
Why do proton conducting polybenzimidazole phosphoric acid membranes perform well in high-temperature PEM fuel cells?†
Jan-Patrick
Melchior
a,
Günter
Majer
b and
Klaus-Dieter
Kreuer
*a
aMax-Planck-Institut für Festkörperforschung, Stuttgart, Germany. E-mail: kreuer@fkf.mpg.de
bMax-Planck-Institut für Intelligente Systeme, Stuttgart, Germany
First published on 5th December 2016
Abstract
Transport properties and hydration behavior of phosphoric acid/(benz)imidazole mixtures are investigated by diverse NMR techniques, thermogravimetric analysis (TGA) and conductivity measurements. The monomeric systems can serve as models for phosphoric acid/poly-benzimidazole membranes which are known for their exceptional performance in high temperature PEM fuel cells. 1H- and 31P-NMR data show benzimidazole acting as a strong Brønsted base with respect to neat phosphoric acid. Since benzimidazole's nitrogens are fully protonated with a low rate for proton exchange with phosphate species, proton diffusion and conduction processes must take place within the hydrogen bond network of phosphoric acid only. The proton exchange dynamics between phosphate and benzimidazole species pass through the intermediate exchange regime (with respect to NMR line separations) with exchange times being close to typical diffusion times chosen in PFG-NMR diffusion measurements (ms regime). The resulting effects, as described by the Kärger equation, are included into the evaluation of PFG-NMR data for obtaining precise proton diffusion coefficients. The highly reduced proton diffusion coefficient within the phosphoric acid part of the model systems compared to neat phosphoric acid is suggested to be the immediate consequence of proton subtraction from phosphoric acid. This reduces hydrogen bond network frustration (imbalance of the number of proton donors and acceptors) and therefore also the rate of structural proton diffusion, phosphoric acid's acidity and hygroscopicity. Reduced water uptake, shown by TGA, goes along with reduced electroosmotic water drag which is suggested to be the reason for PBI–phosphoric acid membranes performing better in fuel cells than other phosphoric-acid-containing electrolytes with higher protonic conductivity.
Introduction
Neat phosphoric acid (H3PO4) is the compound with the highest intrinsic proton conductivity,1 and it is the main constituent of proton conducting membranes for high temperature polymer electrolyte membrane (PEM) fuel cells operating in the T = 130–160 °C range. Such membranes comprise a basic polymer, such as poly-benzimidazole2–8 forming an adduct with phosphoric acid. The high conductivity of the pure acid is mainly the consequence of its “frustrated” hydrogen bond network (there is a severe imbalance of potential proton donors and acceptors) and the strength of its highly polarizable hydrogen bonds. The underlying proton conduction mechanism (structural diffusion) comprises very rapid intermolecular proton transfer and hydrogen bond formation reactions within the highly viscous environment of pure phosphoric acid as revealed by a recent ab initio molecular dynamics study.9 The proposed mechanism explains in a natural way that for the series phosphoric acid – phosphonic acid – phosphinic acid decreasing viscosity goes along with decreasing proton conductivity.10 This striking observation is at odds with the so-called Walden rule11 relating viscosity and equivalent conductivity and underlines the peculiar nature of proton conductivity in neat phosphoric acid. It also questions viscosity arguments for explaining conductivity trends in phosphoric acid based systems.12 Relative to the very high conductivity of the neat acid, the conductivity of phosphoric acid's complexes with poly-benzimidazole (PA–PBI) is significantly reduced even for high acid concentrations,2–8 which raises the question why they still perform so well in high temperature PEM fuel cells. In the present work, we show that most of the conductivity reduction is a consequence of the chemical interaction between the two constituents, and that is also the reason why mixtures of phosphoric acid and (benz)imidazole can serve as models for phosphoric acid/poly-benzimidazole membranes. For these model systems, we clarify whether the protic sites of (benz)imidazole participate in proton diffusion trajectories (as suggested before5,12,13) and in which way acid/base interactions affect proton conductivity in this system. We also elucidate to which extent and why these interactions affect the system's hygroscopicity. This is of particular interest because PA–PBI membranes are exposed to varying relative humidity in fuel cells, and water is the only additive which is known to increase phosphoric acid's high conductivity.13–15A recent study of the nature of this conductivity increase disclosed that hydrogen bond network “frustration” decreases with the addition of water through proton transfer from phosphate to aqueous species.16 Hydrogen bonding between phosphate and aqueous species is strong for low water contents but weakens rapidly with hydration level leading to a progressive decoupling of the diffusion of aqueous species (H2O, H3O+) from that of phosphate species. In addition, the acidity of phosphoric acid is significantly increased at low water contents. As a consequence, fast diffusion of a high concentration of protonated aqueous species (vehicle mechanism17) accounts for the increasing ionic conductivity with increasing water content. Since this emerging conductivity contribution is on the expense of phosphoric acid's genuine structural diffusion of protons, any effect of phosphoric acid's affinity towards water also effects the nature of its ionic conductivity (which is in contrast to recent claims18,19). Finally, we will demonstrate that this has important implications for the use of such materials as electrolyte in high temperature PEM fuel cells, and we explain why PA–PBI is performing well despite its aforementioned reduced conductivity.
Methodology
A combination of proton nuclear magnetic resonance (1H-NMR) techniques offers the unique possibility to characterize systems comprising of two differently diffusing subsystems exchanging protons at a certain rate. Diffusion coefficients and exchange processes are actually related in that the latter affects not only the shape of NMR spectra in the so-called intermediate and fast exchange regimes20,21 but also the apparent diffusion coefficients as obtained by pulsed field gradient (PFG) NMR.22,23 Analysis of the NMR lineshape is an established technique to determine the exchange rate of protons between different chemical sites,20,21 while only a few examples exist employing PFG-NMR to quantify exchange rates between two subsystems with known diffusion coefficients.24,25 In this study, NMR lineshape analysis and PFG-NMR are combined to obtain accurate diffusion coefficients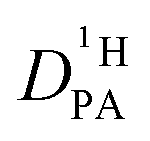 of protons in the phosphoric acid part (PA) of mixtures consisting of phosphoric acid and (benz)imidazole.
of protons in the phosphoric acid part (PA) of mixtures consisting of phosphoric acid and (benz)imidazole.
In a PFG-NMR experiment, the diffusion coefficient D is usually determined from the dependence of the echo attenuation
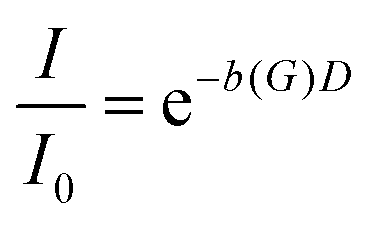 | (1) |
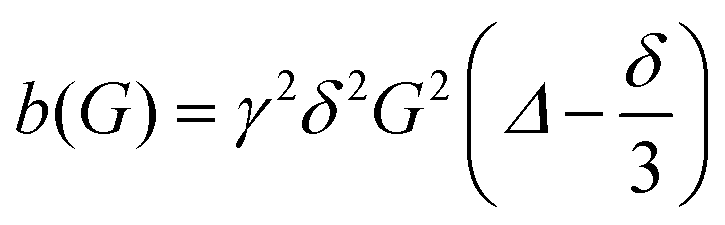 is obtained for a stimulated-echo sequence with rectangular gradient pulses of length δ and strength G. The diffusion time Δ is given by the separation of the leading edges of the gradient pulses and γ denotes the gyromagnetic ratio of the proton.
is obtained for a stimulated-echo sequence with rectangular gradient pulses of length δ and strength G. The diffusion time Δ is given by the separation of the leading edges of the gradient pulses and γ denotes the gyromagnetic ratio of the proton.
PFG-NMR has been widely used for determining proton diffusion coefficients in systems containing phosphorus oxoacids (neat phosphoric,16,28 phosphonic,29 and phosphinic acid,10 di-phosphoric acid,16,30 phosphoric acid/water mixtures,16,19,31 PA–PBI at various water contents32,33 and phosphoric acid-containing gels34). In some cases the effects arising from proton exchange between species of different diffusion coefficient are not critical, or the information, which is lost through these effects, is regained by measuring the diffusion coefficient of different types of nuclei (e.g.31P and 17O in the case of phosphoric acid–water mixtures16). For instance, the reactants of condensation and hydrolysis reactions in phosphoric acid systems (e.g. H4P2O7, H3PO4) are so long-lived35 that their separate 31P-NMR lines allow for a separate determination of the diffusion coefficient of each species (low exchange limit).16,30,31 In this case, the echo attenuation for each line is measured separately by PFG-NMR. On the other hand, very fast proton exchange in the same systems gives rise to a single 1H-NMR line which is generally used to determine an average proton diffusion coefficient (fast exchange limit).16,19,31
At intermediate exchange rates, the decay of echo intensity contains additional information on the populations of both subsystems, the diffusion coefficients in these systems and the exchange rate between them. In the present work we consider the exchange of protons between H3PO4 (PA) and protons attached to benzimidazole's nitrogen atoms (BI) with mean residence times τPA and τBI. The corresponding rate 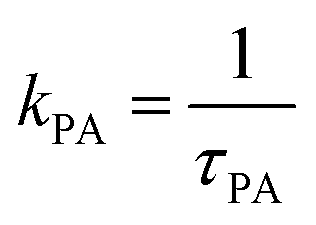 denotes the transfer from PA to BI and
denotes the transfer from PA to BI and 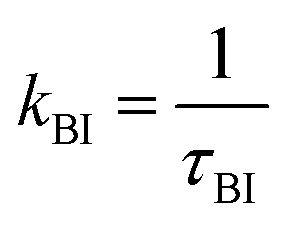 that from BI to PA. There is a detailed balance between protons on PA with population pPA and protons on BI with population pBI yielding
that from BI to PA. There is a detailed balance between protons on PA with population pPA and protons on BI with population pBI yielding 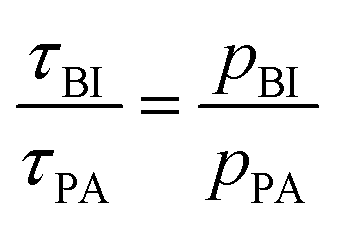 for the ratio of the mean residence time at PA and at BI.
for the ratio of the mean residence time at PA and at BI.
