
Spectral pointillism of enhanced Raman scattering for accessing structural and conformational information on single protein
Jean-Emmanuel
Clément†
,
Aymeric
Leray†
*,
Alexandre
Bouhelier
and
Eric
Finot
Laboratoire Interdisciplinaire Carnot de Bourgogne, UMR 6303 CNRS, Université Bourgogne Franche-Comté, 21000 Dijon, France. E-mail: aymeric.leray@u-bourgogne.fr; eric.finot@u-bourgogne.fr
First published on 22nd November 2016
Abstract
In this contribution, we provide new insights on the temporal fluctuations of surface enhanced Raman spectra (SERS) of large single molecules such as proteins. Because they can only fit partly into small active volume, SERS analysis is referred to spectral pointillism where only protein subdomains are shined and the whole protein landscape is built from the dynamics of successive individual spectra. By applying our approach on bovine serum albumin, we show that single protein subdomains are mostly comprised of three distinct amino acids. Surface amino acids such as lysine are preferentially detected in the open form of the protein. The investigation of the tryptophan Fermi doublet in the single protein regime is highly instructive on the protein conformation. We finally demonstrate that spectral pointillism enables to correlate individual amino acids with structural information.
Introduction
Over this past decade, a large number of papers have been published to report that surface enhanced Raman spectroscopy (SERS) is able to enhance spontaneous Raman scattering signals with several orders of magnitude (at least 106) at the proximity of metallic nanoparticles.1–3 This huge enhancement factor originates from localized surface plasmon resonance excited by the interaction of light with metallic nanoparticles.4 This interaction generates coherent electron cloud oscillations with a resonant frequency dependent on the size, shape and material properties of the nanoparticle and on the surrounding medium. These collective oscillations produce finally a strong evanescent electromagnetic field on the nanoparticle surface and thus amplify Raman signals.4 SERS measurements down to the single molecule regime have been demonstrated many times for small molecules.1,5,6For larger molecules such as proteins, a wide variation between enhanced spectra have been reported in the literature.7–10 This variability usually interpreted as a lack of SERS substrate reproducibility is thought to come from the enhancement fluctuations since the plasmon resonance, the active volume and the enhancement factor are dramatically dependent on the size and shapes of metallic nanostructures.4,11 Despite many efforts at elaborating reproducible SERS active substrate, temporal fluctuations of SERS spectra still exist.12–14 It is now recognized that these temporal fluctuations stem from the diffusion of single molecule which are likely to occupy several conformations and orientations on the probing site;15,16 this diffusion could be thermally activated and/or coupled with photo-induced electron transfer. As already pointed out,1,12 large molecule such as protein can only fit partly into small enhanced active volume (around nm3). In this case, SERS measurements may probe only few building blocks, namely amino acids, thereby leading to unavoidable spectral fluctuations according to the area under scrutiny.
In this work, we propose to exploit these temporal fluctuations that contain precise information on the protein building blocks. We develop an innovative concept which enables to retrieve the whole protein spectrum by probing successively various domains composing the protein. We use the term “spectral pointillism” to refer to the acquisition of individual SERS spectra at several distinct areas of the protein. Our approach requires (i) the fast acquisition of spectra in the single molecule regime ensuring both with a microfluidic platform where proteins circulate individually (low concentration) and with an optimized SERS substrate and (ii) the adapted signal processing of the corresponding numerous fluctuating spectra.
We apply the spectral pointillism to bovine serum albumin (BSA), a protein largely investigated in both spontaneous Raman17–19 and SERS experiments.7–10 BSA is the most widespread protein in plasma (almost 60% of all plasma proteins) and it serves to transport a large number of physiological and non-physiological ligands. It is built from 583 amino acids residues forming three homologous domains (named I, II, and III) stabilized by 17 disulfide bonds. In a physiological environment, it is organized in a heart-shaped structure of size 14 × 4 × 4 nm. This structure can be denaturated by adding various kinds of molecules20–22 or by varying temperature or pH.8,17,19 For 2.80 < pH < 4.40, a reversible conformational change occurs in two steps.19 During the first step called N → F transition (for 3.75 < pH < 4.40), the domain III is unfolded; and then in the pH range from 2.80 to 3.60, BSA undergoes another expansion with the unfolding of domain I and II (F → E transition). The size of the major axis of this unfolded state is increased to 23 nm.23
This structural modification of BSA was already monitored with both spontaneous Raman and SERS spectroscopy.8,17,19,20 In order to obtain reproducible spectra, previous studies were performed by acquiring Raman signal emitted from large surface (tens of μm2) and integrated during a long time (tens of s) enabling to probe a large number of molecules. Raman spectrum contains then the molecular vibrational information of an ensemble of proteins. The measured intensity at one Raman shift is thus a linear combination of the individual components intensity, which is directly proportional to their concentration. Complex spectral multivariate analysis are usually necessary for decomposing Raman spectra and extracting the proportion of each component.24
Our spectral pointillism avoids this tedious spectral analysis treatment. By probing successively various areas of a single BSA and by analysing these numerous individual SERS spectra, we can not only retrieve results obtained from average spectrum, but also have directly access to precious information about the protein structure (i.e. polypeptide backbone) and composition (i.e. amino acids), which are not simply accessible by spontaneous Raman spectroscopy.
Material and methods
Acquisition of BSA spectra
For investigating protein conformation, BSA was studied in physiological (pH 7) and acidic environment (pH 3). At pH 7, BSA was diluted into a phosphate-buffered saline (PBS) solution to a concentration of 1.5 × 10−6 mol L−1. At pH 3, BSA was diluted into an acidic solution obtained by mixing 90 mL of sodium acetate (at a concentration of 0.2 mol L−1) and 10 mL of acetic acid (c = 0.2 mol L−1). The final concentration of BSA in acidic environment is also 1.5 × 10−6 mol L−1.For SERS measurements, a raspberry-like gold nanoparticles template deposited on a glass coverslip and capped with microfluidics channels was used (fabrication steps are described in ref. 25).
