 to A. R. P. Varela, SFRH/BD/104205/2014 to A. E. Ventura, SFRH/BD/88194/2012 to A. C. Carreira, Compromisso para a Ciência 2008 and Investigador FCT 2014 (IF/00437/2014) to L. C. Silva. A. H. Futerman is The Joseph Meyerhoff Professor of Biochemistry at the Weizmann Institute of Science.References
- A. R. P. Varela, A. M. P. S. Goncalves da Silva, A. Fedorov, A. H. Futerman, M. Prieto and L. C. Silva, Biochim. Biophys. Acta, 2013, 1828, 1122–1130 CrossRef CAS PubMed.
- M. J. Hope, M. B. Bally, G. Webb and P. R. Cullis, Biochim. Biophys. Acta, 1985, 812, 55–65 CrossRef CAS.
- S. N. Pinto, L. C. Silva, R. F. M. de Almeida and M. Prieto, Biophys. J., 2008, 95, 2867–2879 CrossRef CAS PubMed.
- M. J. Sarmento, M. Prieto and F. Fernandes, Biochim. Biophys. Acta, 2012, 1818, 2605–2615 CrossRef CAS PubMed.
- L. C. Silva, R. F. M. de Almeida, B. M. Castro, A. Fedorov and M. Prieto, Biophys. J., 2007, 92, 502–516 CrossRef CAS PubMed.
- R. F. M. de Almeida, A. Fedorov and M. Prieto, Biophys. J., 2003, 85, 2406–2416 CrossRef CAS PubMed.
- A. R. P. Varela, A. S. Couto, A. Fedorov, A. H. Futerman, M. Prieto and L. C. Silva, Biophys. J., 2016, 110, 612–622 CrossRef CAS PubMed.
- B. M. Castro, M. Prieto and L. C. Silva, Prog. Lipid Res., 2014, 54, 53–67 CrossRef CAS PubMed.
- B. M. Castro, L. C. Silva, A. Fedorov, R. F. M. de Almeida and M. Prieto, J. Biol. Chem., 2009, 284, 22978–22987 CrossRef CAS PubMed.
- B. M. Castro, R. F. M. de Almeida, L. C. Silva, A. Fedorov and M. Prieto, Biophys. J., 2007, 93, 1639–1650 CrossRef CAS PubMed.
- B. M. Castro, R. F. M. de Almeida, A. Fedorov and M. Prieto, Chem. Phys. Lipids, 2012, 165, 311–319 CrossRef CAS PubMed.
- G. Legler, Methods Enzymol., 1977, 46, 368–381 CAS.
- T. Parasassi, E. K. Krasnowska, L. Bagatolli and E. Gratton, J. Fluoresc., 1998, 8, 365–373 CrossRef CAS.
- A. R. P. Varela, A. M. P. S. Gonçalves da Silva, A. Fedorov, A. H. Futerman, M. Prieto and L. C. Silva, Langmuir, 2014, 30, 4094–4104 CrossRef CAS PubMed.
- J. Sot, M. Ibarguren, J. V. Busto, L.-R. Montes, F. M. Goni and A. Alonso, FEBS Lett., 2008, 582, 3230–3236 CrossRef CAS PubMed.
- J. B. Massey, Biochim. Biophys. Acta, 2001, 1510, 167–184 CrossRef CAS.
- A. B. Garcia-Arribas, H. Ahyayauch, J. Sot, P. L. Lopez-Gonzalez, A. Alonso and F. M. Goni, Langmuir, 2016, 32, 9053–9063 CrossRef CAS PubMed.
- S. Maunula, Y. J. E. Bjorkqvist, J. P. Slotte and B. Ramstedt, Biochim. Biophys. Acta, 2007, 1768, 336–345 CrossRef CAS PubMed.
- M. J. Ruocco and G. G. Shipley, Biophys. J., 1984, 46, 695–707 CrossRef CAS PubMed.
- Y. J. E. Bjorkqvist, T. K. M. Nyholm, J. P. Slotte and B. Ramstedt, Biophys. J., 2005, 88, 4054–4063 CrossRef PubMed.
- B. Westerlund and J. P. Slotte, Biochim. Biophys. Acta, 2009, 1788, 194–201 CrossRef CAS PubMed.
- P. G. Nyholm and I. Pascher, Biochemistry, 1993, 32, 1225–1234 CrossRef CAS PubMed.
- P. Dewick, Essentials Of Organic Chemistry: For Students Of Pharmacy, Medicinal Chemistry And Biological Chemistry, John Wiley & Sons, Inc., 2006, pp. 121–167 Search PubMed.
- A. D. Petelska, M. Naumowicz and Z. A. Figaszewski, Langmuir, 2012, 28, 13331–13335 CrossRef CAS PubMed.
- U. Bernchou, H. Midtiby, J. H. Ipsen and A. C. Simonsen, Biochim. Biophys. Acta, 2011, 1808, 2849–2858 CrossRef CAS PubMed.
- J. P. F. Doux, B. A. Hall and J. A. Killian, Biophys. J., 2012, 103, 1245–1253 CrossRef CAS PubMed.
- M. Fuller, T. Rozaklis, M. Lovejoy, K. Zarrinkalam, J. J. Hopwood and P. J. Meikle, Mol. Genet. Metab., 2008, 93, 437–443 CrossRef CAS PubMed.
- L. Astudillo, N. Therville, C. Colacios, B. Ségui, N. Andrieu-Abadie and T. Levade, Biochimie, 2016, 125, 267–280 CrossRef CAS PubMed.
- M. Jmoudiak and A. H. Futerman, Br. J. Haematol., 2005, 129, 178–188 CrossRef CAS PubMed.
- E. B. Vitner, A. Vardi, T. M. Cox and A. H. Futerman, Expert Opin. Ther. Targets, 2015, 19, 321–334 CrossRef CAS PubMed.
- G. van Meer, J. Wolthoorn and S. Degroote, Philos. Trans. R. Soc. London, Ser. B, 2003, 358, 869–873 CrossRef CAS PubMed.
- M. C. Messner and M. C. Cabot, in Sphingolipids as Signaling and Regulatory Molecules, ed. C. Chalfant and M. Del Poeta, Springer New York, New York, NY, 2010, pp. 156–164 Search PubMed.
- D. J. Sillence, V. Puri, D. L. Marks, T. D. Butters, R. A. Dwek, R. E. Pagano and F. M. Platt, J. Lipid Res., 2002, 43, 1837–1845 CrossRef CAS.
- J. V. Busto, A. B. Garcia-Arribas, J. Sot, A. Torrecillas, J. C. Gomez-Fernandez, F. M. Goni and A. Alonso, Biophys. J., 2014, 106, 621–630 CrossRef CAS PubMed.
- J. V. Busto, J. Sot, J. Requejo-Isidro, F. M. Goni and A. Alonso, Biophys. J., 2010, 99, 1119–1128 CrossRef CAS PubMed.
- M. A. Surma, C. Klose and K. Simons, Biochim. Biophys. Acta, 2012, 1821, 1059–1067 CrossRef CAS PubMed.
- D. J. Sillence and F. M. Platt, Semin. Cell Dev. Biol., 2004, 15, 409–416 CrossRef CAS PubMed.
- D. J. Sillence, Mol. Genet. Metab., 2013, 109, 194–200 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1 table and 3 figures. See DOI: 10.1039/c6cp07227e |
| ‡ Equally contributed authors. |
| This journal is © the Owner Societies 2017 |