
A QM/MM study of the reaction mechanism of human β-ketoacyl reductase†
Fabiola E.
Medina
 ,
Rui P. P.
Neves
,
Maria J.
Ramos
and
Pedro A.
Fernandes
*
,
Rui P. P.
Neves
,
Maria J.
Ramos
and
Pedro A.
Fernandes
*
UCIBIO, REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre s/n, 4169-007, Porto, Portugal. E-mail: pafernan@fc.up.pt
First published on 17th November 2016
Abstract
Human fatty acid synthase (hFAS) is a multifunctional enzyme involved in a wide diversity of biological functions. For instance, it is a precursor of phospholipids and other complex processes such as the de novo synthesis of long chain fatty acid. Human FAS is also a component of biological membranes and it is implicated in the overexpression of several types of cancers. In this work, we describe the catalytic mechanism of β-ketoreductase (KR), which is a catalytic domain of the hFAS enzyme that catalyzes the reduction of β-ketoacyl to β-hydroxyacyl with the concomitant oxidation of the NADPH cofactor. The catalysis by KR is an intermediate step in the cycle of reactions that elongate the substrate's carbon chain until the final product is obtained. We study and propose the catalytic mechanism of the KR domain determined using the hybrid QM/MM methodology, at the ONIOM(B3LYP/6-311+G(2d,2p):AMBER) level of theory. The results indicate that the reaction mechanism occurs in two stages: (i) nucleophilic attack by a NADPH hydride to the β-carbon of the substrate, together with an asynchronous deprotonation of the Tyr2034 by the oxygen of the β-alkoxide to hold the final alcohol product; and (ii) an asynchronous deprotonation of the hydroxyl in the NADP+'s ribose by Tyr2034, and of the Lys1995 by the resulting alkoxide in the former ribose to restore the protonation state of Tyr2034. The reduction step occurs with a Gibbs energy barrier of 11.7 kcal mol−1 and a Gibbs reaction energy of −10.6 kcal mol−1. These results have provided an understanding of the catalytic mechanism of the KR hFAS domain, a piece of the heavy hFAS biosynthetic machinery.
1. Introduction
Fatty acid synthase (FAS) is a multi-enzyme involved in the synthesis of palmitic acid from acetyl-CoA, malonyl-CoA and NADPH.1,2 The mechanism consists of seven cycles of sequential reactions in which the acetyl-CoA substrate successively incorporates malonyl-CoA molecules to elongate its carbon chain until the palmitic acid product is formed. The growing substrate is kept covalently linked to a phosphopantetheine (PNS) prosthetic group, throughout the whole process, which in turn is covalently linked to a serine in an acyl carrier protein (ACP) domain of FAS. This domain is responsible for the sequential transport of the substrate through the remaining domains where each reaction will occur: malonyl-acetyl transferase (MAT), β-ketoacyl synthase (KS), β-ketoreductase (KR), dehydratase (DH) and enoyl reductase (ER).3 At the end of this process, in which the cycle is repeated seven times, the fatty acyl product is released by the chain-terminal thioesterase (TE) domain. Fig. S1 in the ESI† illustrates this very complex catabolic process. Most of the biosynthesis pathways are carried out by successive independent enzymes with the intermediates freely diffusing from one to another. Why nature has created this machine to synthesize palmitic acid is still unclear. In several human cancer cells, overexpressed levels of FAS have been detected, indicating that it contributes to the growth of cancer cells. Some examples are breast, lung and ovarian carcinomas, among others.4–7 It is also a potential target to control bacterial infections.8 In addition to this role in oncogenicity, FAS has been considered as a possible target for the treatment of obesity too.7Thus, different human FAS inhibitors with anticancer activity have been studied. However these inhibitors are non-specific, or it is not clear in which hFAS catalytic domain they act.9 Nevertheless, in the search for novel inhibitors, a family of inhibitors that could be selective towards the KR domain has been found (Hardwicke et al. 2014 and Vázquez et al. 2008).10,11 In our study, we present the catalytic mechanism of the KR domain of the hFAS, providing atomistic insight into the way in which the β-ketoacyl moiety of the substrate is reduced to β-hydroxyacyl by a NADPH cofactor (Scheme 1), providing the essential transition state structures for the rotational discovery optimization of inhibitors.
 | ||
| Scheme 1 Schematic representation of the general reaction mechanism. | ||
The KR domain has a characteristic Rossmann-fold and it exhibits close interactions with the pseudo-methyltransferase (ΨME), and pseudo-ketoreductase (ΨKR) non-catalytic domains. According to Maier et al.,3 the ΨME domain is probably a consequence of a previously functional enzyme (methyltransferase) from a polyketide synthase;12 currently, together with ΨKR, these domains could contribute to maintaining the structural conformation of the KR domain by forming a compact structure. Nevertheless, none of them shows a direct interaction with the environment of the catalytic residues; the distance between the Cα-terminal of the non-catalytic domains and the Cβ of the substrate is ∼30 Å.2,3 Hence, these should not be very important for the chemical reaction.
