
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
VUV photochemistry of the H2O⋯CO complex in noble-gas matrices: formation of the OH⋯CO complex and the HOCO radical
Sergey V.
Ryazantsev
ab,
Luís
Duarte
 b,
Vladimir I.
Feldman
a and
Leonid
Khriachtchev
*b
b,
Vladimir I.
Feldman
a and
Leonid
Khriachtchev
*b
aDepartment of Chemistry, Lomonosov Moscow State University, Moscow 119991, Russia
bDepartment of Chemistry, University of Helsinki, P. O. Box 55, FI-00014 Helsinki, Finland. E-mail: leonid.khriachtchev@helsinki.fi
First published on 17th November 2016
Abstract
Vacuum ultraviolet (VUV, 130–170 nm) photochemistry of the H2O⋯CO complex is studied by matrix-isolation infrared spectroscopy. The H2O⋯CO complexes in Ne, Ar, Kr, and Xe matrices are generated by ultraviolet (UV, 193 and 250 nm) photolysis of formic acid (HCOOH). VUV photolysis of the H2O⋯CO complexes is found to lead to the formation of the OH⋯CO radical–molecule complexes and trans-HOCO radicals. It is shown that the matrix material, local matrix morphology, and possibly the H2O⋯CO complex geometry strongly affect the VUV photolysis pathways. The intrinsic reactivity of the matrix-isolated OH⋯CO complex resulting in the formation of trans-HOCO is directly demonstrated for the first time. This reaction occurs in Ar, Kr, and Xe matrices upon annealing above 25 K and may proceed over the barrier. The case of a Ne matrix is very special because the formation of trans-HOCO from the OH⋯CO complex is observed even at the lowest experimental temperature (4.5 K), which is in sharp contrast to the other matrices. It follows that quantum tunneling is probably involved in this process in the Ne matrix at such a low temperature. Infrared light also promotes this reaction in the Ne matrix at 4.5 K, which is not the case in the other matrices. The last findings show the effect of the environment on the tunneling and infrared-induced rates of this fundamental chemical reaction.
Introduction
Non-covalent interactions play an important role in a great variety of chemical processes. Light-induced chemical transformations in weakly-bound intermolecular complexes represent an intriguing field of supramolecular photochemistry with a remarkable impact on life science, nanotechnology, and chemical synthesis.1,2 The matrix-isolation technique has been extensively used for the investigation of non-covalent interactions, particularly, involving radicals and other unstable species.3–13 Recent studies have demonstrated an effective approach for the preparation of matrix-isolated complexes in high concentrations using the photolysis of appropriate molecular precursors (see ref. 9 for a review). For example, UV photolysis of formic acid (HCOOH, FA) leads to the formation of the H2O⋯CO complexes.14 Furthermore, one can use matrix isolation to investigate the photochemistry (or, in a wider context, radiation-driven chemistry) of intermolecular complexes. The photoinduced transformations of some complexes have been studied previously under matrix-isolation conditions.15–21 In general, such reactions can lead to the assembly of new species or “cold synthesis” under the conditions of frozen molecular mobility, which may be particularly significant to model and understand the processes in interstellar and cometary ices.Water and carbon monoxide are the two most abundant molecules in interstellar ices,22 and these species also play an important role in the atmospheric chemistry. The intermolecular complex H2O⋯CO has been studied previously both theoretically and experimentally.14,23–34 The reaction of this complex with an H atom produces the radical–molecule complex HCO⋯H2O.35 Another related radical–molecule complex, OH⋯CO, is expected to be the key primary product of the photoinduced transformation of H2O⋯CO. The OH⋯CO complex has been extensively examined by ab initio calculations as an intermediate in the OH + CO → CO2 + H reaction, which is significant in combustion and atmospheric processes.36–43 Lester et al. reported an experimental detection of the OH⋯CO complex in the gas phase having the OH stretching overtone at 6941.8 cm−1 (shifted by −29.6 cm−1 with respect to the OH monomer).37 The electronic excitation spectrum and the intermolecular vibrational frequencies of this complex have also been characterized experimentally.44 Very recently, Brice et al. have reported the OH fundamental frequency of the OH⋯CO complex in He droplets at 3551 cm−1 (shifted by −17 cm−1 with respect to the OH monomer).45 Nevertheless, full experimental characterization of the fundamental IR absorptions of the OH⋯CO complex is still lacking.†
Another species that is relevant to this system is the carboxyl radical (HOCO) that is separated from the OH⋯CO complex by a low energy barrier (a few hundred cm−1).37–40,42 HOCO is an important intermediate in combustion and atmospheric chemistry and it is also assumed to be involved in the formation of prebiotic molecules in the interstellar medium.46–50 This radical has two conformers, the trans form being lower in energy than the cis form by about 500 cm−1. It has been shown that trans-HOCO can be produced in low-temperature matrices upon photolysis or radiolysis of some CO- or CO2-containing intermolecular complexes.19a,21,51 This species was also detected in the VUV- and electron-irradiated H2O/CO solid mixtures.52 The rotational spectra of the two conformers of HOCO and their deuterated analogs in the gas phase were obtained by FTMW spectroscopy and the millimeter-wave double resonance technique.53
The aim of the present work is to study vacuum ultraviolet (VUV) photochemistry of the H2O⋯CO complex under matrix-isolation conditions and identify the OH⋯CO radical–molecule complex. This study is also related to the photochemical preparation of the carboxyl radical HOCO. UV photolysis of matrix-isolated FA is used to produce a sufficient concentration of the H2O⋯CO complexes in noble-gas matrices.14
Experimental details
The experiments were carried out using FA/Ng mixtures (Ng = Ne, Ar, Kr, and Xe) with typical ratios of 1/1000–1/2000. FA (Merck, >98%) was degassed by several freeze–pump–thaw cycles. Ne (99.999%), Ar (99.9999%), Kr (99.999%), and Xe (99.999%) were used as purchased (Linde). The gas FA/Ng mixtures were made in a glass bulb by using standard manometric procedures. Since FA is easily adsorbed on glass surfaces, the bulb was passivated with FA vapors by fill–keep–evacuate cycles prior to the mixture preparation. The FA/Ng matrices were deposited onto a CsI substrate held at 4, 15, 20, and 30 K for Ne, Ar, Kr, and Xe matrices, respectively, in a closed-cycle helium cryostat (RDK-408D2, SHI). The FA/Ng matrices (Ng = Ne, Ar, and Kr) were photolyzed with an excimer laser (MSX-250, MPB) operating at 193 nm (∼10 mJ cm−2) to produce the H2O⋯CO complexes. Since 193 nm photons decompose water in a Xe matrix,54 in this case we used 250 nm radiation (∼5 mJ cm−2) of an optical parametric oscillator (OPO, Sunlite, Continuum). After UV photodecomposition of FA, the matrix was exposed to VUV light (130–170 nm) of a Kr lamp (Opthos). During VUV photolysis, the space between the lamp and the cryostat optical window (MgF2) was flushed with argon. Reference experiments were also made with FA/Ng matrices initially photolyzed with VUV radiation of a Kr lamp, i.e. without preliminary UV photolysis. Selective excitation in the IR region was performed by narrowband light of an OPO (LaserVision). The IR absorption spectra in the 4000–600 cm−1 range were measured at 4.5 K using an FTIR spectrometer (Vertex 80, Bruker) with 1 cm−1 resolution and 500 scans.Results
In these experiments, the H2O⋯CO complex was generated in noble-gas matrices by photodecomposition of FA. The deposited FA/Ng matrices (Ng = Ne, Ar, Kr, and Xe) contained mostly FA monomers (in the more stable trans form)14 with small amounts of FA dimers (tt1 and tt2)55,56 and trans-FA⋯H2O complexes.56,57 The obtained spectra of trans-FA are in full agreement with the previous data. UV irradiation of the deposited matrices (∼5000 pulses at 193 nm for Ng = Ne, Ar, and Kr and ∼20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pulses at 250 nm for Ng = Xe) resulted in efficient decomposition of FA (typically >70%). The difference IR spectra showing the result of UV photolysis in different matrices are presented in Fig. 1. The increase of the absorbance of H2O⋯CO bands correlates with the FA photodecomposition. The fundamental frequencies of the H2O⋯CO complex isolated in noble gas matrices are given in Table 1. The spectra of the H2O⋯CO complex observed in our experiments in Ar, Kr, and Xe matrices are in good agreement with previous works,14,27c however, some additional weak features of the perturbed H2O fundamentals are also detected. These weaker bands correlate with the other H2O⋯CO absorption bands. We suppose that the absence of these features in the earlier work14 may be due to higher temperatures in their experiments (15 K). The IR absorptions of the H2O⋯CO complex in the Ne matrix have been previously reported only for ν2(H2O) and ν(CO).58 In our work, we also observed the ν1 and ν3 bands of water in this complex.