BSA solutions were injected into the microfluidic channels with a flow of 2 μL min−1 driven by a peristaltic pump and the SERS signal was detected by a custom-built inverted confocal Raman microscope. Position of the microfluidic chip was ensured by a piezoelectric translation stage. A 785 nm laser diode focused with a 60× water immersion objective (NA = 1.2, Nikon) was used for excitation. Back scattered enhanced Raman signal is collected at a frequency of 2 Hz by a spectrometer composed of a 600 lines per millimeter grating associated with a cooled CCD camera (1024 × 256 pixels). Many records of 600 s have been performed on different hot spots with the same laser power (around 0.2 mW at the objective back pupil plane).
For each pH, we have recorded more than 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 spectra corresponding to almost 15 temporal series. At the beginning and at the end of these kinetics, no SERS spectra were usually observed confirming that no fragment or/and other molecules stack to the nanostructure and disturb the measurement.
000 spectra corresponding to almost 15 temporal series. At the beginning and at the end of these kinetics, no SERS spectra were usually observed confirming that no fragment or/and other molecules stack to the nanostructure and disturb the measurement.
Spectra analysis
Experimental data were analysed using a home made program developed with MATLAB and Statistics toolbox (version R2014B, The MathWorks Inc.). SERS spectra are first background corrected by subtracting an average offset extracted from a spectral band of low intensity. In the single molecule regime, the majority of the acquired spectra are flat due to the absence of protein. Exploitable spectral data are then selected by retaining only SERS spectra whose signal to noise ratio is superior to 100. They represent respectively 1607 and 2229 spectra in physiological and acidic environment. From these spectra, Raman peaks were determined computationally with a standard algorithm finding local maxima. These Raman peaks are finally compared with the Raman bands of: amide I and III (cf.Table 1), the secondary structures (indicated in Fig. 2), the disulfide bridges (cf.Table 2) and the amino acids (reported in Table 3). When they match with a precision of ±6 cm−1, corresponding spectra are identified and counted.| Δν (cm−1) | Occ. at pH 7 (%) | Occ. at pH 3 (%) | |
|---|---|---|---|
| Amide I | 1630–1690 | 10 | 17 |
| Amide III | 1230–1300 | 62 | 78 |
| Conformation | Δν (cm−1) | Occ. at pH 7 (%) | Occ. at pH 3 (%) |
|---|---|---|---|
| ggg | 510 | 30 | 33 |
| ggt or tgg | 525 | 50 | 32 |
| tgt | 540 | 20 | 35 |
| Amino acid | Raman bands (in cm−1) |
|---|---|
| Aspartic acid | 747 779 871 939 1082 1261 1338 1425 |
| Cysteine | 499 678 764 787 860 932 1343 1414 |
| Glutamic acid | 530 764 871 915 1079 1115 1342 1419 |
| Leucine | 728 852 890 962 1135 1252 1345 1453 |
| Lysine | 779 849 1064 1139 1321 1360 1414 1447 |
| Methionine | 653 700 723 755 875 1261 1343 1428 |
| Phenylalanine | 618 750 821 1005 1032 1210 1589 1607 |
| Tryptophan | 759 879 1013 1344 1363 1430 1463 1554 |
| Tyrosine | 643 785 831 850 1175 1263 1355 1610 |
Results and discussion
In order to differentiate protein conformations at a single molecule level, we investigate solutions of BSA diluted in a micromolar concentration at two pH: 3 for the open conformation and 7 for the globular one.Cumulative spectrum at pH 7 shown in Fig. 1A is smooth and poorly resolved with numerous broad bands. It results from the superposition of numerous well-resolved Raman spectra characterized by a limited number of narrow peaks (some examples are shown in Fig. 1C) and thus from the accumulation of plenty vibration frequencies. Extraction of each individual molecular vibration proportion does not require complex statistical treatment; it is simply performed by analysing the temporal fluctuations of the SERS spectra measured from individual proteins.
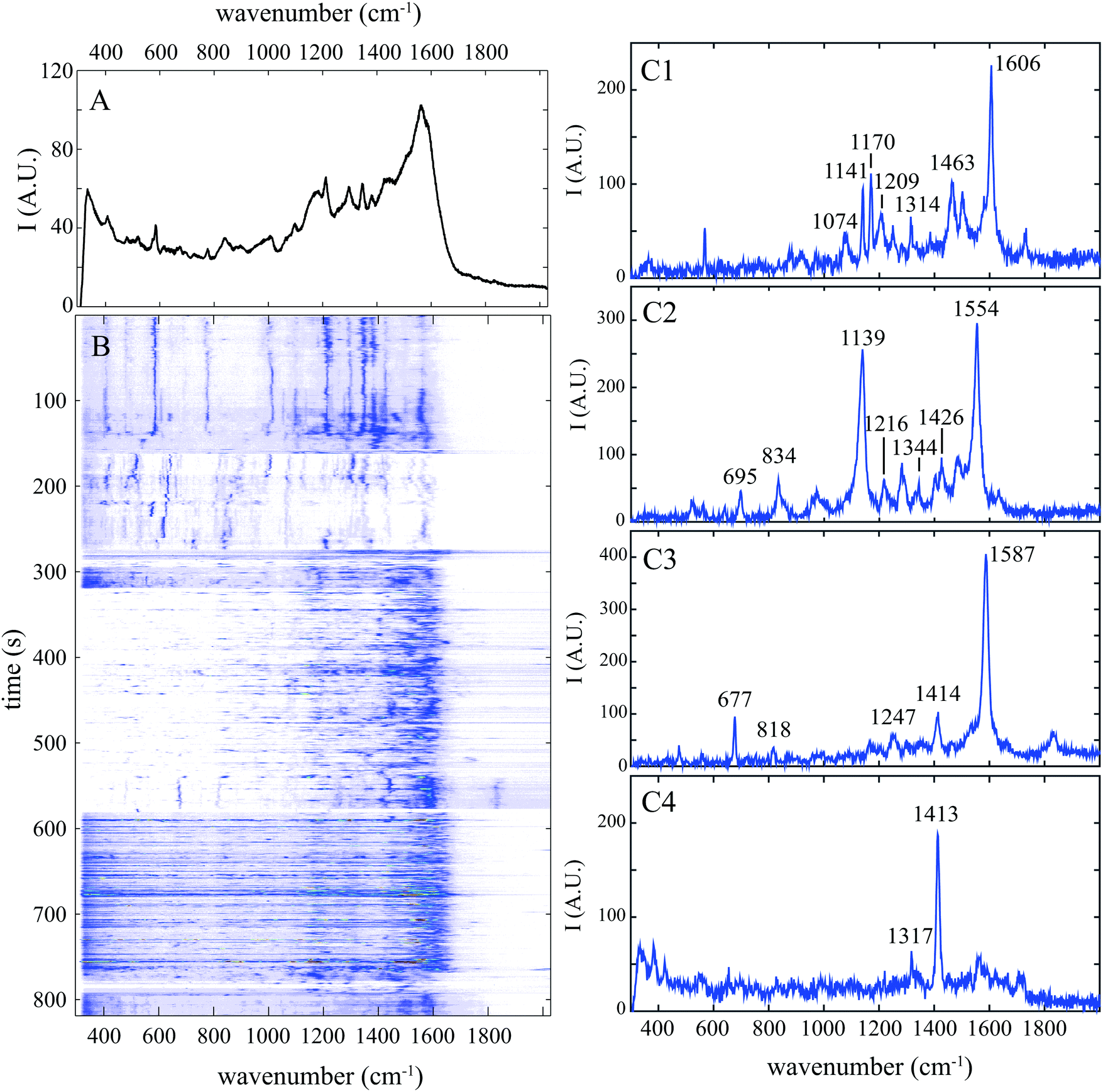 | ||
| Fig. 1 Surface enhanced Raman spectrogram of single BSA protein at ph 7. Spectrum was acquired every 0.5 s. (A) Cumulative SERS spectrum acquired during 800 s. (B) Spectrogram showing the temporal evolution of the spectrum intensity. (C1–C4) Raw individual SERS spectra extracted from the time series at 440, 538, 564 and 606 s (B). Wavenumber of the most intense Raman bands is indicated. | ||