If the values for the mean time of residence in both phases are of the same order of magnitude as the diffusion time Δ (in the range of milliseconds), the simple eqn (1) is no longer applicable. The generalized description of the echo decay in a system of two subsystems with different diffusion coefficients is given by Kärger's equations:22–24
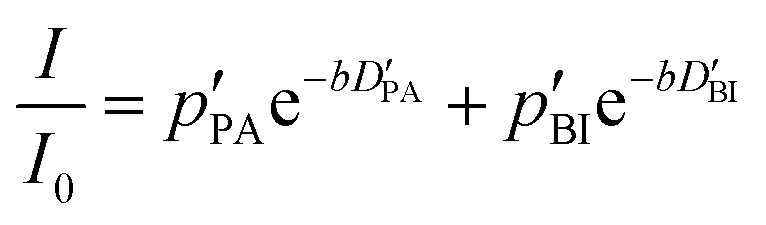 | (2) |
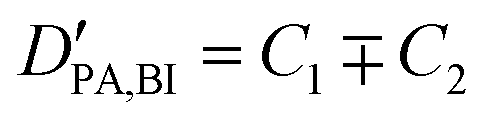 | (2a) |
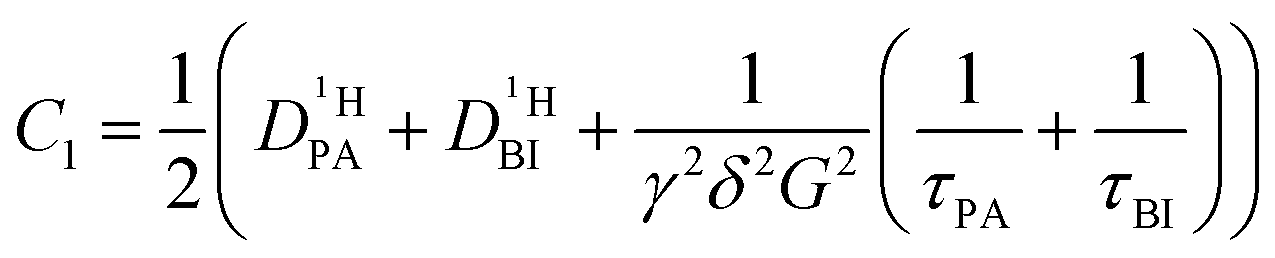 | (2b) |
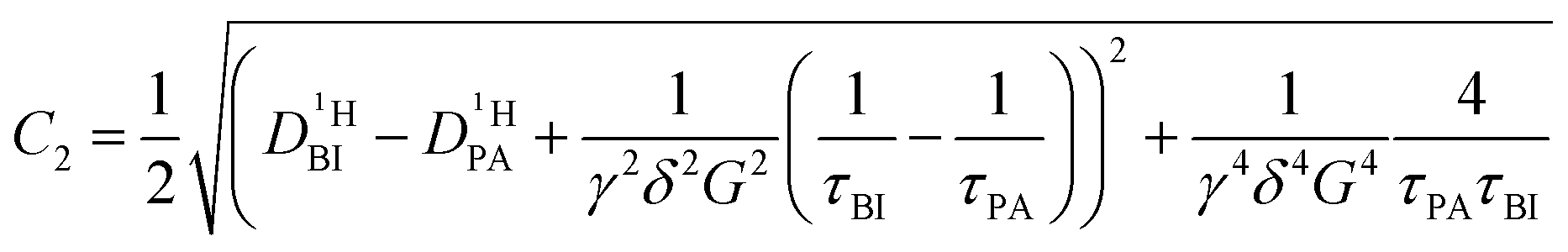 | (2c) |
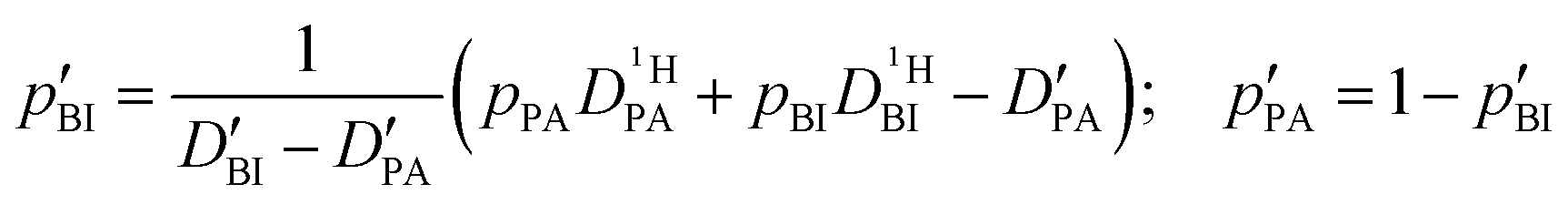 | (2d) |
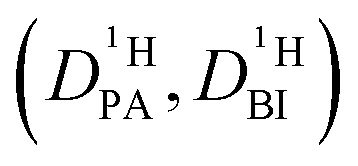 of the protons in the subsystems (PA, BI) and is quasi double-exponential. This behavior is not always immediately evident from the experimental data and can be easily overlooked. For low
of the protons in the subsystems (PA, BI) and is quasi double-exponential. This behavior is not always immediately evident from the experimental data and can be easily overlooked. For low 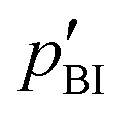 or a large difference between the diffusion coefficients, for example, the decay will appear almost mono-exponential, as the onset of the second exponential is disguised in signal noise, or the maximum gradient strength G is not high enough to observe the onset of the second exponential decay. In such cases, apparent diffusion coefficients obtained from mono-exponential fits of the Stejskal–Tanner equation (eqn (1)) strongly depend on experimental settings, e.g., the diffusion time Δ and particularly the maximum gradient strength Gmax, and a simple analysis of the recorded echo intensities may yield highly inaccurate values for the diffusion coefficients (see ESI†).
or a large difference between the diffusion coefficients, for example, the decay will appear almost mono-exponential, as the onset of the second exponential is disguised in signal noise, or the maximum gradient strength G is not high enough to observe the onset of the second exponential decay. In such cases, apparent diffusion coefficients obtained from mono-exponential fits of the Stejskal–Tanner equation (eqn (1)) strongly depend on experimental settings, e.g., the diffusion time Δ and particularly the maximum gradient strength Gmax, and a simple analysis of the recorded echo intensities may yield highly inaccurate values for the diffusion coefficients (see ESI†).
Intermediate exchange effects are also observable in NMR spectra if the exchange rates are similar to the difference of the resonance frequencies of protons at PA and BI. At low temperature, i.e., low exchange rate, those lines are clearly separated with Δν > 0.5 kHz and their intensity ratio allows for determination of the population ratio 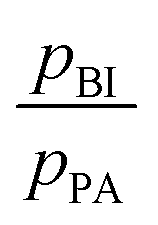 . At higher temperatures, when the exchange rate increases, the lines coalesce and the shape of the spectrum depends on the exchange rates and the proton populations.21 In this work, exchange rates have been obtained for all samples through analysis of the coalescing 1H-NMR spectra using a two-site exchange model (MEXICO 3.0 routines).20
. At higher temperatures, when the exchange rate increases, the lines coalesce and the shape of the spectrum depends on the exchange rates and the proton populations.21 In this work, exchange rates have been obtained for all samples through analysis of the coalescing 1H-NMR spectra using a two-site exchange model (MEXICO 3.0 routines).20
Obtaining population and exchange rate from spectral analysis, actually reduces the 4 independent parameters 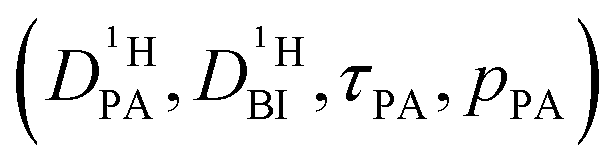 in the Kärger equation (eqn (2)) to two parameters
in the Kärger equation (eqn (2)) to two parameters 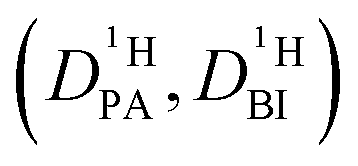 . Additionally in the system (benz)imidazole–phosphoric acid the benzimidazole diffusion coefficient
. Additionally in the system (benz)imidazole–phosphoric acid the benzimidazole diffusion coefficient 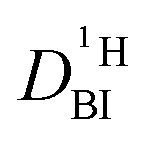 is obtained from the echo decay of non-exchanging C–H proton resonances, which further reduces the number of parameters in the Kärger equation to one free parameter (
is obtained from the echo decay of non-exchanging C–H proton resonances, which further reduces the number of parameters in the Kärger equation to one free parameter (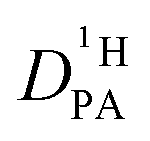 ). This fitting parameter, the diffusion coefficient of protons in the phosphoric acid part of the mixture
). This fitting parameter, the diffusion coefficient of protons in the phosphoric acid part of the mixture 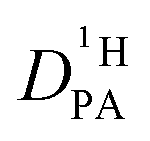 , can thus be obtained at high accuracy (see Experimental). It needs to be emphasized that the described way of reducing the number of free parameters in the Kärger equations is only feasible as the intermediate exchange regimes in spectral analysis and PFG-NMR overlap. This overlap is a special case, as in both experiments the occurrence of intermediate exchange effects depend on independent parameters: the frequency difference Δν for the spectral analysis and the diffusion time Δ for PFG-NMR.
, can thus be obtained at high accuracy (see Experimental). It needs to be emphasized that the described way of reducing the number of free parameters in the Kärger equations is only feasible as the intermediate exchange regimes in spectral analysis and PFG-NMR overlap. This overlap is a special case, as in both experiments the occurrence of intermediate exchange effects depend on independent parameters: the frequency difference Δν for the spectral analysis and the diffusion time Δ for PFG-NMR.
Experimental
Sample preparation
Nominally dry phosphoric as obtained by centrifuging crystalline phosphoric acid (Aldrich, >99.999%)16,28 and dry benzimidazole (Fluka, purum > 98%) or imidazole (Merck, >99%) powder were mixed by weight in a glovebox under dry nitrogen atmosphere. The samples were mixed under slight heating (50–80 °C) in closed glass viols. The resulting viscous liquids were either filled in a Teflon crucible for thermogravimetric analysis, impedance cells for conductivity measurements, or in 5 mm NMR tubes with screw lids (Deutero Duran S300). Dead volume above the sample was filled with a glass filler and the NMR tube was closed with its lid inside the nitrogen atmosphere and subsequently heat sealed outside the glove box.Thermo-gravimetrical analysis (TGA)
Equilibrium thermo gravimetric analysis (TGA) was carried out by using a balance (Mettler AT20) magnetically coupled (Rubotherm) to a PTFE crucible containing the sample. This technique allowed for weighing even at high temperature T and high and very low relative humidity RH, as described before.36 Changes of the buoyancy were compensated by a two parameter (temperature of sample and humidifier) baseline correction. Equilibration times of up to several days (especially at low T and RH) were chosen in order to ensure thermodynamic equilibrium. Reversibility was cross-checked through comparing the weights recorded during heating and cooling runs which also allowed excluding the uptake and release of species other than water. Nominally dry phosphoric acid (crystalline ortho-phosphoric acid) was used for mixtures with benzimidazole, and their weights were taken as reference (λ = 3). TGA crucibles were filled with such mixtures (1 g) under dry nitrogen atmosphere before transferring to the balance under the same dry gas.Impedance spectroscopy
Temperature-dependent dc-conductivities were derived from impedance spectra measured by a HP-ac-impedance analyzer (4192A LF) in the frequency range 10 Hz – 1 MHz. A closed conductivity cell made from glass (Duran, sample volume 0.3–0.6 ml) with platinum electrodes (diameter ∼ 6 mm), as previously described,16 has been used for measuring conductivities in the temperature range T = 20–160 °C. The maximum temperature was limited by the reactivity of phosphoric acid with glass. Heating and cooling runs were performed to ensure reversibility. The cell constant was determined by calibration with standard KCl solutions of different normalities (0.1 N and 0.01 N CertiPUR® Merck).NMR spectroscopy
NMR measurements have been performed with a Bruker Avance III 400 spectrometer equipped with a diff60 gradient probe in the temperature range T = 70 to 160 °C.31P-NMR spectra clearly resolve H3PO4, H4P2O7 and H5P3O10 lines16,31,37,38 and the molar concentrations xi of the phosphoric acid (P1: H3PO4, H2PO4−, H4PO4+; P2: H3P2O7−; P3: H3P3O102−) and aqueous (aq: H2O, H3O+) species are calculated from peak intensities as described previously.16
Diffusion coefficients of the different species are measured by 31P PFG-NMR. For this, the stimulated echo PFGSTE sequence with spoiler gradients,39 sinoidal gradient shape, effective gradient time δ = 1 ms to and a diffusion time Δ = 20 ms were used,26,40 for which 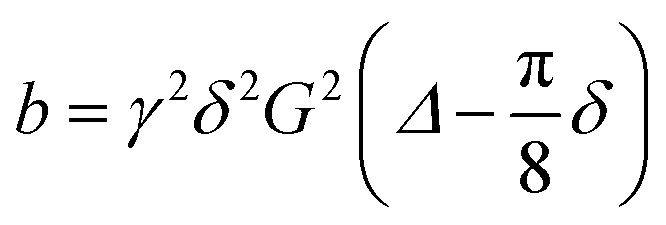 .40–42 The obtained diffusion coefficient were found to be independent of the diffusion time Δ which was varied in the range Δ = 20–40 ms.
