
A systematic study of 25Mg NMR in paramagnetic transition metal oxides: applications to Mg-ion battery materials†
Jeongjae
Lee
 a,
Ieuan D.
Seymour
a,
Andrew J.
Pell‡
a,
Siân E.
Dutton
b and
Clare P.
Grey
*a
a,
Ieuan D.
Seymour
a,
Andrew J.
Pell‡
a,
Siân E.
Dutton
b and
Clare P.
Grey
*a
aDepartment of Chemistry, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: cpg27@cam.ac.uk
bCavendish Laboratory, J. J. Thomson Avenue, Cambridge CB3 0HE, UK
First published on 11th November 2016
Abstract
Rechargeable battery systems based on Mg-ion chemistries are generating significant interest as potential alternatives to Li-ion batteries. Despite the wealth of local structural information that could potentially be gained from Nuclear Magnetic Resonance (NMR) experiments of Mg-ion battery materials, systematic 25Mg solid-state NMR studies have been scarce due to the low natural abundance, low gyromagnetic ratio, and significant quadrupole moment of 25Mg (I = 5/2). This work reports a combined experimental 25Mg NMR and first principles density functional theory (DFT) study of paramagnetic Mg transition metal oxide systems Mg6MnO8 and MgCr2O4 that serve as model systems for Mg-ion battery cathode materials. Magnetic parameters, hyperfine shifts and quadrupolar parameters were calculated ab initio using hybrid DFT and compared to the experimental values obtained from NMR and magnetic measurements. We show that the rotor assisted population transfer (RAPT) pulse sequence can be used to enhance the signal-to-noise ratio in paramagnetic 25Mg spectra without distortions in the spinning sideband manifold. In addition, the value of the predicted quadrupolar coupling constant of Mg6MnO8 was confirmed using the RAPT pulse sequence. We further apply the same methodology to study the NMR spectra of spinel compounds MgV2O4 and MgMn2O4, candidate cathode materials for Mg-ion batteries.
1 Introduction
Mg transition metal (TM) compounds are important materials for future energy technology, as they can potentially be used as cathodes for Mg-ion batteries1 that offer high volumetric capacity, low cost, and improved safety compared to the prevalent Li-ion batteries.2,3 In addition, they can, for example, be potentially used as carbon capture materials4 and electrochemical catalysts for water oxidation.5Solid-state Nuclear Magnetic Resonance (NMR) spectroscopy is a valuable tool to aid the development of such materials, as it can give information about local disorder which is difficult to analyze with diffraction methods that probe long range order. Solid-state NMR has been successfully used to study a range of technologically important materials, such as battery electrodes, fuel cells, zeolites, and supercapacitors.6–9 Utilizing the same approach for Mg, however, is challenging because of the low gyromagnetic ratio (approximately 1/20 of 1H), low natural abundance (10%), and significant quadrupolar moment (I = 5/2, Q = 0.2 barns) of 25Mg. Only recently has the development of sophisticated NMR equipment, such as high-field magnets and magic-angle spinning (MAS) probes, enabled high resolution 25Mg NMR spectra to be acquired.10,11 With the development and application of novel pulse sequences, such as rotor-assisted population transfer (RAPT)12 to enhance the sensitivity and magic-angle turning (MAT)13 to separate the spinning-sideband manifolds from the isotropic resonances, such studies of challenging quadrupolar nuclei are expected to become more common.
On top of the experimental difficulties described above, studying battery materials with solid-state NMR is often made more complicated by the presence of unpaired electron spins in the systems, resulting in large paramagnetic NMR shifts which are difficult to measure and interpret. Due to the inherent difficulty of 25Mg paramagnetic NMR experiments, computational predictions are invaluable aids for recording and interpreting the spectra.
Recently, Kim et al. reported 25Mg NMR spectra of paramagnetic materials;14 two 25Mg resonances were seen in the spectrum of MgMn2O4, but no detailed investigation was performed to determine the origin of the two peaks. To our knowledge, no previous study of the 25Mg paramagnetic NMR parameters involved both experimental and computational approaches. To address this, we study four Mg TM oxides which have different crystal structures: Mg6MnO8 (defect rocksalt), MgCr2O4, MgV2O4 (cubic normal spinel), and MgMn2O4 (tetragonal normal spinel). First, we report the results of their structural and magnetic characterization, which is followed by 25Mg NMR experiments. Ab initio calculations for magnetic data and paramagnetic 25Mg NMR shifts follow. First principles results are compared to experimental spectra acquired using various state-of-the-art methods. Finally, we discuss the observed NMR shifts in terms of Fermi contact interaction and decompose the value of shifts in terms of the individual TM–O–Mg spin transfer pathways.
2 Experimental
2.1 Sample preparation
Mg6MnO8, MgCr2O4, and MgV2O4 samples were prepared via solid-state synthesis from stoichiometric amounts of MgO (Sigma-Aldrich, 99.99%), V2O3 (Sigma-Aldrich, 99.7%), Cr2O3 (Sigma-Aldrich, 99.99%), and MnO2 (Sigma-Aldrich, 99.99%). Mixtures were ball milled, pressed into pellets, and calcined according to Table 1. The MgMn2O4 sample was synthesized from anhydrous citric acid (Breckland Scientific, 99%), Mg(NO3)2·6H2O (Sigma-Aldrich, 99%), and Mn(NO3)2·4H2O (Sigma-Aldrich, 97%) through a citrate sol–gel method.15 The organic matter was burned off at 453 K and final calcination was performed at 773 K with an intermediate grinding step. Synthesized samples were checked using a PANalytical Empyrean powder X-ray diffractometer (Cu Kα = 1.5406 Å) and the structures were refined from their diffraction patterns by using the Rietveld method16 as implemented in the X'pert Highscore Plus software.| Sample | Thermal profile | Atmosphere |
|---|---|---|
| MgV2O4 | 1273 K 24 h | 5% H2 in Ar |
| MgCr2O4 | 1073 K 12 h–1473 K 24 h | Air |
| Mg6MnO8 | 1173 K 12 h | Air |
| MgMn2O4 | 453 K 12 h–773 K 36 h | Air |
2.2 Magnetic measurements
Magnetization measurements were performed with Quantum Design Material Property Measurement System (MPMS). Magnetic moments of zero field-cooled samples were measured at temperatures from 2 K up to 301 K to obtain the dependence of magnetic susceptibility χ(T) = dM/dH ≈ M/H on temperature. The Weiss constant Θ was extracted by fitting the Curie–Weiss law χ = C/(T − Θ), where C is the Curie constant. The effective electron magnetic moment μeff was extracted from C by the relation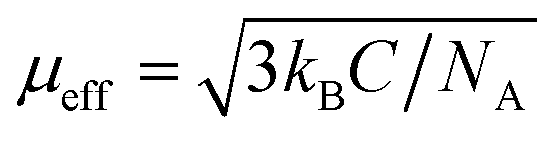 where kB is the Boltzmann constant and NA is the Avogadro constant. The magnetic exchange coupling constant J1 between the nearest neighboring spins (S) was calculated from Θ by the mean-field relation
where kB is the Boltzmann constant and NA is the Avogadro constant. The magnetic exchange coupling constant J1 between the nearest neighboring spins (S) was calculated from Θ by the mean-field relation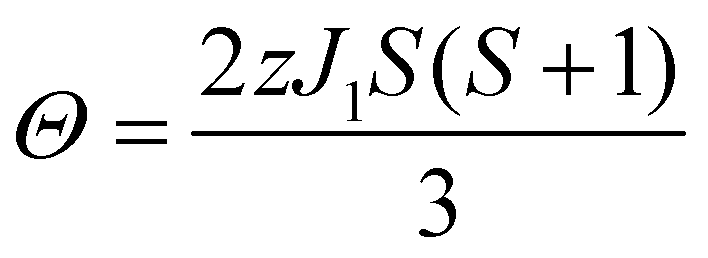 | (1) |
2.3 25Mg NMR
All NMR spectra were acquired on a 16.4 T Bruker Avance III spectrometer operating at a Larmor frequency of 42.9 MHz for 25Mg with conventional Bruker 4 mm and 3.2 mm triple resonance MAS low-γ probes. π/2-pulse amplitudes were calibrated on solid MgO, giving 90° pulse amplitudes of 89 kHz and 108 kHz for the 4 mm and 3.2 mm probes, respectively. All shifts were referenced to MgO at 26 ppm.17 Spectra were acquired with rotor-synchronised spin echo, magic angle turning (MAT),18 and rotor assisted population transfer (RAPT)12 pulse sequences as indicated in the ESI† (Section S2). We have used a X–![[X with combining macron]](https://www.rsc.org/images/entities/char_0058_0304.gif) pulse train to saturate the satellite levels. RAPT modulation frequency νm was calculated from DFT values of CQ. Values of νm = 270 kHz (CQ = 3.6 MHz) and 240 kHz (CQ = 3.2 MHz) were used for Mg6MnO8 and MgMn2O4, respectively. Experimental signal enhancement was measured for varying modulation frequencies for Mg6MnO8. Fitting of the spectra was performed assuming a central transition lineshape using the Bruker Topspin 3.0 software.
pulse train to saturate the satellite levels. RAPT modulation frequency νm was calculated from DFT values of CQ. Values of νm = 270 kHz (CQ = 3.6 MHz) and 240 kHz (CQ = 3.2 MHz) were used for Mg6MnO8 and MgMn2O4, respectively. Experimental signal enhancement was measured for varying modulation frequencies for Mg6MnO8. Fitting of the spectra was performed assuming a central transition lineshape using the Bruker Topspin 3.0 software.
2.4 Ab initio calculations
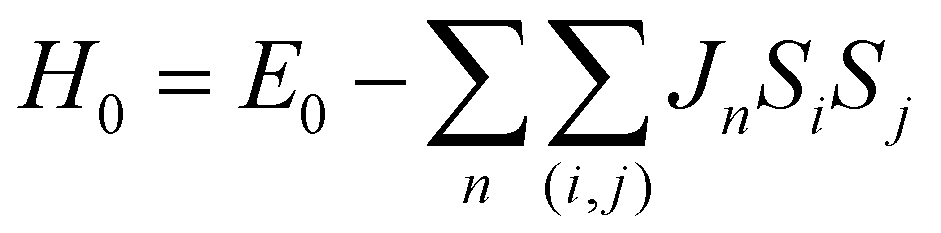 | (2) |
The paramagnetic contribution to the shift tensor originates from the isotropic Aiso and anisotropic Aaniso components of the electron–nuclear hyperfine coupling tensor. Here the isotropic component is ascribed solely to the Fermi contact (FC) mechanism, which contributes to the isotropic shift. The anisotropic (dipolar) component contributes to the shift anisotropy.