Our approach that we named “spectral pointillism” requires both optimized SERS substrate enabling fast single molecule acquisition and the signal processing of a large set of highly fluctuating spectra. These temporal fluctuations in both intensity and Raman shifts are well observed in the spectrogram of Fig. 1B showing the evolution of the spectra during the acquisition.
We will first consider Raman bands related to the polypeptide backbone –Cα–CO–NH–Cα– and bands related to the disulfide bridges. We will show that estimating the occurrences of specific Raman bands is an efficient way to retrieve structural information on protein.
Structural information
Amide I band (between 1630 and 1690 cm−1) corresponding mainly to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration is not observed in the cumulative spectrum (Fig. 1A) and it is detected only in 10% of individual spectra at pH 7, which is significantly lower than the value obtained with spontaneous Raman spectroscopy.17 Contrary to the spontaneous Raman scattered from an ensemble of freely diffusing molecules, SERS signal is exalted only close to gold nanoparticle's surface. The CO stretching vibration (mainly presents in the amide I band) is not enhanced since amino acids and peptides interact more preferably with gold through carboxylic group.27 Because this band is sparsely detected in the individual spectra, it cannot be used as reliable SERS marker of protein.
O stretching vibration is not observed in the cumulative spectrum (Fig. 1A) and it is detected only in 10% of individual spectra at pH 7, which is significantly lower than the value obtained with spontaneous Raman spectroscopy.17 Contrary to the spontaneous Raman scattered from an ensemble of freely diffusing molecules, SERS signal is exalted only close to gold nanoparticle's surface. The CO stretching vibration (mainly presents in the amide I band) is not enhanced since amino acids and peptides interact more preferably with gold through carboxylic group.27 Because this band is sparsely detected in the individual spectra, it cannot be used as reliable SERS marker of protein.
The amide III band (between 1230 and 1300 cm−1) was observed in more than 60% of individual spectra with a medium intensity at pH 7. This band which is predominantly explained by the in-phase combination of N–H in-plane bending and C–N stretching vibrations can be helpful for a better understanding of the protein structure. Indeed, when the polypeptide interacts with the gold surface, its backbone tends to maximize its contact area, through a large number of non covalent bonds that do not block the C–N bending vibration.28 Because unfolded proteins are preferred gold binders, amide III band is statistically more frequently detected at pH 3 (78%).
In addition, this Raman band is highly sensitive to two local sub-structures of the peptide backbone chain named the secondary structure: the α-helix and the β sheets. The α-helix is a structure where the polypeptide backbone follows a right-handed helical path; it is identified with the 1268–1286 cm−1 range. The β sheets consist of an arrangement of stretched polypeptide chain in an extended conformation (called β strand) that are connected by several hydrogen bonds, forming a generally twisted, pleated sheet; they correspond to the 1228–1231 cm−1 wavenumbers range.18,26
For extracting the proportion of each secondary structure, complex spectral analysis is necessary. In spontaneous Raman spectroscopy, it is usually performed by decomposing the acquired spectrum into a sum of functions whose parameters are determined with fitting algorithm (such as linear or non-linear least square methods) for minimizing errors between the recorded spectrum and a mathematical model.24
In this work, spectral decomposition of cumulative SERS spectrum (see Fig. 2B1) is simply performed by counting the identified components in individual spectra (see Fig. 2B2). The calculated occurrences of secondary structures (α-helix, β sheets or random structures) at pH 7 and pH 3 are reported in Fig. 2A.