The biological assembly of the KR domain has been characterized for Homo sapiens (hKR – PDB code 4PIV),10Sus scrofa (pFAS – PDB code 2VZ9)3,13 and Mycobacterium tuberculosis (mtFAS – PDB code 3V1U)14 through X-ray crystallography. Structure alignments of the KR domains (Fig. 1) reveal that the main residues that interact with the β-ketoacyl substrate (KAC) and the nicotinamide ring of the NADPH cofactor are similarly placed and are the same for the different organisms. Furthermore, there are also four residues that have been considered to be critical for KR activity, and are present in all organisms.2,3,10,15 These are Ser2021(347), Tyr2034(360), Lys1995(364) and Asn2038(319) [the number in round brackets is the residue number in mtFAS]; however Lys364 and Asn319 are interchanged in the bacterial homologs (Fig. 1).
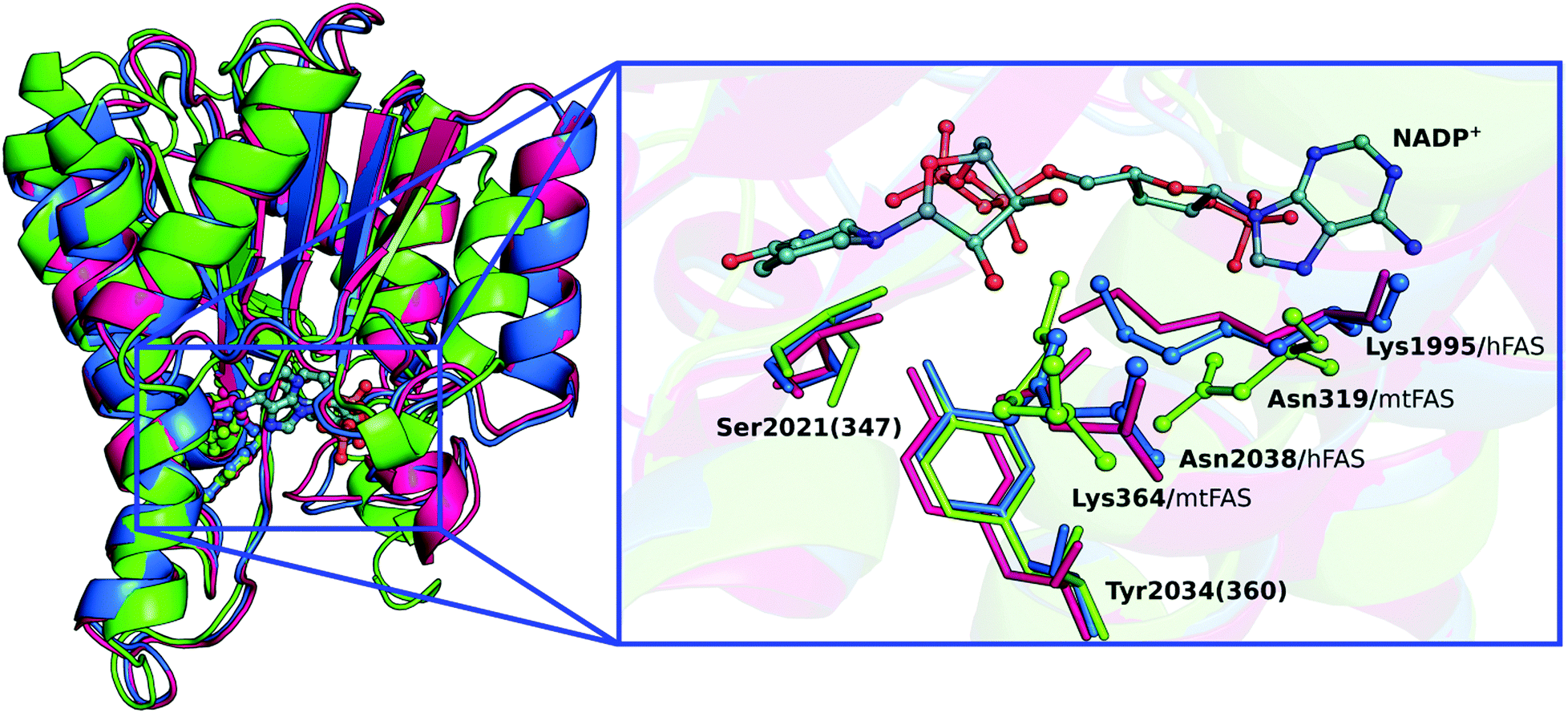 | ||
| Fig. 1 (left) Cartoon representation for the alignment of three KR domains; hKR (blue), pKR (magenta) and mtKR (green), with the active site and the NADP+ cofactor depicted in ball and stick representation. (right) NADP+ cofactor and disposition of catalytic residues in the three KR domains. The residues that differ between the hFAS and the mtFAS (Lys and Asn) are highlighted in ball and stick representation. | ||
In view of the highly conserved nature of the overall folding and catalytic residues, the KR domains of pFAS, hFAS and polyketide synthase will likely exhibit the same mechanism of action proposed for Mycobacterium tuberculosis and Escherichia coli.14–16 According to these studies, the catalysis by hKR should occur in two stages: (i) a nucleophilic attack of a hydride from NADPH to the β-carbon (Cβ) of the substrate and (ii) a deprotonation of an acid by the alkoxide moiety of the β-carbon, to hold the final alcohol product (Scheme 2).