As 25Mg is a quadrupolar nucleus (I = 5/2), the overall observed isotropic shift δiso under MAS conditions can be expressed as a sum of Fermi contact component δFC and second-order quadrupolar component δQ. Paramagnetic NMR parameters were calculated using the scaling approach of Kim et al.20 assuming the FC is the dominant shift mechanism. This method takes Aiso and Aaniso from a ferromagnetic calculation performed at 0 K, and then scales it to paramagnetic case using a Curie–Weiss type factor Φ:
| δiso = δFC + δQ | (3) |
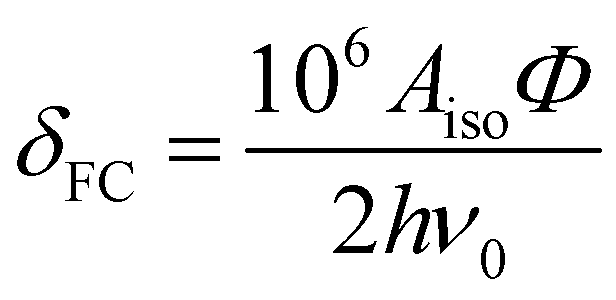 | (4) |
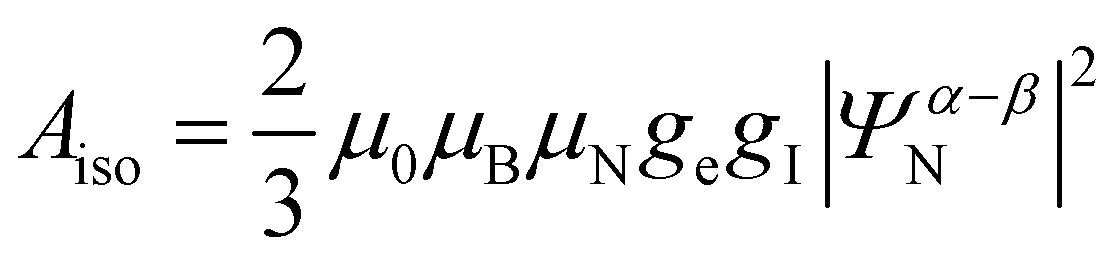 | (5) |
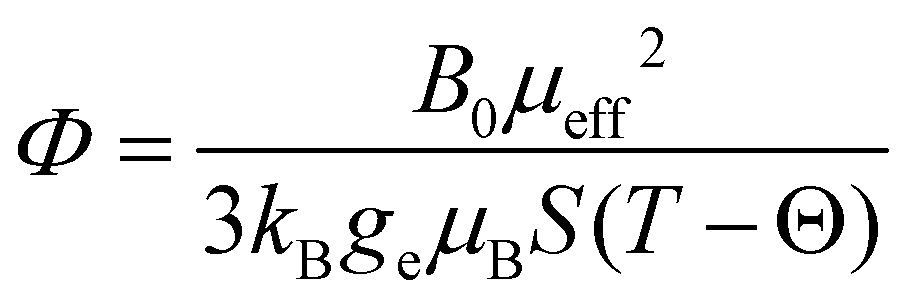 | (6) |
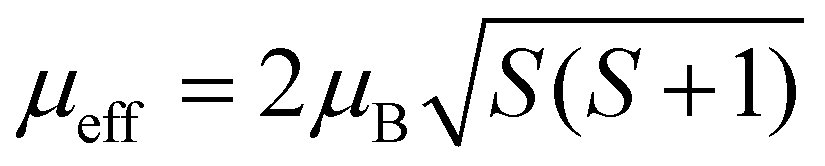 (spin-only value).
(spin-only value).
In the above formalism, shift anisotropy can also be computed with the electron–nuclear dipolar interaction tensor Tij and scaled in the same way to obtain the anisotropic components of the shift tensor δij:
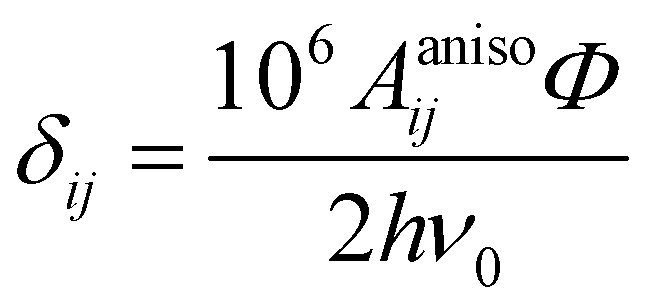 | (7) |
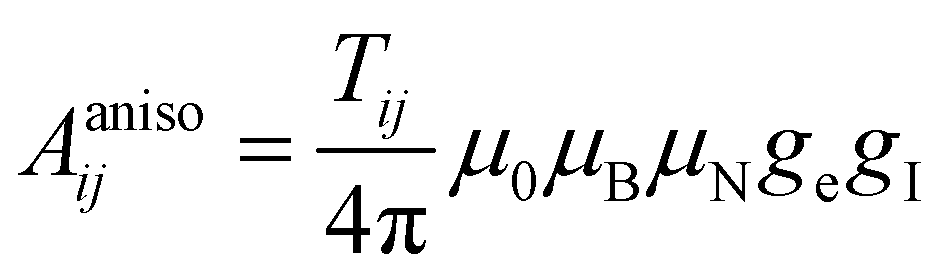 | (8) |
After this δij can be diagonalized to obtain the principal components δii which can be used to calculate the shift anisotropy. In this study, we employ the convention of Herzfeld and Berger,21 where the span Ω and skew κ are used to represent the anisotropy. Following the approach of Middlemiss et al., the contributions of each Fermi contact pathway to the total spin density Δ∣Ψα–βN∣2 were also calculated by reversing the direction of electronic spins on each TM site in turn.22 In addition, the total spin density ∣Ψα–βN∣2 was calculated from experimental shift by eqn (5) and (7).
The second-order quadrupolar shift δQ can be obtained from the quadrupolar frequency νQ from the expression23
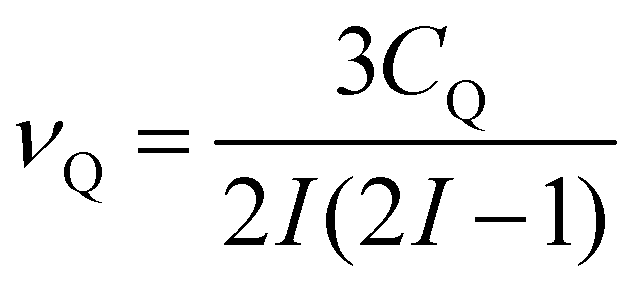 | (9) |
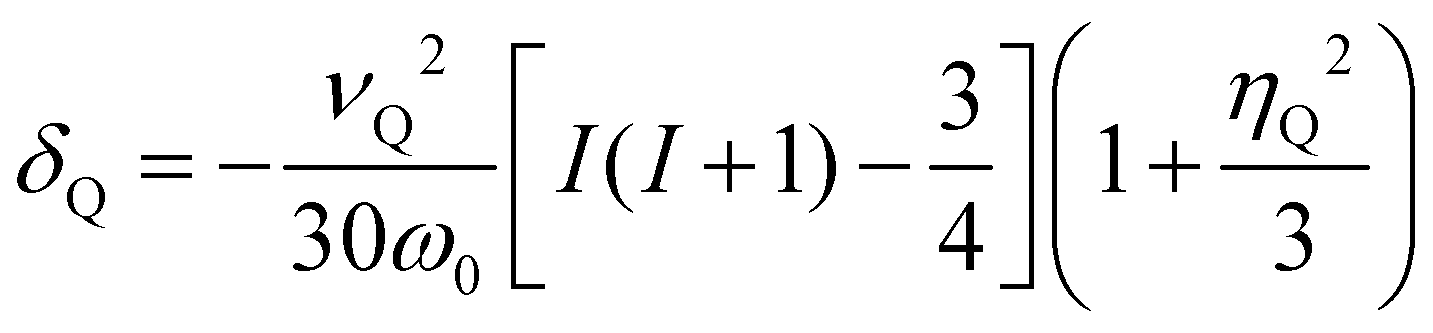 | (10) |
For systems with a large shift anisotropy tensor and quadrupolar coupling tensor such as Mg6MnO8 (see results), the relative orientation of the two tensors is also important in accurate fitting of the spinning sideband manifolds. This can be represented by three Euler angles α, β, and γ, where the angles represent rotations of the electric field gradient principal axes to match the Aanisoii principal axes. Hyperfine and electric field gradient tensor principal axes are ordered such that ∣A22∣ ≤ ∣A11∣ ≤ ∣A33∣ and ∣V22∣ ≤ ∣V11∣ ≤ ∣V33∣. The ZYZ convention and counterclockwise (positive) rotation are assumed. The full rotation matrix is shown in Section S4 (ESI†).
All calculations were performed using the spin-polarized B3LYP functional.29,30 Recent results indicate that using 35% Hartree Fock (HF) exchange gives better results for solid-state magnetic calculations.31,32 Previous ab initio studies on 7Li, 23Na, and 31P paramagnetic shifts show that values obtained using 20% and 35% provide the upper and lower bounds for the experimental shifts.20,22,24 Hence the original B3LYP (termed ‘Hyb20’) and a modified B3LYP with 35% of HF exchange (termed ‘Hyb35’) were both used in calculations.
Convergence of energy and spin density was checked with the number of sampled points in the reciprocal space, and 4 × 4 × 4 Monkhorst–Pack33 sampling scheme was used for all systems. Self-consistent field (SCF) cycles were converged to an energy difference of 10−7 hartree.