000 pulses at 250 nm for Ng = Xe) resulted in efficient decomposition of FA (typically >70%). The difference IR spectra showing the result of UV photolysis in different matrices are presented in Fig. 1. The increase of the absorbance of H2O⋯CO bands correlates with the FA photodecomposition. The fundamental frequencies of the H2O⋯CO complex isolated in noble gas matrices are given in Table 1. The spectra of the H2O⋯CO complex observed in our experiments in Ar, Kr, and Xe matrices are in good agreement with previous works,14,27c however, some additional weak features of the perturbed H2O fundamentals are also detected. These weaker bands correlate with the other H2O⋯CO absorption bands. We suppose that the absence of these features in the earlier work14 may be due to higher temperatures in their experiments (15 K). The IR absorptions of the H2O⋯CO complex in the Ne matrix have been previously reported only for ν2(H2O) and ν(CO).58 In our work, we also observed the ν1 and ν3 bands of water in this complex.
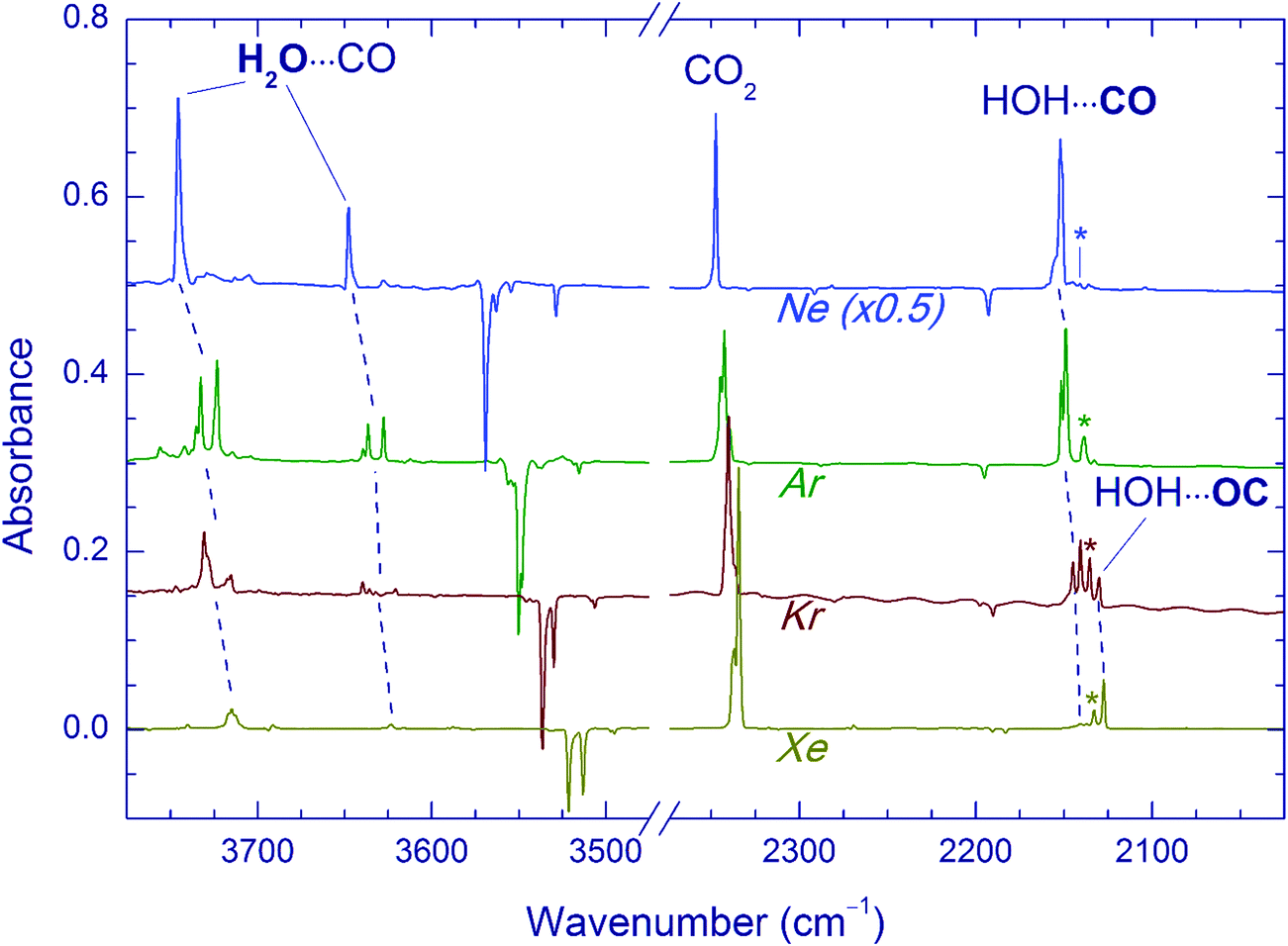 | ||
| Fig. 1 FTIR spectra of the H2O⋯CO complex in different matrices. All traces are difference spectra of FA/Ng matrices (Ng = Ne, Ar, Kr, and Xe) showing the result of UV photolysis of FA (193 nm in Ne, Ar, and Kr matrices; 250 nm in the Xe matrix). The negative bands correspond to decomposed FA. The bands marked with asterisks are due to the CO monomer. | ||
| Mode | Ne | Ar | Kr | Xe |
|---|---|---|---|---|
| a New assignments are in bold; absorptions attributed to unrelaxed matrix sites are in italics; br – broad. b Corresponds to the HOH⋯CO structure. c Corresponds to the HOH⋯OC structure. d Weak feature, virtually unchanged upon annealing. Assignment to the relaxed site is made according to the data from ref. 14a. | ||||
| ν 3(H2O) | 3745.6 | 3735.3 | 3730.8 | 3716.5 |
| 3732.6 | 3728.8 | 3715.0 | ||
| 3723.3 | 3717.2 | 3712.9 | ||
| 3715.3 | ||||
| ν 1(H2O) | 3647.9 | 3639.4 | 3639.6 | 3623 br |
| 3636.5 | 3635.8 | |||
| 3627.7 | 3632.1 | |||
| 3620.7 | ||||
| ν(CO) | 2151.8b | 2151.7 | 2145.0b | 2141.0b |
| 2149.0b | 2140.7 | 2137.2 | ||
| 2130.2 | 2127.5 | |||
| ν 2(H2O) | 1599.5 | 1600.3 | 1596.5 | 1591.5d |
| 1595.4 | 1592.9 | 1588.3 | ||
| 1590.7 | 1589.9 | 1586.6 | ||
The H2O⋯CO complex can have two different structures, HOH⋯CO and HOH⋯OC, in which a water hydrogen interacts with different atoms of carbon monoxide (C and O, respectively). These two structures can be distinguished spectroscopically by the CO stretching shift with respect to CO monomer. The computationally more stable HOH⋯CO complex has a blue shift of this mode, whereas the HOH⋯OC complex shows a red shift.14,27 In full agreement with the previous results,14 we observed a matrix-specific ratio of these complexes produced by UV photolysis of FA. In fact, only the HOH⋯CO structure is observed in Ne and Ar matrices; both HOH⋯CO and HOH⋯OC structures are found in a Kr matrix, and the HOH⋯OC form dominates in a Xe matrix. It is worth noting that in Kr and Xe matrices, the bands of water in the H2O⋯CO complex are quite structured, which is most probably due to several matrix sites with overlapping absorptions. This fact prevents us from definite assignment of these bands to the two complex structures (HOH⋯CO and HOH⋯OC). In addition to the H2O⋯CO complex, CO2 (possibly complexed with H2) and monomeric CO are also produced by UV photolysis of FA. The relative yield of CO (both complexed and monomeric) with respect to CO2 decreases with the increase of the matrix dielectric constant, which is discussed elsewhere.14
The effect of Kr-lamp radiation (130–170 nm) on matrices containing the H2O⋯CO complexes is shown in Fig. 2. This irradiation resulted in gradual bleaching of the H2O⋯CO bands, an increase of the band of the CO monomer (2140.9, 2138.5, 2135.5 and 2133.0 cm−1 in Ne, Ar, Kr, and Xe, respectively)21,23,51,56 and a noticeable growth of the CO2 bands (668.0, 2347.4, 3611.0, and 3713.2 cm−1 in Ne; 662.5, 2344.8/2342.7 with a shoulder at 2339.7, and 3706.6/3703.8 with a shoulder at 3699.8 cm−1 in Ar; 660.5, 2340.5 with a shoulder at 2336.5, 3595.6, and 3699.6 cm−1 in Kr; 659.7, 2334.7, 3587.9, and 3691.6 cm−1 in Xe).51,56 The minor isotopologues of CO2 were also observed (13CO2: 2281.8, 2274.2/2277.2 with a shoulder at 2279.3, 2275.1, and 2269.5 cm−1 for Ne, Ar, Kr, and Xe respectively; 16O12C18O: 2330.5, 2327.7/2325.6 with a shoulder at 2323.0, 2323.3, and 2317.8 cm−1 for Ne, Ar, Kr, and Xe respectively).51,56trans-HOCO (Table 2), Ng2H+ ions (Ng = Ar, Kr, and Xe),59 and trace amounts of HCO35,51,60 and HCO⋯H2O35,56 were also detected. In addition, a new absorber with bands at 3546.5 and 2159.8 cm−1 in a Ne matrix, at 3526.5 and 2152.5 cm−1 in an Ar matrix, and at 3521.9 and 2147.9 cm−1 in a Kr matrix was observed. These bands are assigned in this work to the OH⋯CO complex as discussed below. In this series of experiments, no candidates for the OH⋯CO absorptions were found after VUV photolysis in the Xe matrix even though the trans-HOCO bands were quite evident. It should be stressed that trans-HOCO and the OH⋯CO complex were not observed during the initial UV photolysis. It is also worth noting that no evidence for the stabilization of the higher energy cis-HOCO conformer has been found in these studies although it was reported previously in N2 and CO matrices as well as in solid H2O–CO mixtures.19a,52 To our knowledge, cis-HOCO has never been observed in noble-gas matrices.