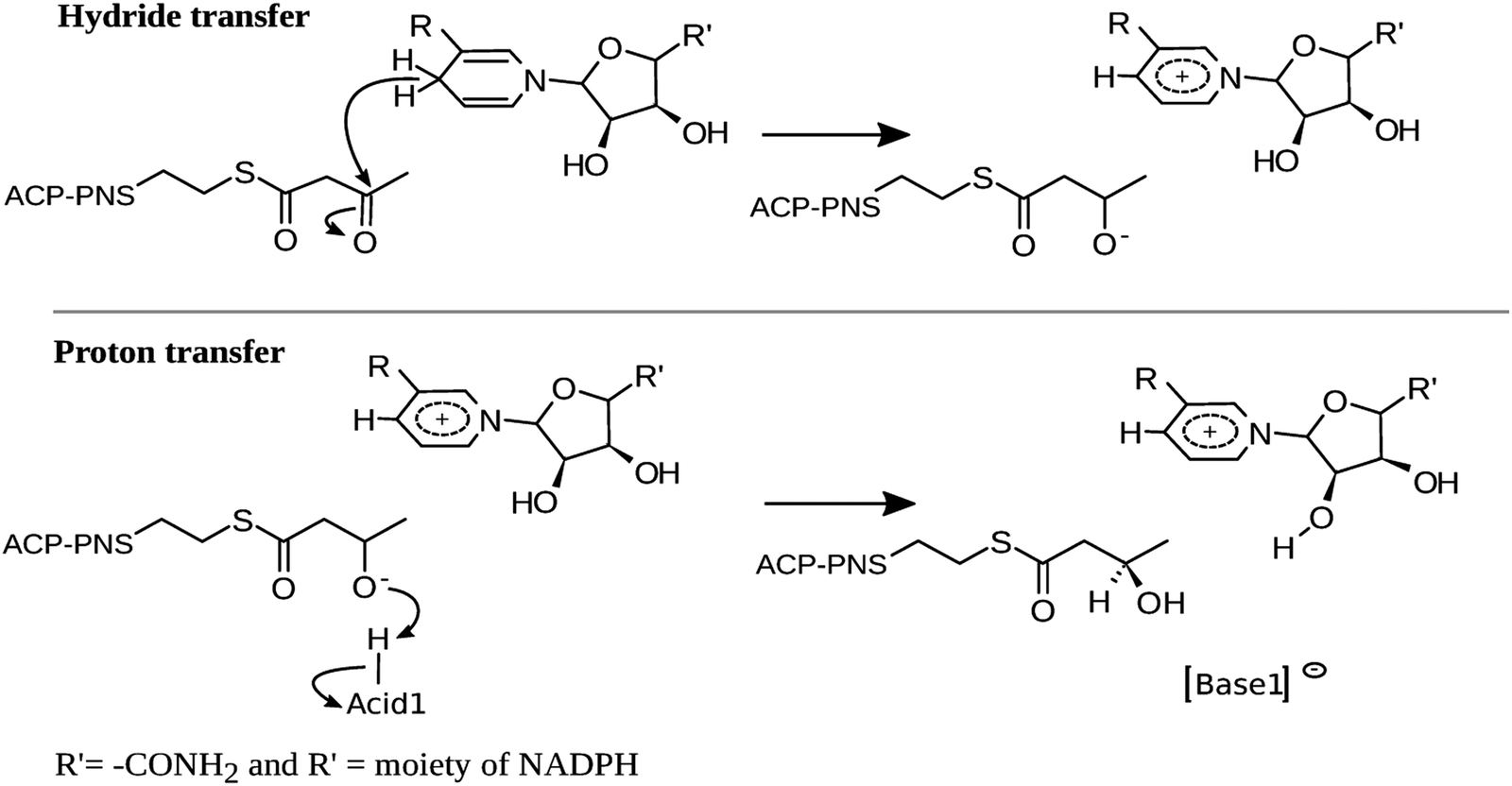 | ||
| Scheme 2 General mechanism proposed. | ||
In order to unveil the role of the four residues depicted in Fig. 1: Ser2021, Tyr2034, Lys1995 and Asn2038, proposed to be essential to perform the NADP+-dependent reduction of KAC to β-hydroxyacyl (HAC) in the hKR domain, we studied this problem using quantum mechanics/molecular mechanics (QM/MM) methods.17 This is an extensively validated methodology in reaction mechanisms of biological systems,18–22 where the stationary points of the reaction are determined from the study of the corresponding potential energy surface (PES) of the reaction coordinates for the enzyme's catalytic mechanism.