Experimental cell geometries were expanded into supercells and were optimized in CRYSTAL09 with all cell symmetries removed, except for the MgV2O4 case where the cubic cell was used with the available space group symmetry due to instabilities in the SCF cycles. All geometry optimizations were performed under the CRYSTAL default convergence criteria. Results from DFT calculations are reported in Table 2 and are also discussed in detail in the results section.
| Mg6MnO8 | MgCr2O4 | MgV2O4 | MgMn2O4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hyb20 | Hyb35 | Expt | Hyb20 | Hyb35 | Expt | Hyb20 | Hyb35 | Expt | Hyb20 | Hyb35 | Expt | ||
| Magn. | J 1/K | −0.7 | −0.5 | −0.7 | −19.8 | −14.8 | −30.4 | See text | −50 | −45.0 | −46.2 | — | |
| Θ/K | −22.5 | −16.4 | −21.9 | −297.3 | −222.2 | −456.7 | −600 | −448.3 | −466.8 | −452.5 | |||
| NMR | ∣Ψα–βN∣2/10−3 × Bohr−3 | 19.5 | 17.4 | 17.0 | 40.4 | 34.1 | 37.9 | 41.2 | 36.1 | 36.1 | 43.0 | 34.1 | 34.9 |
| Δ∣Ψα–βN∣2/10−3 × Bohr−3 | 9.5 | 8.5 | — | 3.3 | 3.0 | — | 3.4 | 3.0 | — | 4.0/2.6 | 3.3/1.8 | — | |
| δ iso/ppm | 3291 | 3152 | 2994 | 3842 | 3690 | 2862 | 2105 | 1840 | 1845 | 3901 | 3021 | 3128 | |
| δ FC/ppm | 3337 | 3199 | 3041 | 3842 | 3690 | 2862 | 2105 | 1840 | 1845 | 3938 | 3054 | 3178 | |
| δ Q/ppm | −46 | −47 | −47 | 0 | 0 | 0 | 0 | 0 | 0 | −37 | −33 | −50 | |
| Ω/ppm | 2110 | 2297 | 1930 | 0 | 0 | 840 | 0 | 0 | 672 | 698 | 632 | 1340 | |
| κ | −0.84 | −0.82 | −0.84 | — | — | 1.0 | — | — | −0.75 | −1.0 | −1.0 | 1.0 | |
| C Q/MHz | 3.64 | 3.69 | 3.70* | 0 | 0 | 0 | 0 | 0 | 0 | 3.41 | 3.22 | 3.40* | |
| η Q | 0.40 | 0.38 | 0.38* | — | — | — | — | — | — | 0.0 | 0.0 | 1.0* | |
| α | 270°* | — | — | 0°* | |||||||||
| β | 90°* | — | — | 0°* | |||||||||
| γ | 90°* | — | — | 42°* | |||||||||
3 Results and discussion
3.1 Diffraction and magnetic characterization
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group symmetry (Fig. 2). The structure of Mg6MnO8 exhibits ordered cation vacancies □, where a vacancy is formed according to the following defect reaction written in Kroger–Vink notation:
m space group symmetry (Fig. 2). The structure of Mg6MnO8 exhibits ordered cation vacancies □, where a vacancy is formed according to the following defect reaction written in Kroger–Vink notation: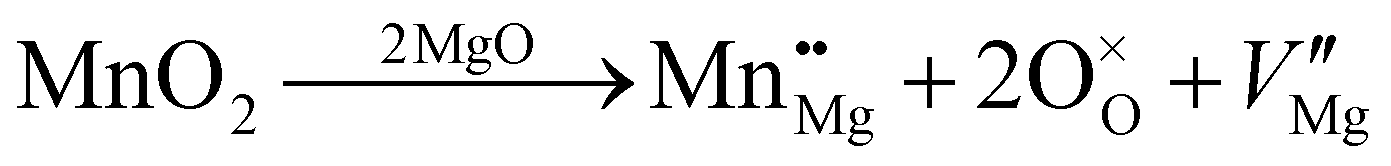 | (11) |
The oxygen sublattice is also slightly distorted from the ideal cubic rocksalt structure due to the smaller ionic radius of Mn4+ (0.53 Å) compared to Mg2+ (0.72 Å).35 The Mg site in this structure shows m.mm site symmetry, reflecting the distorted octahedra as opposed to the undistorted octahedra in the parent MgO structure. This should give a nonvanishing quadrupolar coupling constant, CQ, and quadrupolar asymmetry parameter, η, for 25Mg.
The powder X-ray diffraction pattern of Mg6MnO8 and the Rietveld refinement result are shown in Fig. S3 (ESI†). The product is single phase, with a diffraction pattern that matches to the previously reported result of Mg6MnO8, showing a cubic Fmm symmetry.34
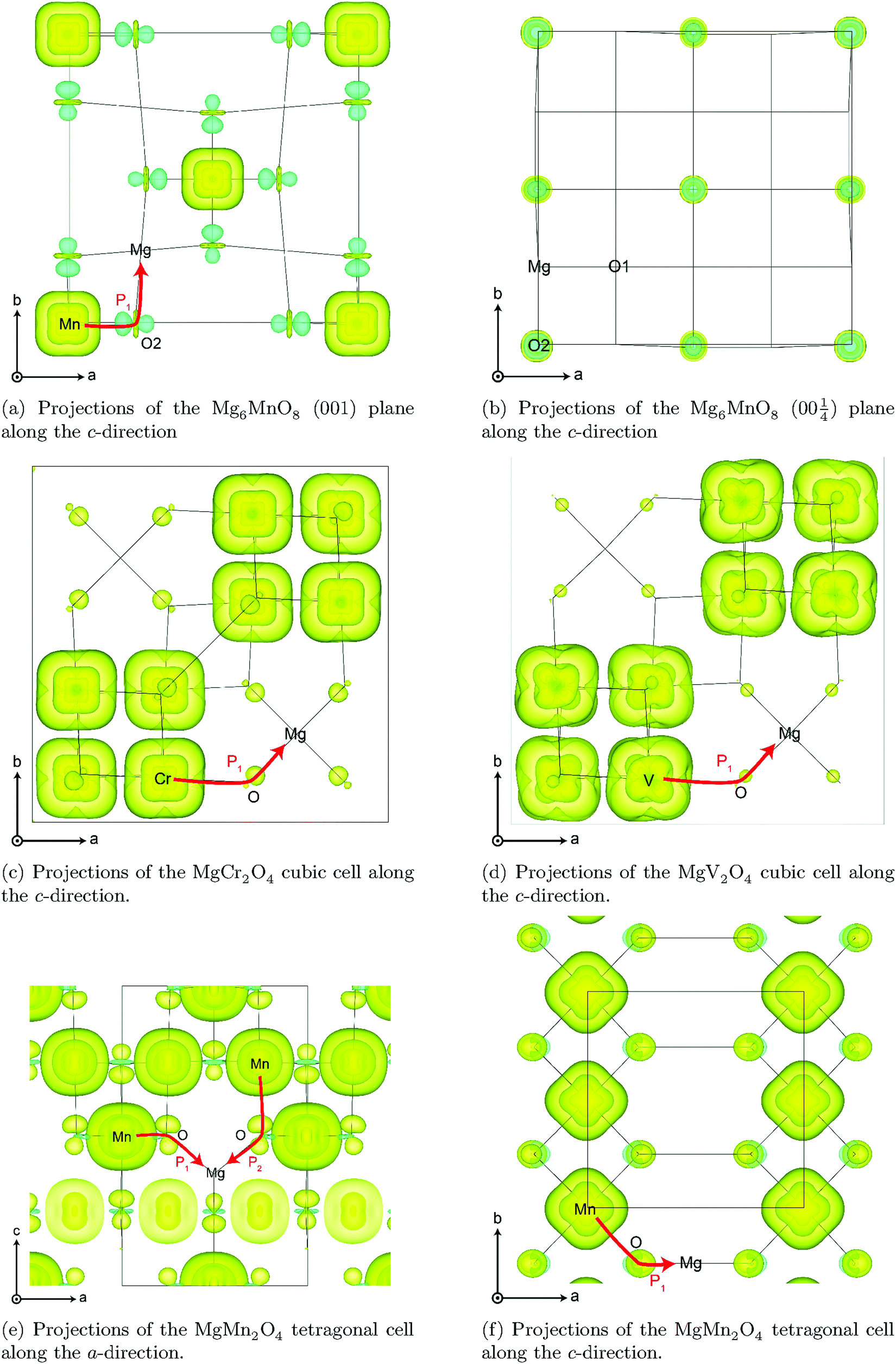
Transition metal (TM) sites in Mg6MnO8 are separated by Mn–O–Mg–O–Mn bond pathways, resulting in an extended superexchange interaction between the unpaired electronic spins of Mn4+ (Fig. 1a). The nature (ferromagnetic or antiferromagnetic) and magnitude of this interaction can be represented by an exchange coupling parameter J1, where we are only considering the nearest neighboring interactions (12 for each Mn4+). At temperatures T > 20 K, the magnetic susceptibility data of Mg6MnO8 (Fig. S7, ESI†) show the weak Curie–Weiss paramagnetism, with an antiferromagnetic ordering transition at the Néel temperature of TN = 5 K. Fitting to the Curie–Weiss law, 35 < T < 300 K, yielded the Weiss constant Θ = −21.9 ± 0.4 K and J1 = −0.73 ± 0.01 K. Due to the long Mn–Mn distance (6 Å), weak exchange coupling is expected; in this case, the coupling is antiferromagnetic (J < 0) as expected for the extended superexchange interactions.36 The obtained Weiss constant is in excellent agreement with the published experimental result of −20 ± 5 K.37 In addition, the effective electron magnetic moment μeff = 3.99 ± 0.01 μB obtained from the Curie constant shows good agreement with the spin-only value of 3.87 μB.
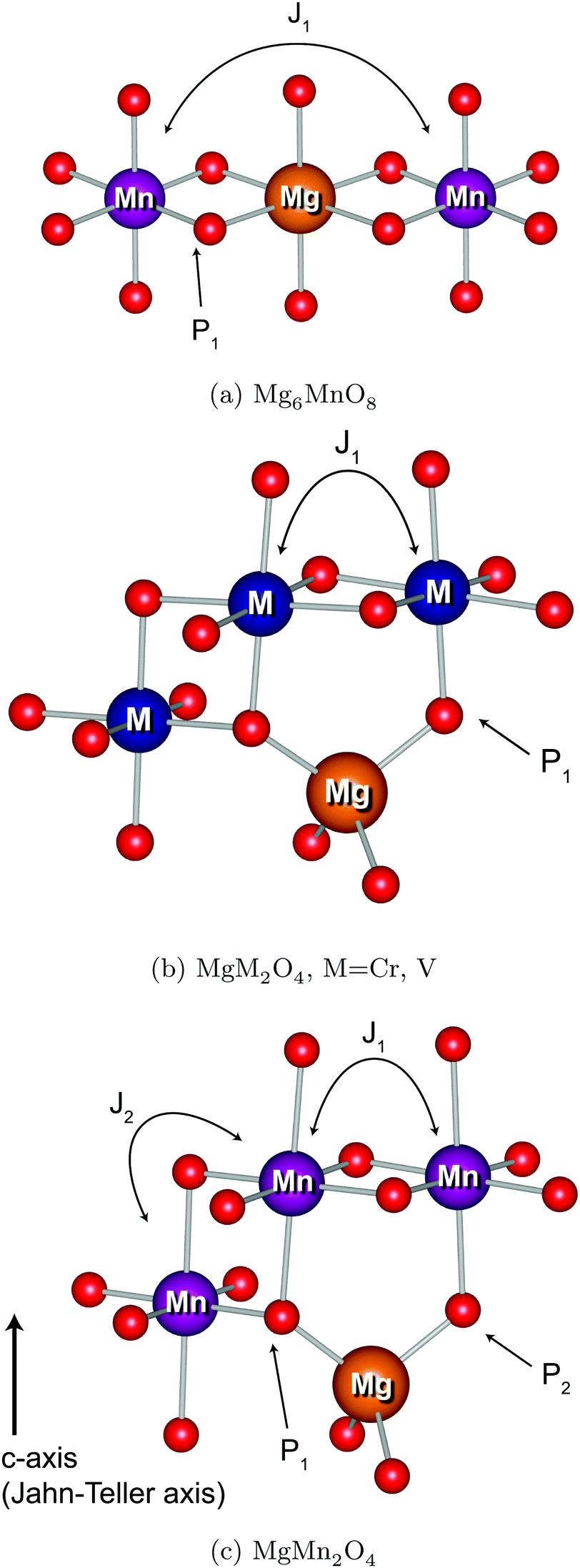 | ||
| Fig. 1 25Mg bond pathway (P) contributions and TM–TM J couplings. | ||
m space group symmetry and Mg2+ ions in the tetrahedral (8a) sites, the Cr/V occupying the octahedral (16d) metal sites. The thermodynamic factors controlling normal versus inverse spinel formation are well described in the literature.38–41 In both these materials, the crystal field stabilization energy (CFSE) of the TM ions are responsible for their preference for occupancy of the 16d site. In the MgV2O4 case, the orbital degree of freedom in V3+ ion also favors the normal spinel structure.40
The powder X-ray diffraction patterns of MgCr2O4 and MgV2O4 are shown in Fig. S4 and S5 (ESI†), respectively. As expected from the structural discussion above, both spinels show cubic Fdm symmetries and are normal spinels. 15.4 ± 0.4% by weight of unreacted V2O3 is also seen in the diffraction pattern. This behavior is known to occur due to the reduction of MgO and sublimation of Mg metal at high temperatures under a reducing atmosphere of H2.42 In contrast, no impurity phase is detected in the MgCr2O4 sample.