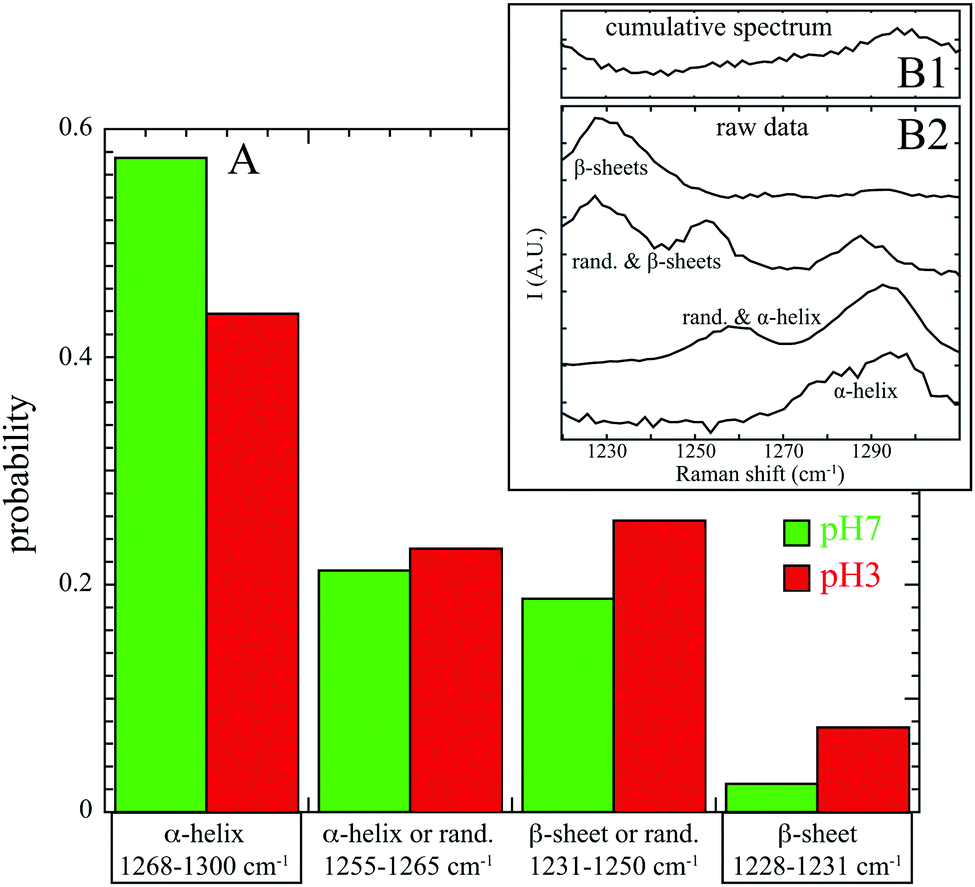 | ||
| Fig. 2 (A) Influence of the pH on the normalized occurrence of secondary structures: α-helix, β sheets or random structures with corresponding Raman shifts (amide III band). Cumulative SERS spectrum at pH 7 is reported in (B1) and selected individual experimental spectra are indicated in (B2). | ||
At pH 7, α-helix secondary structures are mainly detectable in the SERS spectra (57% in α-helix and 21% in random structures and α-helix) and the β sheets are almost missing (2% only). These results are in good agreement with structural information obtained by X-ray crystallography which concludes that albumin structure is mostly α-helical (67%) with no β-sheets.29 Our experimental analysis shows few β sheet structures, which may be partially explained by the fact that BSA adsorption onto substrate induces conformational changes and β-sheets formation.30 Furthermore, this confirms that protein interacts with gold preferentially through β-sheets (and not α-helix) in maximizing the backbone contact with Au nanoparticle.28
At pH 3, two major differences are visible: a decrease in pure α-helix structures (44% instead of 57%) and a significant increase of β sheets or random structures (26% compared to 19% and 8% compared to 2%). Our results at the single protein level confirm again the BSA denaturation below pH 5 already observed by spontaneous Raman spectroscopy of an ensemble of proteins.17
In spontaneous Raman spectroscopy, the ggg conformation of the disulfide bridges (near 510 cm−1) is commonly observed, as in X-ray crystallographic studies of human serum albumin (HSA).29 Other conformations of disulfide bridges have been visualized by other authors for BSA in solution18,19,21,32 and a majority of ggt or tgg conformation has also been see by other groups.21,32
In the present study, in single protein at pH 7, we measure a majority of ggt or tgg conformations (50%) and almost 30% of ggg conformation and a minority of tgt (20%). In an acidic environment (pH 3), the proportion of the different conformations of the disulfide bridges is almost identical (33% for ggg, 32% for ggt or tgg and 35% for tgt). The proportion of tgt conformation of disulfide bridges is increased in comparison with pH 7. We guess that a greater number of thiol groups will be blocked20 when the BSA is adsorbed onto the gold substrate in an unfolded conformation at pH 3. We remark also that the ratio ggg/ggt is larger at pH 3 (=1) than pH 7 (=0.6), in agreement with previous study.19 At pH 7, about six disulfide bridges adopt the energetically unfavorable ggt (or tgg) conformation. At pH 3, the domains I and II of BSA isomerize and the domain III is unfolded. They conclude that the disulfide bridges in domain III may convert from ggt (or tgg) conformation to ggg conformation which is more energetically stable.
Amino acids are identified by comparing Raman bands extracted from previous works33,34 (reported in Table 3) with experimental peaks determined computationally from individual SERS spectra. We consider that at least two specific experimental Raman bands must be identified to ensure reliable detection of this amino acid. Each SERS spectrum was composed by one or more amino acids as shown by the distribution of the number of amino acids detected in SERS spectrum at pH 7 and pH 3 (Fig. 3A). The fact that mostly 3 amino acids can be detected simultaneously proves that the individual spectrum is the fingerprint of a BSA subdomain, and not the whole protein. The broad distribution in the proportion of each amino acid (Fig. 3B1–B9) was found over a large set of individual spectra.