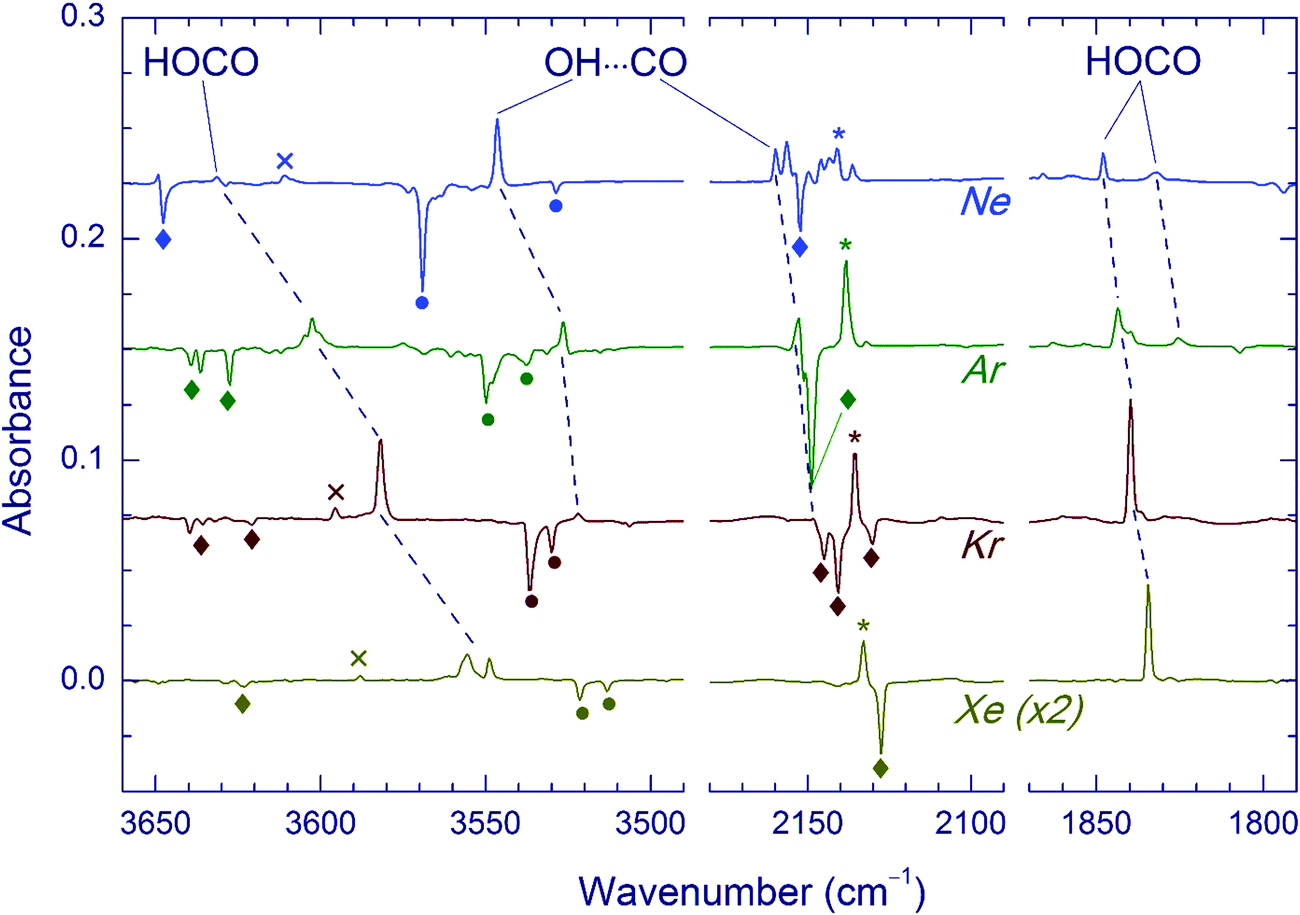 | ||
| Fig. 2 VUV photolysis (130–170 nm) of the H2O⋯CO complex in different matrices. All traces are difference spectra showing the result of VUV photolysis (for ∼70 min) of initially UV-photolyzed FA/Ng matrices (Ng = Ne, Ar, Kr, and Xe). The bands marked with diamonds, asterisks, and circles are due to the H2O⋯CO complex, CO monomer, and residual FA, respectively. The weak features marked with crosses are the (2ν2 + ν3) absorptions of CO2 (this peak in the Ar matrix is masked by the band of trans-HOCO). | ||
| Mode | Ne | Ar | Kr | Xe |
|---|---|---|---|---|
| a Our data are in good agreement with the previous studies;21,51,56,58,61 new assignments are in bold; sh – shoulder. | ||||
| v 1 | 3631.3 | 3604.8 | 3581.9 | 3555.5 |
| 3627.6 | 3602.6 | 3548.9 | ||
| 3601 sh | ||||
| 2v4 | 2083.9 | 2108.7 | 2112.8 | 2107.6 |
| 2081.1 | 2109.2 | 2104.9 | ||
| v 2 | 1847.9 | 1843.5 | 1839.7 | 1834.4 |
| 1832.0 | 1839.8 | 1836.8 | ||
| 1825.4 | ||||
| v 3 | 1210.5 | 1211.1 | 1210.9 | 1209.9 |
| 1208.8 | 1205.9 | 1202.4 | ||
| 1205.6 | 1202 sh | |||
| v 4 | 1052 sh | 1066 sh | 1066 sh | 1062.9 |
| 1050.6 | 1064.5 | 1064.9 | ||
The formation of trans-HOCO and OH⋯CO tends to saturate for substantial consumption of H2O⋯CO, whereas the production of CO2 continues. This behavior can be interpreted as an interplay between primary (production of OH⋯CO and trans-HOCO) and secondary (production of CO2) channels of VUV photolysis of the H2O⋯CO complex. The direct photoproduction of CO2 from H2O⋯CO is also possible.
After the first (UV) photolysis, the matrices contain some remaining amounts of FA. As seen in Fig. 2, FA is partially decomposed by VUV irradiation. In order to estimate the contribution of VUV photolysis of FA to the formation of OH⋯CO and trans-HOCO, we performed reference experiments with initial VUV photolysis of FA in Ar and Kr matrices. Although both OH⋯CO and trans-HOCO were also observed in these experiments, their amounts (normalized to the amount of decomposed FA) were markedly lower as compared to the case of VUV photolysis performed after UV photolysis. Moreover, the H2O⋯CO complex was also produced upon VUV photolysis of FA, and its photodecomposition evidently occurred. Thus, we conclude that both OH⋯CO and trans-HOCO are mainly the products of VUV photolysis of the H2O⋯CO complex. However, as discussed below, the direct photodissociation channel HCOOH → HOCO + H neither can be excluded.
In a separate series of experiments, we annealed the matrices after UV photolysis (prior to VUV photolysis). The annealing temperatures were 25, 35, and 40 K for Ar, Kr, and Xe matrices respectively. Ne matrices were not treated in this way since they are stable only below 12 K. This annealing led to some changes in the matrix sites of the H2O⋯CO complex. An example of the Ar matrix is shown in Fig. 3. Based on the annealing-induced behavior, we distinguish the IR absorptions of the H2O⋯CO complex in unrelaxed (decreasing upon annealing) and relaxed (increasing upon annealing) matrix sites irrespective of the geometry of the complex (see Table 1). The absorptions attributed to the unrelaxed sites are broader and/or more structured compared to the ones of the relaxed sites. It should be noted that the complete conversion to the relaxed matrix site (as in the case of the Ar matrix) was not achieved in the Kr matrix. In the Xe matrix, the weak band at 2141.0 cm−1 slightly increased in intensity, whereas no candidates for the relaxed site were found in the water stretching region. To remind, the yield of the H2O⋯CO complex in the Xe matrix was lower than in the other matrices. The observed effect of annealing on the H2O⋯CO bands is in good agreement with the previous observations.14a We have also found that annealing of the UV-irradiated FA/Ng matrices leads to the formation of HCO (Ar: 1863.4 cm−1; Kr: 2467.1 and 1860.3 cm−1; Xe: 2442.4, 1856.6, and 1076.2 cm−1), HCO⋯H2O (Ar: 1853.8 cm−1; Kr: 2530.5, 1858.8, 1853.3, and 1850.6 cm−1; Xe: 3710.4, 3707.5, 2516.4, 2510.7, 1850.0, 1847.6, 1092.8, and 1090.8 cm−1),35,56 and trace amounts of trans-H2COOH (Ar: 967.3 and 964.9 cm−1; Kr: 964.6 and 962.2 cm−1; Xe: 959.6 and 957.8 cm−1).56,62 In the Xe matrix, relatively intense bands of HXeH (1166.5 and 1181.4 cm−1)63 are observed after annealing. Since the used annealing temperatures activate the mobility of H atoms,51,64 these results clearly indicate that UV photolysis of FA produces considerable amounts of H atoms.