.40–42 The obtained diffusion coefficient were found to be independent of the diffusion time Δ which was varied in the range Δ = 20–40 ms.
At low temperature, 1H-NMR spectra show clearly resolved lines for H3PO4, protons attached to (benz)imidazole nitrogens atoms (N–H) and (benz)imidazole benzene ring protons (C–H). At higher temperature the first two lines coalesce, indicating exchange of protons on the millisecond timescale. Exchange life-times and proton populations of N–H and H3PO4 were analyzed through fits with the MEXICO 3.0 fitting routine.20,21
The parameters describing proton distribution between benzimidazole and phosphoric acid (populations and life times) together with diffusion coefficients of (benz)imidazole obtained from 1H-PFG-NMR of C–H protons are then used as input parameter for fitting the echo attenuation of the common 1H signal of N–H and O–H protons in PFG-NMR experiments (256 gradient steps, maximum gradient of Gmax = 29 T m−1, sinoidal gradient shape, diffusion time Δ = 20 and 40 ms, and effective gradient duration δ = 1 ms). For this, the Kärger equation for intermediate exchange (eqn (2)) is used with the diffusion coefficient of protons within the phosphoric acid phase 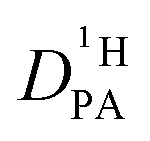 as the only free parameter. This diffusion coefficient is not affected by contributions from the diffusion of benzimidazole protons exchanging with protons in the phosphoric acid phase. It is this diffusion coefficient which is the used for discussing proton conduction mechanisms.
as the only free parameter. This diffusion coefficient is not affected by contributions from the diffusion of benzimidazole protons exchanging with protons in the phosphoric acid phase. It is this diffusion coefficient which is the used for discussing proton conduction mechanisms.
1H T1 relaxation times were measured by inversion recovery to set the re-magnetization times. Their temperature dependence exhibits clear BPP behavior with a 1/T1 maximum in the investigated temperature range.43 A detailed analysis of the data together with data from quasi-elastic neutron scattering (QENS) will be presented in a separate publication.
Results and discussion
Phosphoric acid–benzimidazole: a model system for PA–PBI
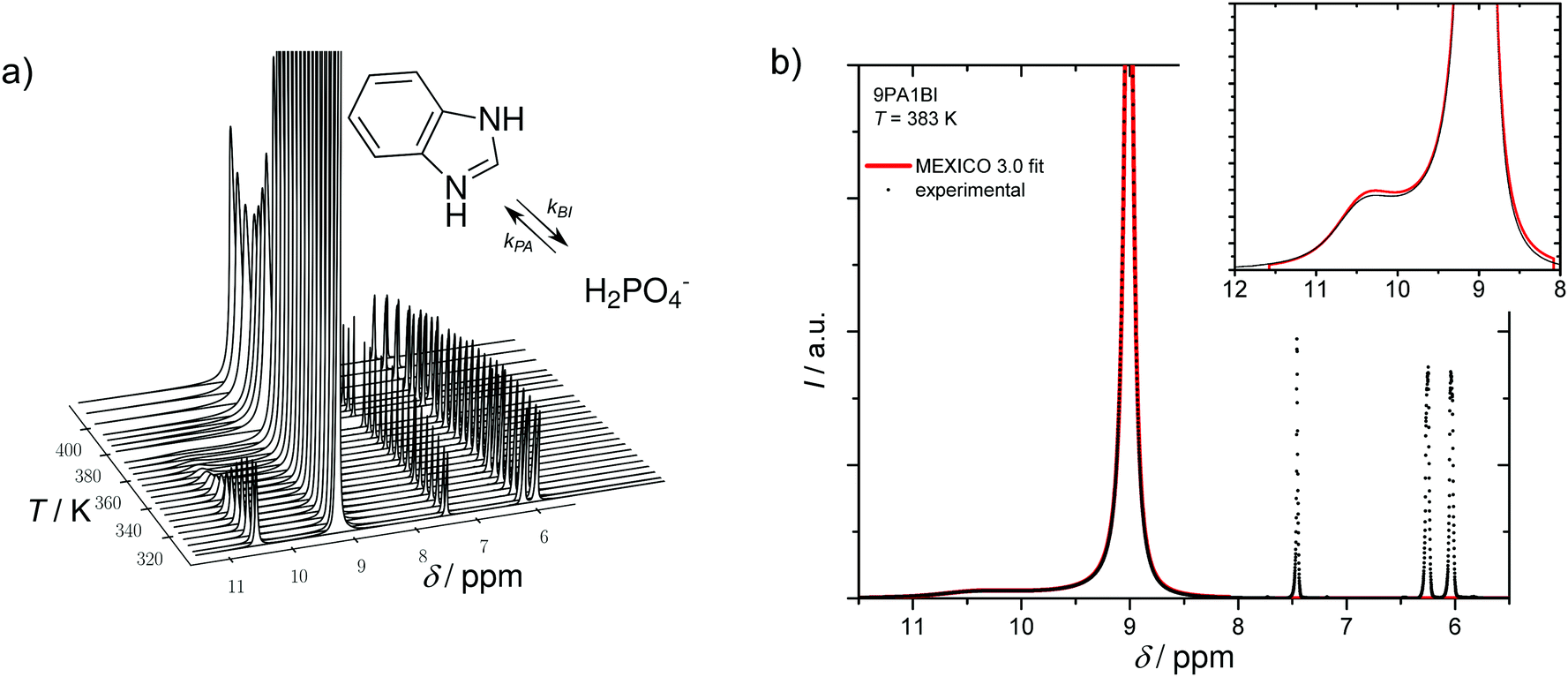
Let us first prove that phosphoric acid–benzimidazole is a suitable model system for studying transport in PA–PBI adducts. For this, we compare the ionic conductivity decrease for identical ratio of phosphoric acid per equivalent benzimidazole for the monomeric and the polymeric system AB-PBI.44–47 For the monomeric model system, such data have been measured in the present work, but for PA–PBI adducts, conductivity data are recorded for given relative humidity RH only.45However, hydration isotherms measured in this work (Fig. 1) allow identifying values for RH which correspond to certain water contents λ. The nominally dry complex 3PA·1BI (λ = 3) exists at RH ∼ 12% almost independent of temperature. We therefore compare the conductivity of 3PA·1BI with the conductivity of 3PA·1ABPBI recorded at RH = 12%45 (Fig. 2). For both cases the conductivity decrease compared to the high conductivity of pure phosphoric acid is similar (more than an order of magnitude). Obviously, the detrimental effect on conductivity is essentially the consequence of the chemical interaction of the benzimidazole unit with phosphoric acid rather than steric effects associated with the polymeric nature of PBI. These chemical effects can be studied more easily for the monomeric system, and for this the hygroscopicity is significantly reduced compared to the very high hygroscopicity of pure phosphoric acid (Fig. 1). Since hygroscopicity and enhanced acidity of pure phosphoric acid are most likely related to hydrogen bond network frustration,16,43 this observation points towards a reduction of network frustration. This will be discussed in the following section followed by the central sections on transport discussing the implications on proton diffusion and conductivity including the underlying transport mechanisms.
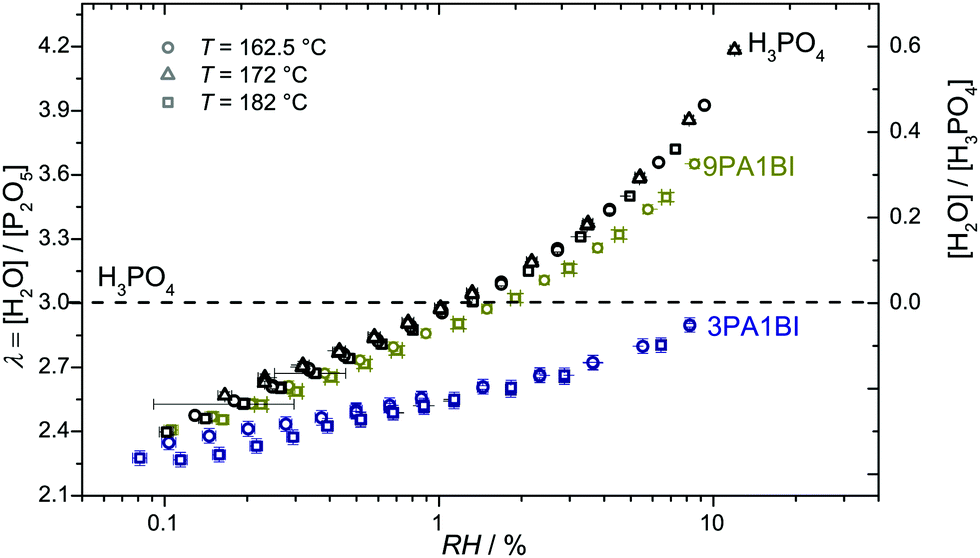 | ||
| Fig. 1 Hydration number λ = [H2O]/[P2O5] for neat H3PO4 and mixtures of H3PO4 and benzimidazole (9PA1BI = 9 mol H3PO4 per 1 mol benzimidazole; 3PA1BI = 3 mol H3PO4 per 1 mol benzimidazole). The water uptake at constant relative humidity (RH) is reduced with increasing benzimidazole content. | ||
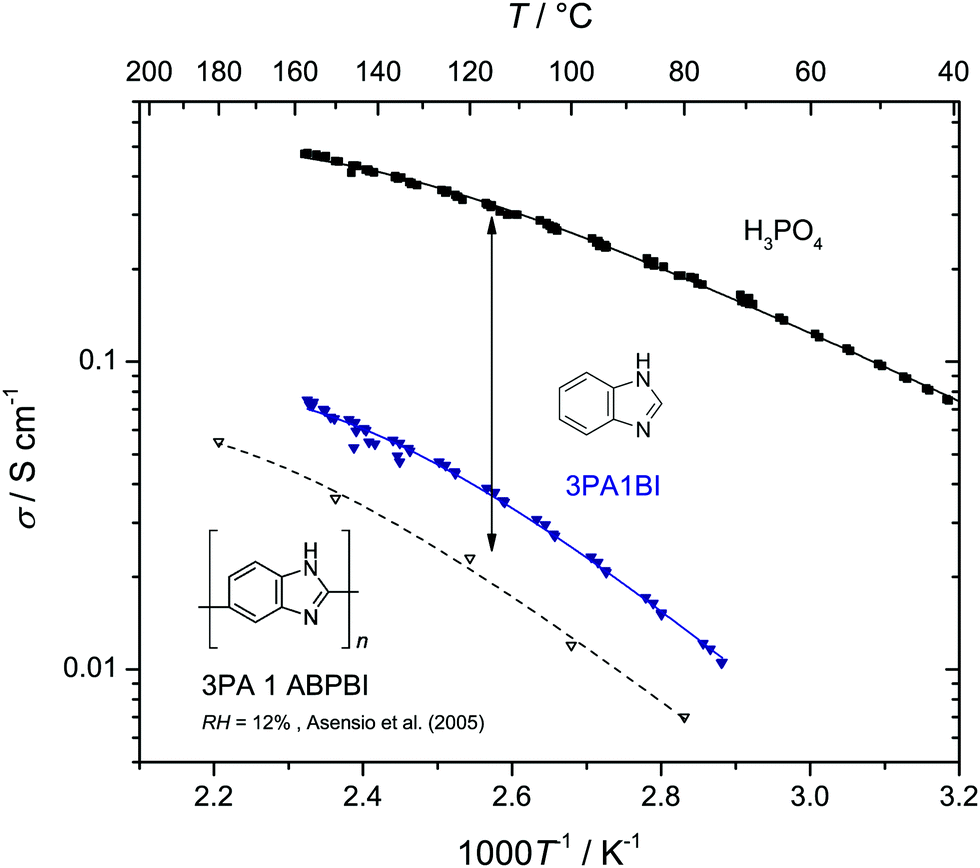 | ||
| Fig. 2 Ionic conductivity of nominally dry phosphoric acid (H3PO4), liquid 3PA·1BI and polymeric AB-PBI–phosphoric acid membranes with the same PA/BI ratio (recorded at RH = 12%,45 see text). The conductivity reduction of the monomeric system almost compasses the one observed for the polymeric membrane. Note that the ionic conductivity of the liquid 3PA·1BI has some contribution from the diffusion of benzimidazolium H–BI+ (see below). | ||
Proton distribution between benzimidazole and phosphoric acid
OH groups which accept a hydrogen bond from a proton donor while being prevented from donating its covalently bound hydrogen to another proton acceptor within frustrated hydrogen bond networks constitute local high energy states.9,16,30 These reactive states tend to relax if adequate pathways are available. In the case of pure ortho-phosphoric acid (H3PO4) frustration of the hydrogen bond network relaxes through condensation reactions:| 3H3PO4 ⇌ H3P2O7− + H4PO4+ + H2O | (3) |
| 3H3P2O7− + H3PO4 ⇌ 2H3P3O102− + H4PO4+ + H2O | (4) |
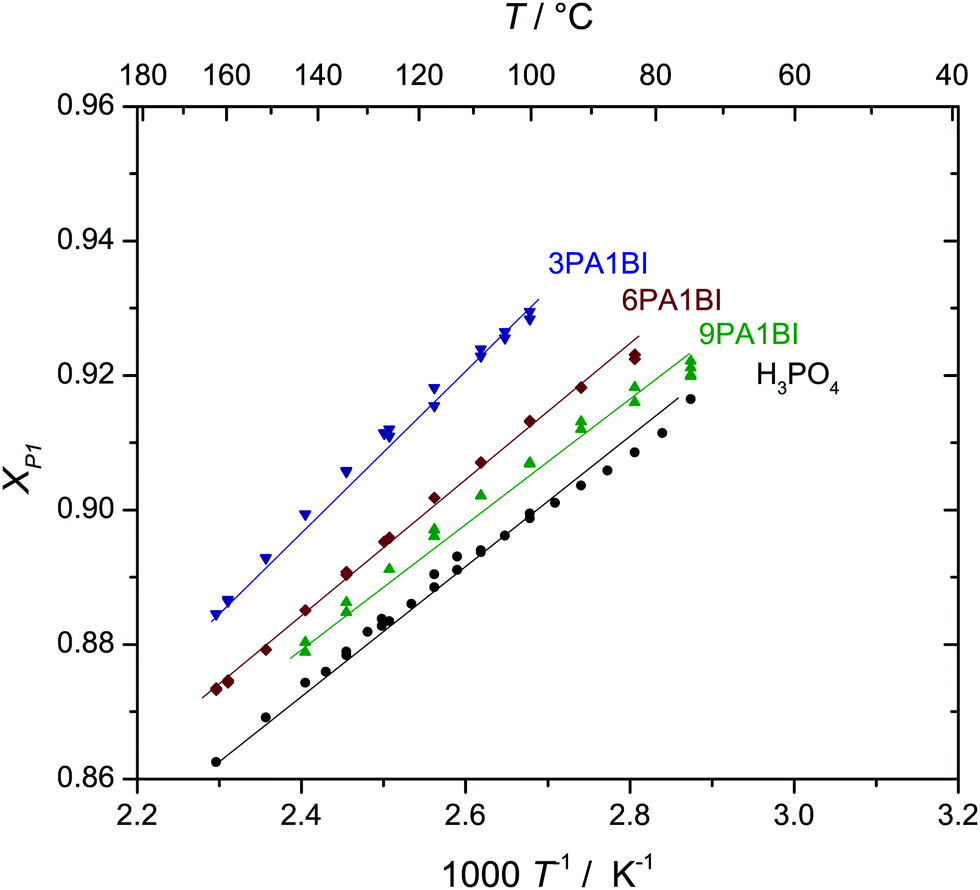 | ||
| Fig. 3 Mole fraction xP1 of ortho-phosphoric acid species (P1: H3PO4, H2PO4−, H4PO4+) in nominally dry phosphoric acid benzimidazole mixtures increases with the addition of the Brønsted base. The corresponding concentrations of di-phosphoric acid (P2: H3P2O7−) and aqueous species (aq: H2O, H3O+) decrease (see reactions (3) and (4) and text). Mole fraction xP1 in nominally dry phosphoric acid imidazole mixtures are virtually identical (see ESI†). | ||
