
DNA ionogel: Structure and self-assembly†
Pankaj Kumar
Pandey
a,
Kamla
Rawat
 *bc,
V. K.
Aswal
d,
J.
Kohlbrecher
e and
H. B.
Bohidar
*ab
*bc,
V. K.
Aswal
d,
J.
Kohlbrecher
e and
H. B.
Bohidar
*ab
aSchool of Physical Sciences, Jawaharlal Nehru University, New Delhi 110067, India
bSpecial Center for Nanosciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: bohi0700@mail.jnu.ac.in; kamla.jnu@gmail.com; Fax: +91 11 2674 1837; Tel: +91 11 26742091
cInter University Accelerator Centre, New Delhi 110067, India
dState Physics Division, Bhabha Atomic Research Centre, Mumbai 400085, India
eLaboratory for Neutron Scattering, Paul Scherrer Institut, Villigen, Switzerland
First published on 8th November 2016
Abstract
DNA dissolved in ionic liquid (IL) solution (1-ethyl-3-methylimidazolium chloride, [C2mim][Cl]) showed a transition to the gel phase ([DNA] ≥ 1% (w/v)). The gelation time was 400 s for the 1% [IL] sample which reduced to 260 s for 5% [IL] concentration. Gelation times, obtained from the viscosity and ergodicity breaking from the dynamic structure factor data, were remarkably identical to each other. Correspondingly, the gelation temperature which was ∼60 °C increased to 67 °C with [IL] content. The small angle neutron scattering (SANS) structure factor profile revealed the presence of the following three distinct length scales: (a) mesh size, ξ ≈ 3 ± 0.5 nm for ionogels, and ≈0.73 ± 0.06 nm, for sol; (b) cross-sectional radius of DNA strand, Rc ≈ 1.6 ± 0.1 nm; and (c) the characteristic inter-cluster distance ≈33 ± 5 nm. Physical conformation of the DNA–IL complexes remained close to the Gaussian coil definition. It was observed that without IL, in the sol phase, the system was completely ergodic and did not gel, while on addition of IL a sudden transition to the non-ergodic (arrested) gel phase occurred. This was due to the formation of an amorphous network of DNA–IL complexes preceding gelation. In summary, it is shown that the DNA ionogels can be prepared with a tunable gel strength (27–70 Pa) and gelation temperature (60–67 °C). Further, the relaxation dynamics was found to be hierarchical in IL content of the gel, revealing considerable self-organization.
1. Introduction
Natural biopolymers have been utilized for pharmaceutical applications for the last few decades due to their unique properties such as versatility, biocompatibility and bio-absorbability. Beside this, most biopolymers have good film forming ability along with better stability, which make them suitable for biomedical applications.1,2 The presence of abundant functional groups on the biopolymer chains provides excellent interfacial properties, resulting in fast communication between the attached molecules for the development of biosensors.3,4 Generally, the active sites of biopolymers are embedded in the core, which affects the direct binding with its receptor molecules, resulting in the hindrance of direct electron communication. However, ILs create an interface resulting in an enhanced fast electron transfer rate between the electrolyte and the underlying electrode for sensor and biosensor applications.5,6Nucleic acids constitute an important class of biomolecules that are a repository of genetic information. The aqueous dispersion behavior of these biopolymers is rich in soft matter phases and their self-assembly gives rise to a wide range of morphologies.7–9 DNA gels have attracted much attention recently due to their potential applications as drug and gene delivery vehicles.10–12 Therefore, tailoring of their physical properties is required. In order to generate a new class of intelligent biomaterials involving DNA, it is necessary to explore its dispersion behavior in IL, and other organo-solutions.
Organic salts having a melting point of less than 100 °C are called ionic liquids. These are thermally stable, non-flammable and highly conductive.13–16 The ILs are found to be good stabilizers for biopolymers due to the availability of nitrogen-containing cations, such as imidazolium, and inorganic or organic anions. Ionic liquids as electrolytes or in the form of composites have been used for electrochemical based sensor applications. IL based DNA ionogels consist of two types of ILs, short and long chain {1-ethyl-3-methylimidazolium chloride [C2mim][Cl]} and {1-octyl-3-methyl imidazolium chloride [C8mim][Cl]} which are used in drug delivery and tissue engineering applications. Many reports reveal that DNA encapsulation can be used either in gene therapy or chitosan microsphere based delivery vehicles.17,18 Moran et al. have studied the swelling and dislocation behaviour of DNA gel particles.10
ILs are introduced into polymer/biopolymer solutions to from ionogels with physically or chemically cross-linked networks that can absorb large amounts of water without being dissolved.19,20 It was observed that these ionogels originated from a homogeneous sol due to the availability of imidazolium head groups which strongly interact with biopolymer molecules via hydrogen bonding, electrostatic and π–π interactions.21 Ionogels are used as chemiresistive transducer materials for the detection of volatile organic compounds (VOCs). Different types of ILs, when encapsulated in silica gel networks produced specific resistive responses due to the different molecular interactions between ionic liquids and VOCs.22 The use of DNA hydrogels in controlled drug delivery, tissue engineering, 3D cell culture, and cell transplant therapy has been reported in the literature.23 Lee et al. have used DNA hydrogels to create an electrical circuit where water acts as a switch.24 Xiong et al. have extensively discussed the applications of hydrogels in the development of sensors and logic gates among others.25
Angelova et al. (2011) demonstrated lipidopolymeric self-assembled nanocarriers with tunable nanochannel size, morphology, and hierarchical inner organization that provided a potential vehicle for the predictable loading and release of therapeutic proteins, peptides, or nucleic acids. They also studied the structure and dynamics of functionalized self-assembled lipid nanosystems during stimuli-triggered liquid-crystalline phase transformations.26 The influence of the topology and internal structure of PEGylated lipid nanocarriers on neuronal transfection was investigated using synchrotron radiation SAXS and cryo-TEM studies.27 Angelov et al. (2012) described sterically stabilized DNA functionalized lipid nanocarriers using SAXS.28 Angelov et al. (2013) discussed the millisecond structural dynamics of DNA/fusogenic lipid nanocarrier assembly.29 In a separate investigation the significance of large channels in cationic PEGylated cubosome nanoparticles was explored by the same authors.30
In most of the reported studies, DNA was present with a complementary surfactant, or polymer which favored formation of inter-polymer complexes largely through Coulombic interactions. We do not specifically emphasize hydrophobic interactions.31–33 In our case, DNA gels were prepared in the ionic liquid solutions, and we focus on enhancement of the gel properties of these new biomaterials. Since ionogels have a wider window of temperature stability, these are good substitutes for hydrogels.
