
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzimidazolylquinoxalines: novel fluorophores with tuneable sensitivity to solvent effects†
Timur I.
Burganov
a,
Nataliya A.
Zhukova
a,
Vakhid A.
Mamedov
a,
Christoph
Bannwarth
b,
Stefan
Grimme
b and
Sergey A.
Katsyuba
 *a
*a
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Centre of the Russian Academy of Sciences, Arbuzov str. 8, 420088 Kazan, Russia. E-mail: katsyuba@iopc.ru; skatsyuba@yahoo.com
bMulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie der Universität Bonn, Beringstr. 4, 53115 Bonn, Germany
First published on 31st January 2017
Abstract
We report on the photophysical properties, conjugation, conformational behavior, intra- and intermolecular hydrogen bonds (HBs) of a series of novel fluorophores, consisting of 3-arylquinoxaline and benzimidazole moieties linked by a single CC bond. Computations employing density functional theory (DFT) reveal that conjugation between these moieties stabilizes syn-conformers with two HB centers located on the same side of the molecule. Anti-conformers form stronger intermolecular HBs with DMSO and DMF than syn-conformers, and this influences the energy gap between syn- and anti-forms, especially upon excitation of the molecules to the S1 state. Substituents introduced in various positions of the molecules modify their conformational behavior, and mutual disposition of excited singlet states relative to the ground states. Various substitution patterns produce very different effects on relative quantum yield of luminescence: from a moderate increase in polar DMSO and DMF relative to 1,2-dichloroethane solutions to complete quenching of emission which is observable in polar media. The observed behavior is understood with the aid of DFT and time-dependent DFT calculations. The tuneability of the spectroscopic range of the luminescence and especially of its sensitivity to environmental effects allows rational design of the novel fluorophores of this family for various applications.
Introduction
Bifunctional azaaromatic compounds, containing a hydrogen bond (HB) donor and acceptor located in separate moieties linked by a single bond, attract considerable attention as fluorophores often demonstrating a remarkable sensitivity to environmental effects, which makes them suitable for fluorescence sensing in a wide range of chemical, biochemical and biophysical applications (see, for example, ref. 1 and 2 and references cited herein). Such a topology often leads to a solvent-dependent appearance of two conformers, syn and anti, with the two HB centers located either on the same or on the opposite sides of the molecule, respectively (Fig. 1). It is believed that an intramolecular HB stabilizes the syn conformer, which usually dominates in nonpolar solvents. In polar solvents, where formation of intermolecular HBs with HB donor and/or acceptor sites is possible, the fraction of the more polar anti conformer may become significant. In spite of the vast number of studies of these conformationally flexible systems, they were limited to systems, comprising either a pyridyl or a naphtyridine moiety as a HB acceptor and a pyrrole, pyrazole or indole unit as an intramolecular HB donor (see, for example, pyridylpyrroles,3 -pyrazoles4,5 and -indoles,5 or indole-naphtyridines7). These latter units typically contain the only heteroatom in the immediate vicinity of the C–C bond linking the HB donor and acceptor groups (Fig. 1).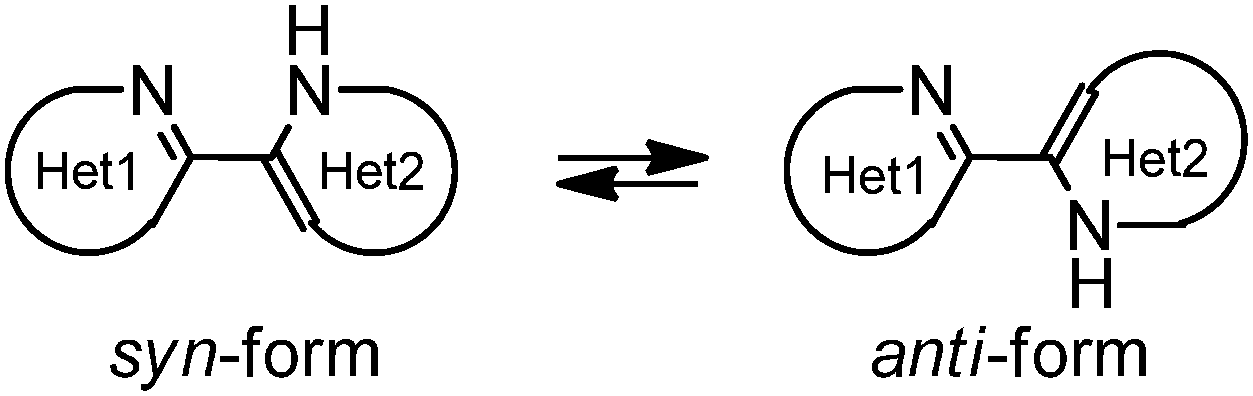 | ||
| Fig. 1 Syn- and anti-conformers of bifunctional azaaromatic compounds. | ||
Recently we have synthesized a series of 2-(benzimidazol-2-yl)-3-arylquinoxalines8 (Fig. 2), where the benzimidazole moiety, being the HB donor owing to its NH group, also includes another nitrogen atom immediately adjacent to the C–C link to the HB acceptor, i.e., the 3-arylquinoxaline moiety.
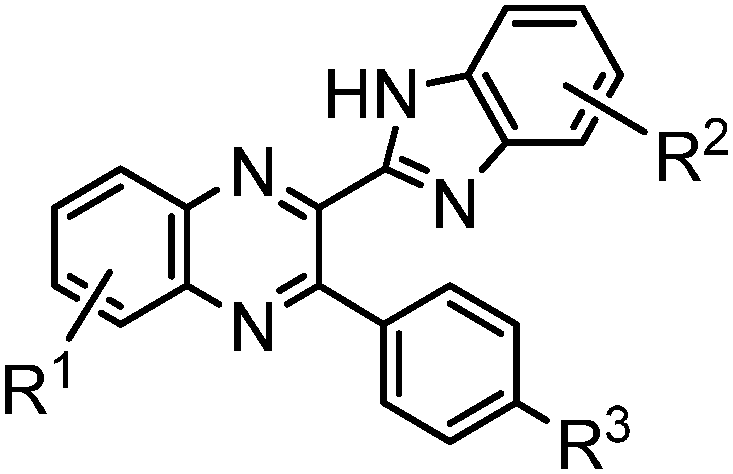 | ||
| Fig. 2 General formula of novel 2-(benzimidazol-2-yl)-3-arylquinoxalines. | ||
This structural peculiarity may result in inversion of relative polarity of syn and anti forms as compared with other bifunctional azaaromatic compounds mentioned above. In the present work we study internal rotation, conjugation, HB and photophysical characteristics of these novel compounds in polar and nonpolar solvents. The results demonstrate that absorption and emission of the benzimidazolylquinoxalines (BQ) and environmental effects influencing their luminescence are strongly dependent on substituents introduced in various positions of the aromatic moieties of these compounds.
Experimental and computational details
All compounds considered were synthesized according to the protocols described earlier.8,9 Electronic absorption (UV-Vis) and steady state fluorescence spectra were recorded at room temperature on a Perkin-Elmer Lambda 35 spectrometer and a Cary Eclipse Fluorescence Spectrophotometer (Varian), respectively, using 10 mm quartz cells. Absorption spectra were registered with a scan speed of 480 nm min−1, using a spectral width of 1 nm. All samples were prepared as solutions in three solvents: 1,2-dichloroethane (DCE), dimethylsulfoxide (DMSO), and dimethylformamide (DMF), with the concentrations ranging from ∼10−5 to ∼10−4 mol L−1. The absorbance at excitation wavelength was less than 0.1 to avoid the “inner filter effect”. Fluorescence quantum yields were measured using aqueous solution of quinine sulfate as the standard (λex = 355 nm, φ = 0.57 in 0.1 M H2SO4). Appropriate corrections were made for the optical density of the solutions and the refractive index of the medium.10All density functional theory (DFT) calculations were performed with the Turbomole 6.4 program package.11 In the ground state self-consistent field (SCF) calculations, the resolution-of-the-identity approach for Coulomb and exchange integrals (RI-JK)12–15 was employed. In general the spherical def-TZVP atomic orbital (AO) basis set was used.16 All structures were optimized with the hybrid PBE0 functional.17 In all geometry optimizations the D3 approach18 to describe the London dispersion interactions together with the Becke–Johnson (BJ) damping function19 was employed as implemented in the TURBOMOLE program. Stationary points were characterized as minima by frequency analyses. The same frequencies were used in the computations of the free energy within the framework of a modified rigid rotor, harmonic oscillator statistical treatment (ΔGRRHO).20 The optimized geometries were further used for the single-point calculations of the electronic energies by applying the PW6B95 meta-hybrid density functional21 in combination with the quadruple-zeta def2-QZVP Ahlrich's basis set.22,23 The computed free energies are then obtained from ΔGgas = ΔE(PW6B95/def2-QZVP) + ΔGRRHO(PBE0/def-TZVP). Solvent effects on the thermochemical properties have been obtained by the COSMO-RS method24 (COSMOtherm software package25) based on BP86/TZVP26 calculations (parameterization from 2012). Solvation contributions to free energies at 298.15 K are computed from the gas phase structures obtained at the abovementioned levels of theory. The computed free energies are then obtained by ΔGsolution = ΔGgas + ΔδGCOSMO-RS.