The TM sites in cubic spinels have six nearest neighboring TM sites connected through 90° TM–O–TM bonds, resulting in magnetic exchange which can be represented with J1 (Fig. 1b). First we consider the MgCr2O4 sample. From the Curie–Weiss fit (100 < T < 301 K) to the experimental magnetic susceptibility data (Fig. S8, ESI†), Néel temperature of TN = 13 K, Weiss constant of Θ = −456.7 ± 3.4 K, and J1 = −30.4 ± 0.2 K are obtained. The effective electron magnetic moment μeff = 4.25 ± 0.03 μB obtained from the Curie constant is slightly larger than the spin-only value of 3.87 μB, which originates from the fact that there are still significant short-range fluctuations over the temperature range of fitting (Θ > 300 K). The observed Weiss constant is of the same order of magnitude as the previously reported value of −433 K.43
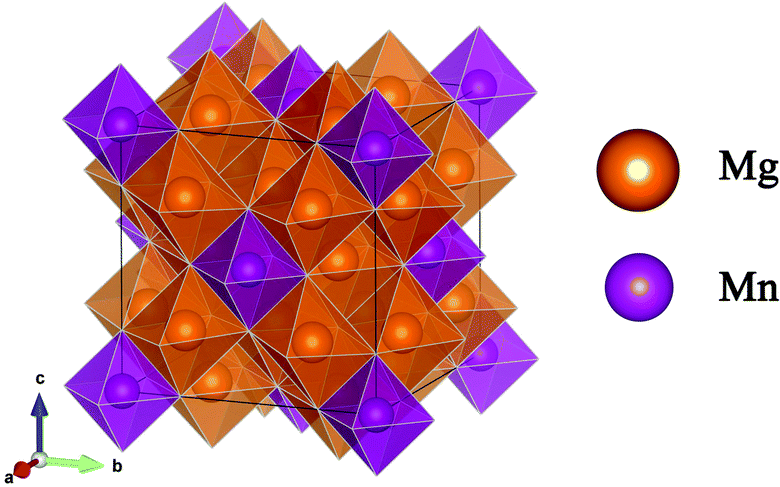 | ||
| Fig. 2 Structure of Mg6MnO8, shown with oxygen polyhedra around metal ions. | ||
In the case of MgV2O4, an experimental measurement of magnetic susceptibility was not performed due to the presence of the V2O3 secondary phase. Detailed discussion on the magnetism follows in the DFT section below.
| Mg2+(tet) + 2Mn3+(oct) → Mg2+(oct) + Mn4+(oct) + Mn2+(tet) | (12) |
This inversion behavior is known to occur around 1073 K,44,45 resulting in the following cation distribution:
| [Mg2+1−xMn2+x]tet[Mg2+xMn2+xMn4+2−2x]octO4 | (13) |
It is known experimentally that low-temperature synthesis of this material through a coprecipitation or sol–gel method suppresses this inversion, resulting in an ordered normal spinel.15,46 We also note that this material has been previously shown to be a promising Mg-ion battery cathode material, being able to reversibly de-insert Mg2+ ions in an aqueous electrolyte system.14
As discussed above, we have attempted the synthesis of an ordered, normal MgMn2O4 spinel through a citrate sol–gel method at a relatively low temperature of 773 K. The X-ray diffraction pattern of this sample is shown in Fig. S6 (ESI†), alongside the results from the Rietveld refinement. Refinement of the sample shows a tetragonal spinel, with 7.9 ± 0.3% of Mg6MnO8 secondary phase by weight. This impurity phase could not be eliminated despite attempts to make a homogeneous mixture of Mg and Mn in the gel, and repeated calcination attempts with intermediate grinding. Previous syntheses of MgMn2O4 through sol–gel and solid-state methods have also reported the presence of this secondary phase.14,15,47 This suggests that excess Mn is present in the tetrahedral (4a) site of the spinel structure in a divalent form, Mn2+. From the starting Mg to Mn ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, the stoichiometry of this sample is calculated as (Mg2+0.81Mn2+0.19)Mn3+2O4. However, refinement of the site occupancies shows a composition of (Mg2+0.92±0.02Mn2+0.08±0.02)Mn3+2O4, which suggests overall Mn deficiency in the starting composition. Due to the low calcination temperature, the peaks are inherently broad and good fit could not be obtained.
:2, the stoichiometry of this sample is calculated as (Mg2+0.81Mn2+0.19)Mn3+2O4. However, refinement of the site occupancies shows a composition of (Mg2+0.92±0.02Mn2+0.08±0.02)Mn3+2O4, which suggests overall Mn deficiency in the starting composition. Due to the low calcination temperature, the peaks are inherently broad and good fit could not be obtained.
Evidence for presence of this tetrahedral Mn2+ comes from the magnetic susceptibility data (Fig. S9, ESI†). In an ordered normal MgMn2O4 spinel, the tetragonal Jahn–Teller distortion of Mn3+ ions results in four nearest neighbor interactions J1 along the a and b axes and two second nearest neighbor interactions J2 along the c axis (Fig. 1c). Despite the fact that these values are not straightforward to obtain from experiments without a good model for the magnetism, we should expect a maximum below the magnetic ordering transition in the inverse susceptibility (1/χ) versus temperature plot, since the exchange coupling in the octahedral sublattice is antiferromagnetic. Zero-field cooled susceptibility data, however, clearly shows presence of a ferrimagnetic component with a minimum in the plot. This behavior is similar to Mn3O4, where an antiferromagnetic coupling between the tetrahedral Mn2+ and octahedral Mn3+ causes ferrimagnetic ordering at low temperatures.48 In addition, the fitted effective electron magnetic moment μeff = 5.93 ± 0.06 μB is clearly larger than the spin-only value of 4.90 μB for the Mn3+ ion (d4), suggesting the presence of Mn2+ ion (d5). The calculated spin-only magnetic moments μeff of the (Mg2+0.81Mn2+0.19)Mn3+2O4 and (Mg2+0.91Mn2+0.09)Mn3+2O4 (determined from composition and refinement, respectively) are 5.00 and 4.95 μB, respectively. As for the MgCr2O4 case, the large discrepancy is likely due to the short-range fluctuations at T < Θ.
The magnetic properties of the (MgxMn1−x)Mn2O4 solid solution have previously been studied at T < 60 K.48 The magnetic susceptibility is shown to be small (<0.1 emu mol−1) and qualitatively similar for MgMn2O4 and Mg0.75Mn0.25Mn2O4 at 60 K. In this work, we are interested in χ at 320 K (MAS frictional heating) and so will approximate our sample to MgMn2O4. Finally, we note that the experimental Weiss constant Θ = −452.5 K is consistent with a previously reported value of −500 K,49 with the deviation between the two values potentially arising due to the excess Mn as discussed above.