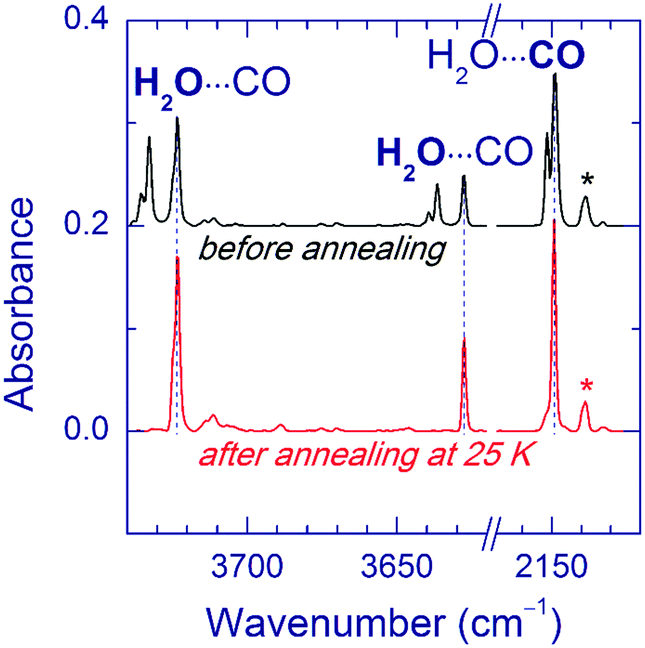 | ||
| Fig. 3 Effect of annealing on matrix sites of the H2O⋯CO complex in an Ar matrix. The upper trace is a FTIR spectrum of a FA/Ar matrix after 193 nm photolysis. The lower trace is a FTIR spectrum of the same matrix after annealing at 25 K for 10 min. The band of monomeric CO is marked by an asterisk. | ||
Annealing of the matrices prior to VUV photolysis significantly affects the branching ratio of the photoproducts. In fact, the ratio of the OH⋯CO and trans-HOCO amounts markedly increased in the annealed matrices. Fig. 4 illustrates this result for the Ar matrix. A similar trend was observed in the Kr matrix. In the Xe matrix annealed after UV photolysis, VUV photolysis produced a weak band at 2145.0 cm−1, which was attributed to the CO absorption of the OH⋯CO complex; however, no band of this complex in the OH stretching region was found in this experiment.
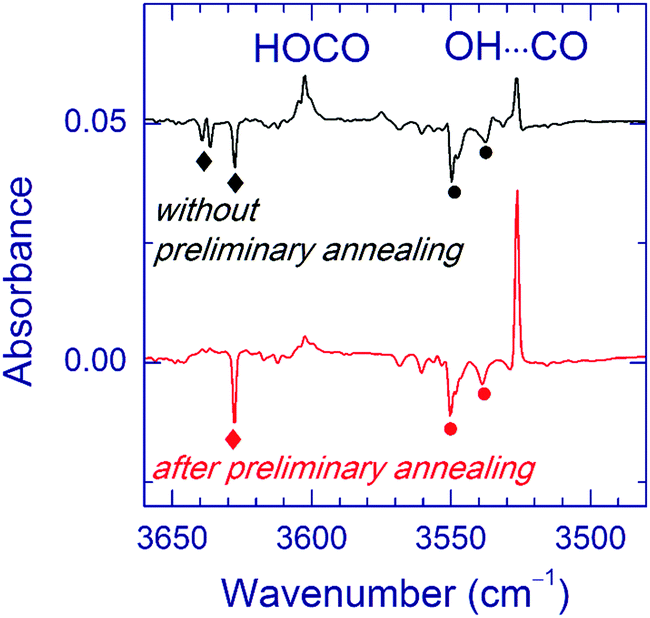 | ||
| Fig. 4 Effect of annealing on the products of VUV photolysis in an Ar matrix. The upper trace is a difference FTIR spectrum showing the result of VUV photolysis (30 min) of an initially UV-photolyzed FA/Ar matrix (without annealing). The lower trace is an analogous spectrum of a FA/Ar matrix annealed at 25 K after UV photolysis (prior to VUV photolysis). The bands marked with diamonds belong to the H2O⋯CO complex, and the ones marked with circles are due to residual FA. | ||
Annealing of the matrices after VUV photolysis results in a decay of the OH⋯CO complex as illustrated for the Ar matrix in Fig. 5a. In Ar and Kr matrices, the OH⋯CO bands almost disappear after annealing at 30–35 K. Fig. 5b shows the effect of annealing on the OH⋯CO decay and the formation of trans-HOCO and HCO⋯H2O in the Ar matrix. First, there is no correlation between the formation of HCO⋯H2O, which is due to the H + CO⋯H2O reaction35 and the OH⋯CO decay. This fact evidences that the main decomposition channel of OH⋯CO is not connected with mobile H atoms. Second, there is a good overall correlation between the decomposition of OH⋯CO and the formation of trans-HOCO, particularly above 25 K, which features the OH⋯CO → trans-HOCO reaction. It should be understood that the strict correlation between these processes is difficult to expect due to a number of additional channels. For example, some of the OH and CO fragments can be shortly separated in the matrix after photolysis and recombine upon annealing. Agglomeration of species upon annealing is another complicating factor. The probable reaction of the OH⋯CO complex with H atoms can also change the temperature dependences. In this respect, we can mention that some increase of the amount of the H2O⋯CO complex is systematically observed after annealing. In the Xe matrix, the amount of OH⋯CO is very small, and it is difficult to study this process in detail. However, this annealing allowed us to detect the OH fundamental of the OH⋯CO complex in the Xe matrix (at 3520.0 cm−1), which was otherwise masked by the absorption of residual FA (see Fig. 6). In Kr and Xe matrices, annealing at 35 and 45 K, respectively, also resulted in a slight decrease of the trans-HOCO bands (see the lower trace in Fig. 6). A similar behavior was observed previously and may indicate the reaction of trans-HOCO with mobile H atoms.56
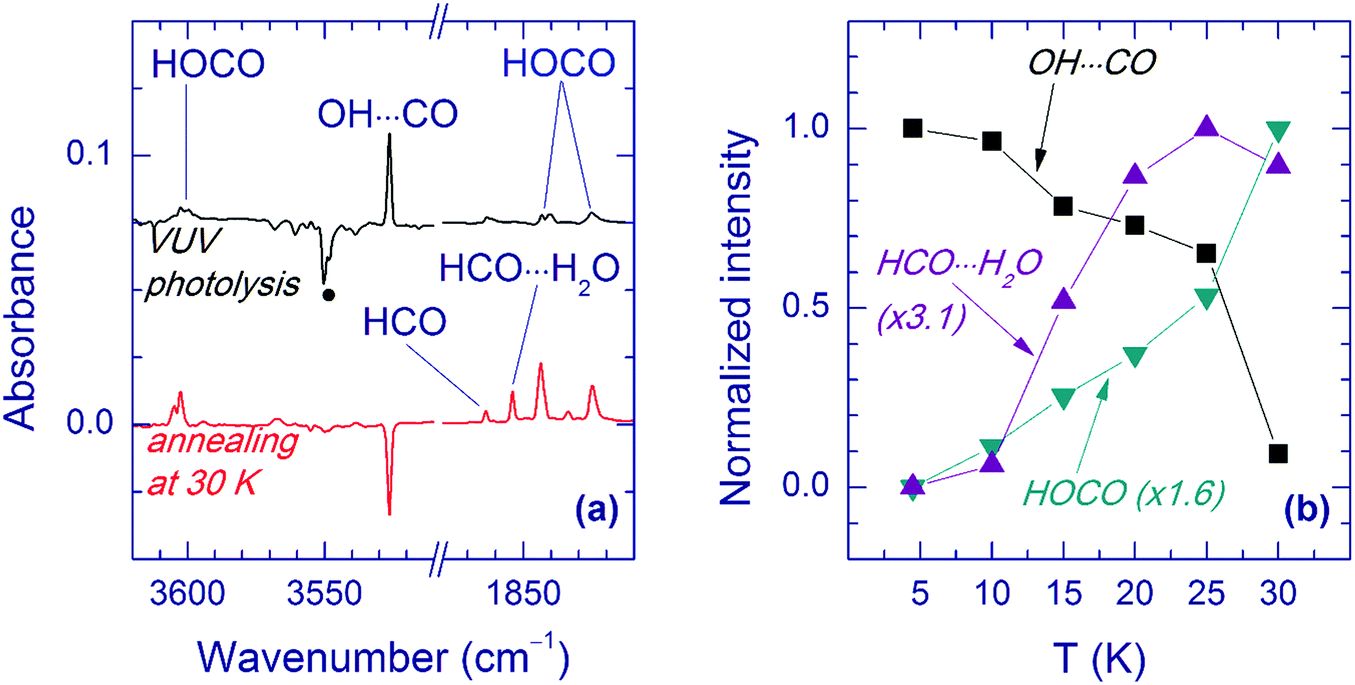 | ||
| Fig. 5 Annealing-induced decay of the OH⋯CO complex in an Ar matrix. (a) The upper trace is a difference FTIR spectrum showing the result of VUV photolysis of a FA/Ar matrix initially UV-photolyzed and then annealed at 25 K. The lower trace is a difference spectrum showing the result of annealing of this matrix at 30 K. The band marked with a circle is due to residual FA. (b) The effect of annealing (5 min at each temperature) on the decay of OH⋯CO and the formation of trans-HOCO and HCO⋯H2O in an Ar matrix. The initial intensities for trans-HOCO and HCO⋯H2O are subtracted. The normalization factors are presented in parentheses. | ||
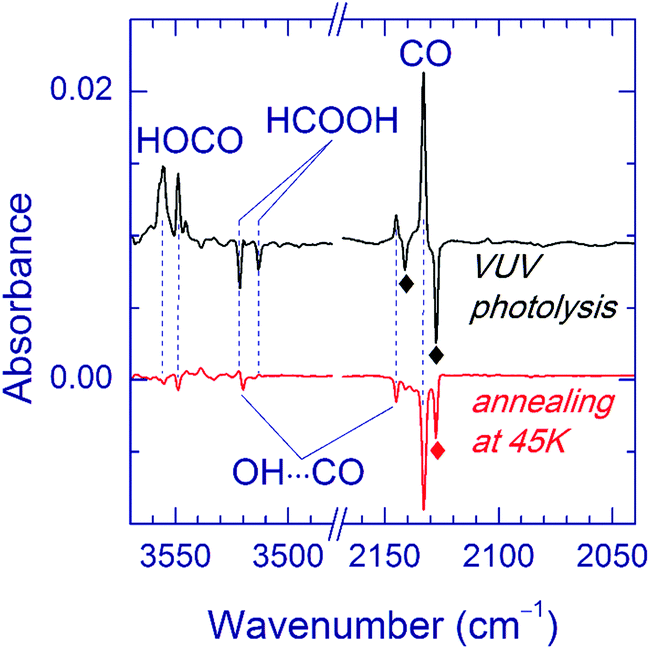 | ||
| Fig. 6 FTIR spectrum of the OH⋯CO complex in a Xe matrix. The upper trace is a difference spectrum showing the result of VUV photolysis of a FA/Xe matrix initially UV-photolyzed and annealed at 40 K. The weak OH band of OH⋯CO is masked by the FA absorptions. The lower trace is a difference spectrum showing the result of annealing of the same matrix at 45 K. Annealing-induced decay of the OH⋯CO complex makes it possible to distinguish the weak OH band. The bands marked with diamonds are due to the H2O⋯CO complex. | ||