Time-dependent density functional response theory (TD-DFT)27–29 has been employed to compute the vertical excitation energy (i.e., absorption and emission wavelengths) and oscillator strength on the ground state and first excited state geometries, each optimized in the gas phase as well as in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with DMSO molecules. The equilibrium geometries of the lowest excited singlet states have been determined at the PBE0-D3(BJ)/def-TZVP level, making use of Tamm–Dancoff approximated TD-DFT30 for calculation of the vertical excitation energies. In most cases, the spectra were broadened by Gaussian functions with a full-width at 1/e height of 0.4 eV. No energy shift has been applied. The dipole length representation is used to calculate oscillator strengths discussed in the present paper. The hybrid B3LYP31,32 functional in combination with the 6-31+G* basis set33–36 was used for computation of the Raman spectra according to our previous experience in studies of conjugational effects.37 Molecular orbitals based on PBE0/def-TZVP were visualized in the ChemCraft 1.6 program38 with a 0.03 contour value.
:1 complexes with DMSO molecules. The equilibrium geometries of the lowest excited singlet states have been determined at the PBE0-D3(BJ)/def-TZVP level, making use of Tamm–Dancoff approximated TD-DFT30 for calculation of the vertical excitation energies. In most cases, the spectra were broadened by Gaussian functions with a full-width at 1/e height of 0.4 eV. No energy shift has been applied. The dipole length representation is used to calculate oscillator strengths discussed in the present paper. The hybrid B3LYP31,32 functional in combination with the 6-31+G* basis set33–36 was used for computation of the Raman spectra according to our previous experience in studies of conjugational effects.37 Molecular orbitals based on PBE0/def-TZVP were visualized in the ChemCraft 1.6 program38 with a 0.03 contour value.
Results and discussion
Conformations in the ground (S0) and excited (S1) states
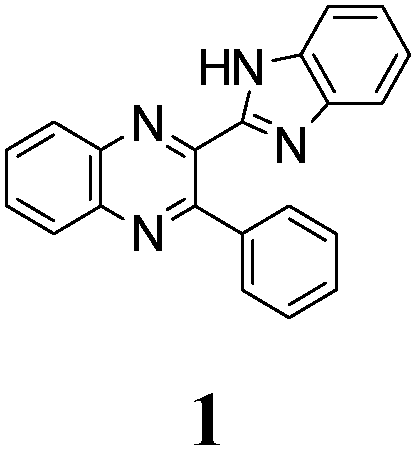
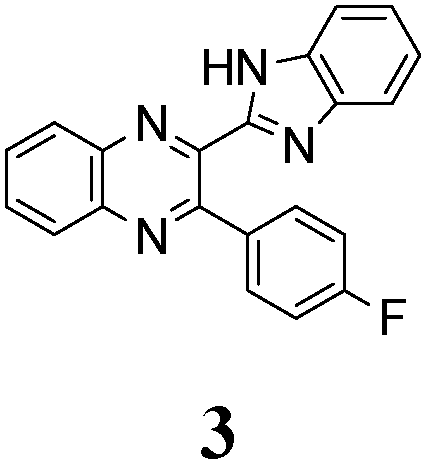
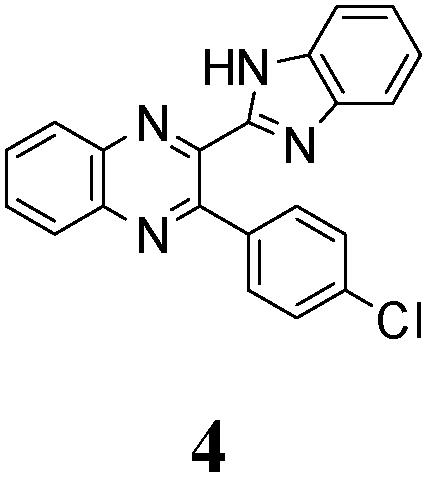
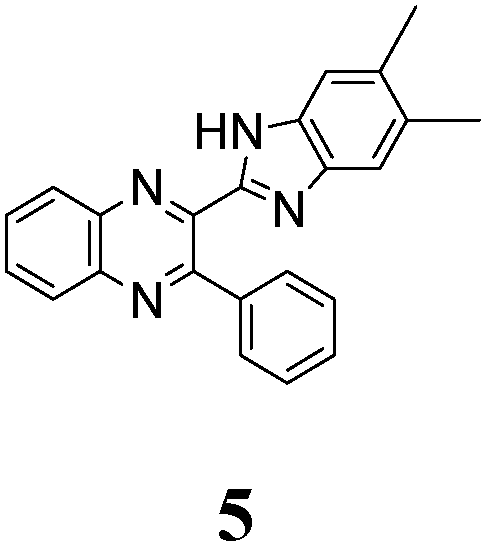
The quinoxaline moiety (Q) of the studied molecules is linked by CQ–CB and CQ–CPh single bonds with benzimidazole (B) and phenyl (Ph) moieties, respectively. According to quantum chemical computations, internal rotation about the CQ–CPh bond results in the only stable conformation with a dihedral angle of ca. 40–50° between the Q and Ph planes, depending on the specific molecule and the conformation about the CQ–CB bond. In spite of non-coplanarity of the Q and Ph rings, both aromatic moieties are conjugated, which is clearly indicated by ca. a ten- to fifteen-fold increase of the Raman intensity of the band of the phenyl ν8a mode at ca. 1600 cm−1 (I8a) relative to the corresponding band of toluene (for 1, 2 and 5, see Table 1 for numbering of compounds) or para-fluoro- (for 3) and para-chloro-toluenes (for 4 and 6).37,39 These toluenes are regarded as “external standard” molecules, where the aromatic ring does not conjugate with the methyl group.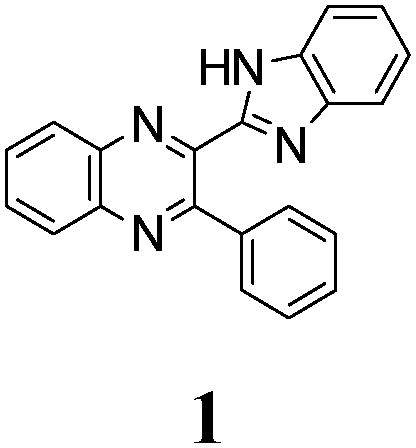