2. Methods
Enzyme:substrate complex
Our initial structure of the hKR domain was modeled from its human form (PDB code: 4PIV), as published by Hardwicke et al.10 This structure contains the ΨME–ΨKR–KR domains with NADPH and an inhibitor.10 The inhibitor is placed in the active site of the KR domain:3,15 it contains a carbonyl group on its β-carbon, similar to the KAC substrate, and forms hydrogen bonds with two catalytic residues: Ser2021 and Tyr2034 (as it is also expected for KAC).2,3 Following this reasoning, the KAC substrate was directly modeled from the inhibitor. In the ESI† (Fig. S2) we show the KAC alignment with the inhibitor. The PNS prosthetic group was included in the model, since it remains covalently bonded to KAC during the whole reaction.2,15 Hence, the PNS moiety was modeled from the hexanoyl-CoA that mimics a fatty acyl substrate covalently bonded to ACP, in the bacterial homologue of the KR domain (PDB code 3V1U).14 The parameters for the KAC substrate and the PNS prosthetic group were determined from optimized structures at the HF/6-31G(d) level of theory. These were obtained with the Antechamber tool, as implemented in AMBER12,23 using the GAFF force field24 to derive the intramolecular and Lennard-Jones parameters, and the RESP (restrained electrostatic potential) method25 to derive atomic charges at the HF/6-31G(d) level of theory. The NADP+ cofactor parameters were those determined by Ryde,26,27 the model enzyme was parameterized using the AMBER FF99SBildn force field,28,29 and all hydrogen atoms were added using the Xleap module of the AMBER12 software.23 TIP3P water molecules30 were added within 12.0 Å from the modeled system, to fill a rectangular box.All molecular mechanics/molecular dynamics calculations were performed using the AMBER12 software.23 To perform the classical molecular dynamics (cMD) simulations, we started by performing an energy minimization of the water molecules, hydrogen bonds and finally of the overall system. The SHAKE algorithm was used during all MD simulations, to constrain bonds involving hydrogen atoms. A progressive heating was performed from 0 to 310 K, in 18 ps, followed by another 32 ps at 310 K, in the NVT ensemble. The cMD production was performed for 20 ns, within a constant pressure and temperature (NPT) ensemble, at 310 K and 1 bar.
QM/MM model
We built our QM/MM model from the minimized structure at the FF99SBildn level of theory, including the whole hKR domain and all water molecules within 6 Å of the catalytic residues proposed in the literature, and the NADPH, KAC and PNS molecules (a total of 3978 atoms, with an overall charge of −3 and a singlet spin multiplicity). In the QM/MM methodology, it is necessary to define the MM and DFT layers. The DFT layer comprehended 82 atoms; it included the side chains of Ser2021, Tyr2034, Asn2038 and Lys1995 residues, the nicotinamide and ribose rings of NADPH, KAC and the methylthio-moiety of PNS (see the right-hand side of Fig. 2). In the MM layer we included the rest of the protein, water molecules and the remaining of NADPH and PNS (Fig. 2). Furthermore, we used three different hydrogen-bond networks in the QM layer to study the catalytic mechanism of hKR. These conformations were prepared upon hydrogen re-orientations in polar residues of the active site, to explore the effects of different hydrogen bond networks in the reduction of KAC (Fig. S3, ESI†). We also took into account water molecules within the active site of this domain. None of these water molecules was in the X-ray structure to start with. Nevertheless, we studied the influence of the one water molecule that is nearby the Lys1995 by including it in the QM layer. We have tested the reduction of KAC by NADPH, and observed that it did not change the most favorable reaction pathway (see Table S1, ESI†). | ||
| Fig. 2 (left) Cartoon representation of the hKR domain, water molecules (spheres), NADPH, PNS and KAC (sticks representations). (right) QM layer with the catalytic residues, nicotinamide and ribose ring of the NADPH moiety, PNS moiety and KAC substrate (sticks representations). The black arrow indicates the link-atoms' positions. | ||
QM/MM calculations and reaction coordinates
The QM/MM calculations were performed using the electrostatic embedding scheme of the ONIOM method,31,32 available in Gaussian 09.33 For the DFT layer, the truncated bonds in the QM frontier were completed by the link-atom method (Fig. 2),23,34 using hydrogen atoms.All optimizations and linear transit scans were performed at the B3LYP/6-31G(d):FF99SB level of theory.35–37
