
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A common mechanism for coenzyme cobalamin-dependent reductive dehalogenases†
Linus O.
Johannissen
*a,
David
Leys
ab and
Sam
Hay
 *ab
*ab
aSYNBIOCHEM, Manchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. E-mail: linus.johannissen@manchester.ac.uk; sam.hay@manchester.ac.uk
bSchool of Chemistry, University of Manchester, 131 Princess Street, Manchester M1 7DN, UK
First published on 6th February 2017
Abstract
Distinct mechanisms have been proposed for the biological dehalogenation catalyzed by cobalamin-dependent enzymes, with two recent crystallographic studies suggesting different mechanisms based on the observed interaction between the organohalide substrate and cobalamin. In one case, involving an aromatic dibromide substrate in NpRdhA, a novel CoII–Br interaction was observed using EPR, suggesting a mechanism involving a [Co⋯X⋯R] adduct. However, in the case of trichloroethylene in PceA, a significantly longer Co–Cl distance was observed in X-ray crystal structures, suggesting a dissociative electron transfer mechanism. Subsequent DFT models of these reactions have not reproduced these differences in binding modes. Here, we have performed molecular docking and DFT calculations to investigate and compare the interaction between different organohalides and cobalamin in both NpRdhA and PceA. In each case, despite differences in binding in the CoII state, the reaction likely proceeds via formation of a [Co⋯X⋯R] adduct in the CoI state that weakens the breaking carbon–halide bond, suggesting this could be a general mechanism for cobalamin-dependent dehalogenation.
Introduction
Organohalide-respiring bacteria are responsible for the biological degradation of a range of organohalide pollutants such as polychlorinated biphenyls and dioxins.1–4 Reductive dehalogenases (RDase) overcome the inherent difficulty of direct nucleophilic substitution of halide from aryl or vinyl substrates by concomitantly reducing the substrate using a base-off cobalamin cofactor. Two [4Fe–4S] clusters are responsible for reducing the cob(II)alamin resting state to the active cob(I)alamin.5 Two recent X-ray crystallographic studies of the NpRdhA and PceA enzymes reveal the RDase active site contains a conserved Tyr ideally positioned above the cobalamin for protonation of the substrate during the reaction, facilitated by an adjacent positively charged Arg or Lys residue that stabilises the deprotonated Tyr.6,7 However, differences in the position of the substrate/product bound within the active site of the resting states of NpRdhA and PceA have led to distinct mechanistic proposals for each enzyme, and subsequent DFT calculations used to analyse these mechanisms has not reproduced these distinct binding modes.8,9A crystallographic study of PceA by Bommer et al. suggested a dissociative electron transfer (eT) from cobalamin to the substrate trichloroethylene (TCE) during the reaction (Scheme S1 in the ESI†).6 This forms a dichlorovinyl radical, and with free cobalamin in solution this would recombine with the resulting cob(II)alamin forming a Co–C bond.10 However, in the enzyme this recombination is likely sterically hindered, leading to the proposal that the proximal RDase [4Fe–4S] cluster facilitates a second eT, generating a transient carbanion species which is protonated through the active site Tyr. On the other hand, it has been suggested based on electrochemical data, that an outer sphere eT mechanism is unlikely, and that the mechanism probably involves a “more intimate interaction” between cobalamin and substrate.11 Along these lines, a mechanism involving a cobalamin–substrate adduct was proposed by Payne et al. for the debromination of 3,5-dibromo-4-hydroxybenzoic acid (DBHB; Scheme S2, ESI†) by NpRdhA. In this case, a direct CoII–Br interaction was observed by EPR, although only substrate-free and halide-bound X-ray crystal structures of the enzyme were obtained.7 The mechanism was proposed to follow either homo- or heterolytic C–Br bond cleavage. The heterolytic cleavage mechanism requires that the substrate be sufficiently activated after adduct formation (step 2A in Scheme S2, ESI†) to directly deprotonate the active site Tyr (step 2B). If this is not the case, further reduction of the substrate, via reduction of cobalamin by the proximal [4Fe–4S] cluster or directly by eT from the proximal [4Fe–4S] cluster, may be required.