As to the case of the Ne matrix, a slow decay (on a timescale of more than 10 h) of the OH⋯CO complex accompanied by the formation of trans-HOCO was observed even in the dark at 4.5 K (the lowest temperature of our experiment) (see Fig. 7). It is worth mentioning that no other products of OH⋯CO decay (e.g. CO2) were found in these experiments, except for small amounts of the H2O⋯CO complex. According to our kinetics measurements, the decay of the OH⋯CO complex in the dark is not a single exponential but slows down in time, which is common for solid-state reactions.65 The decay of the OH⋯CO complex is accelerated by temperature; however, the narrow temperature range of stability of the Ne matrix and the complicated decay curve prevented further studies on this matter. In addition, it was found that this process was accelerated by broadband IR light of the globar and selective excitation of the OH mode by narrow-band radiation of OPO (3546.5 cm−1). IR light seemed to decrease the trans-HOCO yield (compared to the process in the dark), but additional channels were not observed. It should be stressed that no dark OH⋯CO → trans-HOCO reaction was detected in other matrices at 4.5 K as well as there was no efficient effect of IR light.
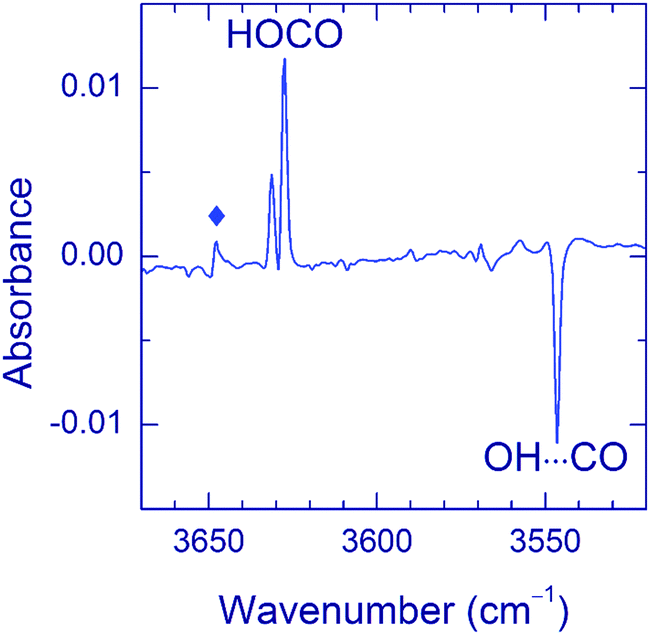 | ||
| Fig. 7 Difference FTIR spectrum showing the result of an overnight stay (in the dark) of a Ne matrix containing the OH⋯CO complexes. The weak band marked with a diamond is due to the H2O⋯CO complex. | ||
Finally, we examined the photostability of the OH⋯CO and trans-HOCO species in the Kr matrix. It was found that ∼250 pulses at 193 nm decomposed trans-HOCO almost completely, whereas the OH⋯CO bands remained virtually unchanged. The 193 nm photolysis of trans-HOCO yielded CO2, but the amount of CO monomer did not change.
Discussion
Light-induced decomposition of water is known to occur via the H2O → H + OH channel and the energy of Kr-lamp photons is enough to dissociate water in matrices.66 Therefore, one may expect the formation of the OH⋯CO complex upon photolysis of the H2O⋯CO complex. In the present work, we observed two bands, correlating with the VUV-induced decomposition of H2O⋯CO, which are assigned to the fundamental vibrations of the OH⋯CO complex. The OH and CO absorption bands appear at 3546.5 (Ne), 3526.5 (Ar), 3521.9 (Kr), and 3520.0 cm−1 (Xe) and at 2159.8 (Ne), 2152.5 (Ar), 2147.9 (Kr), and 2145.0 cm−1 (Xe), respectively.The available calculations predict two structures for this complex, which can be represented as OH⋯CO and OH⋯OC (both linear), the OH⋯CO structure being energetically more favorable (by 140–180 cm−1).37–39,42,43 According to the calculations, the experimentally observed complex has the OH⋯CO structure, in which the hydrogen atom of OH interacts with the carbon atom of CO. Table 3 shows the experimental and calculated shifts of the OH⋯CO bands with respect to the OH and CO monomers. The experimental values are in good agreement with the theory. We did not observe any candidates for the less stable OH⋯OC structure. This species should have a blue-shifted OH absorption and a red-shifted CO one, i.e. the directions of the shifts are opposite in comparison with those of OH⋯CO.38,43,45 Thus, it seems that the less stable OH⋯OC structure is not stabilized under our experimental conditions. It is worth noting that this result is in agreement with the studies in the gas-phase and He droplets, where only the OH⋯CO structure was detected.37,45
| Mode | Experiment (this work) | Theoryb | |||||
|---|---|---|---|---|---|---|---|
| Ne | Ar | Kr | Xe | Ref. 38 | Ref. 43 | Ref. 45 | |
| a Experimental frequencies of the OH monomer in noble-gas matrices are taken from ref. 51 and 71. b For the more stable OH⋯CO structure. c Calculations at the CCSD(T)/cc-pVTZ level of theory. d Calculations at the CCSD(T)/Def2-TZVP level of theory. | |||||||
| ν(OH) | −26.5 | −21.5 | −16.7 | −11.4 | −12 | −27 | −19 |
| ν(CO) | +18.9 | +14.0 | +12.4 | +12.0 | +16 | +20 | — |
The decrease of the complexation-induced shift with the increase of the matrix dielectric constant is an interesting observation (see Table 3). This trend is presumably due to a weaker matrix effect on vibrations of the complex units compared to the monomers. It was previously discussed that the complexation-induced shift is worth counting from the monomer in a vacuum rather than from that in the matrix.21 A Ne matrix is presumably the best approximation of a vacuum among the practical noble gases and the very good agreement of the experiment in the Ne matrix with calculations of ref. 43 is remarkable.
trans-HOCO is another product observed upon VUV photolysis of the H2O⋯CO complex. The previous matrix-isolation studies have shown that this radical can be produced in different ways. First, it is known that trans-HOCO can be formed in low-temperature matrices via the OH + CO → trans-HOCO reaction. In this case, the CO molecule should be in close proximity to the species that served as a precursor of the OH radical. This reaction pathway has been demonstrated in a number of experimental works, where trans-HOCO was generated upon VUV photolysis of H2O in solid CO,52a UV photolysis of the CO⋯H2O2 complex in noble-gas matrices,21 near-UV photolysis of the CO⋯HONO complex in a N2 matrix,19a and X-ray radiolysis of the H2O⋯CO2 complex in noble-gas matrices.51 Upon X-ray radiolysis of matrix-isolated FA, the direct dissociation channel HCOOH → HOCO + H occurs,56 which was not found previously in UV photolysis. In the present work, trans-HOCO was efficiently produced in the “two-color” photochemical experiments with FA/Ng matrices, i.e. when UV photolysis of the matrix was followed by VUV photolysis.