A vibrational analysis was carried out for each minimized structure to confirm the absence of imaginary frequencies in the reactant, intermediate and product states, thus confirming that they are real minima. On the other hand, to characterize transition states (TSs), we looked for one imaginary frequency in the DFT layer, which represents a minimum in energy with respect to all coordinates of the system except that of the reaction coordinate. The values of zero-point-energy and thermal corrections were computed to estimate Gibbs energy. In order to improve the accuracy of the calculations, we performed single-point energy calculations on the previously optimized stationary states at the B3LYP/6-311+G(2d,2p):FF99SB level, and also the DFT-D3 dispersion corrections were introduced for the reaction.38,39 The relevance of the dispersion correction energy term for stationary states is more significant in the first transition state (decrease 2.10 kcal mol−1). For the remaining stationary states the energies varied around 0.20 kcal mol−1. In the ESI† (Table S2) we compare the PESs obtained with B3LYP, M06-2X, ωB97XD, MPW1K and MPWB1K, to discuss the effect of different density functionals in the characterization of the stationary points of the studied catalytic path. The energy barriers provided by the five density functionals compared with each other showed variations within ∼2.0 kcal mol−1. The accurate reaction path was also studied for each transition state, by following the intrinsic reaction coordinate (IRC)40 of the reaction. The Hirshfeld41–43 atomic charges were calculated to discuss the charge rearrangement along the reaction path.
3. Results and discussion
In this section, we present and discuss the results from the QM/MM calculations performed to elucidate the catalytic mechanism of the hKR. The first section presents the conformations used to explore the effects of different hydrogen bond networks in the reaction mechanism of KAC by hKR. The second section presents the atomic charge distribution and the change in the bond lengths in the QM region in the reaction path. The energy profiles for the reaction are presented in the last section.3.1 Hydrogen-bonding networks in the DFT layer
In Fig. 3 and Fig. S3 (ESI†), we show the three different conformations for which we have studied the PES of the catalytic mechanism by hKR. | ||
| Fig. 3 Hydrogen bond networks from the Tyr2034 and the O2′-ribose in the three different conformations used for the DFT layer. A corresponds to Conf-1, B to Conf-2 and C to Conf-3. | ||
Conf-1 exhibits a hydrogen bond between the Tyr2034-hydroxyl and the O2′ atom of the ribose ring (1.8 Å).44 We observe that during the reduction of the Cβ of KAC, the Cβ–alkoxide is protonated by the nearby Ser2021 (Fig. 2). This event exhibits an electronic energy barrier of 28.0 kcal mol−1 (with a reaction electronic energy of 27.2 kcal mol−1). In Conf-2, the hydrogen of the Tyr2034 is oriented towards the Cβ–carbonyl of KAC, favoring hydrogen bond interactions. The reduction of KAC is accompanied by the protonation of the Cβ–alkoxide by Tyr2034, exhibiting an electronic energy barrier of 16.6 kcal mol−1 and an electronic energy of reaction of 3.2 kcal mol−1. These energy values are much more favorable for the reaction to occur; however tyrosinate does not exhibit interactions that can either stabilize it or restore its protonation state.
Finally, the PES for Conf-3 exhibits two subsequent reaction events that corresponded to: step (i) the nucleophilic attack of the Cβ by a hydride of the NADPH cofactor, quickly followed by the deprotonation of the nearby Tyr2034 by the alkoxide group in the Cβ; and step (ii) the deprotonation of Lys1995, via the ribose of NADP+, by Tyr2034. The corresponding energy barriers are 14.8 and 4.4 kcal mol−1, respectively, and the reaction energy is −12.1 kcal mol−1. Conf-3 was used as a relative minimum electronic energy to compare the PESs for the three models (Fig. 4).
 | ||
| Fig. 4 Energy comparison between three conformations used: Conf-1 (green bar), Conf-2 (blue dark bar) and Conf-3 (blue). The dashed line shows the energy values relative to Conf-3. | ||
The results that we will discuss hereinafter will refer to Conf-3 as our definite DFT layer (see Fig. 5b and the full_model.pdb file in the ESI†).