Liao et al. have since employed DFT modelling to investigate the mechanism in both NpRdhA8 and PceA9 for a range of halides and proposed that for both enzymes dehalogenation occurs by a heterolytic C–X cleavage mechanism after reduction of CoII to CoI, via a [Co⋯X⋯R] adduct (with bonding between Co and the transferring halide), and that this is concerted with the proton transfer from the active site Tyr. However, these DFT calculations did not reproduce the different experimentally observed CoII binding modes, and resulted in cobalamin–substrate adducts in their PceA/TCE models (with Co–X distances of 2.79 to 2.96 Å depending on the substrate orientation in the model) as well as NpRdhA/2,6-dibromo-4-methylphenolate (DBMP) (3.16 Å). In this study we employ a computational method that qualitatively reproduces the observed cobalamin–substrate interactions in the CoII oxidation state of both enzymes, and investigate the nature of this interaction, its effect on the dehalogenation mechanism and the factors that affect it.
Computational methods
Docking
Computational docking was performed in Autodock vina,12 using AutoDock Tools 1.5.6 to assign hydrogens as described previously for NpRdhA.7 PDB IDs for the structures used are 4RAS for NpRdhA and 4UR0 for PceA. For NpRdhA the DBMP analogue 3,5-dibromo-4-hydroxybenzoic (DBHB) was used, and four active site residues (F291, Y426, K488 and R552) were made flexible.7 The conformations with the shortest C–X distance were selected, with energies 0.8 and 5.9 kJ mol−1 higher than the lowest energy conformations for TCE and DBHB, respectively.DFT calculations
Using the docked conformations shown in Fig. 1 as starting points, active site models were created that incorporate first shell amino acids and cobalamin with the peripheral cobalamin substituents facing away from the substrate trimmed to methyl groups (Fig. 2 and Fig. S1, S2 in the ESI†). The amino acids included are: F291, F310, S422, Y426, K488 and R552 for NpRdhA and F38, Y246, R305, W376 and Y382 for PceA. Calculations were carried out in Gaussian09 rev. d.13 The TPSS functional14 with the Def2-TZVP basis sets15–18 and Grimme's D3 function19,20 is suitable for cobalamin calculations.21 Since this is computationally expensive, we applied the Def2-TZVP basis sets to all atoms of the cobalamin, halide substrate and active site tyrosine, and the Def2-SVP basis sets16,18 to the remaining amino acids. This approach qualitatively reproduces the CoII binding modes for NpRdhA/DBMP and PceA/TCE. During each calculation, the Co and the Cβ of each amino acid side chain were kept fixed. Transition states were obtained by relaxed potential energy scans along the distance between the C bound to the transferring X and the H of the active site Tyr, with a step size of no more than 0.03 Å near the maximum. Gibbs free energies for the