The addition of the strong Brønsted base (benz)imidazole to phosphoric acid provides another direct pathway for reducing the proton concentration within the frustrated hydrogen bond network of the ortho-phosphate species by simple proton transfer to the basic species as evidenced by IR spectroscopy:48,49
| BI + H3PO4 → H–BI+ + H2PO4− | (5) |
In a recent ab initio molecular dynamics simulation50 of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 phosphoric acid imidazole (2PA1Imi) mixture imidazole was fully protonated without any proton exchange events taking place on the ps time scale of the simulation. The present NMR study, however, clearly shows that there are very rare proton exchange events between BI and PA which lead to coalescing 1H-NMR lines for NH and OH protons (Fig. 4a). For the mixture 9PA1BI, e.g., the NMR lines of NH (δ ∼ 10.5 ppm) and OH protons (δ ∼ 9.2 ppm) are well separated (∼1.3 ppm corresponding to 520 Hz) at T = 320 K with the expected intensity ratio for full nitrogen protonation but start to coalesce with increasing temperature (Fig. 4a). Coalescence occurs when the rate of proton exchange between the two chemically different environments is close to the spectral separation of the two 1H-NMR signals Δν ∼ 600 Hz. Therefore, proton exchange between benzimidazole and phosphoric acid must be very slow with a rate about nine orders of magnitude lower compared to the THz scale where proton exchange reactions within the phosphoric acid part take place. While previous reports have argued that exchange of protons between benzimidazole and phosphoric acid might occur on fast timescales relevant for proton conductivity,5,12,13 this observation indicates that exchanging protons are virtually trapped at benzimidazole's nitrogen with very rare excursions to phosphate species limiting the rate of successful proton exchange events. Average populations and exchange rates are quantified through fits with MEXICO 3.0 routines20 confirming full protonation of benzimidazole's nitrogen (100% within the error bars of about ±4%) and exchange rates on the kHz scale which are compiled in Fig. 5 as a function of temperature for different PA/BI ratios.
:1 phosphoric acid imidazole (2PA1Imi) mixture imidazole was fully protonated without any proton exchange events taking place on the ps time scale of the simulation. The present NMR study, however, clearly shows that there are very rare proton exchange events between BI and PA which lead to coalescing 1H-NMR lines for NH and OH protons (Fig. 4a). For the mixture 9PA1BI, e.g., the NMR lines of NH (δ ∼ 10.5 ppm) and OH protons (δ ∼ 9.2 ppm) are well separated (∼1.3 ppm corresponding to 520 Hz) at T = 320 K with the expected intensity ratio for full nitrogen protonation but start to coalesce with increasing temperature (Fig. 4a). Coalescence occurs when the rate of proton exchange between the two chemically different environments is close to the spectral separation of the two 1H-NMR signals Δν ∼ 600 Hz. Therefore, proton exchange between benzimidazole and phosphoric acid must be very slow with a rate about nine orders of magnitude lower compared to the THz scale where proton exchange reactions within the phosphoric acid part take place. While previous reports have argued that exchange of protons between benzimidazole and phosphoric acid might occur on fast timescales relevant for proton conductivity,5,12,13 this observation indicates that exchanging protons are virtually trapped at benzimidazole's nitrogen with very rare excursions to phosphate species limiting the rate of successful proton exchange events. Average populations and exchange rates are quantified through fits with MEXICO 3.0 routines20 confirming full protonation of benzimidazole's nitrogen (100% within the error bars of about ±4%) and exchange rates on the kHz scale which are compiled in Fig. 5 as a function of temperature for different PA/BI ratios.
 | ||
| Fig. 4 (a) 1H-NMR spectra of 9PA·BI as a function of temperature showing the coalescing lines of NH (δ = 10.5 ppm) and OH (δ = 9.2 ppm) protons. (b) Example of a fit with MEXICO 3.0 routines from which populations and exchange rates (see Fig. 5) are obtained. | ||
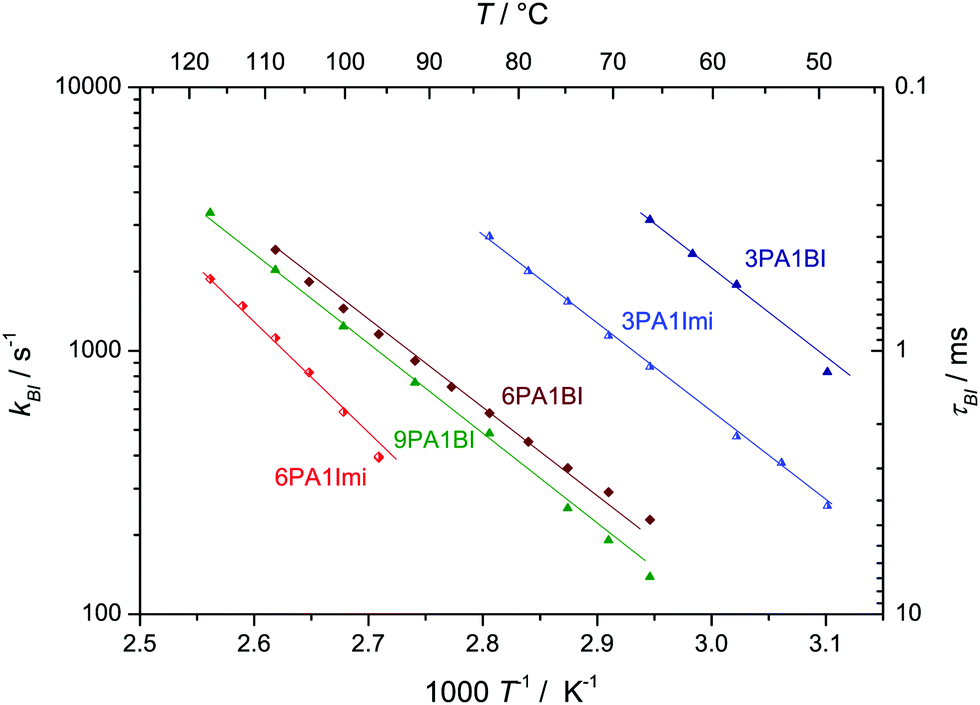 | ||
| Fig. 5 Exchange rate kBI and lifetime τBI of protons exchanging between (benz)imidazole and phosphoric acid. Note, that life times are of the same order as diffusion times typically chosen for PFG-NMR experiments. | ||
The observed proton distribution has several important implications in the context of the present work. Benzimidazole's nitrogen sites are strong traps for protons, which reduces frustration within phosphoric acid's hydrogen bond network, and excludes the nitrogen sites from any fast proton diffusion pathway. (Previously proposed proton exchange between different benzimidazole molecules is effectively suppressed by the full protonation of benzimidazole, i.e., a lack of proton accepting sites. In fact, proton transport between NH sites through the structural diffusion mechanism of pure heterocycles,51 has already been shown to break down upon addition of acid.52,53 Exchange of protons between benzimidazole and phosphoric acid is too slow to contribute to any fast proton diffusion or structural conductivity.) However, proton exchange between phosphoric acid and benzimidazole is still fast enough for inducing mixing of NH (BI) and OH (PA) 1H-NMR signals. As will be shown in the next section this effect needs to be included into the quantitative evaluation of PFG-NMR data, from which diffusion coefficients of protons in the phosphoric acid part are reliably obtained.