3.2 25Mg NMR
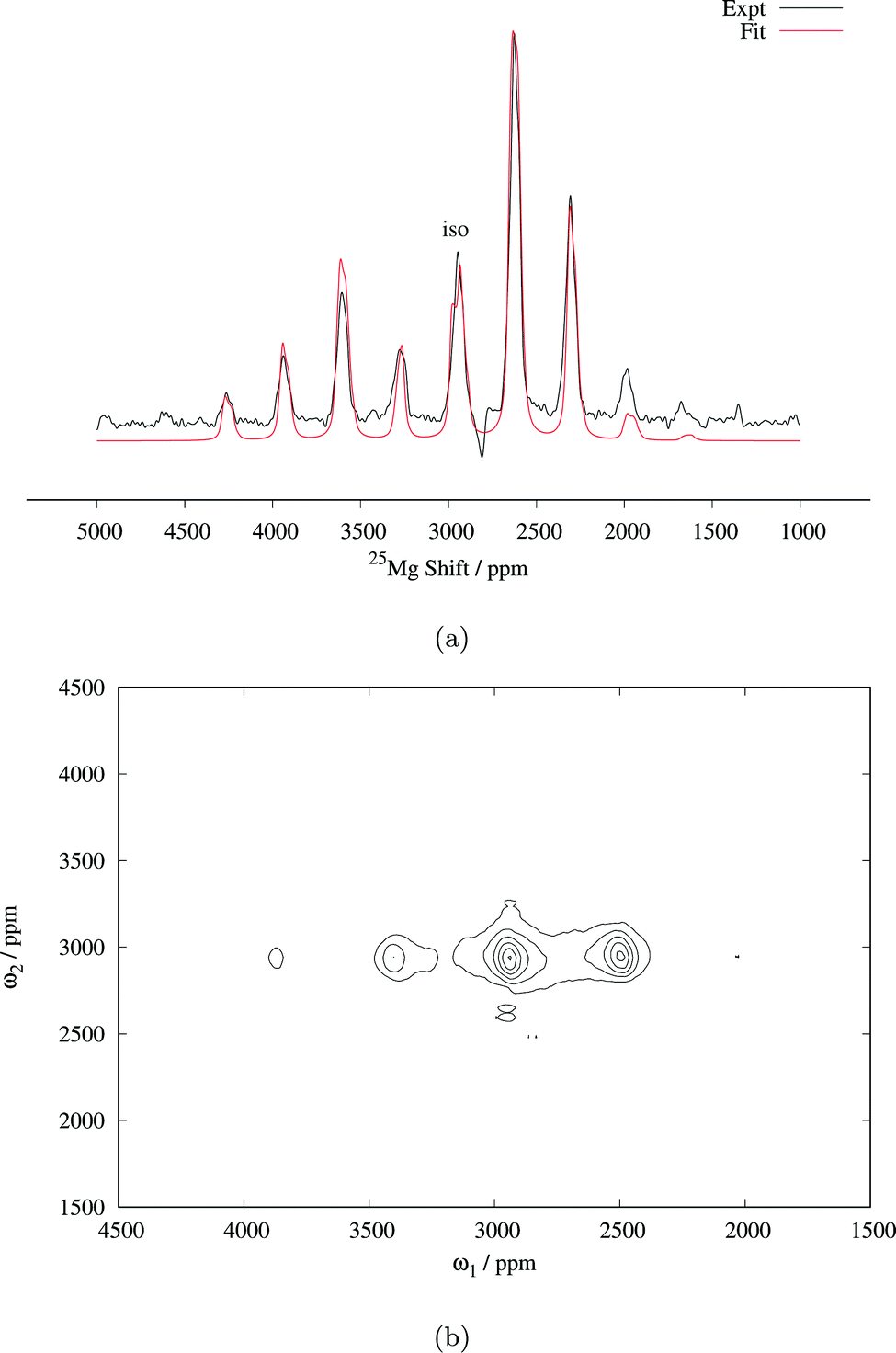 | ||
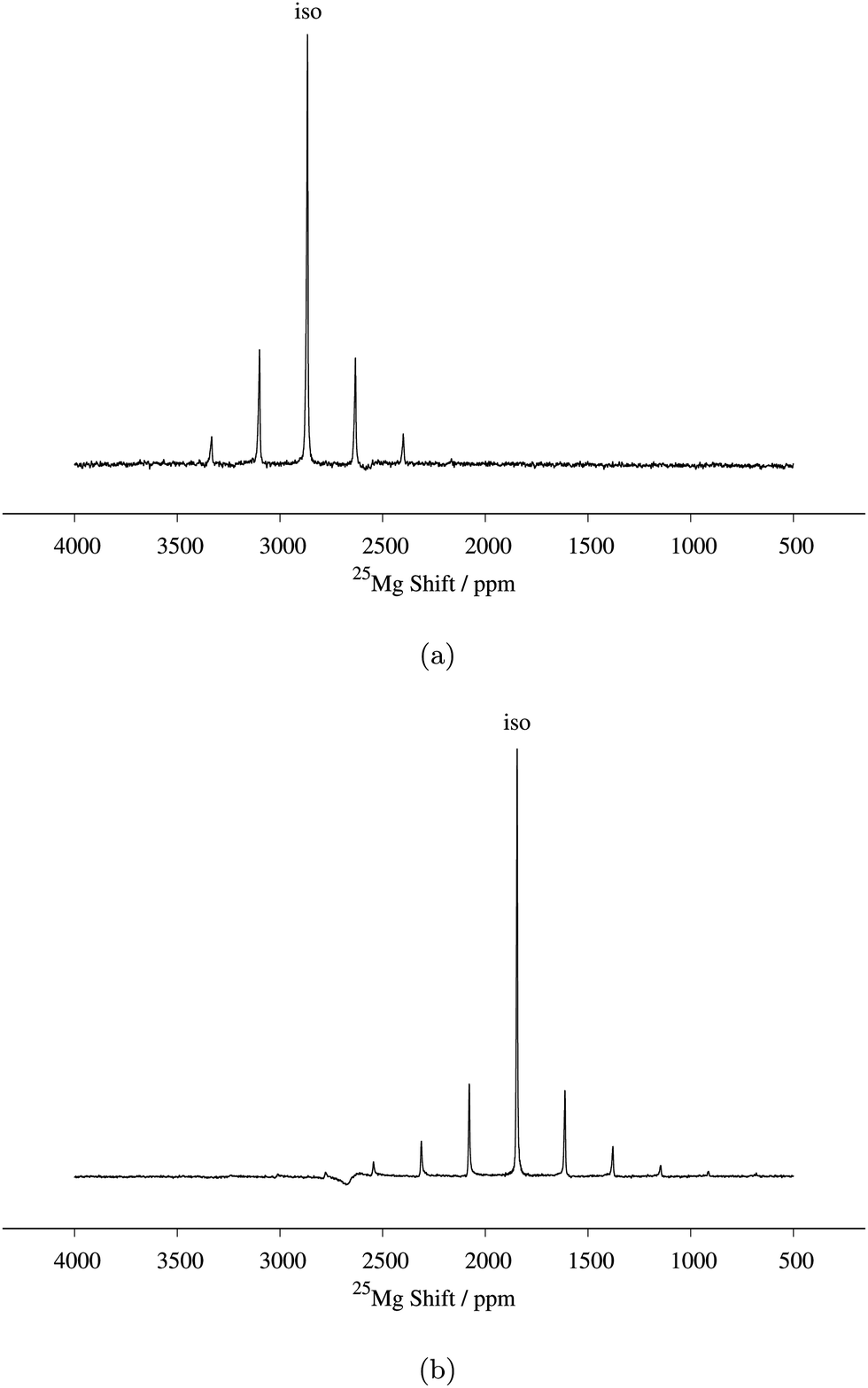
| Fig. 3 (a) 25Mg spin echo spectrum of Mg6MnO8 (14 kHz MAS, 81712 transients), with fitted central transition lineshape including contributions from both the paramagnetic shift anisotropy and the quadrupolar interaction (parameters are listed in Table 2). (b) Magic angle turning (MAT) spectrum of Mg6MnO8 (20 kHz MAS, 128 slices in the F1 dimension with 2.23 μs delay increment, 3072 transients acquired in each slice). RAPT pulses were applied before the MAT pulses to enhance the signal-to-noise ratio. | ||
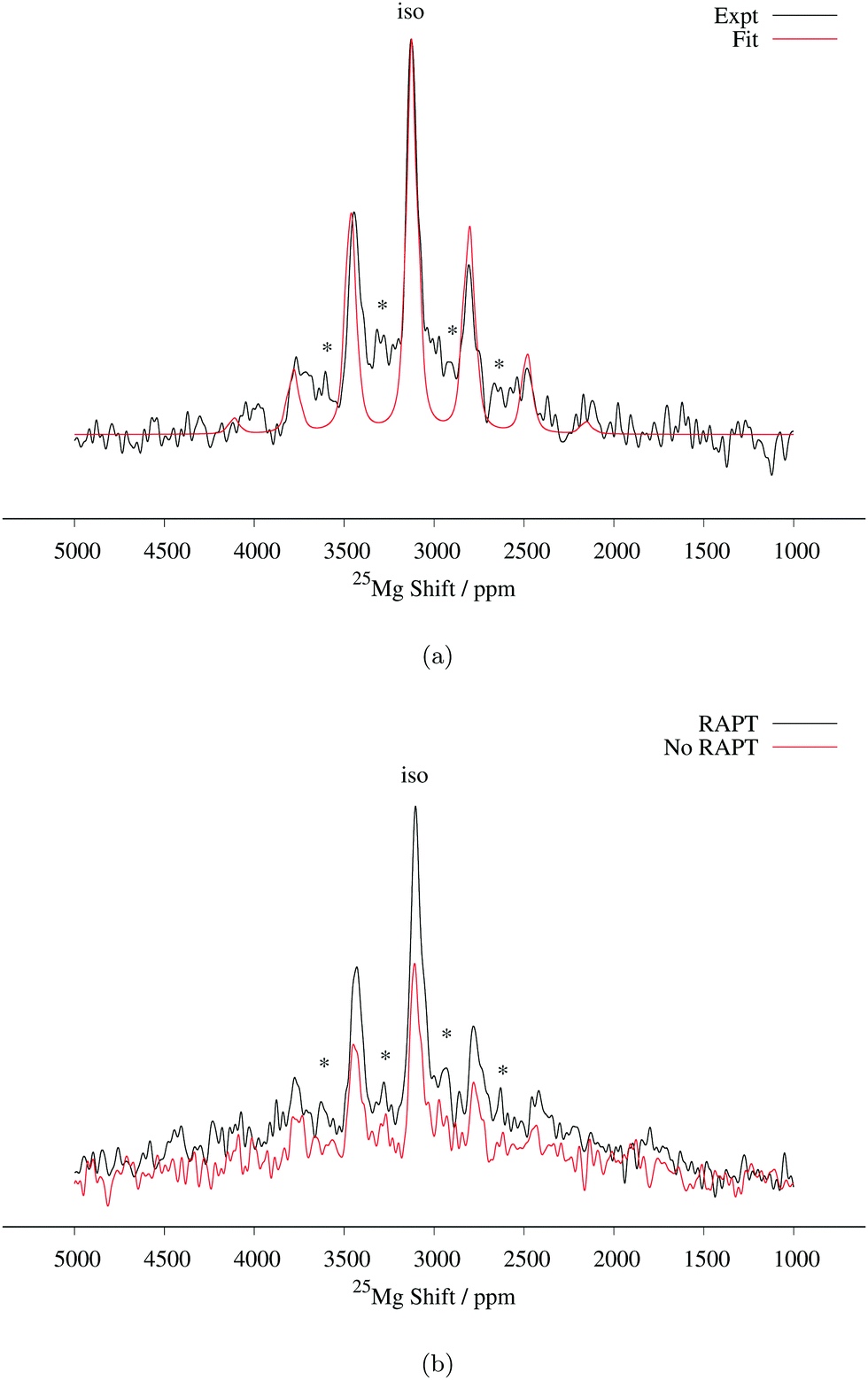
Determination of quadrupolar parameters from paramagnetic NMR spectra is considered to be difficult, as the characteristic quadrupolar patterns are often smeared out due to paramagnetic broadening of the spectra. We used the RAPT pulse sequence, which was previously used to estimate 27Al quadrupolar parameters in diamagnetic samples,50 to estimate the quadrupolar coupling constant, CQ, of the Mg site in Mg6MnO8. For optimum enhancement, the modulation frequency νm of saturating pulse trains alternating in phase (+π/2, −π/2) should match the value of νQ/2, where νQ is the quadrupolar frequency of the observed nucleus given by eqn (9).12 This condition is given by νm = 3CQ/40 for I = 5/2. By plotting the enhancement in integrated signal intensity versus the offset frequency, we see that the maximum enhancement occurs between νm = 250–300 kHz (Fig. 4a). This is consistent with the DFT prediction of νm = 277 kHz, or the quadrupolar coupling constant CQ = 3.69 MHz (Table 2).