reactions (ΔG‡) were obtained from frequency calculations on the optimised ground and transition states and energies were corrected from single point calculations using a polarisable continuum model (PCM) with a dielectric constant ε = 5.7 to mimic the enzyme environment.22–24 Note, however, that for the purposes of this study the precise energetics of the reactions are not as critical as the trends in the energies and the geometries.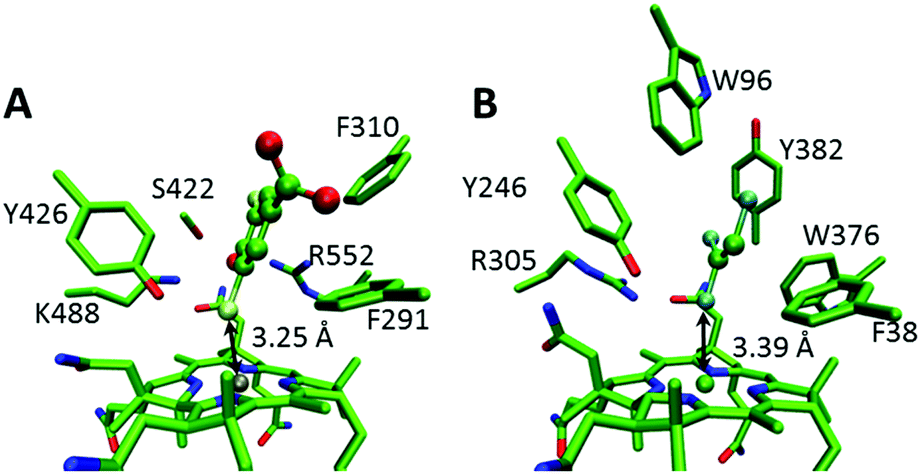 | ||
| Fig. 1 Docked structures of (A) DBHB in NpRdhA and (B) TCE in PceA. Key active site residues and the Co–X distances are labeled and the substrates are rendered in ball and stick mode. | ||
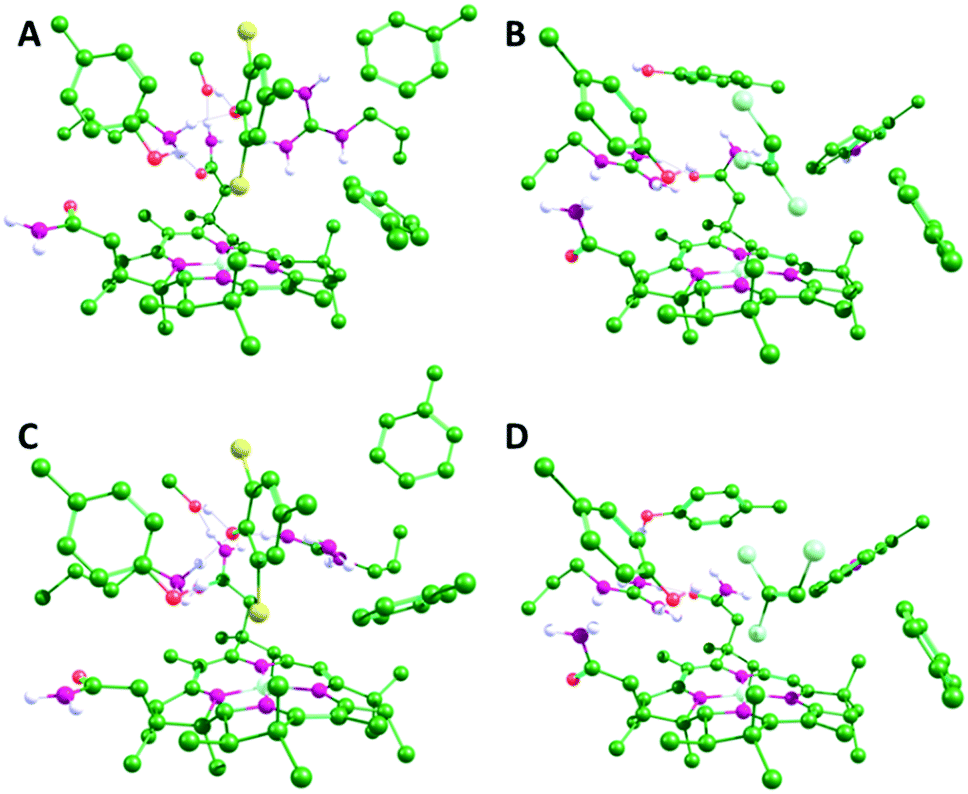 | ||
| Fig. 2 Optimized geometries of the NpRdhA/DBMP (A and C) and PceA/TCE (B and D) active site models in the CoII (A and B) and CoI (C and D) oxidation states. Only polar hydrogens are shown. Equivalent structures for all four enzyme/substrate models are shown in Fig. S2 (ESI†). | ||
Molecular orbitals, charges and spin densities were obtained from single point calculations on truncated models that include the cobalamin and substrate. Charges were calculated both from natural bonding orbitals analyses and the Merz–Kollman method, which maps point charges onto the electrostatic potential.25,26
Results and discussion
There are obvious differences in the active sites of NpRdhA and PceA. In particular, the active site of PceA is more occluded than that of NpRdhA.6,7 Nevertheless, molecular docking of TCE and DBHB into the active sites of PceA and NpRdhA, respectively, suggests that similar binding modes are possible (Fig. 1). Further, docking DBHB into NpRdhA required the inclusion of four flexible residues, so it appears easier to achieve binding with TCE in PceA than with DBHB in NpRdhA. Sterically, there is therefore no obvious reason why these enzymes should bind the halogenated substrates in fundamentally different ways, or why TCE-binding in PceA should be incompatible with adduct formation.To investigate the organohalide–cobalamin interactions in NpRdhA and PceA, DFT calculations were performed on active site models in both the CoI and CoII states. Active site models included the bound PceA substrates TCE and its analogue tribromoethylene (TBE) and the NpRdhA DBHB substrate analogue DBMP and its chlorinated equivalent 2,6-dichloro-4-methylphenolate (DCMP). These models are illustrated in Fig. S1 (ESI†), along with the substrates and the truncated cobalamin model used. In all cases, the substrates remained bound within the active site, with the Co–X distance shorter, and substrate–X bonds longer, in the CoI oxidation state (Fig. 2 and Table 1). The CoII–Br ‘bond’ distance in NpRhA/DBMP is also shorter than the CoII–Cl bond distance in PceA/TCE, consistent with EPR7 and crystallographic6 data for these systems.