The previous works on UV photolysis of matrix-isolated FA showed only two primary channels, HCOOH → H2O + CO and HCOOH → H2 + CO2.14 The present results suggest the existence of additional channels. First, UV photolysis of matrix-isolated FA efficiently produces H atoms, which is evidenced by annealing-induced reactions. It follows that some radical products are formed under these conditions. Second, trans-HOCO is rapidly decomposed by UV radiation most probably yielding CO2 and H atoms. It is possible that the direct dissociation of FA to trans-HOCO and the H atom occurs upon UV photolysis, similarly to X-ray radiolysis,56 but trans-HOCO is not observed because of its very efficient photodecomposition. In addition, we cannot exclude the production of H atoms via the photodecomposition of HCO. Indeed, the reaction HCOOH → HCO + OH is a main channel in the gas-phase photolysis of FA.67 In this situation, the photodecomposition of HCO and possibly HOCO may be responsible for the production of the CO monomer observed in significant amounts in Kr and Xe matrices (see Fig. 1). Thus, the UV photochemistry of matrix-isolated FA probably deserves further studies.
In our experiments with VUV irradiation, trans-HOCO is mainly formed from the H2O⋯CO complex although the direct dissociation channel HCOOH → HOCO + H is also possible. The present results cannot directly prove whether the H2O⋯CO → trans-HOCO photoreaction (VUV) is a single-step process or it occurs via the OH⋯CO intermediate. It is difficult to distinguish these two channels based on the kinetic measurements because the VUV-induced degradation of both trans-HOCO and OH⋯CO (yielding CO2) significantly complicates this analysis. Nevertheless, the possibility of the two-step mechanism H2O⋯CO → OH⋯CO → trans-HOCO is indirectly supported by the observed transformation of OH⋯CO to trans-HOCO, and this process is discussed in more detail below.
The gas-phase reaction between OH and CO leading to trans-HOCO has been suggested to occur via the OH⋯CO complex.37,68,69 Our observations provide valuable information about the reactivity of the OH⋯CO complex. The annealing-induced decay of OH⋯CO occurring in Ar and Kr matrices at ∼30 K correlates with the formation of trans-HOCO. As mentioned above, the lack of a strict correlation between these processes observed in the Ar matrix below 25 K (see Fig. 5b) features additional reactions. As suggested previously,51 the recombination of shortly-separated (non-complexed) pairs OH + CO → trans-HOCO may be responsible for the formation of trans-HOCO at temperatures between 10 and 25 K. The short-range recombination with H atoms recovering the H2O⋯CO complex can contribute to the decay of OH⋯CO at ∼15 K. In the Ne matrix, the OH⋯CO decay in the dark is also accompanied by the formation of trans-HOCO. Thus, we conclude that trans-HOCO is the main product of decomposition of the OH⋯CO complex under our experimental conditions and an additional minor channel may be the OH⋯CO + H → H2O⋯CO reaction. The annealing-induced reaction OH⋯CO → trans-HOCO in Ng matrices (Ng = Ar, Kr, and Xe) can be an over-barrier process. Indeed, the Arrhenius equation yields the characteristic reaction time of ca. 10 s at 30 K and 2 × 1021 years at 10 K, for the reaction barrier of 665 cm−1 and the pre-exponential factor of 7.4 × 1012 s−1.‡
In the Ne matrix, the OH⋯CO decay and the formation of trans-HOCO occur in the dark even at 4.5 K. This observation can be considered as a signature of quantum tunneling because the over-barrier reaction at such a low temperature can be excluded. The OH⋯CO → trans-HOCO reaction induced by IR light also takes place in the Ne matrix. It is interesting that in other matrices, the OH⋯CO → trans-HOCO process was not found at 4.5 K. Concerning the possible tunneling mechanism, this difference may be connected with the details of the matrix environment (solvation), in agreement with the known observations.70 It is more difficult to understand the lacking efficiency of the IR-induced process in other noble-gas matrices. We can speculate that the vibrational energy relaxation of OH⋯CO is faster for heavier matrix atoms, and the required reorganization of the complex does not occur.
Finally, we discuss the effects of the matrix material (see Fig. 2) and matrix annealing (see Fig. 4) on the branching ratio of the OH⋯CO and trans-HOCO channels of VUV photolysis of the H2O⋯CO complex. First, this ratio sufficiently decreases in the Kr matrix as compared to Ar and Ne matrices. In the Xe matrix, the OH⋯CO formation is very inefficient. Second, the relative yield of the OH⋯CO production can be sufficiently increased by annealing of the matrices after initial UV photolysis (prior to VUV-photolysis). These trends qualitatively correlate with the matrix site splitting of the H2O⋯CO absorptions. The ratio between the H2O⋯CO amounts in the relaxed and unrelaxed matrix sites after UV photolysis of FA decreases from Ne to Xe. We believe that the bands of H2O⋯CO in the Ne matrix correspond to the relaxed matrix site because they are narrow and unstructured. Matrix annealing stimulates conversion of the unrelaxed site to the relaxed one. (In general, annealing leads to relaxation of matrix morphology to a lower energy.) These correlations seem to show that VUV photolysis of the H2O⋯CO complex in an unrelaxed environment produces mainly trans-HOCO. In contrast, a more efficient formation of the OH⋯CO complex occurs in a relaxed environment, e.g., after annealing of the matrix. The exact structure and energetics of these matrix sites are unknown.
In addition to matrix morphology, the geometry of the parent H2O⋯CO complex can affect its VUV photoproducts. It is possible that the photochemical channels are different for the HOH⋯CO and HOH⋯OC structures. For example, the low yield of the OH⋯CO complex in the Xe matrix may be connected with the dominating HOH⋯OC structure of the precursor. However, to examine this effect, one needs to obtain matrices with different structures of the H2O⋯CO complex (HOH⋯CO only and HOH⋯OC only). In the present work, we failed to find experimental conditions providing these matrices and this question remains unanswered.
Conclusions
In the present work, we have studied VUV photolysis (130–170 nm) of the H2O⋯CO complexes in noble-gas matrices (Ne, Ar, Kr, and Xe). The H2O⋯CO complexes were generated by UV photolysis (193 and 250 nm) of formic acid (FA) in noble-gas matrices. In agreement with the previous observations,14 the matrix material has a significant effect on both the yield and the geometry of the H2O⋯CO complex. As a new finding, UV photolysis of FA is observed to produce considerable amounts of H atoms. The formation of H atoms possibly includes the photolysis of trans-HOCO and/or HCO directly produced from FA. This observation shows that UV photochemistry of FA in noble-gas matrices is not fully understood.VUV photolysis of the H2O⋯CO complex in noble-gas matrices leads to the formation of the OH⋯CO radical–molecule complex, in which the hydrogen atom of OH interacts with the carbon atom of CO. All fundamental absorptions of this complex were found in the four matrices. The red shifts of −26.5, −21.5, −16.7, and −11.4 cm−1 (with respect to OH monomer) were observed for the OH stretching mode in Ne, Ar, Kr and Xe matrices, respectively, whereas the blue shifts of +18.9, +14.5, +12.4 and +12.0 cm−1 were found for the corresponding CO stretching vibrations. The obtained data are in good agreement with the calculations reported previously.38,43,45 No evidence for the stabilization of the less stable OH⋯OC structure was found. The formation of the OH⋯CO complex is quite efficient in Ne and Ar matrices, and it is hardly visible in the Xe matrix. The yield of the OH⋯CO complex increased if the matrices containing the H2O⋯CO complexes were subjected to annealing prior to VUV photolysis. These observations suggest that the local matrix morphology and possibly also the H2O⋯CO complex geometry affect the photolysis pathways.
The trans-HOCO radicals are efficiently formed by VUV photolysis of the matrix-isolated H2O⋯CO complexes. Several new assignments are suggested for this species. We have to note that the multi-step production of trans-HOCO from FA via the H2O⋯CO complex is a promising approach for further matrix-isolation studies on this important species. It is worth noting that neither the OH⋯CO complexes nor the trans-HOCO radicals appear after UV photolysis of FA in the noble-gas matrices; thus, the VUV photolysis has a clear advantage in this case.