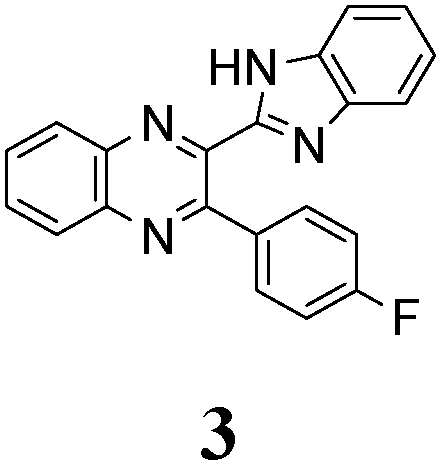
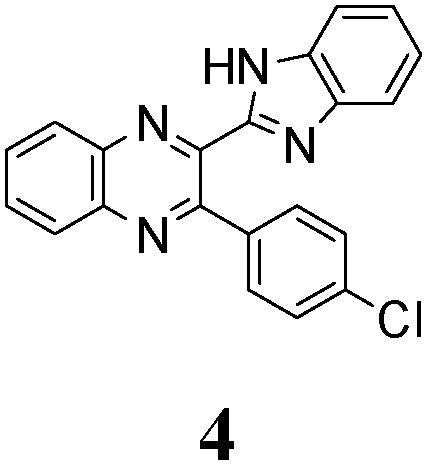
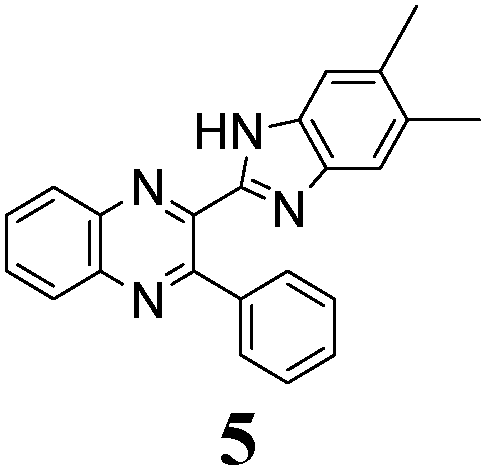
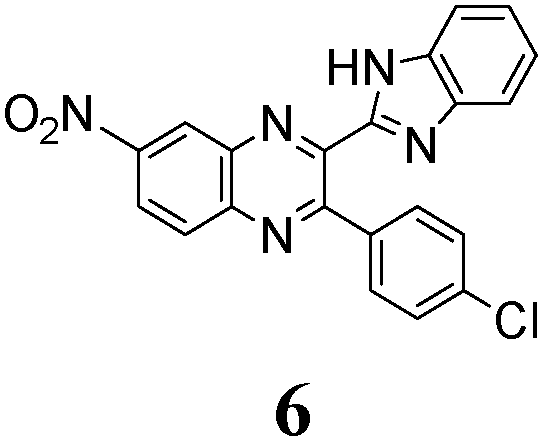
Internal rotation about the CQ–CB bond results in two stable conformations: syn and anti (vide supra). Q and B moieties lie practically in the same plane in the syn-conformation (Fig. 1S, ESI†), but are not coplanar in the anti-conformation for steric reasons. Conjugation between the almost coplanar Q and B moieties in the syn-conformers is more pronounced than in the non-planar anti-conformers, which is suggested by the highest occupied molecular orbital (HOMO) pictures (Fig. 3) and red shift of the electronic absorption maxima in the TDDFT calculated spectra of the syn-conformers relative to the anti-forms (Fig. 4).
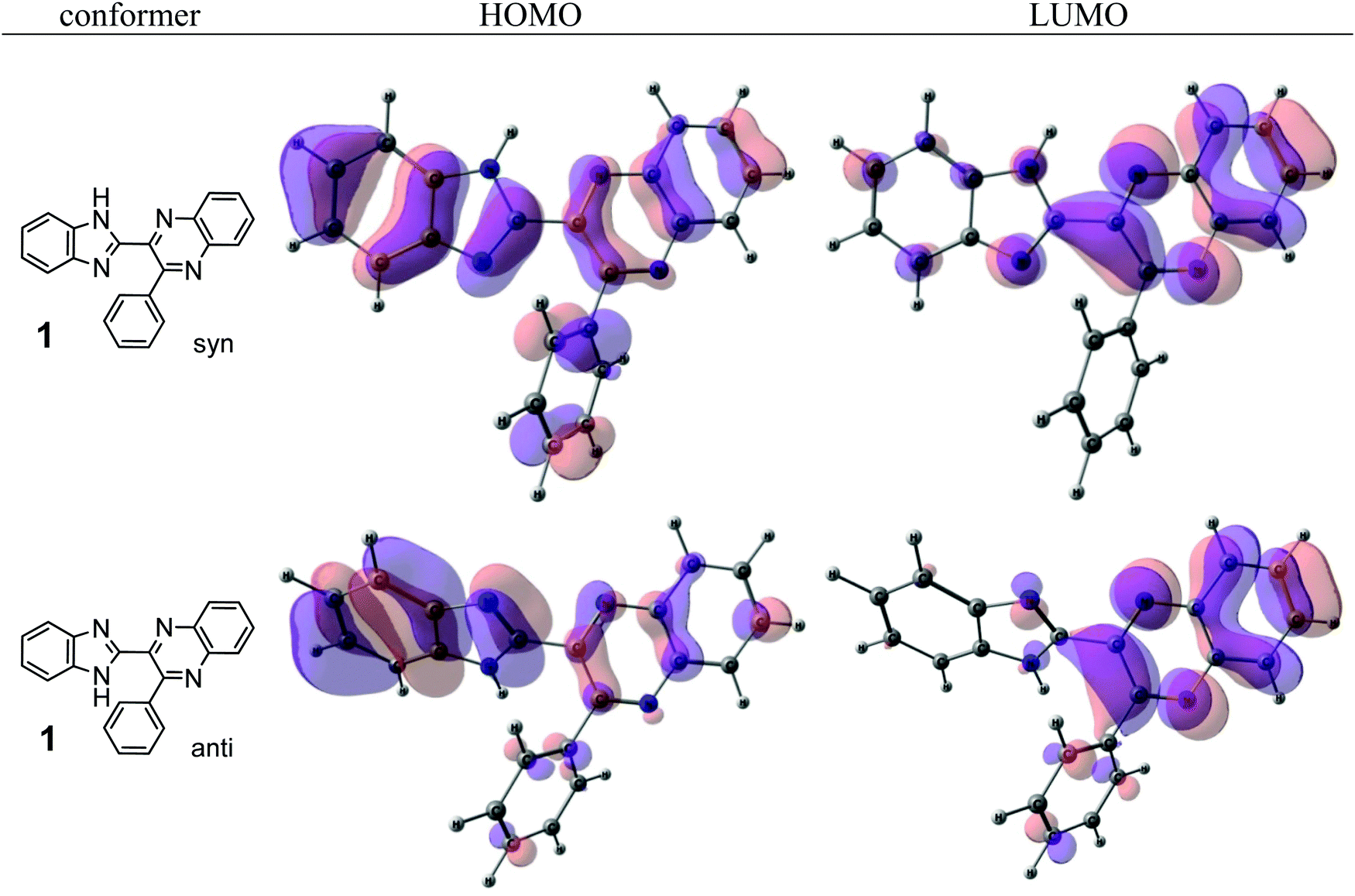 | ||
| Fig. 3 The highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals of the syn- and anti-conformers of 1. | ||
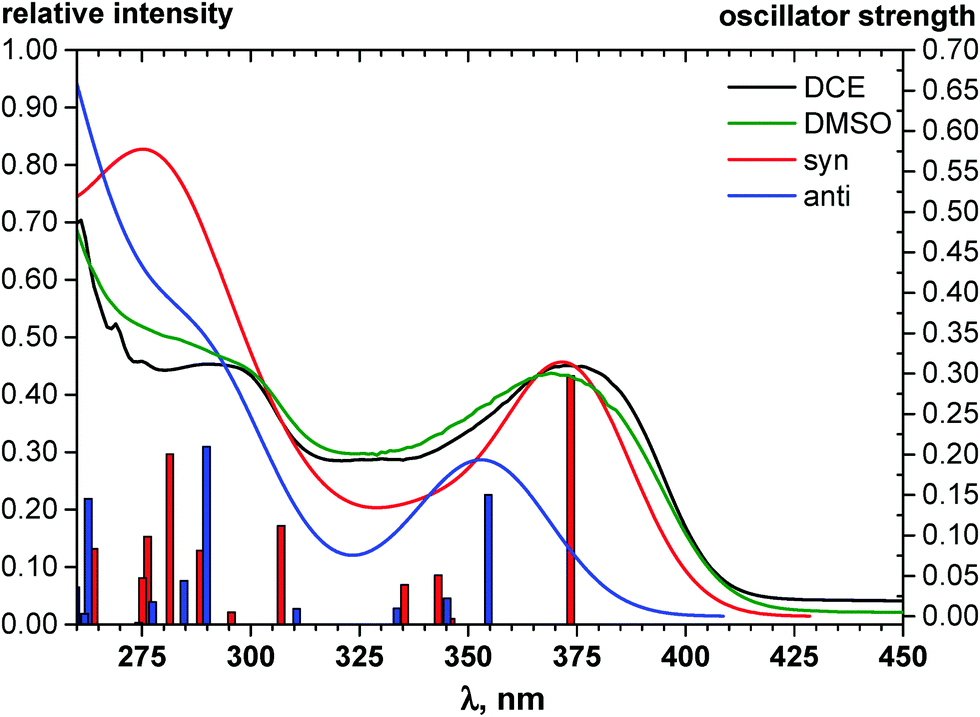 | ||
| Fig. 4 TDDFT simulated absorption spectra of syn- (red lines) and anti-conformers (blue lines) of 3 in comparison with the experimental spectrum registered for 3 solutions in DCE (black line) and DMSO (green line). The heights of the vertical straight lines correspond to the calculated oscillator strengths of the corresponding vertical transitions. Red and blue envelope curves are obtained by broadening of the vertical straight lines by Gaussian functions with a full-width at 1/e height of 0.4 eV. | ||
To estimate the possible role of intramolecular HB NH⋯N in stabilization of syn-conformers relative to the anti-conformers we have compared structural, spectroscopic and electron density characteristics of the conformers. The intramolecular HB can be quantitatively characterized by elongation of the N–H bond of the syn-conformer relative to the anti-conformer, ΔNHsyn–anti, and a red shift and an increase of the intensity of the infrared (IR) band of NH stretching vibrations ΔνNHsyn–anti and ΔI(νNH)syn–anti, respectively.40 Almost negligibly small values found for all three characteristics of HB strength (Table 1S, ESI†) suggest that NH⋯N intramolecular H-bonding is either very weak or absent. E.g., ΔNHsyn–anti = 0.00123 Å in the case of 1. Following the Bader analysis of the electronic density (see, e.g., ref. 41 and references cited herein) a stabilizing HB could be indicated by a NH⋯N bond critical point. Nevertheless, the critical points at the NH⋯N intramolecular contacts were localized in none of the syn-conformers of molecules 1, 3–6. This fact is additional evidence of the absence or the insignificant role of intramolecular HBs in conformational behavior of the studied compounds. Hence, the energetic preferability of syn-conformers (Table 1) results from the other factors, e.g., the abovementioned difference in conjugation of syn- and anti-conformers, as well as steric congestion in the anti-conformation.