| Model | R(Co–X)/Å | R(Ca–X)/Å | R(C–Hb)/Å | |
|---|---|---|---|---|
| a C refers to the C bound to the transferring X. b H refers to the proton transferred from the active site Tyr. c Active site model with neutralised Arg552 residue. | ||||
| PceA/TCE | CoII | 3.83 | 1.72 | 2.81 |
| CoI | 2.77 | 1.79 | 2.65 | |
| PceA/TBE | CoII | 2.62 | 1.92 | 2.45 |
| CoI | 2.57 | 2.17 | 2.15 | |
| NpRdha/DBMP | CoII | 3.60 | 1.93 | 2.48 |
| CoI | 2.84 | 2.02 | 2.33 | |
CoII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
3.39 | 1.93 | 2.52 | |
| CoIc |
2.96 | 1.98 | 2.47 | |
| NpRdha/DCMP | CoII | 3.70 | 1.77 | 2.51 |
| CoI | 3.10 | 1.78 | 2.37 | |
There is no transfer of electron density between molecular orbitals (MOs) from the substrate and cobalamin for PceA/TCE, while inspection of these MOs confirms that an adduct is formed in the other three models, although the specifics of the interaction differs between the two enzymes (Fig. 3 and Fig. S3, ESI†). For the NpRdhA models, electron density is transferred from the substrate into the singly occupied CoII dz2 orbital, so that the lowest unoccupied MOs (LUMOs) consist of orbitals form the highest occupied MO (HOMO) of the substrate, leading to elevated charges and spin densities on DBMP and DCMP (Table 2). For PceA/TBE, the substrate HOMO is delocalised with several bonding orbitals of the cobalamin (Fig. S3, ESI†), so that the LUMO is formed by delocalisation of the CoII dz2 orbital with a C–Br anti-bonding orbital. For PceA/TCE there is no delocalisation of orbitals from the substrate and cobalamin, and no transfer of electron density between substrate and cobalamin. Thus, a cob(II)alamin–halide adduct is formed in each model except that for PceA/TCE.
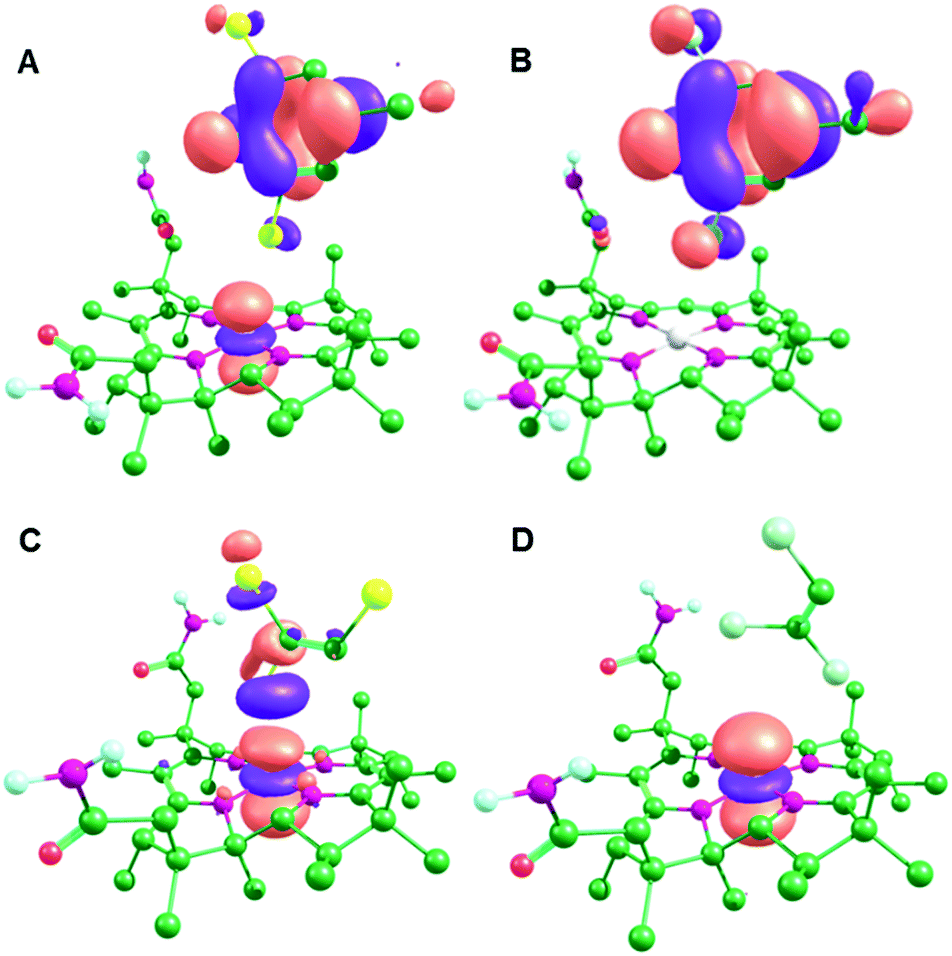 | ||