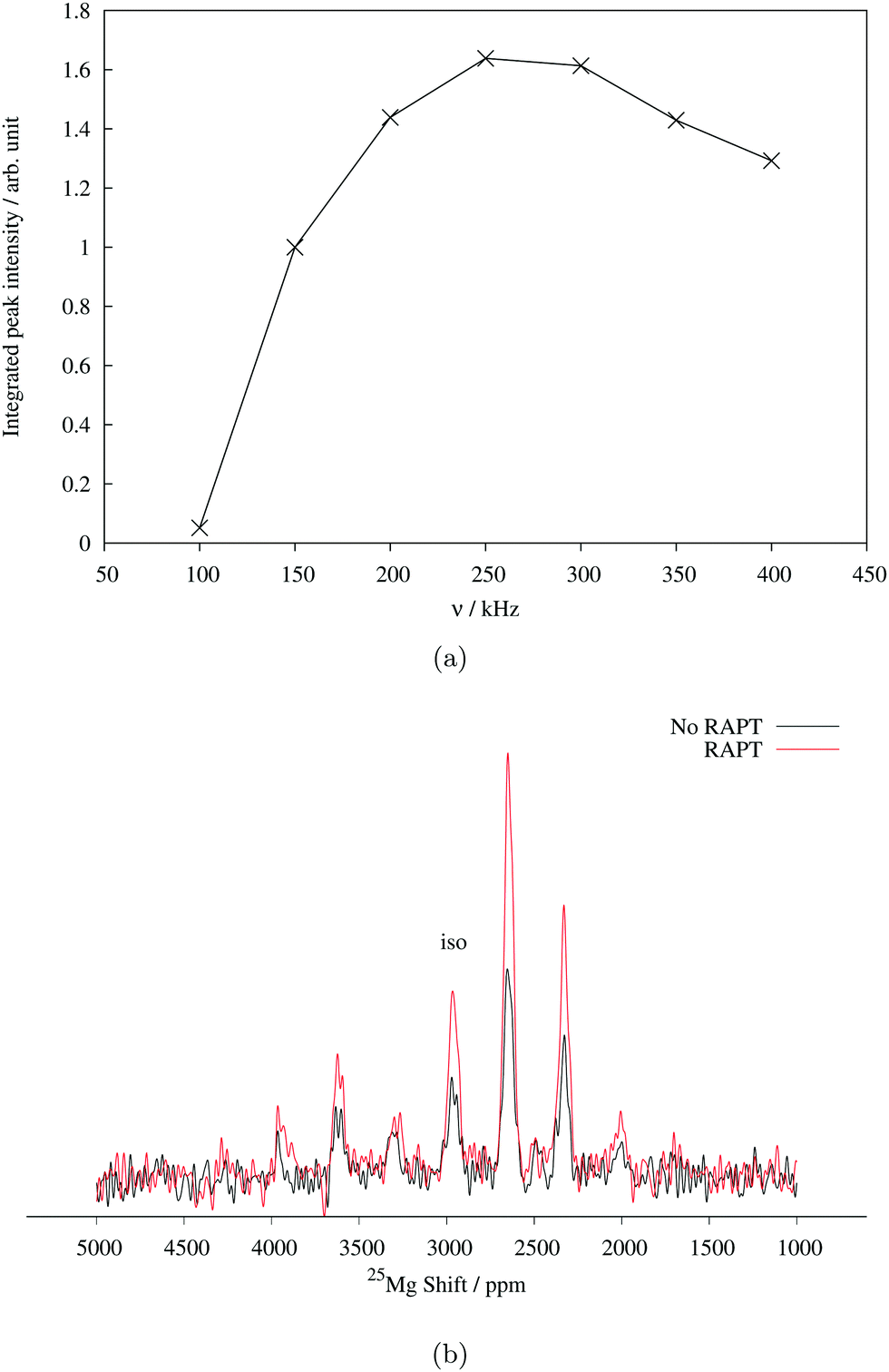 | ||
| Fig. 4 (a) 25Mg spin echo signal intensity of Mg6MnO8 with increasing offset frequency of saturating pulse trains in RAPT experiment. (b) Enhancement of spin echo signal intensity using the RAPT pulse. Saturating pulses were applied at modulation frequency of 270 kHz. Both experiments were performed at MAS spin rate of 14 kHz. 1024 transients were acquired in each spectrum. | ||
Starting from these values of δiso and CQ, a spectral fitting was performed with quadrupolar parameters fixed to the Hyb35 calculated values. The MAS lineshape could be fit assuming paramagnetic shift anisotropy and a quadrupolar interaction and the fitted parameters are shown in Table 2. Good fitting was obtained with the quadrupolar parameters from DFT calculations, and no further attempts were made to fit the quadrupolar parameters. The spectrum is dominated by the central transition (transition between spin levels 1/2 ↔ −1/2), which is only affected to second-order by the quadrupolar coupling, and hence (other than the linewidth of the individual peaks within the spinning sideband manifold) is relatively insensitive to the size of the quadrupolar interaction. The paramagnetic shift anisotropy, arising from the dipolar coupling to the Mn4+ ions, which gives rise to a lineshape identical to that observed from the chemical shift anisotropy, is clearly visible. We note that the fitted δiso = 2994 ppm is larger than the δiso = 2960 ppm obtained from the MAT experiment above. As the slower MAS rate results in lower sample temperatures, an increase in the paramagnetic shift is expected in the 14 kHz MAS case when compared to 20 kHz MAS spectrum (i.e. the MAT experiment).
From the RAPT pulse sequence, a maximum signal-to-noise enhancement by a factor of close to 2 is observed (Fig. 4b). Under ideal conditions, the RAPT pulse sequence should result in an enhancement factor of I + 1/2 = 3 for 25Mg. However, due to the difficulty of saturating multiple satellite levels in a polycrystalline I = 5/2 system, it is not uncommon to observe enhancement factors lower than the theoretical maximum (for instance, an enhancement factor of 2 is reported for polycrystalline 27Al (I = 5/2) spectrum in diamagnetic albite NaAlSi3O8).12,51 Despite the added difficulty of enhanced relaxation of the spin polarization in paramagnetic materials, the observed enhancement factor of 2 clearly shows that the RAPT pulse sequence can be used for significant improvement in signal-to-noise ratio in paramagnetic materials.
In addition to the RAPT pulse sequence, we have also attempted a Double Frequency Sweep (DFS) enhancement scheme52 also frequently used for quadrupolar nuclei. Only a signal-to-noise enhancement factor of around 1.5 is observed (Fig. S2, ESI†), as opposed to 2 times enhancement in RAPT. This could be explained in terms of faster paramagnetic relaxation effects present in the sample, as DFS pulses are usually longer (around 2000 μs) to satisfy adiabaticity, whereas RAPT pulses are shorter (around 110 μs optimized for Mg6MnO8).
Based on the DFT (see below) and 25Mg NMR results of Mg6MnO8, we propose that Mg6MnO8 can be conveniently used as a model compound for paramagnetic 25Mg NMR studies owing to a number of attractive properties: (1) it has six Mg atoms per formula unit and good NMR sensitivity, allowing signals to be observed with only 1024 scans at 16.4 T field; (2) it has nonvanishing quadrupolar coupling, meaning that we can study the quadrupolar behavior of Mg atoms, which is likely to be the case for many technologically relevant materials showing non-zero quadrupolar couplings; (3) the d3 electron configuration of Mn4+ in an octahedral environment eliminates the need to consider spin–orbit coupling effects in ab initio calculations to a good approximation; (4) the large distance (6 Å) between Mn atoms makes it a weak, well-defined paramagnet, which reduces the possible error in the paramagnetic scaling approach as taken in this work; (5) it is easy to prepare as a pure phase through solid-state reaction of corresponding oxides in air, although previous works have reported a sol–gel route53 or calcination of carbonates under oxygen atmosphere.34 It is also interesting to note that this compound was theoretically predicted to be a good carbon capture and storage material through a large-scale screening study.54
 | ||
| Fig. 5
25Mg spin echo spectrum of (a) MgCr2O4 and (b) MgV2O4, both at a MAS spin rate of 10 kHz. 20480 and 505600 transients were acquired in each case. The broad feature seen around 2700 ppm is due to probe background. | ||
Tetrahedral coordination also dictates that the anisotropic (dipolar) electron–nuclear spin interaction be zero. Despite this, small nonvanishing dipolar interactions were observed (Ω = 840 ppm for MgCr2O4 and 672 ppm for MgV2O4). This discrepancy may be attributed to (i) the bulk magnetic susceptibility effect which originates from inhomogeneous magnetic field due to random crystallite orientations, as noted in previous paramagnetic MAS NMR works,55,56 or (ii) local defects present in the open spinel structure, resulting in breaking of the local symmetry. With the current data, it is difficult to ascertain which of these is going to be dominant; it is likely that both are responsible for the sideband intensities.
 | ||
| Fig. 6
25Mg spin echo spectrum of MgMn2O4 at a MAS spin rate of 14 kHz. Mg6MnO8 secondary phase signals are shown with asterisks (*). 3550928 transients were acquired. (a) Comparison between experimental and fitted spectrum. Fitted parameters are listed in Table 2. (b) Enhancement of spin echo signal intensity with RAPT pulse sequence. RAPT modulation pulses were applied at 240 kHz. 1024000 transients were acquired in each spectrum. | ||
We now compare this result to the work of Kim et al., who have recently reported a 25Mg NMR spectrum of this compound.14 Their reported NMR spectrum at a MAS frequency of 24 kHz shows two peaks at 2980 and 2850 ppm, which they assigned to MgMn2O4, the two peaks being assigned to discontinuities of a second-order quadrupolar lineshape with a CQ of 5.4 MHz. Considering the inverse temperature dependence of Fermi contact shifts (eqn (6)), the MgMn2O4 resonance frequency should occur at higher shifts in the case of the 14 kHz MAS used here. For example, assuming representative rotor temperatures of 340 K and 320 K for 24 kHz and 14 kHz MAS (from our previous temperature calibration measurements), respectively, and using experimental value of Θ, our observed shift of δiso = 3128 ppm at 14 kHz MAS translates to δiso = 3047 ppm at 24 kHz MAS for our MAS probes (eqn (3)). Thus, we assign their 2980 ppm peak to the Mg in MgMn2O4 structure, the lower shift originating from a lower sample temperature and/or slight differences in magnetic properties between samples. Considering the twicefold (i.e. the maximum attainable enhancement observed in the Mg6MnO8 case) enhancement in signal intensity using the RAPT pulse parameters calculated from CQ = 3.2 MHz, the extra peak at 2850 ppm does not appear to arise from a MAS quadrupolar lineshape with CQ of 5.4 MHz as suggested in their paper. Alternatively, we suggest that these peaks represent Mg sites in different coordination environments to the Mn in different oxidation states, arising from the antisite defect (eqn (12)). We tentatively assign the peak at 2850 ppm to Mg neighboring one Mn4+ and eleven Mn3+, as opposed to the 2980 ppm peak where the Mg is neighboring twelve Mn3+. Detailed investigation on the nature of this site is ongoing.