The effects of acid/base interaction on ionic diffusion and conductivity
The strong acid/base interaction between benz(imidazole) and phosphoric acid is expected to have severe implications for transport within the system. Proton transfer from phosphoric acid to benz(imidazole) (reaction (5)) leads to the formation of additional ionic species (H–BI+ and H2PO4−) and reduces frustration within phosphoric acid's hydrogen bond network. The increased ionicity may increase viscosity, reduce local dynamics and the diffusion coefficient of PA and BI species while the number of ionic charge carriers may increase. In which way these combined effects affect vehicular transport of protons is not clear. The most relevant question, however, is to what extent the reduced network frustration and retarded dynamics reduces the rate of proton structural diffusion. For neat phosphoric acid structural diffusion is the dominant proton transport mechanism, i.e., proton diffusion is significantly faster than phosphate diffusion and structural diffusion accounts for 97% of the total ionic conductivity.16,28 Therefore, the severe reduction of proton conductivity in PA/BI mixtures must be closely related to the retardation of structural diffusion within the phosphoric acid part of the system.In the following section, we therefore first evaluate 1H-PFG data (NH and OH 1H-lines, see above) taking into account the effects of proton exchange between phosphoric acid and (benz)imidazole's nitrogen. With the 1H-diffusion coefficient in the phosphoric acid part and the hydrodynamic background, also measured through (31P, 1H)-PFG-NMR, the rate of structural proton diffusion is readily obtained. In a separate section we then translate diffusion data into conductivity contributions which are discussed on the background of experimental conductivity data.
Proton diffusion
The proton diffusion coefficient within the phosphate part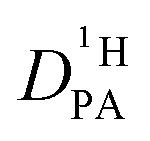 is buried in the echo attenuation of the combined NH and OH 1H-NMR signal in PFG-NMR experiments (see above). As expected for the intermediate exchange case (eqn (2)), quasi double exponential decays are observed (Fig. 6), which depend on the proton exchange rate and the proton population of the involved species (PA and BI), the diffusion coefficient of H–BI+ (benzimidazolium) as a whole and the diffusion coefficient of rapidly exchanging protons within the PA part. Since benzimidazolium diffusion coefficients are readily obtained by PFG-NMR making use of the C–H proton line (see Fig. 9ff, ESI†), and exchange rates (Fig. 5) and populations are available through the coalescence analysis of the two NMR lines (see above), the proton diffusion coefficient
is buried in the echo attenuation of the combined NH and OH 1H-NMR signal in PFG-NMR experiments (see above). As expected for the intermediate exchange case (eqn (2)), quasi double exponential decays are observed (Fig. 6), which depend on the proton exchange rate and the proton population of the involved species (PA and BI), the diffusion coefficient of H–BI+ (benzimidazolium) as a whole and the diffusion coefficient of rapidly exchanging protons within the PA part. Since benzimidazolium diffusion coefficients are readily obtained by PFG-NMR making use of the C–H proton line (see Fig. 9ff, ESI†), and exchange rates (Fig. 5) and populations are available through the coalescence analysis of the two NMR lines (see above), the proton diffusion coefficient 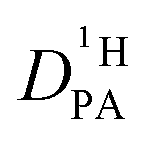 is left as the only free parameter in the expression for the echo-attenuation (eqn (2)). The fact that recorded echo attenuations are well fitted with one parameter only (Fig. 6) and are reproduced for two different diffusion times (Δ = 20 ms and 40 ms) provide confidence in the assumptions and the reliability of the input data. From the proton diffusion coefficients
is left as the only free parameter in the expression for the echo-attenuation (eqn (2)). The fact that recorded echo attenuations are well fitted with one parameter only (Fig. 6) and are reproduced for two different diffusion times (Δ = 20 ms and 40 ms) provide confidence in the assumptions and the reliability of the input data. From the proton diffusion coefficients 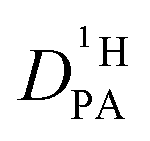 obtained in this way for several PA/BI ratios as a function of temperature (Fig. 7a, the corresponding data for PA/Imi mixtures are shown in Fig. 16a, ESI†) the proton structural diffusion contribution is extracted by subtracting the hydrodynamic background, i.e., proton diffusion stemming from the diffusion of phosphate species as a whole. Since there are not only diffusion contributions from ortho-phosphate species (P1) but also from condensates (P2, P3), especially at higher temperature (see also Fig. 3), we have measured
obtained in this way for several PA/BI ratios as a function of temperature (Fig. 7a, the corresponding data for PA/Imi mixtures are shown in Fig. 16a, ESI†) the proton structural diffusion contribution is extracted by subtracting the hydrodynamic background, i.e., proton diffusion stemming from the diffusion of phosphate species as a whole. Since there are not only diffusion contributions from ortho-phosphate species (P1) but also from condensates (P2, P3), especially at higher temperature (see also Fig. 3), we have measured 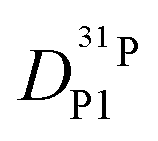 ,
, 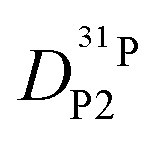 , and
, and 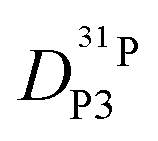 (for reasons of clarity, only
(for reasons of clarity, only 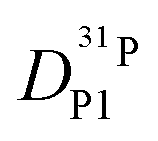 is shown in Fig. 7b) the corresponding data for PA/Imi mixtures are shown in Fig. 16b (ESI†). By simply weighing each diffusion coefficient with the concentrations of the diffusing species and the number of protons they are carrying, the effective hydrodynamic background of proton diffusion is obtained (for details see ref. 16).
is shown in Fig. 7b) the corresponding data for PA/Imi mixtures are shown in Fig. 16b (ESI†). By simply weighing each diffusion coefficient with the concentrations of the diffusing species and the number of protons they are carrying, the effective hydrodynamic background of proton diffusion is obtained (for details see ref. 16).
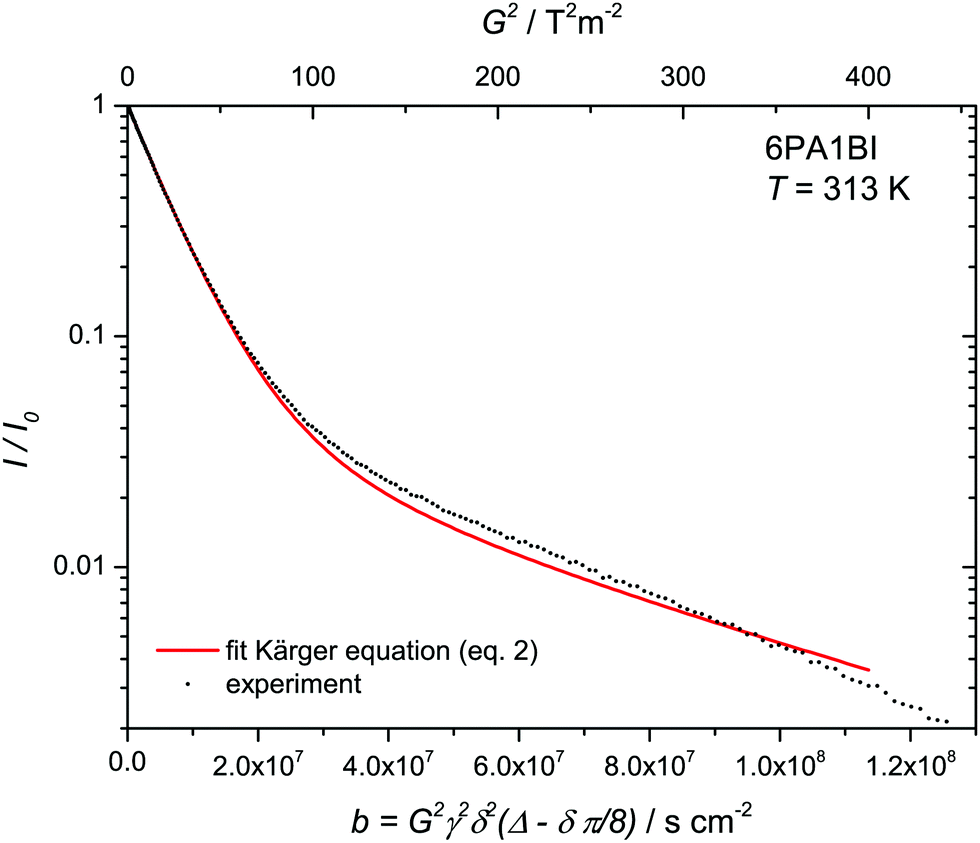 | ||
Fig. 6 Attenuation of normalized echo intensity as recorded by a 1H PFG-NMR experiment (stimulated echo) with the resonance of protons distributed over NH and OH sites of a 6PA·1BI mixture at T = 313 K (see Experimental). The decay is fitted by the Kärger equation (eqn (2)) with proton populations (pPA = 17/19, pBI = 2/19) and exchange rates (τNH = 25 ms) obtained from coalescence analysis and diffusion coefficient of benzimidazolium (H–BI+) recorded by PFG-NMR of CH protons (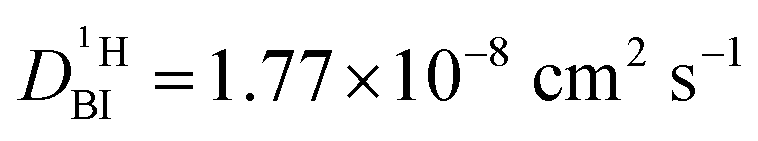 ) leaving the diffusion of rapidly exchanging protons within the phosphate part ) leaving the diffusion of rapidly exchanging protons within the phosphate part 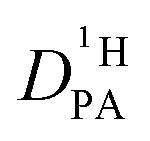 as only free parameter. as only free parameter. | ||
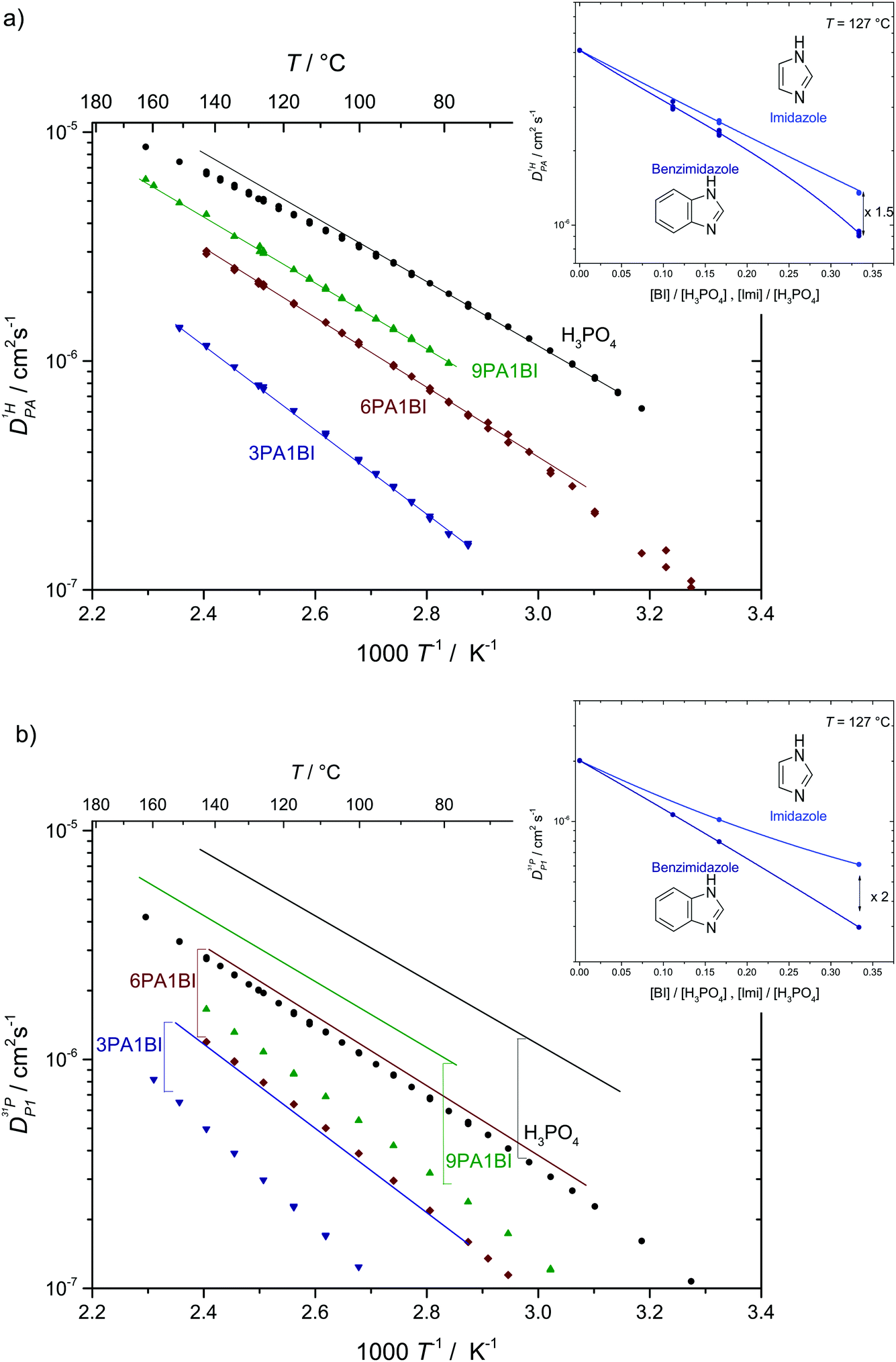 | ||