| Fig. 3 LUMOs for the CoII optimised active site models: (A) NpRdhA/DBMP, (B) NpRdhA/DCMP, (C) PceA/TBE and (D) PceaA/TCE. | ||
| Model | Δ chargea | Spin densityb | |
|---|---|---|---|
| a Difference in charge relative to the isolated substrate (−1 for DBMP and DCMP, 0 for TBE and TCE); charges shown are from natural bond orbital analysis (and the Merz–Kollman method in parentheses). b Spin densities of the CoII (singlet radical) models. | |||
| PceA/TCE | CoII | 0.006 (−0.033) | 0.002 |
| CoI | −0.153 (−0.112) | N/A | |
| PceA/TBE | CoII | 0.179 (0.198) | 0.078 |
| CoI | −0.334 (−0.386) | N/A | |
| NpRdhA/DBMP | CoII | 0.499 (0.522) | 0.490 |
| CoI | 0.003 (0.068) | N/A | |
| NpRdhA/DCMP | CoII | 0.977 (0.961) | 0.989 |
| CoI | 0.018 (0.077) | N/A | |
Despite the differences in the substrate–cobalamin interactions in the (formally) CoII state, the interactions after reduction to CoI are very similar (Fig. 4). In each case, there is transfer of electron density from the Co dz2 orbital into the C–X antibonding orbital for the breaking C–X bond, as previously proposed by Cooper et al.27 This has the effect of activating the substrate towards C–X bond cleavage by stretching the C–X bond and concomitantly decreasing the active site Tyr proton-substrate carbon distance (C–H in Table 1). This effect is strongest for PceA, where a significant amount of charge is transferred onto TCE and TBE, and very little charge transfer is observed for NpRdhA (Table 2). Thus, while these DFT models are consistent with the different binding modes observed experimentally for NpRhA/DBMP and PceA/TCE in the CoII states, they suggest that reduction to CoI leads to very similar [Co⋯X⋯R] adducts (Fig. 4), and that the strongest CoI interaction (based on Co–X distance and degree of charge/spin transfer) corresponds to the weakest CoII interaction.