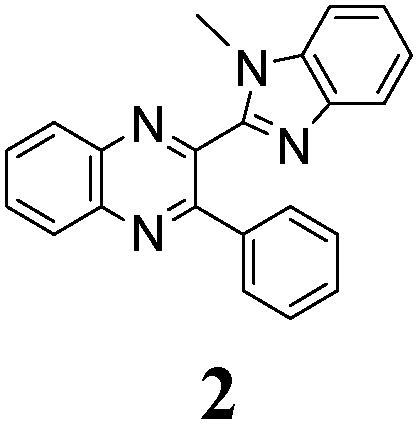
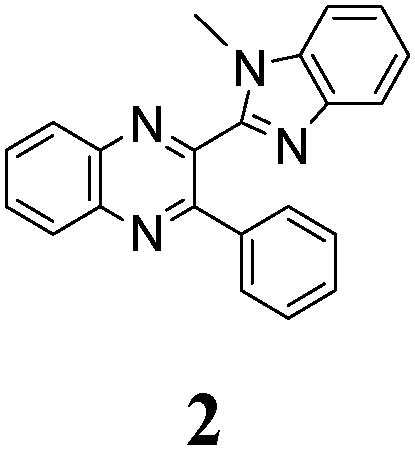
The minimal difference in Gibbs free energy (ΔG) of the anti-conformer relative to the corresponding syn-conformer, calculated for the gas phase, is found to be 3.2 kcal mol−1 for compound 1. The barrier ΔG≠ for syn→anti transformation during internal rotation about the CQ–CB bond amounts to 5.9 kcal mol−1 for 1, while the anti→syn barrier is only 2.8 kcal mol−1. Thus, population of syn-conformations is favorable thermodynamically. A minimal ΔG value is found for molecule 2, where replacement of the N–H moiety of molecule 1 by an N–Me moiety results in steric strain induced by the methyl group, which, in turn, essentially increases non-planarity of the syn-conformer of 2 (dihedral angle between Q and B moieties = 44°) and strongly destabilizes this conformer relative to the anti-form (Table 1).
The above conformational behavior of the isolated molecules suggests that in non-polar media benzimidazolylquinoxalines 1, 3–6 should exist mainly in the syn-form. Solvent effects of the moderately polar aprotic solvent, 1,2-dichloroethane (DCE), on the thermochemical properties obtained by the COSMO-RS24 method do not change the above picture essentially: ΔG (syn–anti) estimated for DCE solutions varies between 1.3 and 2.1 kcal mol−1 (Table 1). To estimate the possible influence of polar solvents, dimethylsulfoxide (DMSO) and dimethylformamide (DMF), on conformational equilibria of the compounds under study, we optimized H-bonded 1:1 complexes of syn- and anti-conformers with the respective solvent molecules (e.g., Fig. 5).
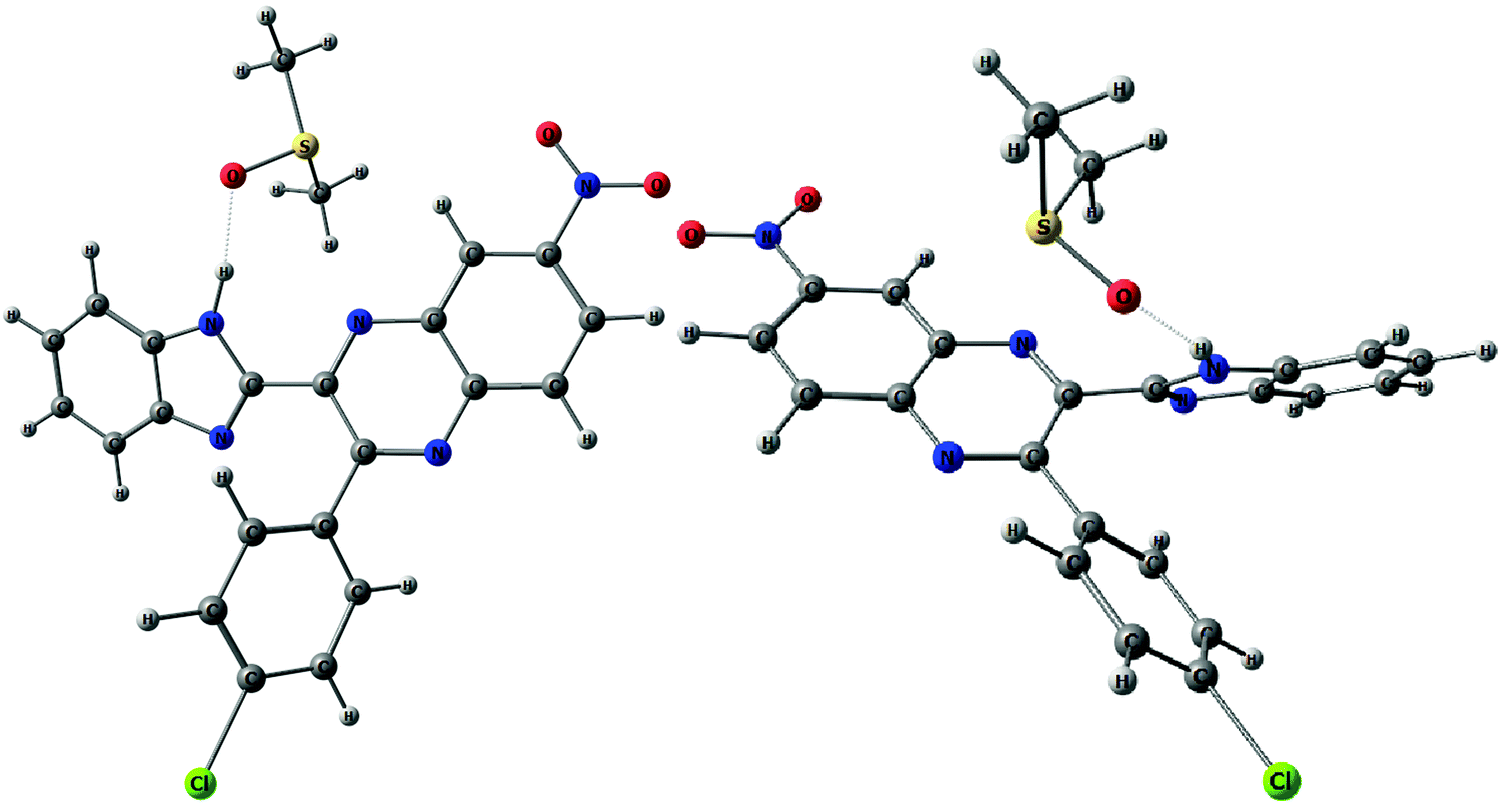 | ||
| Fig. 5 H-bonded 1:1 complexes of syn- (left) and anti-conformers (right) of 6 with DMSO molecules. H-bonds are shown with dotted lines. | ||
According to this explicit solvation approach, HB-complexes formed by anti-conformers with the solvent molecules are stronger than the HBs formed by syn-conformers (Table 1S, ESI†), and this difference results in stabilization of the anti-forms relative to the syn-conformations. It should be noted that the influence of DMSO, taken into account implicitly within the framework of the COSMO-RS24 model, produces very similar thermodynamic effects: even more polar syn-conformers are destabilized in this polar solvent relative to less polar anti-conformers (Table 1). In spite of this decrease of ΔG (syn–anti) values to ca. 1–3 kcal mol−1, the syn-conformers are still expected to dominate even in the polar solutions, though the presence of the anti-conformers cannot be excluded in these cases. The domination of syn-conformers in polar media is confirmed by comparison of the TDDFT simulated absorption spectra of syn- and anti-conformers with the experimental spectra of 1–6 registered for DMF or DMSO solutions (see, e.g., Fig. 4). In contrast, similar comparison for 2 (Fig. 2S, ESI†) suggests domination of anti-conformers both in DCE and in polar solutions.