 | ||
| Fig. 5 (a) Interatomic distances that were followed along the QM/MM calculations. (b) Optimized DFT layer for the reactant state obtained with the B3LYP/6-31G(d):FF99SB level. | ||
3.2 Reaction coordinates and charge distribution for the two concerted stages in the catalytic mechanism
In Fig. 5a (Fig. S4, ESI†), we present the bond lengths that vary the most during the catalysis by hKR. We highlight those that concern the two concerted steps to describe the reaction path: the breaking Cre–H bond in the nicotinamide moiety of the NADPH (r6) and the Cβ–H being formed in KAC (Cβ) (r1), and the breaking N–H bond in Lys1995 (r10) and the corresponding O–H bond formation at C2 of the ribose ring in NADP+ (r5).| React | TS1 | I | TS2 | P | |
|---|---|---|---|---|---|
| r 1 | 2.77 | 1.32 | 1.10 | 1.10 | 1.10 |
| r 2 | 1.72 | 1.44 | 1.00 | 0.98 | 0.98 |
| r 3 | 1.91 | 1.67 | 1.76 | 1.80 | 1.82 |
| r 4 | 1.82 | 1.72 | 1.53 | 1.02 | 0.99 |
| r 5 | 2.32 | 2.19 | 2.11 | 1.35 | 1.00 |
| r 6 | 1.10 | 1.36 | 2.64 | 2.71 | 2.72 |
| r 7 | 0.99 | 1.06 | 1.73 | 1.95 | 1.92 |
| r 8 | 0.97 | 0.99 | 0.98 | 0.98 | 0.98 |
| r 9 | 0.98 | 0.99 | 1.03 | 1.62 | 1.78 |
| r 10 | 1.05 | 1.05 | 1.04 | 1.21 | 1.93 |
 | ||
| Fig. 6 Optimized DFT layer for the TS1 and I structures, for the reduction reaction. The distances are indicated in Å. | ||
From TS1 to I, Ser2021 forms a longer hydrogen bond with the Cβ–oxygen (from 1.67 Å to 1.76 Å) (Table 1 and Fig. 6). Moreover, the change in r2 from 1.44 Å to 1.00 Å also indicates that the Tyr2034-hydroxyl is being deprotonated by the Cβ–oxygen of KAC to form the hydroxyacyl (HAC) product. As observed from the analysis of the TS1, the deprotonation of Tyr2034 is concerted, but asynchronous, with the nucleophilic attack of the hydride from NADPH generating the HAC product. In fact, Table 1 confirms that r2 (1.72 Å; 1.00 Å) and r7 (0.99 Å; 1.73 Å) in the I state are symmetric to those of the reactant state. Moreover, NBO bond order calculations in the TS1 have confirmed that the hydrogen of Tyr2034 is still covalently linked to the hydroxyl group, see Table S3 in the ESI.† Furthermore, our study also supports that of Xie et al. 2016, which has demonstrated that Ser and Tyr are fundamental as catalytic residues.49 In the I state, KAC is reduced to HAC and the Gibbs energy barrier observed for this reaction is 11.7 kcal mol−1. The reaction pathway followed from this transition state by IRC calculations shows that the stationary state and the energy barrier are very similar to those obtained in the study of the PESs (see Fig. S6 in the ESI†). We also observe that r4 shortens in about 0.29 Å, with Tyr2034 significantly approaching the C2-hydroxyl of the ribose ring (r4 = 1.53 Å), and thus favouring the deprotonation of the C2-hydroxyl. At the same time, the atomic charge of the oxygen atom in the C2–ribose ring (Table 2) becomes more negative from TS1 to I.
In this process, the C3–hydroxyl of the ribose ring rotates from 135.5° to 82.5° (C2–C3–O–H in Fig. 7), reorienting its hydroxyl group towards the nitrogen atom of Lys1995 (the distance decreases from 2.83 Å to 2.13 Å, see Table S4 in ESI†). In the Asn2038, the atomic charge remains constant along the reaction path, and only in the last step there is a slight decrease, which probably indicates that it can stabilize the Lys1995.