Finally, the intrinsic reactivity of the matrix-isolated OH⋯CO complex resulting in the formation of trans-HOCO is directly demonstrated. This reaction was observed in Ar and Kr matrices upon annealing at 30–35 K. These results confirm that the OH⋯CO complex can act as an intermediate in the OH + CO → HOCO reaction. The case of the Ne matrix is special because the formation of trans-HOCO from the OH⋯CO complex occurs in the dark even at the lowest experimental temperature (4.5 K), which is in contrast to other matrices. It is possible that quantum tunneling is involved in this process because the over-barrier reaction is improbable at such a low temperature. If it is true, this finding represents a strong matrix effect on the tunneling rate. IR light activates this reaction in the Ne matrix at 4.5 K, and again, this effect is not certainly found in the other matrices, which is not fully understood. The Ne matrix is the closest approximation of the gas phase among the noble-gas solids; thus, the tunneling and IR-induced reactions found in the Ne matrix may also occur in the gas phase.
Acknowledgements
This work was supported by the Academy of Finland (Projects No. 1277993 and No. 1288889).References
- Supramolecular Photochemistry, ed. V. Balzani, Reidel, Dordrecht, 1987 Search PubMed.
- Supramolecular Photochemistry: Controlling Photochemical Processes, ed. V. Ramamurthy and Y. Inoue, Wiley, New York, 2011 Search PubMed.
- A. J. Barnes, J. Mol. Struct., 1983, 100, 259 Search PubMed.
- G. Maes, in Intermolecular Forces: An Introduction to Modern Methods and Results, ed. P. L. Huyskens, W. A. P. Luck and T. Zeegers-Huyskens, Springer, Berlin, 1991, ch. VIII, pp. 195–216 Search PubMed.
- L. Schriver-Mazzuoli, in Molecular Complexes in Earth's, Planetary, Cometary, and Interstellar Atmospheres, ed. A. A. Vigasin and Z. Slanina, World Scientific, Singapore, 1998, ch. 7, pp. 194–237 Search PubMed.
- L. Andrews, Chem. Soc. Rev., 2004, 33, 123 Search PubMed.
- A. J. Barnes and Z. Mielke, J. Mol. Struct., 2012, 1023, 216 Search PubMed.
- N. A. Young, Coord. Chem. Rev., 2013, 257, 956 Search PubMed.
- L. Khriachtchev, J. Phys. Chem. A, 2015, 119, 2735 Search PubMed.
- (a) B. Nelander, J. Phys. Chem. A, 1997, 101, 9092 Search PubMed; (b) T. Svensson and B. Nelander, Chem. Phys., 2000, 262, 445 Search PubMed; (c) T. Svensson, B. Nelander and G. Karlström, Chem. Phys., 2001, 265, 323 Search PubMed; (d) T. Svensson and B. Nelander, Chem. Phys., 2003, 286, 347 Search PubMed; (e) A. Engdahl, G. Karlström and B. Nelander, J. Chem. Phys., 2003, 118, 7797 Search PubMed; (f) A. Engdahl and B. Nelander, Phys. Chem. Chem. Phys., 2004, 6, 730 Search PubMed; (g) A. Engdahl and B. Nelander, J. Chem. Phys., 2005, 122, 126101 Search PubMed.
- (a) A. Lignell, L. Khriachtchev, H. Mustalampi, T. Nurminen and M. Räsänen, Chem. Phys. Lett., 2005, 405, 448 Search PubMed; (b) A. Lignell, L. Khriachtchev, H. Lignell and M. Räsänen, Phys. Chem. Chem. Phys., 2006, 8, 2457 Search PubMed.
- L. Khriachtchev, J. Mol. Struct., 2008, 880, 14 Search PubMed.
- A. Lignell and L. Khriachtchev, J. Mol. Struct., 2008, 889, 1 Search PubMed.
- (a) J. Lundell and M. Räsänen, J. Phys. Chem., 1995, 99, 14301 Search PubMed; (b) J. Lundell and M. Räsänen, J. Mol. Struct., 1997, 436, 349 Search PubMed.
- (a) M. Hawkins, L. Andrews, A. J. Downs and D. J. Drury, J. Am. Chem. Soc., 1984, 106, 3076 Search PubMed; (b) M. Hawkins and L. Andrews, Inorg. Chem., 1985, 24, 3285 Search PubMed; (c) L. Andrews, M. Hawkins and R. Withnall, Inorg. Chem., 1985, 24, 4234 Search PubMed; (d) R. Withnall and L. Andrews, J. Phys. Chem., 1987, 91, 784 Search PubMed; (e) S. D. Allen, M. Poliakoff and J. J. Turner, J. Mol. Struct., 1987, 157, 1 Search PubMed; (f) B. W. Moores and L. Andrews, J. Phys. Chem., 1989, 93, 1902 Search PubMed; (g) R. J. H. Clark and J. R. Dann, J. Phys. Chem., 1996, 100, 532 Search PubMed; (h) H. Clark and J. R. Dann, J. Phys. Chem., 1997, 101, 2074 Search PubMed; (i) R. J. H. Clark, J. R. Dann and L. J. Foley, J. Phys. Chem., 1997, 101, 9260 Search PubMed; (j) M. Bahou, L. Schriver-Mazzuoli, A. Schriver and P. Chaquin, Chem. Phys., 1997, 216, 105 Search PubMed; (k) D. Wakamatsu, N. Akai, A. Kawai and K. Shibuya, Chem. Lett., 2012, 41, 252 Search PubMed.
- (a) T. C. McInnis and L. Andrews, J. Phys. Chem., 1992, 96, 2051 Search PubMed; (b) L. Andrews, T. C. McInnis and Y. Hannachi, J. Phys. Chem., 1992, 96, 4248 Search PubMed; (c) K. Johnsson, A. Engdahl, P. Ouis and B. Nelander, J. Phys. Chem., 1992, 96, 5778 Search PubMed.
- (a) A. de Saxce, N. Sanna, A. Schriver and L. Schriver-Mazzuoli, Chem. Phys., 1994, 185, 365 Search PubMed; (b) A. Pieretti, N. Sanna, A. Hallou, L. Schriver-Mazzuoli and A. Schriver, J. Mol. Struct., 1998, 447, 223 Search PubMed.
- M. Fushitani, T. Shida, T. Momose and M. Räsänen, J. Phys. Chem. A, 2000, 104, 3635 Search PubMed.
- (a) Z. Mielke, A. Olbert-Majkut and K. G. Tokhadze, J. Chem. Phys., 2003, 118, 1364 Search PubMed; (b) A. Olbert-Majkut, Z. Mielke and K. G. Tokhadze, J. Mol. Struct., 2003, 656, 321 Search PubMed; (c) M. Wierzejewska and A. Olbert-Majkut, J. Phys. Chem. A, 2003, 107, 10944 Search PubMed; (d) A. Olbert-Majkut and Z. Mielke, Phys. Chem. Chem. Phys., 2006, 8, 4773 Search PubMed; (e) B. Golec, A. Bil and Z. Mielke, J. Phys. Chem. A, 2009, 113, 9434 CrossRef CAS PubMed; (f) K. Grzechnik and Z. Mielke, J. Mol. Struct., 2012, 1025, 124 Search PubMed; (g) A. Bil, K. Grzechnik, M. Sałdyka and Z. Mielke, J. Phys. Chem. A, 2016, 120, 6753 Search PubMed.
- L. Khriachtchev, M. Pettersson, S. Tuominen and M. Räsänen, J. Chem. Phys., 1997, 107, 7252 Search PubMed.
- J. Lundell, S. Jolkkonen, L. Khriachtchev, M. Pettersson and M. Räsänen, Chem. – Eur. J., 2001, 7, 1670 Search PubMed.
- K. I. Öberg, A. C. A. Boogert, K. M. Pontoppidan, S. van den Broek, E. F. van Dishoeck, S. Bottinelli, G. A. Blake and N. J. I. Evans, Astrophys. J., 2011, 740, 109 Search PubMed.
- H. Dubost and L. Abouaf-Marguin, Chem. Phys. Lett., 1972, 17, 269 Search PubMed.
- D. Yaron, K. I. Peterson, D. Zolandz, W. Klemperer, F. J. Lovas and R. D. Suenram, J. Chem. Phys., 1990, 92, 7095 Search PubMed.
- R. E. Bumgarner, S. Suzuki, P. A. Stockman, P. G. Green and G. A. Blake, Chem. Phys. Lett., 1991, 176, 123 Search PubMed.
- (a) J. Sadlej and V. Buch, J. Chem. Phys., 1994, 100, 4272 Search PubMed; (b) J. Sadlej, B. Rowland, J. P. Devlin and V. Buch, J. Chem. Phys., 1995, 102, 4804 Search PubMed.
- (a) J. Lundell and Z. Latajka, J. Phys. Chem. A, 1997, 101, 5004 Search PubMed; (b) J. Lundell and Z. Latajka, Chem. Phys., 2001, 263, 221 Search PubMed; (c) J. Lundell and Z. Latajka, J. Mol. Struct., 2008, 887, 172 Search PubMed.
- M. D. Brookes and A. R. W. McKellar, J. Chem. Phys., 1998, 109, 5823 Search PubMed.