According to our quantum chemical computations, the conformational behavior of the isolated molecules in the first excited state, S1, is qualitatively similar to that described above for the ground state, S0 (Table 1). Energy differences, ΔE, between syn- and anti-conformers in the S1 state moderately increase relative to the S0 state because syn-conformers are additionally stabilized by the NH⋯N intramolecular HB, which is quite pronounced in the excited state.42 The strengthening of the intramolecular HBs can be ascribed to the electron density redistribution upon excitation, resulting in an increase in the excited state basicity of the proton acceptor and/or an increase in the excited state acidity of the proton donor. Similar strengthening of the NH⋯O intermolecular HBs with the DMSO or DMF solvent molecules in the excited state results in the inversion of relative energetic stability of syn- and anti-conformers of BQs in the S1 state (Table 1).43 As anti-conformers form stronger HBs with DMSO or DMF than syn-conformers (Table 1S, ESI†), the former conformers are more effectively stabilized in the S1 state than the latter. These results suggest that the emission spectra of 1, 3–6 should be assigned mainly to syn-conformers in the case of DCE solutions, while the possible presence of anti-conformers should be taken into account in the case of DMSO and DMF solutions. In contrast, both anti- and syn-conformers of 2 are expected to determine emission of all solutions of this compound. It should be noted though that according to TD-DFT computations, the energies of vertical S1 → S0 transitions of syn- and anti-conformers practically coincide (Table 2S, ESI†).
Electronic absorption and fluorescence spectra
The electronic absorption and fluorescence spectra were recorded in three aprotic solvents: intermediately-polar DCE (ε = 10.36), and polar DMF (ε = 36.7) and DMSO (ε = 46.7). The spectra are compared in Fig. 6 and in Table 2. On the basis of TDDFT computations the lowest-energy absorptions in the spectra (see, e.g., Fig. 4) are assigned mainly to transitions between the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) of syn-conformers of 1, 3–6, and of anti-conformers of 2. Visualization of these orbitals (Fig. 3) demonstrates that the HOMO of 1 is spread over π-systems of all three aromatic rings: B, Q, and Ph. The LUMO of 1 is represented mainly by the π*-orbital of the Q moiety. This visual analysis of frontier MOs suggests that the first vertical transition results in partial charge transfer from the donor B moiety to the acceptor Q moiety. The charge-transfer character of this transition is confirmed also by a ca. 3- to ca. 5-fold increase of dipole moments of the molecules in the S1 state relative to the ground S0 state (Table 3S, ESI†). In spite of this, all main features of the experimental absorption and emission spectra of the compounds under study were nicely reproduced (Table 2) by TDDFT computations at the PBE0/def-TZVP level of approximation.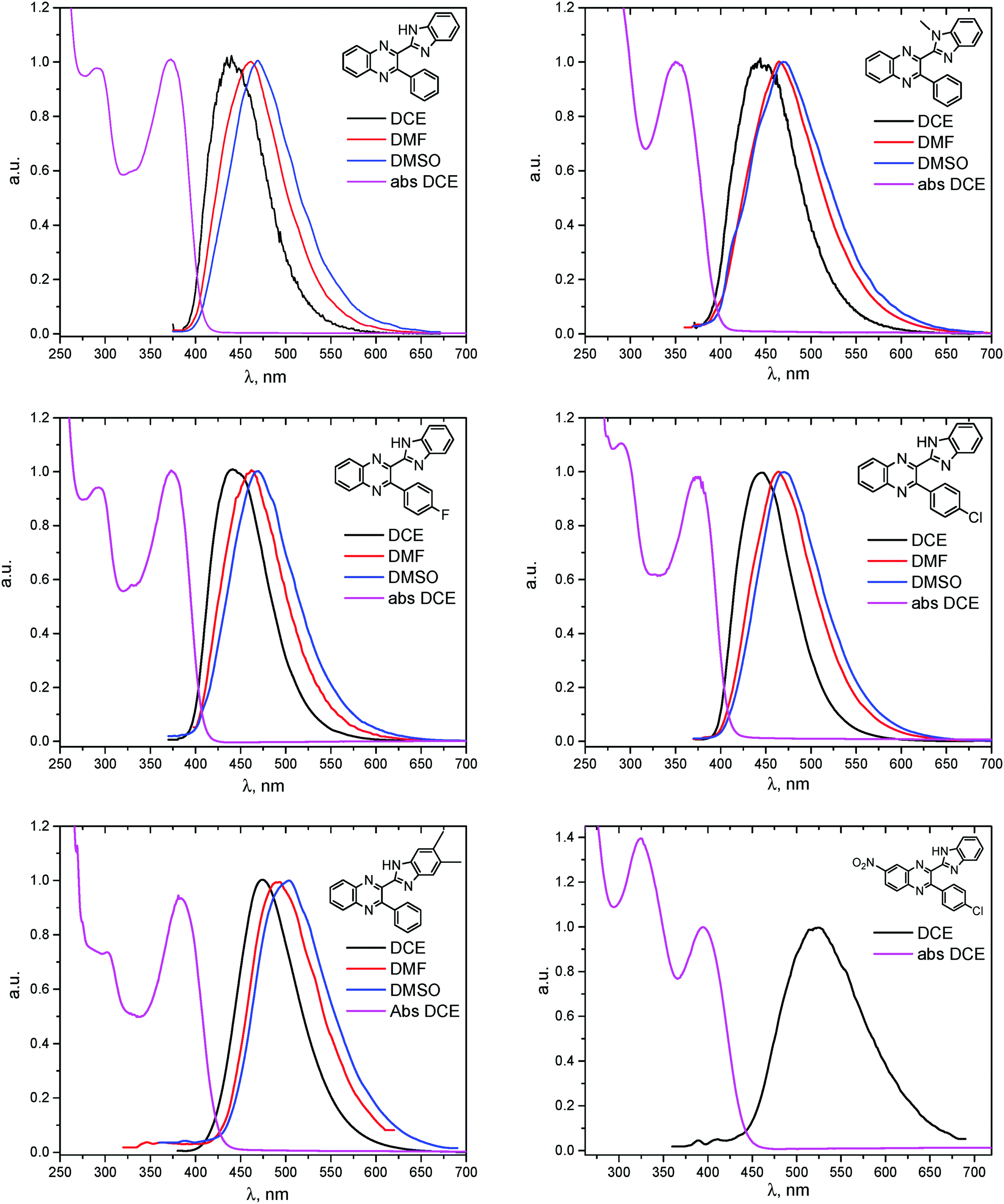 | ||
| Fig. 6 Absorption of BQs 1–6 in DCE (magenta) and fluorescence in DCE (black), DMF (red) and DMSO (blue). | ||
| Compound | Calculationsa | Experimentb | ||||||
|---|---|---|---|---|---|---|---|---|
| λ abs, nm (f) | λ emi, nm (f) |
λ
abs, nm (logε) |
λ emi, nm (φ) | |||||
| DCEc | DMFd | DMSOe | DCEc | DMFd | DMSOe | |||
| a Gas-phase calculations for isolated syn-conformers; f – calculated oscillator strengths (length representation). b λ abs – the longest wavelength absorption maximum; ε – extinction at λabs; λemi – the wavelength of emission maximum; φ – the integral quantum yield of emission. c For solutions in DCE. d For solutions in DMF. e For solutions in DMSO. | ||||||||
|