Fig. 7 (a) Diffusion coefficients for protons in the PA part of PA/BI mixtures 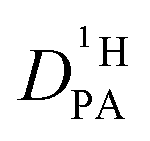 as obtained from 1H-PFG NMR (see text) and (b) diffusion coefficient of ortho-phosphate species as obtained from 1H-PFG NMR (see text) and (b) diffusion coefficient of ortho-phosphate species 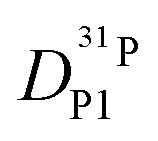 for the same mixtures (for diffusion coefficients of PA/Imi mixtures and for the same mixtures (for diffusion coefficients of PA/Imi mixtures and 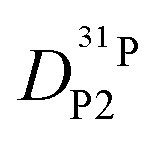 and and 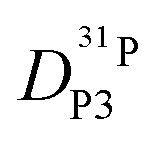 see ESI†). Insets show the same data as a function of base/acid ratio for T = 127 °C. see ESI†). Insets show the same data as a function of base/acid ratio for T = 127 °C. | ||
As the total proton diffusion coefficient within the phosphate part 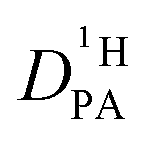 is reduced with the addition of (benz)imidazole also the hydrodynamic background
is reduced with the addition of (benz)imidazole also the hydrodynamic background 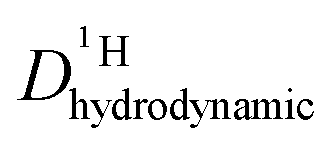 decreases. In the case of PA/BI mixtures this reduction is even more pronounced, especially at low temperature corresponding to a significant increase of the activation enthalpy which is smaller for the proton diffusion coefficient. The retardation of the hydrodynamic diffusion is also noticed as a severe viscosity increase with the addition of BI(Imi). It may be noted that this effect is smaller for the addition of imidazole compared to benzimidazole.
decreases. In the case of PA/BI mixtures this reduction is even more pronounced, especially at low temperature corresponding to a significant increase of the activation enthalpy which is smaller for the proton diffusion coefficient. The retardation of the hydrodynamic diffusion is also noticed as a severe viscosity increase with the addition of BI(Imi). It may be noted that this effect is smaller for the addition of imidazole compared to benzimidazole.
Since the hydrodynamic contribution of proton diffusion remains small also for the mixtures, proton structural diffusion 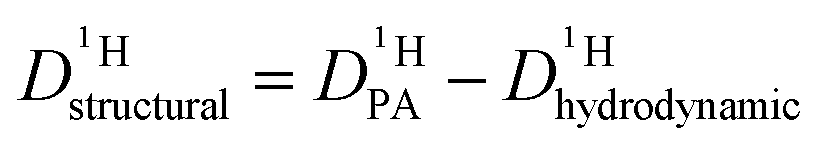 closely follows the evolution of
closely follows the evolution of 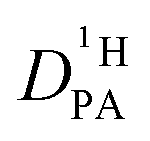 . Fig. 8 shows
. Fig. 8 shows 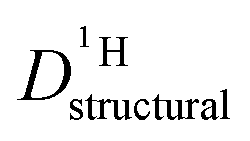 for both series of mixtures in order to demonstrate that the reduction of the rate of this transport mode correlates with the number of basic sites subtracting protons from phosphoric acid's hydrogen bond network. The fact that
for both series of mixtures in order to demonstrate that the reduction of the rate of this transport mode correlates with the number of basic sites subtracting protons from phosphoric acid's hydrogen bond network. The fact that 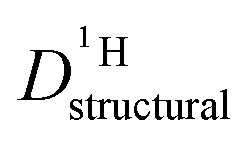 for PA/Imi mixtures is slightly but systematically higher than for the more viscous PA/BI mixtures points towards a small positive effect of increased local dynamics on structural diffusion of protons.
for PA/Imi mixtures is slightly but systematically higher than for the more viscous PA/BI mixtures points towards a small positive effect of increased local dynamics on structural diffusion of protons.
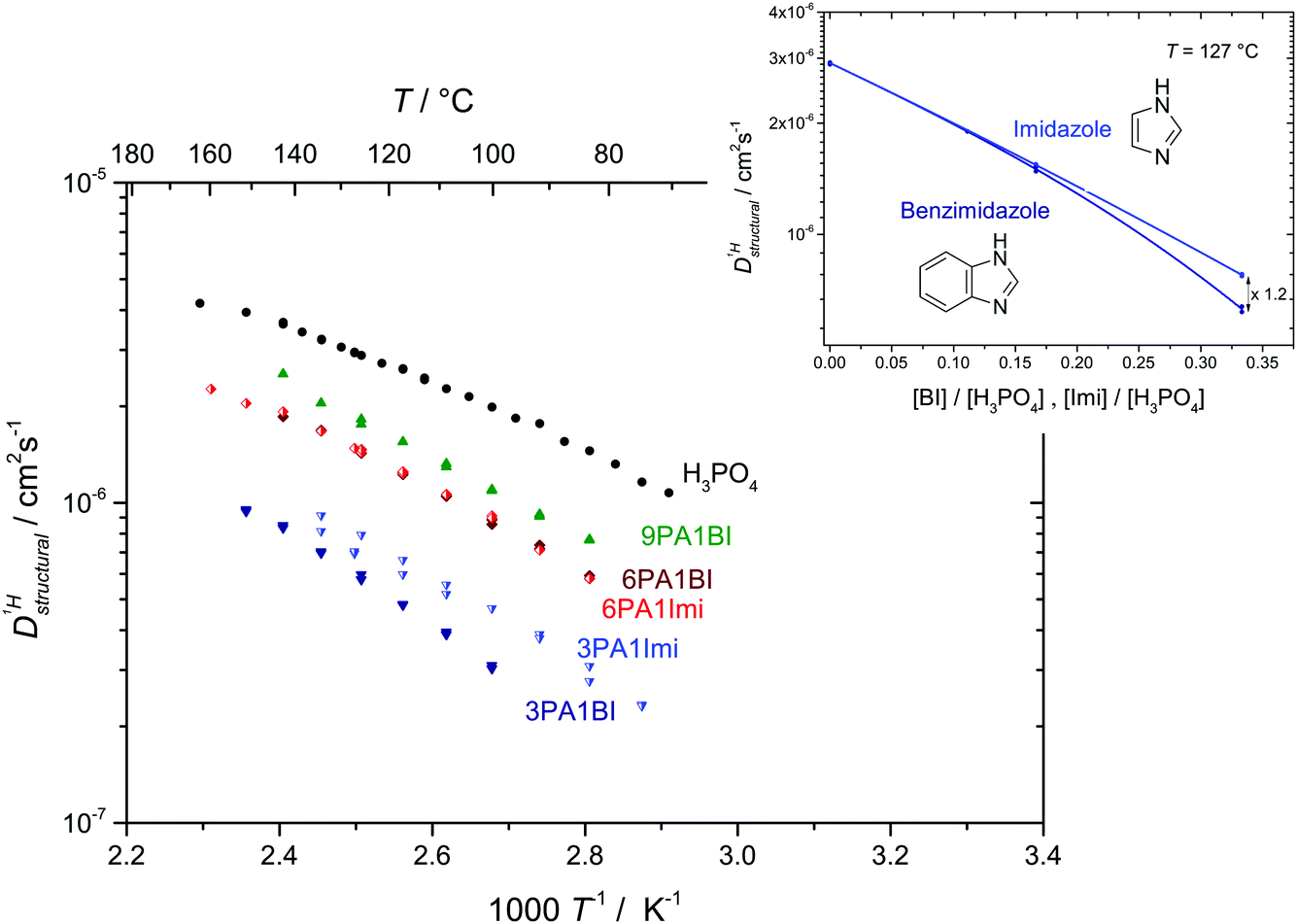 | ||
Fig. 8 Structural diffusion coefficient of protons 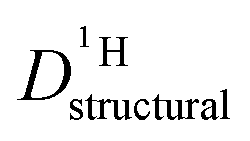 in the phosphoric acid part of (benz)imidazole–phosphoric acid mixtures at different temperatures and mixing ratios. Note, that the data for both types of mixtures are close (see also inset for data at T = 127 °C). in the phosphoric acid part of (benz)imidazole–phosphoric acid mixtures at different temperatures and mixing ratios. Note, that the data for both types of mixtures are close (see also inset for data at T = 127 °C). | ||
It needs to be emphasized that the proton exchange on the millisecond scale between phosphoric acid and benzimidazole, which necessitated the 1H PFG-NMR data treatment described above, is also present in PBI–PA membranes for high-temperature fuel cells. In these membranes, benzimidazole is part of a polymer with diffusion coefficient 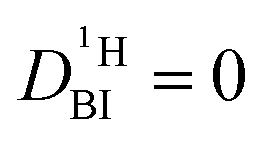 . This special case, in which one of the two diffusion coefficients in eqn (2) is zero, results also in a double exponential decay of the echo intensity, as it has already been discussed by Kärger.22 Implications for measurements of diffusion coefficients in membranes are considered in more detail in the ESI.†
. This special case, in which one of the two diffusion coefficients in eqn (2) is zero, results also in a double exponential decay of the echo intensity, as it has already been discussed by Kärger.22 Implications for measurements of diffusion coefficients in membranes are considered in more detail in the ESI.†
Proton conductivity
Above discussed diffusion coefficients (Fig. 8) are single particle properties (tracer diffusion) their changes directly reflecting changes of the rates of the underlying elementary reactions. In contrast, proton conductivity is a material property which has additional concentration effects. Conductivity is expected to decrease even more than the rate of proton structural diffusion does, since the addition of benzimidazole or imidazole to phosphoric acid not only affects the mobility of protons but also reduces the concentration of exchangeable protons cOH (combined dilution and proton trapping effect).These effects are included into the calculation of the proton structural diffusion contribution of the conductivity by the Nernst–Einstein-relationship:
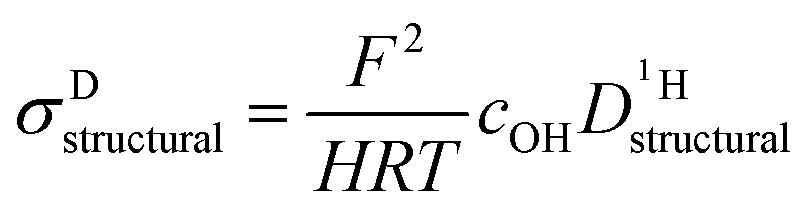 | (6) |
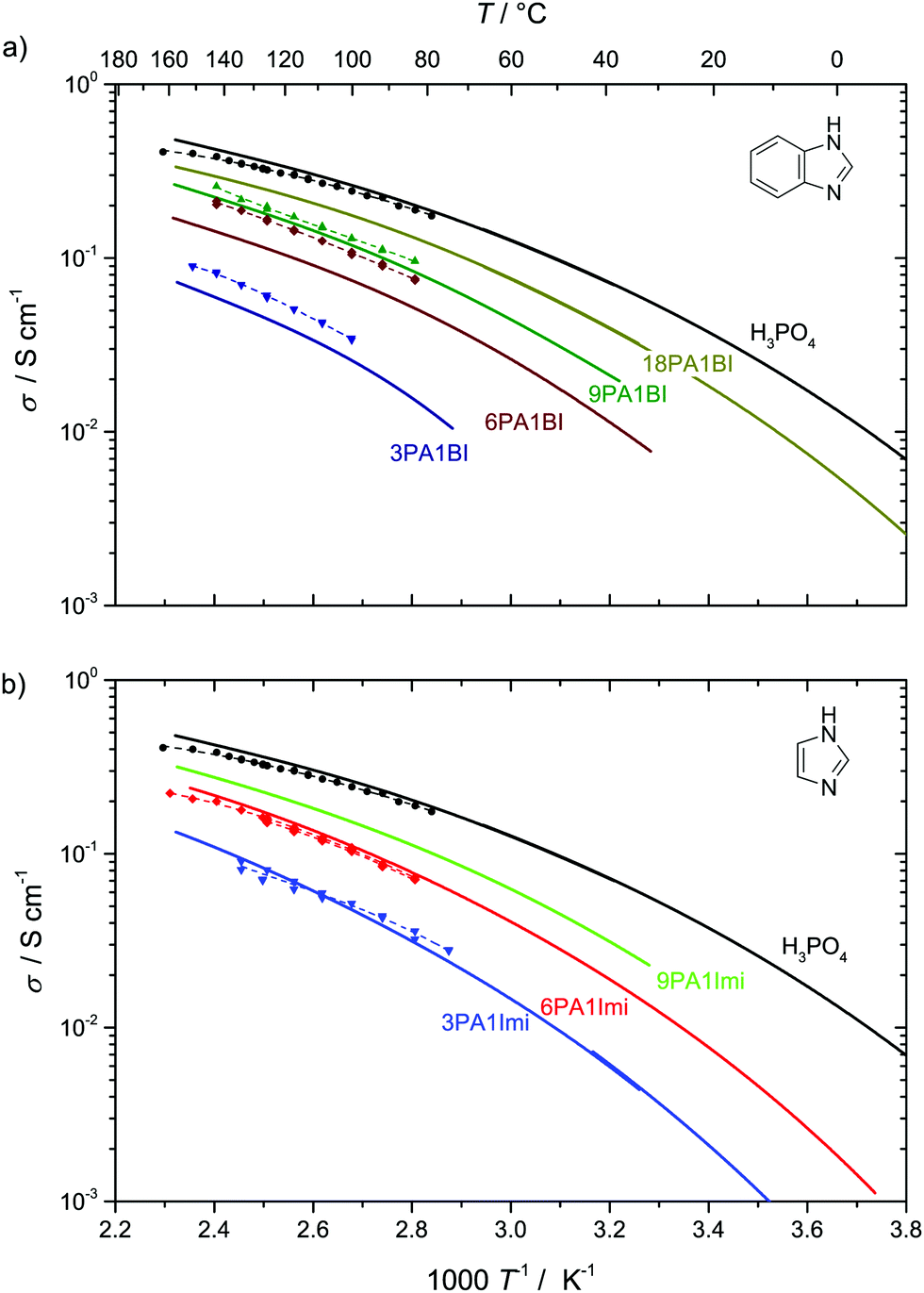 | ||
Fig. 9 Conductivity σDobs (solid lines, see ESI† for data points) and structural conductivity σDstructural (dashed lines, points) as calculated from 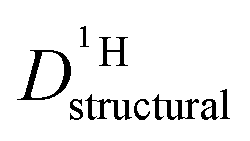 (a) PA–BI (b) PA–Imi (see text). (a) PA–BI (b) PA–Imi (see text). | ||
Implications for the use of PBI–PA membranes in high-temperature PEM-fuel cells
A superficial consideration may suggest that the severe conductivity decrease observed for phosphoric acid when interacting with the basic sites of benzimidazole in PBI–PA membranes is a disadvantage for their use in high-temperature PEM fuel cells. This rational seems to be behind approaches limiting the conductivity decrease by minimizing the polymer part of the membrane (this is possible through the so-called PPA-process3,7,54,55), or by grafting sulfonic acid groups to the polymer,3–6,56,57 both measures increasing the membrane's hydrophilicity.43,58 Water uptake is very well known to increase the conductivity of phosphoric acid based systems, hence such approaches lead to higher conductivity, indeed.2,12,49 Surprisingly, conductivity increases obtained in these ways do not lead to an increase of fuel cell performance. On the contrary, in some cases like Nafion membranes imbibed with phosphoric acid, the very high conductivity of such membranes stands in stark contrast to their bad performance in PEM fuel cells.59,60 The latter seems to be related to severe concentration polarization effects which dramatically increase with increasing current density.Our recent work on the transport properties of phosphoric acid containing extra water clearly indicates that the addition of water severely changes the nature of ionic transport.16 The high hydrogen bond network frustration of neat phosphoric acid makes it a very strong acid at low water contents. In the presence of small amounts of water (less than about two water molecules per phosphoric acid molecule) there is significant proton transfer from phosphoric acid to water leading to the formation of aqueous protonic charge carriers (H3O+). With increasing water content, these species progressively decouple from the phosphoric acid structure leading to an increasing vehicular conductivity contribution (cooperative diffusion of H3O+ and H2O17). Together with the increasing conductivity contribution from H2PO4− (formed in the same reaction) this proton transfer explains the conductivity increase with addition of water. At the same time, proton structural diffusion, i.e., the prevailing conduction mechanism of neat phosphoric acid, decreases eventually dying out at a water content of about [H2O]/[H3PO4] = 2.16