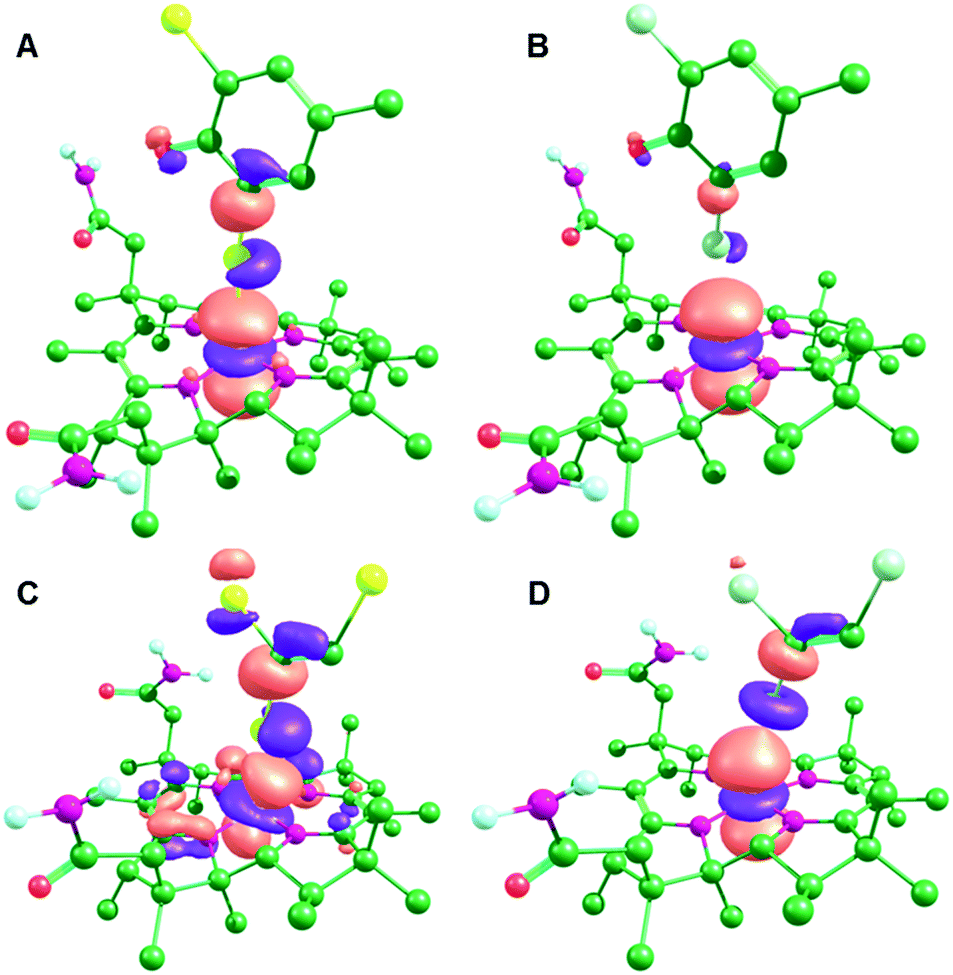 | ||
| Fig. 4 MOs corresponding to the Co dz2 orbital for the CoI optimised active site models: (A) NpRdhA/DBMP (HOMO−2), (B) NpRdhA/DCMP (HOMO−2), (C) PceA/TBE (HOMO) and (D) PceaA/TCE (HOMO). | ||
Consistent with previous models,8,9 dehalogenation after reduction to CoI proceeds via a concerted mechanism for each model (combining steps 2B and 2C in Scheme S2, ESI†), with a single transition state for both C–X bond cleavage and protonation by the active site Tyr (Table 3 and Fig. S4, S5, ESI†) to yield a CoIII–X species which will be rapidly reduced to the experimentally observed CoII–X species.9 For the PceA reactions, the product is cis-1,2-dihaloethene as shown in Fig. S4 (ESI†), which is the experimentally observed product for dehalogenation of TCE. The preference for this regiochemistry is not surprising given that substrate reduction weakens the cleaved C–X bond the most (Fig. S5, ESI†).
| Model | ΔG‡ (ΔE‡)/kJ mol−1 | ν i /cm−1 | R(Co–X)/Å | R(C–X)/Å | R(C–H)/Å |
|---|---|---|---|---|---|
| a Gibbs free energies and gas phase potential energies in (parentheses) relative to the CoI ground state. b In each case, this vibrational mode corresponds principally to motion of the transferring H, with small contributions from C and X. The eigenvalues for H, C and X, respectively, are as follows. PceA/TCE: 0.907, 0.228, 0.049; PceA/TBE: 0.976, 0.108, 0.014; NpRdhA/DBMP: 0.996, 0.0490, 0.0; NpRdhA/DCMP: 0.994, 0.0490, 0.010. | |||||
| PceA/TCE | −11.4 (7.22) | 469.6i | 2.27 | 2.62 | 1.53 |
| PceA/TBE | −14.3 (7.98) | 847.3i | 2.40 | 2.65 | 1.47 |
| NpRdhA/DBMP | 41.1 (53.3) | 1386.7i | 2.47 | 2.68 | 1.39 |
| NpRdhA/DCMP | 49.9 (78.5) | 1412.8i | 2.36 | 2.55 | 1.39 |
For PceA, the gas phase potential energies for the transition states (ΔE‡) are so low that the resulting continuum solvation-corrected Gibbs free energies are negative, suggesting that this reaction is effectively barrierless. Qualitatively, these results are very similar to those of Liao et al., who determined barriers of 30.5 and 2.09 kJ mol−1 for NpRdhA/DBMP8 and PceA/TCE9 (with the TCE orientation most similar to that in this study), respectively. Unsurprisingly, the trend in ΔG‡ is dominated by the gas phase potential energy for the reaction (ΔE‡). The bond dissociation energies (BDEs) for TBE and TCE are much larger than those for the DBMP and DCMP (Table S1, ESI†), but ΔE‡ for both PceA models are much smaller than those for NpRdhaA. This is due to the partial reduction of both PceA substrates prior to C–X bond cleavage, as the substrate BDEs are significantly decreased by reduction (Table S1, ESI†).