368 (0.351) | 438 (0.111) | 373 (4.24) | 367 (4.15) | 367 (4.10) | 440 (0.06) | 460 (0.13) | 470 (0.11) |
|
367 (0.220) | 467 (0.059) | 352 (4.03) | 352 (4.01) | 352 (4.02) | 446 (0.03) | 465 (0.03) | 470 (0.04) |
|
373 (0.321) | 454 (0.133) | 375 (4.20) | 370 (4.13) | 370 (4.18) | 444 (0.03) | 462 (0.16) | 468 (0.12) |
|
373 (0.320) | 442 (0.103) | 375 (4.24) | 371 (4.15) | 371 (4.18) | 446 (0.05) | 468 (0.20) | 470 (0.18) |
|
382 (0.429) | 466 (0.129) | 383 (4.25) | 383 (4.20) | 380 (4.20) | 473 (0.06) | 492 (0.29) | 501 (0.25) |
|
412 (0.262) | 513 (0.133) | 396 (4.16) | 391 (4.12) | 391 (4.09) | 519 (0.03) | — | — |
As can be seen from Table 2, the wavelengths of lowest-energy absorptions in the spectra of various solutions of the BQs vary in the range of 352 to 396 nm (3.55 to 3.13 eV) and practically do not depend on the solvents used. Both visual analysis of the frontier MOs of the molecules (vide supra) and rather large values of the oscillator strengths computed for the vertical S0 → S1 transitions (Table 2) suggest that these absorptions are dominantly of π–π* character.
The fluorescence excitation spectra of the compounds match well the absorption. The wavelengths of the emission maxima vary in the rather broad range of 440 to 519 nm (2.82 to 2.39 eV) and depend on the solvent used: a replacement of DCE by the much more polar DMF and DMSO causes red shifts of ca. 20–25 nm (Table 2). Similar dependencies on solvent polarity were reported also for other bifunctional HB donor/acceptor azaaromatic compounds, see e.g.ref. 3, 5–7, 44 and references cited therein. The above mentioned shifts in the present case can be, at least partly, ascribed to stronger stabilization of the more polar S1 states in polar media relative to the less polar S0 species. Intermolecular HBs N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S or N–H⋯OC with DMSO or DMF molecules, respectively, also decrease the S0–S1 energy gap, as they are stronger in the S1 state than in the ground S0 state (Table 1S, ESI†).43
S or N–H⋯OC with DMSO or DMF molecules, respectively, also decrease the S0–S1 energy gap, as they are stronger in the S1 state than in the ground S0 state (Table 1S, ESI†).43
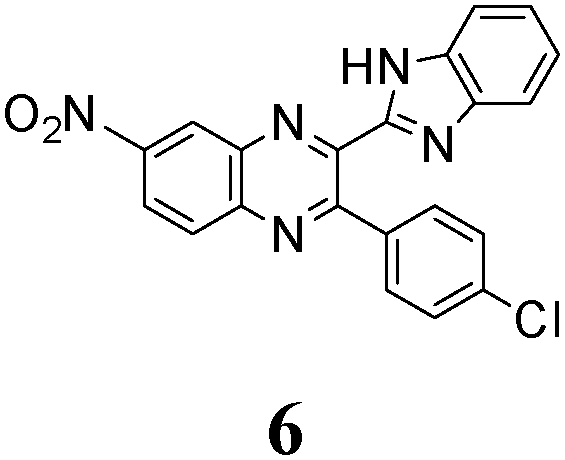
Quantum yields of emission collected in Table 2 vary between 0.03 and 0.29, being quite comparable to the yields reported for the other related azaaromatic chromophores.44–63 A distinguishing feature of the studied compounds is that the intensity of their luminescence varies greatly with the used solvent. In the case of 1, 3–5 (Table 2) it is maximal in the polar DMSO and DMF solvents, while in the less polar DCE the quantum yield of emission drops to ca. 30–70% of its maximal value. In contrast, fluorescence of 6 is easily observable in DCE solutions and is completely quenched in DMF and DMSO. Such a behavior drastically differs from behavior of 1, 3–5 and the other related azaaromatic chromophores.44–63
As no luminescence quenching in the polar solvents is found for other BQs, in particular, for the closely related 4, obviously, the only structural difference between 4 and 6, viz. the NO2 moiety of 6, plays a key role in the above effect. It is known that the presence of nitro groups typically quenches the fluorescence of organic compounds to the level below the detection limit.64 The non-radiative decay pathways may involve internal coordinates of the nitro group.65 A complete mechanistic picture is rather complicated and very high-level single- and multi-reference methods are needed to assure sufficiently accurate theoretical description.65–67 This extremely computationally demanding task is beyond the scope of the present study. Nevertheless, it must be emphasized that the NO2 moiety in molecule 6, indeed, demonstrates remarkable structural changes upon excitation (Fig. 1S, ESI†).