Transference experiments actually showed that each protonic charge carried by vehicle transport is associated with the effective transport of about one water molecule through the combined fluxes of H3O+ and H2PO4− where 17O PFG NMR diffusion data suggest the flux of H3O+ to be significantly higher than the H2PO4− flux.16 In a running fuel cell, these fluxes are expected to lead to water drag from the anode to the cathode. The water flux then should increase with both current density and concentration of excess water. We are not yet able to calculate how the water distribution develops across the membrane, but some water depletion must occur at the anode side. Especially for membranes with additional hygroscopic groups (–SO3H) in which conductivity is even more hydration dependent,43,61 this depletion may lead to a resistance increase at the anode side of the membrane limiting the current across the membrane. At the same time, the increasing water concentration at the cathode side may cause irreversible leaching of phosphoric acid out of the membrane.62 Consistent with our findings of increased vehicle conductivity, i.e., water transport at higher temperatures leaching has indeed already been shown to also increase with temperature and to occur predominantly at the cathode side.62 In any case, enhanced water transport is most likely detrimental for fuel cell performance.63,64
It may be one of the hidden advantages of PBI/PA membranes that their hygroscopicity is reduced through the interaction of phosphoric acid with the basic nitrogen sites of the polymer (see above and Fig. 1). For a relative humidity of RH = 10%, e.g., the phosphoric acid part of the mixture 3PA1BI is nominally dry (Fig. 1) while pure phosphoric acid retains about 0.6H2O per H3PO4 (Fig. 1). Under these conditions, which may be considered to be close to operation conditions of high temperature PEM fuel cells, the conductivity of the model system is more than an order of magnitude lower than the conductivity of hydrated pure phosphoric acid (for data see ref. 16). But the residual conductivity is still high enough for fuel cell applications (σ ∼ 0.1 S cm−1 at T = 160 °C, see Fig. 9), and proton transport is dominated by structural diffusion, i.e., there is very little transport of aqueous and phosphate species. This is in perfect agreement with the absence of any significant electroosmotic water drag in a PBI/PA membrane as measured by Weng et al.61 and appears to be a clear advantage of this type of membranes since water drag is known to cause concentration polarization effects limiting the current in a running fuel cell. On the other hand, the significant water content of pure phosphoric acid under these conditions leads to a water drag of about 0.4 water molecule per proton, which is actually very close to the water drag observed for a Nafion/PA membrane.61 It is worth mentioning that for H3PO4·0.6H2O, the rate of proton structural diffusion is reduced almost by a factor of 1.9, although the total conductivity is a factor of 1.1 higher than the conductivity of “dry” phosphoric acid.
These considerations favor the use of not too high phosphoric acid contents in PBI/PA membranes with good homogeneity maximizing internal acid/base contacts. This is actually different from our earlier approach aiming at stabilizing a phase separated morphology with high conductivity.65 The PPA-process provides a unique route for obtaining a favorably high dispersion of phosphoric acid in the final PBI structure, but the phosphoric acid content is generally high. Reducing the phosphoric acid content while keeping the intimate mix of acid and polymer could be a versatile way to further improve this type of membrane.66 Additional mechanical stabilization, e.g., through cross-linking, may not only improve the membrane's dimensional stability, it may also reduce phosphoric acid loss as a response to external pressure.67 We have already seen several experimental moves in these directions,55,68–72 the present work on model systems providing additional underpinning for these approaches.
Conclusions
The present work, which relies to a large extent on the accuracy of NMR data, allow for conclusions relevant for the application of PFG-NMR techniques in membrane science per se, it provides insight into the proton conduction mechanism in phosphoric acid – based systems containing basic heterocycles such as benzimidazole, and it explains why PBI/phosphoric acid membranes are performing well in high temperature PEM fuel cells.(Benz)imidazole clearly acts as a strong Brønsted base with respect to neat phosphoric acid. Benzimidazole's nitrogen sites are virtually fully protonated with a very slow proton exchange rate with phosphoric acid. The dynamics in such systems constitute very rare cases where proton exchange between two subsystems passes through the intermediate exchange regime in terms of 1H-spectral analysis and is coincidentally close to typical diffusion times chosen in PFG-NMR diffusion measurements (millisecond regime). The resulting effects, described by the Kärger equations (eqn (2)), must then be included into the evaluation of PFG-NMR data for obtaining precise diffusion coefficients.
From the fact that proton exchange between benzimidazole and phosphoric acid is about nine orders of magnitude slower than proton exchange between phosphate species (103 compared to 1012 s−19,50,73) we clearly exclude any proton diffusion trajectory comprising benzimidazole's nitrogen sites. As in the case of pure phosphoric acid, the dominant proton conduction mechanism is structural diffusion within the hydrogen bond network of the phosphoric acid part. However, the subtraction of protons through (irreversible) proton transfer to benzimidazole reduces the degree of frustration and therefore also the rate of structural diffusion as observed in the phosphoric acid/water system.16 As opposed to the latter where aqueous species are highly mobile, the hydrodynamic background (vehicle mechanism) in the system PA/BI is retarded, so that structural diffusion remains the prevailing conduction mechanism.