 | ||
| Fig. 7 Optimized DFT layer for TS2 and P structures. The distances are indicated in Å and the dihedral angle represents the rotation of the C3-hydroxyl around the C2–C3 axis. | ||
3.3 Energy profiles for the complete reaction of hKR
We have calculated the energy barriers of the catalytic mechanism of hKR with different density functionals (B3LYP, M06-2X, ωB97XD, MPW1K and MPWB1K) (Table S2, ESI†). The functionals B3LYP and M06-2X provided energy barriers that are slightly lower than those calculated from other density functionals. Since the energy differences between B3LYP and M06-2X are within an error of ∼2 kcal mol−1, we will discuss our results solely for the B3LYP/6-311+G(2d,2p):FF99SB level of theory, for the sake of consistency with our previous calculations.The Gibbs energy profile for the overall reaction is summarized in Fig. 8. For the first step, the Gibbs activation energy (ΔG‡1) is 11.7 kcal mol−1. Subsequent to the TS1, a stable I is generated exhibiting a Gibbs reaction energy of −3.2 kcal mol−1. At this stage, the Cβ has been reduced by the hydrogen from the NADP+ cofactor, and the Tyr2034 is instantly deprotonated by the formed Cβ–alkoxide. Our results suggest that this is definitely the rate-limiting step of the reaction. For the second stage (structures III and IV of Fig. 7), the energy profile suggests that there is no Gibbs activation energy for this process. This suggests that the I state, which we observe in the first step of the reaction, may in fact be a transient state that is not observed under physiological conditions. The overall catalytic mechanism is an exergonic reaction with a Gibbs energy of −10.6 kcal mol−1. Fig. 8 also shows that energies have the same behavior in the first step with minor variations between the Gibbs and enthalpy energies. However, the barrier registered between the protonation of Tyr2034 and that of the O2′-ribose protonation vanishes (from 1.7 kcal mol−1 to −6.0 kcal mol−1) when the basis set is expanded (−1.0 kcal mol−1), thermal corrections are added (−1.1 kcal mol−1) and dispersion corrections are included (−1.0 kcal mol−1). In the ESI† (Fig. S5), we show the comparison between electronic energies determined with the 6-31G(d) and the 6-311+G(2d,2p) basis sets.
 | ||
| Fig. 8 Thermodynamic profile for the two stages involved in the catalytic reduction of KAC to HAC by hKR: Gibbs energy determined at the ONIOM(B3LYP/6-311+G(2d,2p):FF99SB//B3LYP/6-31G(d):FF99SB) level (blue), and enthalpy determined at the ONIOM(B3LYP/6-311+G(2d,2p): FF99SB) level (orange). | ||
4. Conclusion
The results provide a good description of the different stages of the catalytic mechanism of this hFAS domain: of the three conformations studied, Conf-2 and Conf-3 show the rate-limiting step that is the hydride transfer from the NADPH cofactor to the β-carbon of the substrate. However, Conf-2 suggests that the mechanism is not probable because the fact that Tyr2034 remains as an alkoxide leads to an endothermic reaction. The rate-limiting step occurs with a Gibbs activation energy of 11.7 kcal mol−1; after the TS, the β-alkoxide moiety of the substrate deprotonates a nearby Tyr2034, with its protonation restored by a nearby Lys1995, via the ribose's hydroxyl in NADP+. The overall catalytic mechanism exhibits a Gibbs energy of −10.6 kcal mol−1. Moreover, energy estimates at the B3LYP/6-311+G(2d,2p):FF99SB level, with inclusion of thermal corrections and DFT-D3 dispersion, seem to lead to a suitable description of the catalysis by hKR. In addition, we find that the oxyanion hole contributed for the TS stabilization through the formation of hydrogen bonds that are fundamental to perform the catalytic mechanism with a feasible Gibbs activation energy.The knowledge of the catalytic mechanism of the hKR domain provides useful information for future research related to the design of new therapeutic targets with anticancer activity.
Acknowledgements
This work received financial support from the following institutions: European Union (FEDER funds POCI/01/0145/FEDER/007728) and National Funds (FCT/MEC, Fundação para a Ciência e Tecnologia and Ministério da Educação e Ciência) under the Partnership Agreement PT2020 UID/MULTI/04378/2013; NORTE-01-0145-FEDER-000024, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF); Fundação para a Ciência e a Tecnologia (FCT) through project EXCL-II/QEQ-COM/0394/2012. F.E.M acknowledges the Comisión Nacional de Investigación Científica y Tecnológica - Chile (CONICYT) for a PhD scholarship.References
- P. R. Pandey, W. Liu, F. Xing, K. Fukuda and K. Watabe, Recent Pat. Anti-Cancer Drug Discovery, 2012, 7, 185–197 Search PubMed.
- S. Smith and S.-C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 Search PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 Search PubMed.
- Q. Jin, L. X. Yuan, D. Boulbes, J. M. Baek, Y. N. Wang, D. Gomez-Cabello, D. H. Hawke, S. C. Yeung, M. H. Lee, G. N. Hortobagyi, M. C. Hung and F. J. Esteva, Breast Cancer Res., 2010, 12 Search PubMed.
- S. M. Ueda, K. L. Yap, B. Davidson, Y. Tian, V. Murthy, T.-L. Wang, K. Visvanathan, F. P. Kuhajda, R. E. Bristow, H. Zhang and I.-M. Shih, J. Oncol., 2010, 2010, 12 Search PubMed.