- L. Oudejans and R. E. Miller, Chem. Phys. Lett., 1999, 306, 214 Search PubMed.
- (a) H. Abe and K. M. T. Yamada, J. Chem. Phys., 2001, 114, 6134 Search PubMed; (b) H. Abe and K. M. T. Yamada, J. Chem. Phys., 2004, 121, 7803 Search PubMed.
- A. F. A. Vilela, P. R. P. Barreto, R. Gargano and C. R. M. Cunha, Chem. Phys. Lett., 2006, 427, 29 Search PubMed.
- R. J. Wheatley and A. H. Harvey, J. Chem. Phys., 2009, 131, 154305 Search PubMed.
- M. P. Collings, J. W. Dever and M. R. S. McCoustra, Phys. Chem. Chem. Phys., 2014, 16, 3479 Search PubMed.
- Š. Budzák, P. Carbonniere, M. Medved’ and I. Černušák, Mol. Phys., 2014, 112, 3225 Search PubMed.
- Q. Cao, S. Berski, M. Räsänen, Z. Latajka and L. Khriachtchev, J. Phys. Chem. A, 2013, 117, 4385 Search PubMed.
- K. Kudla, A. G. Koures, L. B. Harding and G. C. Schatz, J. Chem. Phys., 1992, 96, 7465 Search PubMed.
- (a) M. I. Lester, B. V. Pond, D. T. Anderson, L. B. Harding and A. F. Wagner, J. Chem. Phys., 2000, 113, 9889 Search PubMed; (b) M. I. Lester, B. V. Pond, M. D. Marshall, D. T. Anderson, L. B. Harding and A. F. Wagner, Faraday Discuss., 2001, 118, 373 Search PubMed.
- H.-G. Yu, J. T. Muckerman and T. J. Sears, Chem. Phys. Lett., 2001, 349, 547 Search PubMed.
- R. S. Zhu, E. G. W. Diau, M. C. Lin and A. M. Mebel, J. Phys. Chem. A, 2001, 105, 11249 Search PubMed.
- (a) R. Valero and G.-J. Kroes, J. Chem. Phys., 2002, 117, 8736 Search PubMed; (b) R. Valero, M. C. van Hemert and G.-J. Kroes, Chem. Phys. Lett., 2004, 393, 236 Search PubMed.
- Y. He, E. M. Goldfield and S. K. Gray, J. Chem. Phys., 2004, 121, 823 Search PubMed.
- (a) X. Song, J. Li, H. Hou and B. Wang, J. Chem. Phys., 2006, 125, 94301 Search PubMed; (b) J. Li, Y. Wang, B. Jiang, J. Ma, R. Dawes, D. Xie, J. M. Bowman and H. Guo, J. Chem. Phys., 2012, 136, 41103 Search PubMed; (c) J. Li, C. Xie, J. Ma, Y. Wang, R. Dawes, D. Xie, J. M. Bowman and H. Guo, J. Phys. Chem. A, 2012, 116, 5057 Search PubMed.
- S. Du and J. S. Francisco, J. Chem. Phys., 2009, 131, 64307 Search PubMed.
- (a) M. E. Greenslade, M. Tsiouris, R. Timothy Bonn and M. I. Lester, Chem. Phys. Lett., 2002, 354, 203 Search PubMed; (b) M. D. Marshall, B. V. Pond and M. I. Lester, J. Chem. Phys., 2003, 118, 1196 Search PubMed.
- J. T. Brice, T. Liang, P. L. Raston, A. B. McCoy and G. E. Douberly, J. Chem. Phys., 2016, 145, 124310 Search PubMed.
- E. J. K. Nilsson and A. A. Konnov, Energy Fuels, 2016, 30, 2443 Search PubMed.
- (a) J. S. Francisco, J. T. Muckerman and H.-G. Yu, Acc. Chem. Res., 2010, 43, 1519 Search PubMed; (b) C. J. Johnson, R. Otto and R. E. Continetti, Phys. Chem. Chem. Phys., 2014, 16, 19091 Search PubMed.
- C. S. Boxe, J. S. Francisco, R.-L. Shia, Y. L. Yung, H. Nair, M.-C. Liang and A. Saiz-Lopez, Icarus, 2014, 24, 97 Search PubMed.
- D. E. Woon, Astrophys. J., 2002, 571, L177 Search PubMed.
- P. D. Holtom, C. J. Bennett, Y. Osamura, N. J. Mason and R. I. Kaiser, Astrophys. J., 2005, 626, 940 Search PubMed.
- S. V. Ryazantsev and V. I. Feldman, J. Phys. Chem. A, 2015, 119, 2578 Search PubMed.
- (a) D. E. Milligan and M. E. Jacox, J. Chem. Phys., 1971, 54, 927 Search PubMed; (b) C. J. Bennett, T. Hama, Y. S. Kim, M. Kawasaki and R. I. Kaiser, Astrophys. J., 2011, 727, 27 Search PubMed.
- T. Oyama, W. Funato, Y. Sumiyoshi and Y. Endo, J. Chem. Phys., 2011, 134, 174303 Search PubMed.
- M. Pettersson, L. Khriachtchev, J. Lundell and M. Räsänen, J. Am. Chem. Soc., 1999, 121, 11904 Search PubMed.
- K. Marushkevich, L. Khriachtchev, J. Lundell, A. Domanskaya and M. Räsänen, J. Phys. Chem. A, 2010, 114, 3495 Search PubMed.
- S. V. Ryazantsev and V. I. Feldman, Phys. Chem. Chem. Phys., 2015, 17, 30648 Search PubMed.
- K. Marushkevich, L. Khriachtchev and M. Räsänen, J. Phys. Chem. A, 2007, 111, 2040 Search PubMed.
- D. Forney, M. E. Jacox and W. E. Thompson, J. Chem. Phys., 2003, 119, 10814 Search PubMed.
- (a) D. E. Milligan and M. E. Jacox, J. Mol. Spectrosc., 1973, 46, 460 Search PubMed; (b) A. A. Karatun, F. F. Sukhov and N. A. Slovokhotova, High Energy Chem., 1981, 15, 371 Search PubMed; (c) H. Kunttu, J. Seetula, M. Räsänen and V. A. Apkarian, J. Chem. Phys., 1992, 96, 5630 Search PubMed; (d) H. M. Kunttu and J. A. Seetula, Chem. Phys., 1994, 189, 273 Search PubMed.
- C. Pirim and L. Krim, Phys. Chem. Chem. Phys., 2011, 13, 19454 Search PubMed.
- M. E. Jacox, J. Chem. Phys., 1988, 88, 4598 Search PubMed.
- Q. Cao, S. Berski, Z. Latajka, M. Räsänen and L. Khriachtchev, Phys. Chem. Chem. Phys., 2014, 16, 5993 Search PubMed.
- (a) M. Pettersson, J. Lundell and M. Räsänen, J. Chem. Phys., 1995, 103, 205 Search PubMed; (b) V. I. Feldman and F. F. Sukhov, Chem. Phys. Lett., 1996, 255, 425 Search PubMed.
- (a) J. Eberlein and M. Creuzburg, J. Chem. Phys., 1997, 106, 2188 Search PubMed; (b) K. Vaskonen, J. Eloranta, T. Kiljunen and H. Kunttu, J. Chem. Phys., 1999, 110, 2122 Search PubMed; (c) L. Khriachtchev, H. Tanskanen, M. Pettersson, M. Räsänen, V. Feldman, F. Sukhov, A. Orlov and A. F. Shestakov, J. Chem. Phys., 2002, 116, 5708 Search PubMed; (d) L. Khriachtchev, M. Saarelainen, M. Pettersson and M. Räsänen, J. Chem. Phys., 2003, 118, 6403 Search PubMed.
- A. Plonka, Dispersive Kinetics, Springer, Dordrecht, 2001 Search PubMed.
- E. Isoniemi, L. Khriachtchev, J. Lundell and M. Räsänen, Phys. Chem. Chem. Phys., 2002, 4, 1549 Search PubMed.
- D. L. Singleton, G. Paraskevopoulos and R. S. Irwin, J. Phys. Chem., 1990, 94, 695 Search PubMed.
- I. W. M. Smith and A. R. Ravishankara, J. Phys. Chem. A, 2002, 106, 4798 Search PubMed.
- H. Guo, Int. Rev. Phys. Chem., 2012, 31, 1 Search PubMed.
- M. Pettersson, E. M. S. Maçôas, L. Khriachtchev, J. Lundell, R. Fausto and M. Räsänen, J. Chem. Phys., 2002, 117, 9095 Search PubMed.
- J. Goodman and L. E. Brus, J. Chem. Phys., 1977, 67, 4858 Search PubMed.
Footnotes |
| † In ref. 10g, the shift of −21.8 cm−1 is mentioned for the OH fundamental mode of the OH⋯CO complex isolated in an Ar matrix, but no details of the corresponding experimental results are reported. |
| ‡ The calculated value of the barrier height was extracted from ref. 42c and the used pre-exponential factor corresponds to the experimental frequency for the intermolecular bending mode in the OH⋯CO complex (ref. 44b). |
| This journal is © the Owner Societies 2017 |