Reduced hydrogen bond network frustration also reduces phosphoric acid's very high acidity and hygroscopicity, i.e., water uptake for given RH/T conditions is reduced. As a consequence, electroosmotic water drag is reduced, and this is suggested to be the reason why, in fuel cells, PBI–phosphoric acid membranes perform better than other phosphoric acid containing electrolytes with higher protonic conductivity.
Acknowledgements
The authors thank Udo Klock for help with the TGA measurements. JPM thanks Prof. Alex Bain (McMaster University, Ontario) for an introduction to the MEXICO 3.0 routine and Igor Moudrakowski for discussions concerning exchange in NMR. We are also grateful to Robert Usiskin for reading the proofs and kindly acknowledge financial support by the Bundesministerium für Bildung und Forschung and Energie Baden Württemberg EnBW (project PSUMEA-2 No. 03SF0473).References
- K. D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- J. S. Wainright, J. T. Wang, D. Weng, R. F. Savinell and M. Litt, J. Electrochem. Soc., 1995, 142, L121–L123 CrossRef CAS.
- J. Mader, L. Xiao, T. J. Schmidt and B. C. Benicewicz, in Fuell Cells II, ed. G. G. Scherer, Springer, Berlin-Heidelberg, 2008, pp. 63–124 Search PubMed.
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449–477 CrossRef CAS.
- J. A. Asensio, E. M. Sanchez and P. Gomez-Romero, Chem. Soc. Rev., 2010, 39, 3210–3239 RSC.
- E. Quartarone and P. Mustarelli, Energy Environ. Sci., 2012, 5, 6436–6444 CAS.
- M. Molleo, T. Schmidt and B. Benicewicz, in Fuel Cells, ed. K. D. Kreuer, Springer, New York, 2013, pp. 391–431 Search PubMed.
- High Temperature Polymer Electrolyte Membrane Fuel Cells – Approaches, Status, and Perspectives, ed. Q. Li, D. Aili, H. A. Hjuler and J. O. Jensen, Springer International Publishing, Cham, Heidelberg, New York, Dordrecht, London, 2016 Search PubMed.
- L. Vilčiauskas, M. E. Tuckerman, G. Bester, S. J. Paddison and K. D. Kreuer, Nat. Chem., 2012, 4, 461–466 CrossRef PubMed.
- L. Vilčiauskas, C. C. de Araujo and K. D. Kreuer, Solid State Ionics, 2012, 212, 6–9 CrossRef.
- P. Walden, Z. Phys. Chem., 1906, 55, 207–249 CAS.
- Y. L. Ma, J. S. Wainright, M. H. Litt and R. F. Savinell, J. Electrochem. Soc., 2004, 151, A8–A16 CrossRef CAS.
- A. Schechter and R. F. Savinell, Solid State Ionics, 2002, 147, 181–187 CrossRef CAS.
- R. A. Munson and M. E. Lazarus, J. Phys. Chem., 1967, 71, 3245–3248 CrossRef CAS.
- N. N. Greenwood and A. Thompson, J. Chem. Soc., 1959, 3485 RSC.
- J.-P. Melchior, K. D. Kreuer and J. Maier, Phys. Chem. Chem. Phys., 2016 10.1039/c6cp04855b.
- K. D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed. Engl., 1982, 21, 208–209 CrossRef.
- D.-T. Chin and H. Chang, J. Appl. Electrochem., 1989, 19, 95–99 CrossRef CAS.
- S. H. Chung, S. Bajue and S. G. Greenbaum, J. Chem. Phys., 2000, 112, 8515–8521 CrossRef CAS.
- A. D. Bain, D. M. Rex and R. N. Smith, Magn. Reson. Chem., 2001, 39, 122–126 CrossRef CAS.
- A. D. Bain, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 43, 63–103 CrossRef CAS.
- J. Kärger, Ann. Phys., 1969, 479, 1–4 CrossRef.
- J. Kärger, Adv. Colloid Interface Sci., 1985, 23, 129–148 CrossRef.
- J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in Nanoporous Materials, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012 Search PubMed.
- S. So, L. J. Yao and T. P. Lodge, J. Phys. Chem. B, 2015, 119, 15054–15062 CrossRef CAS PubMed.
- J. E. Tanner, J. Chem. Soc., 1970, 52, 2523–2526 CAS.
- E. O. Stejskal and J. E. Tanner, J. Chem. Soc., 1965, 42, 288–292 CAS.
- T. Dippel, K. D. Kreuer, J. C. Lassègues and D. Rodriguez, Solid State Ionics, 1993, 61, 41–46 CrossRef CAS.
- M. Schuster, K. D. Kreuer, H. Steininger and J. Maier, Solid State Ionics, 2008, 179, 523–528 CrossRef CAS.
- R. A. Krueger, L. Vilčiauskas, J.-P. Melchior, G. Bester and K. D. Kreuer, J. Phys. Chem. B, 2015, 119, 15866–15875 CrossRef CAS PubMed.
- Y. Aihara, A. Sonai, M. Hattori and K. Hayamizu, J. Phys. Chem. B, 2006, 110, 24999–25006 CrossRef CAS PubMed.
- J. R. P. Jayakody, S. H. Chung, L. Durantino, H. Zhang, L. Xiao, B. C. Benicewicz and S. G. Greenbaum, J. Electrochem. Soc., 2007, 154, B242–B246 CrossRef CAS.
- S. Suarez, N. K. A. C. Kodiweera, P. Stallworth, S. Yu, S. G. Greenbaum and B. C. Benicewicz, J. Phys. Chem. B, 2012, 116, 12545–12551 CrossRef CAS PubMed.
- K. R. Jeffrey, G. Z. Zukowska, J. R. Stevens and G. Z. Zukowska, J. Chem. Phys., 2003, 119, 2422–2431 CrossRef CAS.
- R. A. Munson, J. Phys. Chem., 1964, 68, 3374–3377 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 2013, 252, 93–101 CrossRef CAS.
- M. M. Crutchfield, C. F. Callis, R. R. Irani and G. C. Roth, Inorg. Chem., 1962, 1, 813 CrossRef CAS.
- A. Schechter, R. F. Savinell, J. S. Wainright and D. Ray, J. Electrochem. Soc., 2009, 156, B283 CrossRef CAS.
- D. Burstein, Concepts Magn. Reson., 1996, 8, 269–278 CrossRef CAS.
- W. S. Price, NMR Studies of Translational Motion – Principles and Applications, Cambridge University Press, Cambridge, 2009 Search PubMed.
- W. S. Price and P. W. Kuchel, J. Magn. Reson., 1991, 94, 133–139 Search PubMed.
- G. Majer and K. Zick, J. Chem. Phys., 2015, 142, 164202 CrossRef CAS PubMed.
- J.-P. Melchior, PhD thesis, Universität Stuttgart, 2015.
- J. A. Asensio, S. Borrós and P. Gómez-Romero, Electrochem. Commun., 2003, 5, 967–972 CrossRef CAS.
- J. A. Asensio and P. Gómez-Romero, Fuel Cells, 2005, 5, 336–343 CrossRef CAS.
- A. L. Gulledge, B. Gu and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 306–313 CrossRef CAS.
- A. L. Gulledge, X. Chen and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 619–628 CrossRef CAS.
- X. Glipa, B. Bonnet, B. Mula, D. J. Jones and J. Rozière, J. Mater. Chem., 1999, 9, 3045–3049 RSC.
- H. Pu, W. H. Meyer and G. Wegner, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 663–669 CrossRef CAS.
- L. Vilčiauskas, M. E. Tuckerman, J. P. Melchior, G. Bester and K. D. Kreuer, Solid State Ionics, 2013, 252, 34–39 CrossRef.
- W. Münch, K. D. Kreuer, W. Silvestri, J. Maier and G. Seifert, Solid State Ionics, 2001, 145, 437–443 CrossRef.
- K. D. Kreuer, A. Fuchs, M. Ise, M. Spaeth and J. Maier, Electrochim. Acta, 1998, 43, 1281–1288 CrossRef CAS.
- K. D. Kreuer, in Solid State Ionics: Science & Technology, ed. B. V. R. Chowdari, et al., World Scientific Publishing Co., New Delhi, 1992, pp. 263–274 Search PubMed.
- L. X. Xiao, H. F. Zhang, E. Scanlon, L. S. Ramanathan, E. W. Choe, D. Rogers, T. Apple and B. C. Benicewicz, Chem. Mater., 2005, 17, 5328–5333 CrossRef CAS.
- S. Yu and B. C. Benicewicz, Macromolecules, 2009, 42, 8640–8648 CrossRef CAS.
- H. Bai and W. S. W. Ho, J. Taiwan Inst. Chem. Eng., 2009, 40, 260–267 CrossRef CAS.
- J. A. Mader and B. C. Benicewicz, Macromolecules, 2010, 43, 6706–6715 CrossRef CAS.
- Q. F. Li, R. H. He, R. W. Berg, H. A. Hjuler and N. J. Bjerrum, Solid State Ionics, 2004, 168, 177–185 CrossRef CAS.
- R. F. Savinell and J. S. Wainright, in High Temperature Polymer Electrolyte Membrane Fuel Cells – Approaches, Status, and Perspectives, ed. Q. Li, D. Aili, H. A. Hjuler and J. O. Jensen, Springer International Publishing, Cham Heidelberg New York Dordrecht London, 2016 Search PubMed.
- D. Aili, R. F. Savinell, J. O. Jensen, L. N. Cleemann, N. J. Bjerrum and Q. Li, ChemElectroChem, 2014, 1, 1471–1475 CrossRef CAS.
- D. Weng, J. S. Wainright, U. Landau and R. F. Savinell, J. Electrochem. Soc., 1996, 143, 1260–1263 CrossRef CAS.
- S. Yu, L. Xiao and B. C. Benicewicz, Fuel Cells, 2008, 8, 165–174 CrossRef CAS.
- U. Reimer, J. Ehlert, H. Janssen and W. Lehnert, Int. J. Hydrogen Energy, 2016, 41, 1837–1845 CrossRef CAS.
- D. Bezmalinovic, S. Strahl, V. Roda and A. Husar, Int. J. Hydrogen Energy, 2014, 39, 10627–10640 CrossRef CAS.
- J. Weber, K. D. Kreuer, J. Maier and A. Thomas, Adv. Mater., 2008, 20, 2595–2598 CrossRef CAS.
- M. A. Molleo, X. Chen, H. J. Ploehn and B. C. Benicewicz, Fuel Cells, 2015, 15, 150–159 CrossRef CAS.
- X. Chen, G. Qian, M. A. Molleo, B. C. Benicewicz and H. J. Ploehn, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 1527–1538 CrossRef CAS.
- Q. F. Li, H. C. Rudbeck, A. Chromik, J. O. Jensen, C. Pan, T. Steenberg, M. Calverley, N. J. Bjerrum and J. Kerres, J. Membr. Sci., 2010, 347, 260–270 CrossRef CAS.
- Y. S. Guan, H. T. Pu, M. Jin, Z. H. Chang and D. C. Wan, Fuel Cells, 2010, 10, 973–982 CrossRef CAS.
- S. Bhadra, N. H. Kim and J. H. Lee, J. Membr. Sci., 2010, 349, 304–311 CrossRef CAS.
- M. Han, G. Zhang, Z. Liu, S. Wang, M. Li, J. Zhu, H. Li, Y. Zhang, C. M. Lew and H. Na, J. Mater. Chem., 2011, 21, 2187–2193 RSC.
- F. Mack, K. Aniol, C. Ellwein, J. Kerres and R. Zeis, J. Mater. Chem. A, 2015, 3, 10864–10874 CAS.
- B. Frick, L. Vilčiauskas, P. P. Deen and S. Lyonnard, Solid State Ionics, 2013, 252, 26–33 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05331a |
| This journal is © the Owner Societies 2017 |