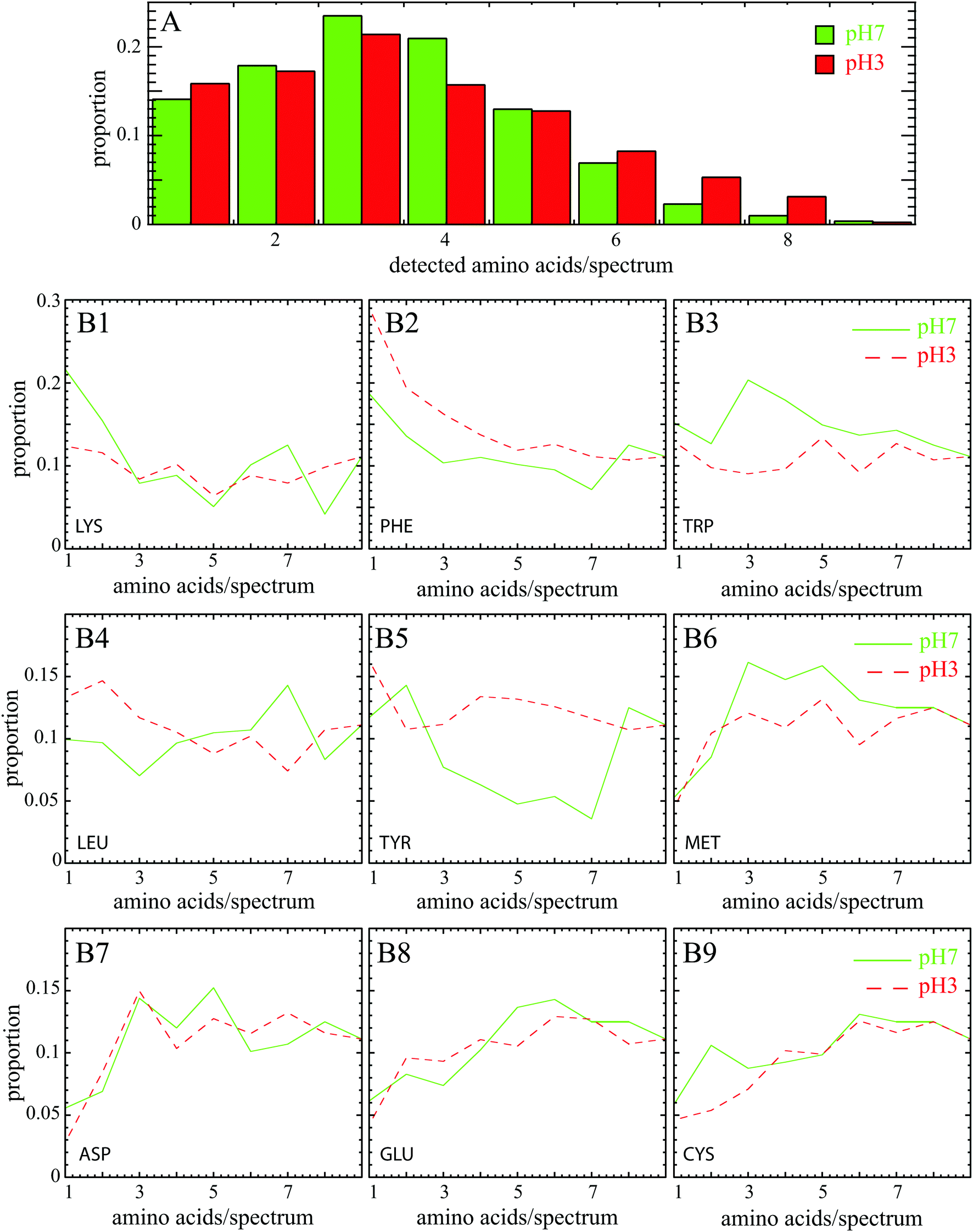 | ||
| Fig. 3 (A) Distribution of the number of amino acids detected per spectrum at pH 7 (in green) and pH 3 (in red). (B1–B9) Proportion of mentioned amino acids at pH 7 (in green solid line) and pH 3 (in red dotted line) as a function of the number of detected amino acids in the individual SERS spectrum. For instance in B1, there is mostly one amino acid per spectrum at pH 7 implying that lysine is mainly at the BSA surface. | ||
Among the potential factors responsible for this variability, we have focused on the impacts of (i) the abundance of amino acids, (ii) their physico-chemical properties (hydrophobic, hydrophilic) related to their position within the protein (surface or core), (iii) their Raman cross sections. All these parameters are reported in Table 4.
| AA | Abund. (%) | Surface (%) | Raman σ | Charge | Hydr. | Exp. pH 7 (%) | Exp. pH 3 (%) |
|---|---|---|---|---|---|---|---|
| LYS | 19 | 73 | + | + | 21 | 12 | |
| PHE | 9 | 22 | +++ | 0 | × | 18 | 29 |
| TRP | 1 | 50 | +++ | 0 | × | 15 | 13 |
| TYR | 6 | 35 | ++ | 0 | 12 | 16 | |
| LEU | 20 | 26 | + | 0 | × | 10 | 13 |
| GLU | 19 | 61 | + | − | 6 | 4 | |
| CYS | 11 | 23 | + | 0 | 6 | 5 | |
| ASP | 13 | 79 | + | − | 5 | 3 | |
| MET | 1 | 25 | + | 0 | × | 5 | 4 |
Note that 4 amino acids (glutamic acid, cysteine, aspartic acid and methionine) are almost never detected alone in experimental SERS spectra but appear in a pool with at least 3 other neighboring amino acids (see Fig. 3). pH has here no impact for one of these three reasons. First, methionine is rare in BSA (only 1%), hence the enhanced scattering signal of this amino acid could be low. However, this argument is not valid for the other amino acids: glutamic acid, cysteine and aspartic acid representing more than 10% of all amino acids. A second reason may be due to the low Raman cross section of these amino acids (cf.Table 4) that are almost not assigned in spontaneous Raman spectroscopy.18 The low SERS signal could also be explained by the remote location of these amino acids from the enhanced area. However, the glutamic acid and aspartic acid are negatively charged polar amino acids that preferentially cover the BSA surface. They also both exhibit one free carboxylic group suitable for interacting with Au nanoparticles.27 The same holds true for the cysteine known to adsorb onto gold surface through Au–S bond.35 One possible explanation could be then that experimental SERS bands are shifted and/or modified by interaction and do not match anymore the reference spontaneous Raman spectra of freely diffusing amino acids; their side chain vibrations are stressed and/or even blocked, thereby changing the selection rules.
The behavior of the Lysine was found to be more understandable. This basic polar amino acid is mainly exposed at the BSA surface and is thus in contact with the environment. Contrary to negatively charged surface amino acids (ASP and GLU), lysine is commonly detected in SERS spectra despite its small Raman cross section. Under the assumption that negatively charged amino acids are preferential anchors to the surface of the nanoparticle, the positively charged Lysine should be close to the active surface area but not directly in contact with gold surface. It is worth noting in Fig. 3 that LYS is mostly present alone at pH 7 whereas it is significantly less visible at pH 3 (12% instead of 21%). Lys preferentially covers the surface of the protein and is thus most accessible when BSA is unfolded (at pH 7) than folded (at pH 3).