3.3 DFT calculation of NMR and magnetic parameters
We now discuss the ab initio results on the compounds studied above. DFT calculated values of magnetic parameters and paramagnetic NMR shifts are shown in Table 2.For MgV2O4, an ab initio prediction of magnetic parameters could not be made due to SCF instabilities in the supercell structure. However, a reasonable estimate of the paramagnetic shift could still be made from a previous experimental measurement of the Weiss constant Θ = −600 K.57 Despite the fact that spin–orbit interactions at the d2 center may influence the shift, calculations involving explicit spin–orbit coupling Hamiltonian could not be performed in the present version of CRYSTAL. Despite this limitation, the shift values obtained without any correction for spin–orbit coupling shows a close match with the experimental result. Mulliken spin population analysis (Table S1, ESI†) and spin density maps (Fig. 7d) of the converged wavefunction shows equal occupation of the three t2g orbitals dxy, dyz, and dzx (which is expected, since we have preserved the cubic symmetry of the experimental cell). This is likely to arise from the fact that the spin–orbit coupling is relatively weak for V3+ ion (the spin–orbit coupling constant is λ = 104 cm−1 for free ion, 95 cm−1 when doped into Al2O3).59 Hence the energy levels split by spin–orbit coupling are equally occupied. It is thought that the ab initio result reflects this average occupation in effect, producing results close to the experimental values.
 | ||
| Fig. 7 3-Dimensional spin density maps. Yellow denotes positive spin; blue denotes negative spin. Bounding box refers to the unit cell and solid lines connecting atoms refer to the metal–oxygen bonds. Bond pathway contributions P to the spin density are shown in red. | ||
Finally, we note that in both cases DFT predicts zero paramagnetic shift anisotropy, which reflects the tetrahedral symmetry of Mg sites. As discussed above, this discrepancy is attributed to both the bulk susceptibility effect and local defects in the open spinel structures.
Paramagnetic NMR shifts calculated using these values also show good agreement with the experimental value, where the experimental shift is between the Hyb20 and Hyb35 calculated values. However, again we see a discrepancy in the anisotropic shift parameter, similar to the result for other spinels MgCr2O4 and MgV2O4. The discrepancy is also attributed to the inhomogeneous magnetic field created by random crystallite orientations and local defects. These effects are more significant in the MgMn2O4 sample, as the low-temperature preparation condition results in (i) smaller crystallites, which increases the magnetic field inhomogeneity, and (ii) higher concentrations of defects present in the sample.
3.4 Shift mechanism and Fermi contact pathways
We now turn our attention to factors determining these large paramagnetic shifts and how they might be rationalized in terms of electronic structures. Paramagnetic NMR shifts in various Li transition metal oxides have been ascribed to a Fermi contact (FC) mechanism.60 In this framework, a superexchange-like mechanism operates between the TM t2g/eg and the Li s orbital, resulting in small amounts of paramagnetic electron spins on the Li nucleus. As a similar kind of mechanism is expected to be responsible for paramagnetic Mg shifts, understanding the shift mechanism is important for rationalizing both the sign and magnitude of shifts.As shown in eqn (4), factors contributing to the Fermi contact shift can be separated into the spin density transfer ∣Ψα–βN∣2 (in units of Bohr−3), and electron paramagnetism. Considering the expression for Θ, we can observe that metal ions with larger S are expected to give larger Fermi contact shifts, due to the μeff factor. This factor only depends on the formal spin of metal ions involved. On the other hand, the Fermi contact spin density transfer depends heavily on the geometry of the TM–O–Mg pathway. Here we can evaluate the contributions of each FC pathway P as Δ∣Ψα–βN∣2, which sums up to ∣Ψα–βN∣2. This is important since pathways with similar bond lengths and angles should contribute similar amounts of spin density to the observed nucleus. With sufficient amount of experimental and ab initio data, a database can be constructed which can give approximate shifts for novel structures without the need for further first principles calculations.
3-Dimensional maps of electron spin density obtained from DFT wavefunctions are shown in Fig. 7. From the spin density maps, it is clear that a delocalization mechanism involving the t2g orbitals is in operation for all four compounds, resulting in positive spin density ∣Ψα–βN∣2 on the nucleus. This is particularly evident in the Mg6MnO8 case, where a 95° interaction along the TM–O–Mg pathway (P1 in Fig. 1a) results in p orbitals with positive spin density pointing directly towards the Mg site (Fig. 7a). We also note that p orbitals with negative spin density all point towards the vacancy site, which would result in a negative shift mediated by a polarization mechanism, had this site been occupied. Also large differences in spin densities are observed between the two crystallographically distinct oxygen sites O1 and O2, where essentially no spin is observed on the O2 site. This originates from the fact that O2 is only surrounded by Mg in its first coordination shell due to the ordered cation arrangements in the defect rocksalt structure (Fig. 7b).
The situation in spinels is more complex, as the TM–O–Mg pathway is no longer near 90°. Here, both delocalization (t2g–pπ–s) and polarization (eg–pσ–s) mechanisms are likely to operate along the P1 pathway (Fig. 1b), resulting in transfer of positive and negative spins on Mg, respectively. Hybridization of the oxygen orbital is evident, as the main lobe points towards the Mg atom in the case of cubic spinels MgCr2O4 and MgV2O4 (Fig. 7c and d, respectively). In both cases where we have no ambiguity in the t2g orbital occupation, it is clear that the delocalization mechanism is dominant, resulting in positive spin density on Mg.
A similar analysis could be done for tetragonal spinel MgMn2O4 where we have a Jahn–Teller distorted Mn3+ ion (d4), but the d orbital occupancies need to be determined. A close examination of the spin density map (Fig. 7e and f) and Mulliken spin population analysis (Table S1, ESI†) reveals that the valence electron configuration for Mn is 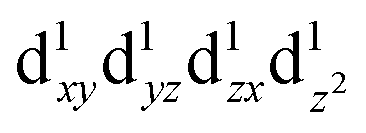 , showing occupation typical of a positive Jahn–Teller elongation. This has important implications for the FC mechanism, as we have two distinct FC pathways P1 and P2 as a result of the Jahn–Teller distortion. P1 lies on the crystallographic a,b-plane and P2 points along the c-axis in the crystal structure (Fig. 1c). In both cases, the occupied dz2 orbital on Mn3+ is likely to contribute positive spin density to the oxygen along the c-axis (delocalization), whereas the empty dx2−y2 orbital will contribute negative spin density to the oxygen along the a-axis (polarization). Again, examination of the spin density map reveals that the dominant shift mechanism is also the delocalization mechanism in both cases, with the oxygen pz orbital playing the most significant role. A large contribution from the dz2–pσ–s pathway is evident. A noticeable negative spin density is observed on oxygen positions (resulting from polarization along the eg–pσ pathway), although this does not contribute significantly to the shift.
, showing occupation typical of a positive Jahn–Teller elongation. This has important implications for the FC mechanism, as we have two distinct FC pathways P1 and P2 as a result of the Jahn–Teller distortion. P1 lies on the crystallographic a,b-plane and P2 points along the c-axis in the crystal structure (Fig. 1c). In both cases, the occupied dz2 orbital on Mn3+ is likely to contribute positive spin density to the oxygen along the c-axis (delocalization), whereas the empty dx2−y2 orbital will contribute negative spin density to the oxygen along the a-axis (polarization). Again, examination of the spin density map reveals that the dominant shift mechanism is also the delocalization mechanism in both cases, with the oxygen pz orbital playing the most significant role. A large contribution from the dz2–pσ–s pathway is evident. A noticeable negative spin density is observed on oxygen positions (resulting from polarization along the eg–pσ pathway), although this does not contribute significantly to the shift.
By obtaining the spin density contributions Δ∣Ψα–βN∣2 from each FC pathways (P), magnitudes of the observed shifts could be rationalized. Comparing the first-order contribution P1 of two d3 ions, Cr3+ in MgCr2O4 and Mn4+ in Mg6MnO8 (Table 2), a significant decrease in spin density transfer could be observed in the former case. In Mg6MnO8, each Mg atom is bonded to two Mn4+ ions via two 95° TM–O–Mg bonds. In terms of spin densities, pathway decomposition shows that each Mn4+ contributes approximately P1 = 9.5 × 10−3 Bohr−3 (Hyb20) or 8.5 × 10−3 Bohr−3 (Hyb35) to Mg. In MgCr2O4 where each Mg is bonded to 12Cr3+, each Cr3+ contributes approximately P1 = 3.3 × 10−3 Bohr−3 (Hyb20) or 3.0 × 10−3 Bohr−3 (Hyb35). This is rationalized by the differences in bond angles: as shown by Carlier et al., 90° interactions between the TM t2g and observed nucleus result in positive spin transfer via a delocalization mechanism, whereas 180° interactions result in negative spin transfer via a polarization mechanism.60 In Mg6MnO8, near 90° interactions dictate that the delocalization mechanism should be the dominant spin transfer mechanism, whereas in MgCr2O4 both delocalization and polarization mechanism should be in operation due to 121° bond angle. The polarization mechanism partially cancels the delocalization mechanism, resulting in small spin density transfer in MgCr2O4.
In MgMn2O4, the Jahn–Teller elongation dictates that c > a. Due to the occupation of the dz2 orbital and bond elongation, we expect P1 > P2. Values of P1 and P2 cannot be gained from experiments and need to be determined ab initio. The DFT calculation shows that the first contribution is roughly 1.5 times larger in terms of spin density contribution (P1 = 4.0 × 10−3versus P2 = 2.6 × 10−3 Bohr−3 for Hyb20 and P1 = 3.3 × 10−3versus P2 = 1.8 × 10−3 Bohr−3 for Hyb35, respectively). As for the case of magnetic exchange coupling (see above), the magnitude of the FC interaction is very sensitive to bond distances, which explains the difference.