One of the key differences between the PceA and NpRdhA active sites, apart from the former being more occluded, is the presence of the additional positively charged residue in the NpRdhA active site, R552. In both enzymes, a charged residue – R305 in PceA and K488 in NpRdhA – stabilizes the deprotonated form of the active site Tyr, and the additional Arg in NpRdhA might serve to enhance binding by stabilising the negatively charged (deprotonated) substrate. However, this additional positive charge adjacent to the substrate is also likely to aid in transfer of electron density from the CoI dz2 orbital into the substrate. To test this hypothesis, a NpRdhA/DBMP model with a neutral R305 analogue was optimized (Fig. S7, ESI†). In this model, the C–X bond is shortened and the Co–X distance increased (Table 1), consistent with a weaker cob(I)alamin–DBMP interaction. In other words, the positive charge on R552 helps activate the substrate towards C–X bond cleavage. Conversely, since the cob(II)alamin–DBMP interaction involves transfer of electron density in the opposite direction, R552 weakens this interaction so that the original model has a longer Co–X bond than the neutral analogue.
The different effect of the enzyme active sites was further analysed by energy minimising an artificial NpRdhA/TCE model (Fig. S8, ESI†). As expected, the positive charge on R552 enhances the degree of eT from cobalamin into TCE in the CoI state, and in this case the C–Cl bond is fully broken to form the dichlorovinyl carbanion. For the neutral R552 analogue, the C–Cl bond is also significantly stretched (2.47 Å) compared to the PceA model, due to the proximity of partial positive charges from the R552 amino groups. NpRdhA enhances the degree of eT relative to PceA because the higher energy LUMOs of DBMP and DCMP compared to TCE or TBE (Table S2, ESI†) makes transfer of electron density into the substrate more difficult.
Conclusions
Comparing the substrate–cobalamin interactions in PceA and NpRdhA reveals that substrate–cobalamin adduct formation in the CoII oxidation state cannot simply be used to infer the likelihood of adduct formation in the CoI state, which has important implications for the use of EPR spectroscopy to study these systems. In fact, for all models examined here, a weaker adduct in the CoII state leads to a stronger adduct in the CoI state. This behaviour arises due to the direction of transfer of electron density between substrate and cobalamin flipping after reduction from CoII to CoI. Nevertheless, despite the differences in active site structure between the NpRdhA and PceA enzymes, and differences in substrate binding in the CoII oxidation state, reduction to CoI results in similar [Co⋯X⋯R] adducts from which dehalogenation proceeds via the same concerted mechanism. This suggests that this is a common mechanism for cobalamin-dependent enzymatic reductive dehalogenation.Acknowledgements
This work was funded by the UK Biotechnology and Biological Sciences Research Council (BBSRC BB/M007316/1 and BB/M017702/1). The authors would like to acknowledge the assistance given by IT Services and the use of the Computational Shared Facility at The University of Manchester.References
- L. Adrian, et al., Bacterial dehalorespiration with chlorinated benzenes, Nature, 2000, 408(6812), 580–583 CrossRef CAS PubMed.
- M. Bunge, et al., Reductive dehalogenation of chlorinated dioxins by an anaerobic bacterium, Nature, 2003, 421(6921), 357–360 CrossRef CAS PubMed.