The three aromatic amino acids (phenylalanine, tyrosine and tryptophan) are also commonly observed in experimental SERS spectra despite their scarcity (<10%) and their position in BSA (mostly buried inside the protein core at pH 7). The primary reason is their large Raman cross sections (see Table 4); as expected from spontaneous Raman spectrum of BSA.18 PHE and TYR are better detected at pH 3 compared to pH 7 (see Fig. 3) since they become more accessible in the unfolded protein at pH 3. More surprisingly, we observe also that they are mostly detected alone. A possible explanation is that these aromatic amino acids emit a strong SERS signal that potentially masks Raman response from other amino acids.
TRP in BSA has been largely studied because it is an intrinsic fluorescent probe under UV excitation.22 Even if there is only 2 residues in BSA (one located on the surface of the protein in domain I, and the other in the core in domain II8), it is frequently detected in SERS experiments both at pH 7 and pH 3 because it is highly Raman active with these two aromatic rings (Raman cross section of 0.483 × 10−27 cm2 sr−1). When it is detected alone (see Fig. 3), no significant difference exists between pH 7 (15%) and pH 3 (13%), probably because there is the same amount of TRP in the core and at the surface of BSA.
It is well known that tryptophan fluorescence is highly sensitive to its local environment leading to changes in both emission spectra and fluorescence lifetimes.22 Raman bands of TRP are also dependent on the local environment. In particular, tryptophan exhibits a doublet with Raman bands at 1340 and 1360 cm−1 whose relative intensities are an indicator of the local environment.18,36 This doublet clearly identified in Fig. 4A1 and B1 probably originates from Fermi resonance between a fundamental band of an in-plane vibration and one or more combination bands of out of plane vibrations.36,37 Raman shifts of the out of plane vibrations due to hydrophobic interactions with solvent may induce small changes in the relative intensity ratio of the doublet.37
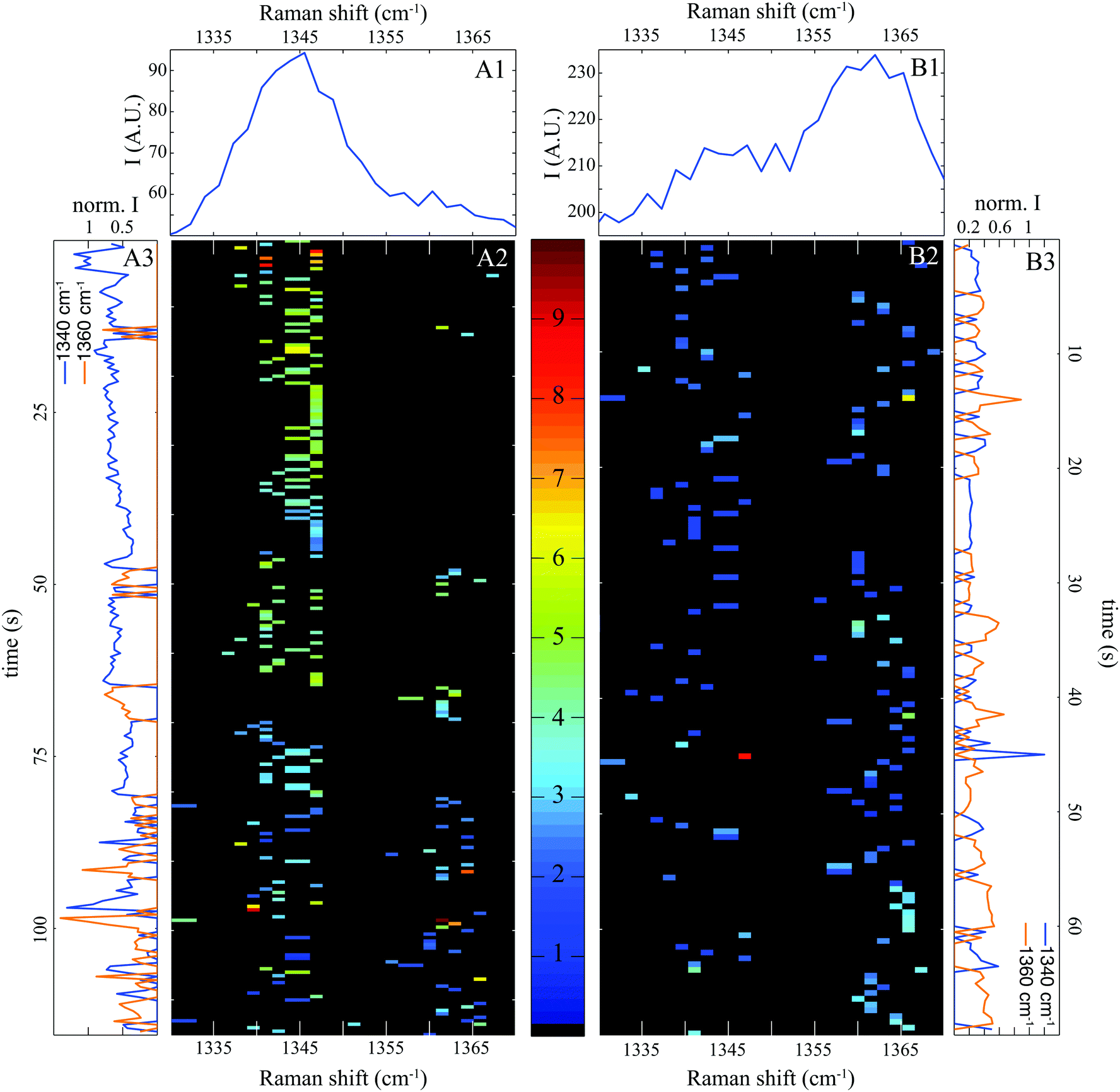 | ||
| Fig. 4 Doublet of tryptophan at 1340 and 1360 cm−1. (A1 and B1) Cumulative SERS spectra at pH 7 and pH 3 respectively. The corresponding spectrograms are indicated in (A2) and (B2) and the temporal evolution of each band is shown in (A3) and (B3). | ||
Note that in individual SERS spectra (represented as spectrograms in Fig. 4A2 and B2), we can detect a band either at 1340 or 1360 cm−1. This is also clearly visible in the temporal evolution of each Raman bands represented in Fig. 4A3 and B3. Two possible explanations for this anti-coincidence can be formulated. First, the in-plane and out-of-plane vibrations implied in the Fermi resonance cannot be excited simultaneously by a linearly polarized laser. Secondly, the molecular vibrations of BSA close to the gold surface are slightly shifted compared to protein freely diffusing in solution. The combination of out of planes modes may be no more satisfied and the Fermi resonance does not occur any more. In any case, this observation confirms that our spectral pointillism probes only single molecule, or even just a subdomain of a single molecule.