There were a few reports showing the dependence of quantum yield of some fluorophores, containing the nitrophenyl group, on solvent polarity, see, e.g.ref. 68 and 69. In contrast, in our case even the addition of small amounts of DMF or DMSO in solution of 6 in DCE, which should not strongly affect the dielectric properties of the solvent, results in a dramatic quenching of the fluorescence (Fig. 7).70
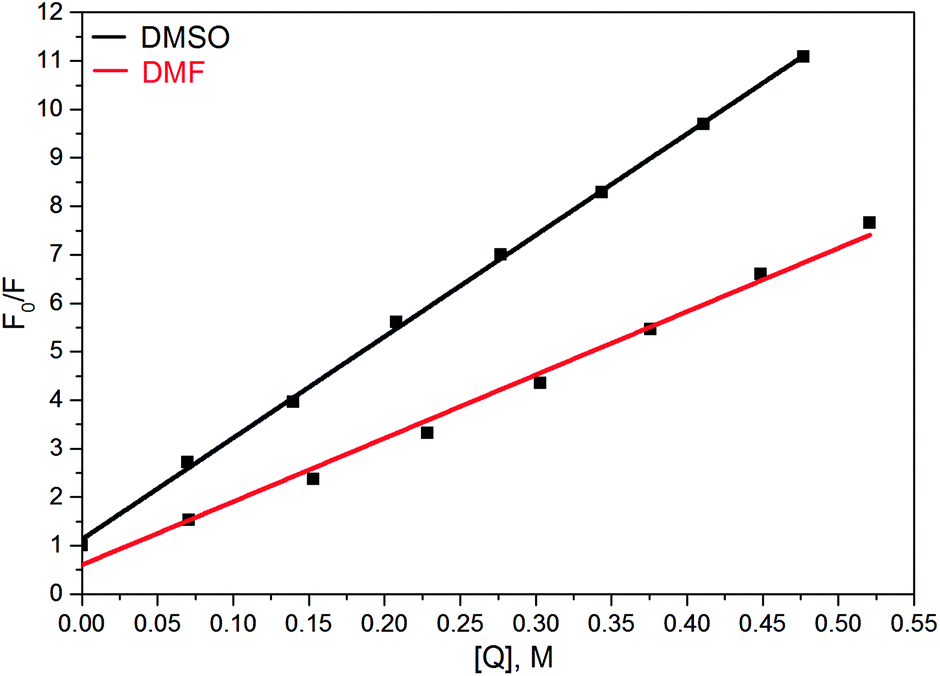 | ||
| Fig. 7 Stern–Volmer plot of fluorescence quenching by DMF and DMSO for solution of compound 6 in DCE. F0 and F are the fluorescence intensities in the absence and presence of quencher [Q], respectively. | ||
This suggests that the polarities of media play only a minor role in the above effect, and instead, HBs of 6 with DMF or DMSO are a key factor of the quenching. The primary effect of H-bonding with DMSO or DMF is an energetic shift of the singly excited states. As π–π* and n–π* states have different sensitivities to H-bonding, the shift results in a change of the energy gap between these states and a decreasing gap can enhance the internal conversion to the ground state.71
Influence of the structure of BQs on their conformational and photophysical properties
The structure of the parent BQ molecule 1 was modified in two ways: first, by the introduction of –Cl (compound 4), –F (3) and –NO2 (6) substituents to the acceptor 3-phenylquinoxaline moiety; second, by the introduction of two methyl groups (compound 5) to the donor benzimidazole moiety. In spite of these modifications, the conformational behavior of all the studied BQs remained qualitatively the same: in the ground state syn-conformers strongly dominate in the gas phase and in intermediately-polar DCE solutions. The energetic preferability of the syn-conformers modestly increases along with the strengthening of the acceptor capacity of the substituted 3-arylquinoxaline moiety (1 < 3 ≈ 4 < 6, according to computations, Table 1), while the modification of the benzimidazole moiety seems to play only a minor role (cf.1 and 5). Replacement of the NH moiety of 1 by the NMe group in 2 increases steric strain in the syn-conformer, and this results in a qualitatively different conformational behavior: the (free) energies of the syn- and anti-conformers of the latter molecule are almost the same.The position of the lowest-energy absorption band of the π–π* transition in the spectra of 1–6 is influenced by both types of structural modifications of the BQs: the stronger the acceptor capacity of the 3-arylquinoxaline moiety or the donor capacity of the benzimidazole moiety, the larger is λabs (Table 2).
The position of the emission band is red-shifted by introduction of donor methyl groups in the case of 5, but the strongest effect is produced by introduction of NO2 groups, which results in a maximal λemi value for 6 (Table 2). In the latter case the nitro group is located in the electron accepting moiety, and demonstrates much more flexibility than the remaining heterocyclic system (Fig. 1S, ESI†). I.e., the NO2 group presence induces pronounced geometry differences between S0 and S1 states and, hence, results in the largest Stokes shift among the whole series. Moreover, the presence of NO2 groups seems to be a key factor responsible for exceptional sensitivity of the quantum yield of luminescence to HBs with solvent molecules (vide supra).
Thus, introduction of various substituents in donor or acceptor parts of the BQ molecules influences their conformational equilibria as well as their photophysical properties. Moreover, the good general agreement of the presented computations with the available experimental data regarding the above mentioned properties suggests that these quantum chemical methods can be of great help in rational tuning of absorption and emission characteristics of these compounds.
Conclusions
We have introduced a series of novel fluorophores, consisting of a 3-arylquinoxaline moiety and a benzimidazole moiety linked by a single bond. Conjugation between these moieties stabilizes the syn-conformers of the BQs relative to the anti-conformers, the former species being strongly thermodynamically preferable in the gas phase and in DCE solutions. Anti-conformers form stronger intermolecular HBs with DMSO and DMF than syn-conformers, and this difference decreases the stability of syn- relative to anti-conformers in these polar solvents. The above effect strengthens upon electronic excitation to the S1 state and may cause an inversion of relative energetic stability of the two conformers in DMF and DMSO solutions. Thus, the absorption spectra of the BQs in all solvents and emission in DCE solutions are dominated by syn-conformers, while the possible presence of anti-forms may be taken into account for the interpretation of the emission spectra of DMF and DMSO solutions. Substituents introduced in various positions of the 3-arylquinoxaline and benzimidazole moieties influence conformational behavior and mutual energetic displacement of excited singlet states relative to their ground state. This in turn strongly affects their luminescence. Various substitution patterns produce very different effects on quantum yield of luminescence: from a moderate increase in polar DMSO and DMF relative to the DCE solutions to complete quenching of emission in these polar media. This tuneability of the BQ's luminescence and especially its sensitivity to environmental effects allows rational design of novel fluorophores of this family for various applications.Acknowledgements
The financial support from scholarship programs of the German Academic Exchange Service (DAAD, personal ref. no. 91577762) and of the government of the Republic of Tatarstan “Algarish” is gratefully acknowledged (SAK).References
- A. Kyrychenko, J. Herbich and J. Waluk, in Tautomerism: Methods and Theories, ed. L. Antonov, Wiley, Weinheim, 2013, pp. 49–78 Search PubMed.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- J. Herbich, M. Kijak, A. Zielinska, R. P. Thummel and J. Waluk, J. Phys. Chem. A, 2002, 106, 2158–2163 CrossRef CAS.
- W. S. Yu, C. C. Cheng, Y. M. Cheng, P. C. Wu, Y. H. Song, Y. Chi and P. T. Chou, J. Am. Chem. Soc., 2003, 125, 10800–10801 CrossRef CAS PubMed.
- V. Vetokhina, K. Dobek, M. Kijak, I. I. Kaminska, K. Muller, W. R. Thiel, J. Waluk and J. Herbich, ChemPhysChem, 2012, 13, 3661–3671 CrossRef CAS PubMed.
- J. Herbich, C.-Y. Hung, R. P. Thummel and J. Waluk, J. Am. Chem. Soc., 1996, 118, 3508–3518 CrossRef CAS.
- B. Golec, M. Kijak, V. Vetokhina, A. Gorski, R. P. Thummel, J. Herbich and J. Waluk, J. Phys. Chem. B, 2015, 119, 7283–7293 CrossRef CAS PubMed.
- V. A. Mamedov, N. A. Zhukova, T. N. Beschastnova, A. T. Gubaidullin, A. A. Balandina and S. K. Latypov, Tetrahedron, 2010, 66, 9745–9753 CrossRef CAS.
- V. A. Mamedov, A. A. Kalinin, A. T. Gubaidullin, A. V. Chernova, I. A. Litvinov, Y. A. Levin and R. R. Shagidullin, Russ. Chem. Bull., Int. Ed., 2004, 53, 164–175 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic, New York, 2nd edn, 1999, pp. 52–54 Search PubMed.
- TURBOMOLE V6.4 2012, a program package developed at the University of Karlsruhe and at the Forschungszentrum Karlsruhe GmbH from 1989–2007, and at TURBOMOLE GmbH since 2007; available from http://www.turbomole.com.
- R. Bauernschmitt, M. Haeser, O. Treutler and R. Ahlrichs, Chem. Phys. Lett., 1997, 264, 573–578 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, J. Chem. Phys., 1996, 104, 9047–9052 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154104 CrossRef PubMed; (b) E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753–12762 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- F. Eckert and A. Klamt, COSMOtherm, Version C3.0, Release 12.01, COSMOlogic GmbH & Co. KG, Leverkusen, Germany, 2012 Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS; A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef; J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- E. U. K. Gross, J. F. Dobson and M. Petersilka, in Density Functional Theory II, Springer Series in Topics in Current Chemistry, ed. R. F. Nalewajski, Springer, Heidelberg, Germany, 1996 Search PubMed.
- M. E. Casida, in Recent Advances in Density Functional Methods, ed. D. P. Chong, World Scientific, Singapore, 1995, vol. 1(27), pp. 155–192 Search PubMed.