- D. Leys, L. Adrian and H. Smidt, Organohalide respiration: microbes breathing chlorinated molecules, Philos. Trans. R. Soc., B, 2013, 368(1616), 20120316 CrossRef PubMed.
- H. Smidt and W. M. de Vos, Anaerobic microbial dehalogenation, Annu. Rev. Microbiol., 2004, 58, 43–73 CrossRef CAS PubMed.
- J. Shey and W. A. van der Donk, Mechanistic studies on the vitamin B-12-catalyzed dechlorination of chlorinated alkenes, J. Am. Chem. Soc., 2000, 122(49), 12403–12404 CrossRef CAS.
- M. Bommer, et al., Structural basis for organohalide respiration, Science, 2014, 346(6208), 455–458 CrossRef CAS PubMed.
- K. A. Payne, et al., Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation, Nature, 2015, 517(7535), 513–516 CrossRef CAS PubMed.
- R.-Z. Liao, S.-L. Chen and P. E. M. Siegbahn, Which Oxidation State Initiates Dehalogenation in the B12-Dependent Enzyme NpRdhA: CoII, CoI, or Co0?, ACS Catal., 2015, 5(12), 7350–7358 CrossRef CAS.
- R.-Z. Liao, S.-L. Chen and P. E. M. Siegbahn, Unraveling the Mechanism and Regioselectivity of the B12-Dependent Reductive Dehalogenase PceA, Chem. – Eur. J., 2016, 22(35), 12391–12399 CrossRef CAS PubMed.
- K. M. McCauley, et al., Properties and reactivity of chlorovinylcobalamin and vinylcobalamin and their implications for vitamin B12-catalyzed reductive dechlorination of chlorinated alkenes, J. Am. Chem. Soc., 2005, 127(4), 1126–1136 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Does catalysis of reductive dechlorination of tetra- and trichloroethylenes by vitamin B12 and corrinoid-based dehalogenases follow an electron transfer mechanism?, J. Am. Chem. Soc., 2005, 127(35), 12154–12155 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2009, 31(2), 455–461 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- J. M. Tao, et al., Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91(14), 146401 CrossRef PubMed.
- A. Schafer, C. Huber and R. Ahlrichs, Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr, J. Chem. Phys., 1994, 100(8), 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- F. Weigend, et al., RI-MP2: optimized auxiliary basis sets and demonstration of efficiency, Chem. Phys. Lett., 1998, 294(1–3), 143–152 CrossRef CAS.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065 RSC.
- S. Grimme, et al., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- U. Ryde, R. A. Mata and S. Grimme, Does DFT-D estimate accurate energies for the binding of ligands to metal complexes?, Dalton Trans., 2011, 40(42), 11176–11183 RSC.
- Z. W. Qu, A. Hansen and S. Grimme, Co-C Bond Dissociation Energies in Cobalamin Derivatives and Dispersion Effects: Anomaly or Just Challenging?, J. Chem. Theory Comput., 2015, 11(3), 1037–1045 CrossRef CAS PubMed.
- S. P. de Visser, et al., Combined experimental and theoretical study on aromatic hydroxylation by mononuclear nonheme iron(IV)-oxo complexes, Inorg. Chem., 2007, 46(11), 4632–4641 CrossRef CAS PubMed.
- D. J. Heyes, et al., Nuclear Quantum Tunneling in the Light-activated Enzyme Protochlorophyllide Oxidoreductase, J. Biol. Chem., 2009, 284(6), 3762–3767 CrossRef CAS PubMed.
- S. Shaik, et al., Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes, Chem. Rev., 2005, 105(6), 2279–2328 CrossRef CAS PubMed.
- U. C. Singh and P. A. Kollman, An Approach to Computing Electrostatic Charges for Molecules, J. Comput. Chem., 1984, 5(2), 129–145 CrossRef CAS.
- E. Sigfridsson and U. Ryde, Comparison of methods for deriving atomic charges from the electrostatic potential and moments, J. Comput. Chem., 1998, 19(4), 377–395 CrossRef CAS.
- M. Cooper, et al., Anaerobic Microbial Transformation of Halogenated Aromatics and Fate Prediction Using Electron Density Modeling, Environ. Sci. Technol., 2015, 49, 6018–6028 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp08659d |
| This journal is © the Owner Societies 2017 |