Fig. 4A1 and B1 show that the Raman band at 1340 cm−1 is maximized at pH 7, and inversely that the band at 1360 cm−1 is maximum at pH 3. By counting the occurrence of bands at 1340 and 1360 cm−1 and calculating the occurrence ratio (1360/1340), we found a ratio of 0.3 at pH 7 and 1.5 at pH 3. These values can be compared to the intensity ratios (1360/1340) observed in spontaneous Raman. Both ratios of the doublet (1360/1340) increase when the environment is strongly hydrophobic.18,36,38 At physiological pH, BSA is folded and highly soluble in water (poorly hydrophobic). Once unfolded at pH 3, protein becomes water insoluble causing a dramatic increase in viscosity (highly hydrophobic).
At a more detailed level, some variations in the Raman shifts of the doublet are observed. They were expected by Takeuchi et al. who assigned them to hydrophobic interactions with solvent molecules.38
Correlated information between amino acids and secondary structures
By studying secondary structures and selected amino acids, we are able to confirm conclusions deduced from spontaneous Raman spectroscopy. However, our SERS spectral pointillism enables to obtain additional information by correlating identification of both secondary structures and amino acids. Based on the individual SERS spectra, our approach allows easily to identify which amino acids participate in the secondary structures. We have represented in Fig. 5 the occurrence of the 4 most frequently observed amino acids into secondary structures (α-helix, β-sheet or random). | ||
| Fig. 5 Probability of presence of 4 selected amino acid in secondary structures at pH 7 (left) and pH 3 (right). | ||
We show that the probability of presence at pH 7 is lower than the occurrence at pH 3 indicating that the BSA secondary structure is not fully accessible at pH 7 probably because the BSA globular structure prevents its access.
At pH 7, hydrophobic amino acids (PHE and TRP) exhibit highest probability of presence in α-helix experimentally (69% and 72%). These results are in agreement with structural study demonstrating that PHE and TRP participate respectively in 90% and 100% of α-helix.29 On the contrary, TYR and LYS exhibit experimentally lowest occurrence in α-helix (54% and 43%), in agreement with structural investigation showing that respectively 85% of TYR and 75% of LYS participate in BSA α-helix.29 From individual SERS spectra, we can then optically retrieve information about the amino acid environment with same trends than crystallographic structure investigation.
At pH 3, no crystallographic structure of BSA have been determined. In Fig. 5, we see that the four most frequently observed amino acids have almost the same occurrences in secondary structures (α-helix and β-sheet), proving that they are probed similarly due to the unfolded conformation of the protein. Because amino acids tend to interact preferentially with gold through their backbone,27 the affinity of the unfolded protein for gold NPs is larger than those of the folded BSA, as illustrated by the higher total occurrences at pH 3. We finally note experimentally for all amino acids a significant increase in β-sheet content, as expected from spontaneous Raman spectroscopy17 and confirming that proteins with β-sheet secondary structure are preferred gold binders.28
Conclusions
In this work, we propose a new concept called spectral pointillism that exploits temporal fluctuations of enhanced spectra for investigating protein in the single molecule regime. This approach requires an optimized SERS substrate for fast SERS acquisition and adapted signal processing for treating numerous temporally fluctuating spectra. We apply it on BSA, the most widespread protein in plasma that have been largely studied in spontaneous Raman spectroscopy.We first investigate polypeptide backbone and secondary structures (α-helix and β-sheets) of two BSA conformations. By analysing individual SERS spectra, we retrieve previous results obtained by spontaneous Raman spectroscopy confirming that numerous successive measurements in the single molecule regime provide the same information as an average acquisition of multiple proteins.
Our spectral pointillism can also be easily applied for decomposing cumulative SERS spectrum by simply counting identified components in individual spectra. We notably used this treatment for determining secondary structure proportions in the amide III band of the polypeptide backbone.
Furthermore, our approach allows to go one step further by giving access to amino acids information such as their identification and proportion. We notably demonstrate that 3 distinct amino acids are mainly observed in our individual experimental SERS spectra, proving that SERS measurements probe only a single protein subdomain. Significant differences between amino acid distributions have been shown following the BSA conformation. Surface amino acids such as LYS are preferentially observed in a physiological environment where the protein is folded.
Investigation of tryptophan Fermi doublet at 1340 or 1360 cm−1 is also extremely instructive. These two bands are never detected simultaneously in individual spectra because either they are never excited together by a linearly polarized laser or out-of-plane vibrations may be shifted and prevent from the Fermi resonance, confirming that SERS experiments probe just a subdomain of single molecule. Furthermore, this doublet gives also information on the BSA conformation since its ratio (1360/1340) is dependent on the hydrophobicity, which is altered when the protein is unfolded.
In addition, by correlating information between amino acids and secondary structures, spectral pointillism gives access to further details on amino acids environment that are usually accessible solely with crystallographic structure measurements.
The potential of our approach was thus demonstrated to explore structural dynamics of proteins at a single-molecule level (and confirmed with the study of disulfide bridges). By probing successively various areas of single BSA, we can gain statistical information about the whole protein. Spectral pointillism enables not only to record individual amino acids but also to identify their role in secondary structure, which is not accessible to date in spontaneous Raman.
In this work, we apply spectral pointillism on BSA. Our concept could be easily adapted to other biomolecules. We thus believe that spectral pointillism is a valuable tool for gaining inestimable insights into single protein conformation.
Acknowledgements
This work was supported by the Labex ACTION program (ANR-11-LABX-0001-01) and the Burgundy Region Council (PARI II NANO2BIO, FABER 2014). This work was performed in the context of the European COST Action MP1302 Nanospectroscopy.References
- H. Xu, E. J. Bjerneld, M. Käll and L. Börjesson, Phys. Rev. Lett., 1999, 83, 4357–4360 CrossRef CAS.