- F. Furche, J. Chem. Phys., 2001, 114, 5982–5992 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, B37, 785–789 Search PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- S. A. Katsyuba, T. I. Burganov, E. E. Zvereva, A. A. Zagidullin, V. A. Miluykov, P. Lönnecke, E. Hey-Hawkins and O. G. Sinyashin, J. Phys. Chem. A, 2014, 118, 12168–12177 CrossRef CAS PubMed.
- http://www.chemcraftprog.com .
- It is well established that conjugation leads to a many-fold increase in the intensity of polarized lines, corresponding to the symmetric stretching vibrations of the bonds participating in conjugation. The effect is discussed in depth in, e.g.: P. P. Shorygin, Russ. Chem. Rev., 1971, 40, 367–392 CrossRef CAS; E. D. Schmid and R. D. Topsom, J. Am. Chem. Soc., 1981, 103, 1628–1632 CrossRef.
- A. V. Iogansen, Spectrochim. Acta, Part A, 1999, 55, 1585–1612 CrossRef.
- S. A. Katsyuba, M. V. Vener, E. E. Zvereva, Z. Fei, R. Scopelliti, J. G. Brandenburg, S. Siankevich and P. J. Dyson, J. Phys. Chem. Lett., 2015, 6, 4431–4436 CrossRef CAS PubMed.
- In contrast to S0 state, critical point is easily localized on N–H⋯N intramolecular HB for syn-conformers in S1 state, except for molecule 6. Strengthening of the N–H⋯N intramolecular HB in S1 state relative to S0 state is also clearly seen from comparison of ΔNHsyn–anti values for S1 and S0 states in Table 1S (ESI†). Moderate difference of ΔNHsyn–anti values for S1 and S0 states (Table 1S, ESI†), pronounced non-coplanarity of B and Q moieties of syn-conformer of 6 in S1 state (Fig. 1S, ESI†), as well as the absence of critical point on N–H⋯N bond, suggest the absence of intramolecular HB even in S1 state of this molecule.
- Present TDDFT computations seem to overestimate this effect, as S1 states of H-complexes formed by DMSO with the BQs are found more energetically stable than in the experiment: e.g., S1 → S0 vertical transition energy computed for the H-complex of DMSO with 1 amounts to 2.10 or 2.05 eV for syn- or anti-conformers, respectively, while 2.64 eV is found experimentally for solution of 1 in DMSO (Table 2).
- M. Kijak, I. Petkova, M. Toczek, G. Viosna-Salyga, A. Zielinska, J. Herbich, R. P. Thummel and J. Waluk, Acta Phys. Pol., A, 2007, 112, 105–119 CrossRef.
- J. Hidalgo, A. Sánchez-Coronilla, M. A. Muñoz, C. Carmona and M. Balon, J. Lumin., 2007, 127, 671–677 CrossRef CAS.
- G. Wiosna, I. Petkova, M. S. Mudadu, R. P. Thummel and J. Waluk, Chem. Phys. Lett., 2004, 400, 379–383 CrossRef CAS.
- P. M. Froehlich and K. Nelson, J. Phys. Chem., 1978, 82, 2401–2403 CrossRef CAS.
- R. W. Ricci and J. M. Nesta, J. Phys. Chem., 1976, 80, 974–980 CrossRef CAS.
- P. Chowdhury, S. Panja, A. Chatterjee, P. Bhattacharya and S. Chakravorti, J. Photochem. Photobiol., A, 2005, 170, 131–141 CrossRef CAS.
- A. Kyrychenko, I. Y. Sevriukov, Z. A. Syzova, A. S. Ladokhin and A. O. Doroshenko, Biophys. Chem., 2011, 154, 8–17 CrossRef CAS PubMed.
- J. Waluk and S. J. Komorowski, J. Mol. Struct., 1986, 142, 159–162 CrossRef CAS.
- J. Waluk and S. J. Komorowski, Chem. Phys. Lett., 1987, 4, 368–372 CrossRef.
- I. Petkova, M. S. Mudadu, A. Singh, R. P. Thummel, I. H. M. van Stokkum, W. J. Buma and J. Waluk, J. Phys. Chem. A, 2007, 111, 11400–11409 CrossRef CAS PubMed.
- S. K. Behera, G. Sadhuragiri, P. Elumalai, M. Sathiyendiran and G. Krishnamoorthya, RSC Adv., 2016, 6, 59708–59717 RSC.
- X. Q. Cao, X. H. Lin, Y. Zhu, Y. Q. Ge and J. W. Wang, Spectrochim. Acta, Part A, 2012, 98, 76–80 CrossRef CAS PubMed.
- A. Kanungo, D. Patra, S. Mukherjee, T. Mahata, P. R. Maulik and S. Dutta, RSC Adv., 2015, 5, 70958–70967 RSC.
- A. Mishra, S. Chaterjee and G. Krishnamoorthy, J. Photochem. Photobiol., A, 2013, 260, 50–58 CrossRef CAS.
- Y. Wu, P. V. Lawson, M. M. Henary, K. Schmidt, J.-L. Brédas and C. J. Fahrni, J. Phys. Chem. A, 2007, 111, 4584–4595 CrossRef CAS PubMed.
- P. E. Kruger, P. R. Mackie and M. Nieuwenhuyzen, J. Chem. Soc., Perkin Trans. 2, 2001, 1079–1083 RSC.
- S. Achelle, C. Baudequin and N. Plé, Dyes Pigm., 2013, 98, 575–600 CrossRef CAS.
- H.-J. Son, W.-S. Han, K.-R. Wee, D.-H. Yoo, J.-H. Lee, S.-N. Kwon, J. Ko and S. O. Kang, Org. Lett., 2008, 10, 5401–5404 CrossRef CAS PubMed.
-
Ca. ten-fold decrease of quantum yield in DMSO solutions relative to CH2Cl2 solutions was reported for two 2-aryl,6-(aryleneethynylene)-1H-indoles: M. Flores-Jarillo, A. Alvarez-Hernandez, R. A. Vazquez-García, E. Arias, I. Moggio and J. R. Torres, Dyes Pigm., 2016, 133, 41–50 CrossRef CAS ; only much less pronounced effect was found for N-alkylated derivatives of the 2-aryl,6-(aryleneethynylene)-1H-indoles, which suggests an important role of intermolecular HB NH⋯OS in the luminescence quenching.
- Ca. 5-fold decrease of quantum yield in DMF solutions relative to CH2Cl2 solutions was reported for some benzimidazole-borondipyrromethenes: Z. Li, L.-J. Li, T. Sun, L. Liu and Zh. Xie, Dyes Pigm., 2016, 128, 165–169 CrossRef CAS.
- B. Valeur, Molecular Fluorescence Principles and Applications, Wiley-VCH Verlag GmbH, 2001, p. 381 Search PubMed.
- J.-M. Mewes, V. Jovanovic, C. M. Marian and A. Dreuw, Phys. Chem. Chem. Phys., 2014, 16, 12393–12406 RSC.
- S. Gozem, F. Melaccio, A. Valentini, M. Filatov, M. Huix-Rottlant, N. Ferre, L. M. Frutos, C. Angeli, A. I. Krylov, A. A. Granovsky, R. Lindh and M. Olivucci, J. Chem. Theory Comput., 2014, 10, 3074–3084 CrossRef CAS PubMed.
- M. Huix-Rottlant, A. Nikiforov, W. Thiel and M. Filatov, Top. Curr. Chem., 2016, 368, 445–476 CrossRef.
- B. Bursa, D. Wróbel, B. Barszcz, M. Kotkowiak, O. Valulyuk, D. Gryko, L. Kolanowski, M. Baraniak and G. Lota, Phys. Chem. Chem. Phys., 2016, 18, 7216–7228 RSC.
- S. Hachiya, K. Asai and G. Konishi, Tetrahedron Lett., 2013, 54, 1839–1841 CrossRef CAS.
- We were not able to find reports of comparable rates of fluorescence quenching by DMSO. Ca. 6-fold decrease of fluorescence intensity of corrole dye substituted with 4-NO2C6H4 group was found at addition of 20% of DMF to toluene solution of the dye (Fig. 5 in ref. 68). In case of compound 6 (Fig. 7) similar effect was achieved at addition of ca. 2% of DMF.
- E. C. Lim, J. Phys. Chem., 1986, 90, 6770–6777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06658e |
| This journal is © the Owner Societies 2017 |