- H. Orita, J. Coulter, C. Lemmon, E. Tully, A. Vadlamudi, S. M. Medghalchi, F. P. Kuhajda and E. Gabrielson, Clin. Cancer Res., 2007, 13, 7139–7145 Search PubMed.
- T. M. Loftus, D. E. Jaworsky, G. L. Frehywot, C. A. Townsend, G. V. Ronnett, M. D. Lane and F. P. Kuhajda, Science, 2000, 288, 2379–2381 Search PubMed.
- B. Q. Liu, Y. Q. Wang, K. L. Fillgrove and V. E. Anderson, Cancer Chemother. Pharmacol., 2002, 49, 187–193 Search PubMed.
- W. X. Tian, Curr. Med. Chem., 2006, 13, 967–977 Search PubMed.
- M. A. Hardwicke, A. R. Rendina, S. P. Williams, M. L. Moore, L. Wang, J. A. Krueger, R. N. Plant, R. D. Totoritis, G. Zhang, J. Briand, W. A. Burkhart, K. K. Brown and C. A. Parrish, Nat. Chem. Biol., 2014, 10, 774–779 Search PubMed.
- M. J. Vázquez, W. Leavens, R. Liu, B. Rodriguez, M. Read, S. Richards, D. Winegar and J. M. Dominguez, FEBS J., 2008, 275, 1556–1567 Search PubMed.
- I. Fujii, N. Yoshida, S. Shimomaki, H. Oikawa and Y. Ebizuka, Chem. Biol., 2005, 12, 1301–1309 Search PubMed.
- N. K. Williams, I. S. Lucet, S. P. Klinken, E. Ingley and J. Rossjohn, J. Biol. Chem., 2009, 284, 284–291 Search PubMed.
- D. Dutta, S. Bhattacharyya, A. Roychowdhury, R. Biswas and A. K. Das, Biochem. J., 2013, 450, 127–139 Search PubMed.
- A. C. Price, Y. M. Zhang, C. O. Rock and S. W. White, Structure, 2004, 12, 417–428 Search PubMed.
- A. T. Keatinge-Clay and R. M. Stroud, Structure, 2006, 14, 737–748 Search PubMed.
- M. W. van der Kamp and A. J. Mulholland, Nat. Prod. Rep., 2008, 25, 1001–1014 Search PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, Chem. Soc. Rev., 2012, 41, 3025–3038 Search PubMed.
- G. Naray-Szabo, J. Olah and B. Kramos, Biomolecules, 2013, 3, 662–702 Search PubMed.
- R. P. P. Neves, P. A. Fernandes and M. J. Ramos, ACS Catal., 2016, 6, 357–368 Search PubMed.
- A. J. M. Ribeiro, L. Yang, M. J. Ramos, P. A. Fernandes, Z.-X. Liang and H. Hirao, ACS Catal., 2015, 5, 3740–3751 Search PubMed.
- A. R. Calixto, N. F. Bras, P. A. Fernandes and M. J. Ramos, ACS Catal., 2014, 4, 3869–3876 Search PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER12, University of California, San Francisco, 2012.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 Search PubMed.
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 Search PubMed.
- U. Ryde, Proteins: Struct., Funct., Genet., 1995, 21, 40–56 Search PubMed.
- U. Ryde, Protein Sci., 1995, 4, 1124–1132 Search PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 Search PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 Search PubMed.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- L. W. Chung, H. Hirao, X. Li and K. Morokuma, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 327–350 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., Wallingford CT, 2009.
- M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700–733 Search PubMed.
- R. Ditchfie, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 Search PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 Search PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 Search PubMed.
- J. P. Ritchie, J. Am. Chem. Soc., 1985, 107, 1829–1837 Search PubMed.
- J. P. Ritchie and S. M. Bachrach, J. Comput. Chem., 1987, 8, 499–509 Search PubMed.
- M. Ahmed, C. Jelsch, B. Guillot, C. Lecomte and S. Domagała, Cryst. Growth Des., 2013, 13, 315–325 Search PubMed.
- M. Fuxreiter and A. Warshel, J. Am. Chem. Soc., 1998, 120, 183–194 Search PubMed.
- A. Warshel, P. K. Sharma, Z. T. Chu and J. Aqvist, Biochemistry, 2007, 46, 1466–1476 Search PubMed.
- Y. K. Zhang, J. Kua and J. A. McCammon, J. Am. Chem. Soc., 2002, 124, 10572–10577 Search PubMed.
- A. Warshel, G. Narayszabo, F. Sussman and J. K. Hwang, Biochemistry, 1989, 28, 3629–3637 Search PubMed.
- X. Xie, A. Garg, A. T. Keatinge-Clay, C. Khosla and D. E. Cane, Biochemistry, 2016, 55, 1179–1186 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp07014k |
| This journal is © the Owner Societies 2017 |