- E. Ringe, B. Sharma, A.-I. Henry, L. D. Marks and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2013, 15, 4110 RSC.
- H. Yockell-Lelièvre, F. Lussier and J.-F. Masson, J. Phys. Chem. C, 2015, 119, 28577–28585 CrossRef.
- E. Le Ru and P. Etchegoin, Principles of Surface-Enhanced Raman Spectroscopy: and Related Plasmonic Effects, Elsevier, 2008 Search PubMed.
- T. Brulé, A. Bouhelier, H. Yockell-Lelièvre, J.-E. Clément, A. Leray, A. Dereux and E. Finot, ACS Photonics, 2015, 2, 1266–1271 CrossRef.
- A. Leray, T. Brulé, M. Buret, G. C. des Francs, A. Bouhelier, A. Dereux and E. Finot, Sci. Rep., 2016, 6, 20383 CrossRef CAS PubMed.
- C. David, N. Guillot, H. Shen, T. Toury and M. Lamy de la Chapelle, Nanotechnology, 2010, 21, 475501 CrossRef PubMed.
- M. Iosin, V. Canpean and S. Astilean, J. Photochem. Photobiol., A, 2011, 217, 395–401 CrossRef CAS.
- J. Zhou, K. Ren, Y. Zhao, W. Dai and H. Wu, Anal. Bioanal. Chem., 2012, 402, 1601–1609 CrossRef CAS PubMed.
- S. Yang, X. Dai, B. B. Stogin and T.-S. Wong, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 268–273 CrossRef CAS PubMed.
- P. Singh, T. Deckert-Gaudig, H. Schneidewind, K. Kirsch, E. M. V. S. Lantman, B. M. Weckhuysen and V. Deckert, Phys. Chem. Chem. Phys., 2015, 17, 2991–2995 RSC.
- A. Otto, J. Raman Spectrosc., 2002, 33, 593–598 CrossRef CAS.
- J. Margueritat, A. Bouhelier, L. Markey, G. C. des Francs, A. Dereux, S. Lau-Truong, J. Grand, G. Lévi, N. Félidj, J. Aubard and E. Finot, J. Phys. Chem. C, 2012, 116, 26919–26923 CrossRef CAS.
- G. Smith, J.-S. Girardon, J.-F. Paul and E. Berrier, Phys. Chem. Chem. Phys., 2016, 18, 19567–19573 RSC.
- H. M. Lee, S. M. Jin, H. M. Kim and Y. D. Suh, Phys. Chem. Chem. Phys., 2013, 15, 5276 RSC.
- G. Nawrocki and M. Cieplak, J. Phys. Chem. C, 2014, 118, 12929–12943 CrossRef CAS.
- V. J. Lin and J. L. Koenig, Biopolymers, 1976, 15, 203–218 CrossRef CAS PubMed.
- A. Synytsya, P. Alexa, J. de Boer, M. Loewe, M. Moosburger, M. Würkner and K. Volka, J. Raman Spectrosc., 2007, 38, 1646–1655 CrossRef CAS.
- K. Nakamura, S. Era, Y. Ozaki, M. Sogami, T. Hayashi and M. Murakami, FEBS Lett., 1997, 417, 375–378 CrossRef CAS PubMed.
- K. Aoki, H. Okabayashi, S. Maezawa, T. Mizuno, M. Murata and K. Hiramatsu, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1982, 703, 11–16 CrossRef CAS.
- C. David, S. Foley, C. Mavon and M. Enescu, Biopolymers, 2008, 89, 623–634 CrossRef CAS PubMed.
- U. Anand and S. Mukherjee, Phys. Chem. Chem. Phys., 2013, 15, 9375–9383 RSC.
- K. J. Freedman, M. Jürgens, A. Prabhu, C. W. Ahn, P. Jemth, J. B. Edel and M. J. Kim, Anal. Chem., 2011, 83, 5137–5144 CrossRef CAS PubMed.
- X. Chen, Y. Lai, X. Chen, Y. Shi and D. Zhu, Analyst, 2016, 141, 5759–5766 RSC.
- T. Brulé, H. Yockell-Lelièvre, A. Bouhelier, J. Margueritat, L. Markey, A. Leray, A. Dereux and E. Finot, J. Phys. Chem. C, 2014, 118, 17975–17982 CrossRef.
- H. Fabian and P. Anzenbacher, Vib. Spectrosc., 1993, 4, 125–148 CrossRef CAS.
- H. Heinz, B. L. Farmer, R. B. Pandey, J. M. Slocik, S. S. Patnaik, R. Pachter and R. R. Naik, J. Am. Chem. Soc., 2009, 131, 9704–9714 CrossRef CAS PubMed.
- M. Hoefling, F. Iori, S. Corni and K.-E. Gottschalk, ChemPhysChem, 2010, 11, 1763–1767 CrossRef CAS PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CrossRef CAS PubMed.
- S. Servagent-Noinville, M. Revault, H. Quiquampoix and M.-H. Baron, J. Colloid Interface Sci., 2000, 221, 273–283 CrossRef CAS PubMed.
- H. Sugeta, A. Go and T. Miyazawa, Chem. Lett., 1972, 83–86 CrossRef CAS.
- M. Iosin, F. Toderas, P. L. Baldeck and S. Astilean, J. Mol. Struct., 2009, 924-926, 196–200 CrossRef CAS.
- G. Zhu, X. Zhu, Q. Fan and X. Wan, Spectrochim. Acta, Part A, 2011, 78, 1187–1195 CrossRef PubMed.
- P. Candeloro, E. Grande, R. Raimondo, D. Di Mascolo, F. Gentile, M. L. Coluccio, G. Perozziello, N. Malara, M. Francardi and E. Di Fabrizio, Analyst, 2013, 138, 7331–7340 RSC.
- J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS PubMed.
- I. Harada, T. Miura and H. Takeuchi, Spectrochim. Acta, Part A, 1986, 42, 307–312 CrossRef.
- S. D. Dieng and J. P. M. Schelvis, J. Phys. Chem. A, 2010, 114, 10897–10905 CrossRef CAS PubMed.
- H. Takeuchi, Biopolymers, 2003, 72, 305–317 CrossRef CAS PubMed.
Footnote |
| † Contributed equally to this work. |
| This journal is © the Owner Societies 2017 |