4 Conclusion
Paramagnetic 25Mg NMR can be a useful tool to study Mg TM compounds, which finds a variety of uses in technologically important materials. However, due to the low sensitivity and paramagnetic nature of 25Mg centers in such compounds, solid-state NMR has been considered difficult and no systematic study is reported to date.In this work, we have successfully combined DFT studies and experimental NMR techniques, such as RAPT and MAT to predict and obtain the paramagnetic NMR spectra of three Mg TM oxides. Experimentally, it is demonstrated that signal-to-noise enhancement is possible in paramagnetic Mg systems with RAPT pulses, allowing us to perform a 2-D MAT experiment on samples with natural abundance 25Mg. While MAT is not strictly necessary to obtain the isotropic shift in the Mg6MnO8 case, we note that MAT experiments were used in the 23Na NMR of related Na-ion battery materials to successfully deconvolute the complex spectra comprising multiple sites and sidebands.24 DFT calculation of 25Mg NMR parameters using the paramagnetic scaling approach is demonstrated with good agreement with the experiment. Comparison of the 25Mg shifts in spinel and Mg6MnO8 compounds shows that TM–O–Mg 90° interaction results in stronger Fermi contact spin transfer, as expected from previous studies.
For spinel compounds MgCr2O4 and MgV2O4, comparison of the experimental results (NMR, SQUID magnetometry) and DFT calculations show that NMR anisotropy parameters are difficult to determine from the experiments due to the bulk magnetic susceptibility effects. In addition, DFT prediction of magnetism is difficult due to the magnetic frustration of these structures. Despite these difficulties, we show that reasonable predictions of 25Mg shifts can be made with the DFT values of spin density and the Weiss constant measured from SQUID magnetometry.
With the aid of DFT calculations and RAPT pulse sequence as shown above, it is possible to predict and acquire paramagnetic 25Mg spectra with signal-to-noise enhancement factors of 2. Good agreements are observed between the DFT predicted values of magnetism and NMR shifts, with the exception of NMR anisotropy parameters. This approach enables us to record and interpret the paramagnetic 25Mg spectra of other Mg compounds, which are more difficult to understand without the aid of DFT calculation. Work is in progress on the local structural characterization of electrochemically cycled cathode materials.
Acknowledgements
Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk). Research was also carried out at the Center for Functional Nanomaterials, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. J. L. acknowledges Trinity College Cambridge for the graduate studentship. I. D. S. acknowledges the Geoffrey Moorhouse Gibson Studentship from Trinity College Cambridge. A. J. P. acknowledges the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy under Contract DE-AC02-05CH11231, under the Batteries for Advanced Transportation Technologies (BATT) Program Subcontract 7057154. We thank Dr Derek Middlemiss for helpful discussions and providing a copy of the mean field magnetism code, and Dr Dinu Iuga for helpful advices on DFS experiments.References
- E. Levi, Y. Gofer and D. Aurbach, Chem. Mater., 2010, 22, 860 CrossRef CAS.
- P. Novák, R. Imhof and O. Haas, Electrochim. Acta, 1999, 45, 351 CrossRef.
- D. Aurbach, Y. Gofer, Z. Lu, A. Schechter, O. Chusid, H. Gizbar, Y. Cohen, V. Ashkenazi, M. Moshkovich and R. Turgeman, J. Power Sources, 2001, 97–98, 28 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 CrossRef PubMed.
- N. Garg, Menaka, K. V. Ramanujachary, S. E. Lofland and A. K. Ganguli, J. Solid State Chem., 2013, 197, 392 CrossRef CAS.
- C. P. Grey and N. Dupré, Chem. Rev., 2004, 104, 4493 CrossRef CAS PubMed.
- F. Blanc, M. Leskes and C. P. Grey, Acc. Chem. Res., 2013, 46, 1952 CrossRef CAS PubMed.
- L. Mafra, J. A. Vidal-Moya and T. Blasco, Annu. Rep. NMR Spectrosc., 2012, 77, 259 CrossRef.
- J. Klinowski, Annu. Rev. Mater. Sci., 1988, 18, 189 CrossRef CAS.
- J. C. C. Freitas and M. E. Smith, Annu. Rep. NMR Spectrosc., 2012, 75, 25 CrossRef CAS.
- I. L. Moudrakovski, Annu. Rep. NMR Spectrosc., 2013, 79, 129 CrossRef CAS.
- Z. Yao, H.-T. Kwak, D. Sakellariou, L. Emsley and P. J. Grandinetti, Chem. Phys. Lett., 2000, 327, 85 CrossRef CAS.
- J. Hu, D. Alderman, C. Ye, R. Pugmire and D. Grant, J. Magn. Reson., Ser. A, 1993, 105, 82–87 CrossRef CAS.
- C. Kim, P. J. Phillips, B. Key, T. Yi, D. Nordlund, Y.-S. Yu, R. D. Bayliss, S.-D. Han, M. He, Z. Zhang, A. K. Burrell, R. F. Klie and J. Cabana, Adv. Mater., 2015, 27, 3377 CrossRef CAS PubMed.
- U. Maitra, B. S. Naidu, A. Govindaraj and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11704 CrossRef CAS PubMed.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- K. J. D. Mackenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Elsevier, 2002, vol. 6, pp. 461–532 Search PubMed.
- I. Hung and Z. Gan, Chem. Phys. Lett., 2010, 496, 162 CrossRef CAS.
- P. J. Pallister, I. L. Moudrakovski and J. A. Ripmeester, Phys. Chem. Chem. Phys., 2009, 11, 11487 RSC.
- J. Kim, D. S. Middlemiss, N. A. Chernova, B. Y. X. Zhu, C. Masquelier and C. P. Grey, J. Am. Chem. Soc., 2010, 132, 16825 CrossRef CAS PubMed.
- J. Herzfeld and A. E. Berger, J. Chem. Phys., 1980, 73, 6021 CrossRef CAS.
- D. S. Middlemiss, A. J. Ilott, R. J. Clément, F. C. Strobridge and C. P. Grey, Chem. Mater., 2013, 25, 1723 CrossRef CAS.
- D. Freude, J. Haase, J. Klinowski, T. Carpenter and G. Ronikier, Chem. Phys. Lett., 1985, 119, 365 CrossRef CAS.
- R. J. Clément, A. J. Pell, D. S. Middlemiss, F. C. Strobridge, J. K. Miller, M. S. Whittingham, L. Emsley, C. P. Grey and G. Pintacuda, J. Am. Chem. Soc., 2012, 134, 17178 CrossRef PubMed.
- M. Catti, G. Valerio, R. Dovesi and M. Causa, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 179 CrossRef.
- M. Catti, G. Sandrone, G. Valerio and R. Dovesi, J. Phys. Chem. Solids, 1996, 57, 1735 CrossRef CAS.
- M. Catti, G. Sandrone and R. Dovesi, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 16122 CrossRef CAS.
- W. Kutzelnigg, U. Fleischer and M. Schindler, NMR-Basic Principles and Progress, Springer-Verlag, 1990, p. 165 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- X. Feng and N. Harrison, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 092402 CrossRef.
- I. d. P. R. Moreira, F. Illas and R. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155102 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. S. Kasper and J. S. Prener, Acta Crystallogr., 1954, 7, 246 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- J. Goodenough, Magnetism and the Chemical Bond, Wiley, 1963 Search PubMed.
- P. Porta and M. Valigi, J. Solid State Chem., 1973, 6, 344 CrossRef CAS.
- H. O'Neill and A. Navrotsky, Am. Mineral., 1983, 68, 181 Search PubMed.
- A. Navrotsky and O. Kleppa, J. Inorg. Nucl. Chem., 1967, 29, 2701 CrossRef CAS.
- K. E. Sickafus, J. M. Wills and N. W. Grimes, J. Am. Ceram. Soc., 1999, 82, 3279 CrossRef CAS.
- E. J. W. Verwey and E. L. Heilmann, J. Chem. Phys., 1947, 15, 174 CrossRef CAS.
- A. T. M. N. Islam, E. M. Wheeler, M. Reehuis, K. Siemensmeyer, M. Tovar, B. Klemke, K. Kiefer, A. H. Hill and B. Lake, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 024203 CrossRef.
- S. E. Dutton, Q. Huang, O. Tchernyshyov, C. L. Broholm and R. J. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 064407 CrossRef.
- R. Manaila and P. Pausescu, Phys. Status Solidi B, 1964, 6, 101 CrossRef.
- R. Manaila and P. Pausescu, Phys. Status Solidi B, 1965, 9, 385 CrossRef CAS.
- S. Yagi, Y. Ichikawa, I. Yamada, T. Doi, T. Ichitsubo and E. Matsubara, Jpn. J. Appl. Phys., 2013, 52, 025501 CrossRef.
- M. Cabello, R. Alcantara, F. Nacimiento, G. Ortiz, P. Lavela and J. L. Tirado, CrystEngComm, 2015, 17, 8728 RSC.
- P. Ghigna, R. De Renzi, M. Mozzati, A. Lascialfari, G. Allodi, M. Bimbi, C. Mazzoli, L. Malavasi and C. Azzoni, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 184402 CrossRef.
- K. Muramori and S. Miyahara, J. Phys. Soc. Jpn., 1960, 15, 1906 CrossRef CAS.
- S. Prasad, H.-T. Kwak, T. Clark and P. J. Grandinetti, J. Am. Chem. Soc., 2002, 124, 4964 CrossRef CAS PubMed.
- H. T. Kwak, S. Prasad, T. Clark and P. J. Grandinetti, Solid State Nucl. Magn. Reson., 2003, 24, 71 CrossRef CAS PubMed.
- A. Kentgens and R. Verhagen, Chem. Phys. Lett., 1999, 300, 435 CrossRef CAS.
- H. Taguchi and M. Nagao, J. Mater. Sci. Lett., 1991, 10, 658 CrossRef CAS.
- M. T. Dunstan, A. Jain, W. Liu, S. P. Ong, T. Liu, J. Lee, K. A. Persson, S. A. Scott, J. S. Dennis and C. P. Grey, Energy Environ. Sci., 2016, 9, 1346 CAS.
- C. P. Grey, C. M. Dobson and A. K. Cheetham, J. Magn. Reson., 1992, 98, 414 CAS.
- M. Stoll and T. Majors, J. Magn. Reson., 1982, 46, 283 CAS.
- H. Mamiya and M. Onoda, Solid State Commun., 1995, 95, 217 CrossRef CAS.
- H. J. Xiang, E. J. Kan, S.-H. Wei, M.-H. Whangbo and X. G. Gong, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 224429 CrossRef.
- W. H. Brumage, C. R. Quade and C. C. Lin, Phys. Rev., 1963, 131, 949 CrossRef CAS.
- D. Carlier, M. Ménétrier, C. Grey, C. Delmas and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 174103 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed first principles calculation, experimental characterization data, and magnetic measurements. See DOI: 10.1039/c6cp06338a |
| ‡ Current address: Department of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius Väg 16 C, SE-106 91 Stockholm, Sweden. |
| This journal is © the Owner Societies 2017 |