In this report, we have comprehensively studied DNA and IL (1-ethyl-3-methyl imidazolium chloride, [C2mim][Cl]) based gels, and evaluated their physical behaviour. Dynamic light scattering (DLS) measurement showed the relaxation dynamics of DNA ionogels, and it was found that relaxation dynamics was hierarchical with IL concentration. These experimental achievements are helpful for our general understanding of the self-organization of DNA ionogels. Considering the utility of nucleic acids in genetic engineering and pharmaceutics, these results are of importance.
2. Material and methods
The salmon testes dsDNA (2000 base pair) sodium salt was purchased from Sigma-Aldrich, USA. It had 41.2% G–C content with a melting temperature of 87 °C in 0.15 M sodium chloride and 0.015 M sodium citrate medium. Its nominal molecular weight was ≈1.3 × 106 g mol−1.34 The IL, 1-ethyl-3-methyl imidazolium chloride, was bought from Sigma-Aldrich, and the heavy water D2O was also purchased from Sigma-Aldrich and used to prepare the stock solutions. All concentrations are in (w/v) unless otherwise stated.The ionic liquid was dissolved in D2O in different amounts to make the stock solutions (1–5%). A stock solution of 1% DNA was made by dissolving the supplied powder material in ionic liquid solutions at 60 °C followed by heating up to 90 °C under stirring to denature the DNA. Our experiments revealed that it was necessary to expose the DNA sols to a heating and cooling cycle to induce gelation in the system. Therefore all the sol samples were systematically heated to 90 °C, and then cooled to room temperature (25 °C). As per literature data at 90 °C, there is cooperative unraveling of the intertwined strands of double helix to single strands.35–42 It should be noted that ssDNA is not of much use in biology per se. However, we describe a DNA based biomaterial with gel-like properties to be possibly used as a matrix for drug and enzyme loading, wound healing and tissue engineering applications.
The SANS measurements were performed at the Swiss Spallation Neutron Source SINQ, PSI, Villigen, Switzerland. The measurements were carried out with a 1.6 nm neutron wavelength and a sample-to-detector distance of 2, 6 and 15 m required to achieve a scattering wavevector in the range of 0.03–3.4 nm−1. See ref. 43 for more details. All the measurements were performed using a 2D position sensitive detector of size 96 cm × 96 cm, a source wavelength of 0.8 nm, a sample-to-detector distance of 2, 8 and 20 m, and a path length of the quartz cuvette was 2 mm.
Viscosity measurement was conducted on a Sine wave Vibro (SV-10, A and D company, Japan) viscometer having two plates, of which one vibrates at 30 Hz frequency; uniform amplitude was maintained by a feedback electrical current. The viscosity value is obtained from the response function of the other plate using the software provided by the manufacturer.
The dynamic light scattering (DLS) technique was used to probe the structure and network dynamics of the samples. DLS experiments were performed on a PhotoCor instrument (USA).7–9 It was operated in a multi-tau mode covering a time scale of 8 decades from 0.5 μs to 10 s. The samples were loaded into a 5 ml borosilicate cell which was housed in a scattering geometry with a thermostatic bath. General discussion on DLS can be found in ref. 44.
To study the viscoelastic properties, rheology measurements were performed with a controlled stress AR 500 rheometer (TA Instruments, Surrey, England) using a cone-plate geometry. See ref. 8 for more instrumental details. General discussion on rheology can be found in ref. 45.
3. Results and discussion
3.1 Scaling and gelation time
The freshly prepared DNA ionosols were examined for their characteristic gelation behavior. Fig. 1 shows the temporal evolution of relative viscosity ηr (ηr = η/η0, η is solution and η0 is solvent viscosity) for different samples. This time-dependent growth could be described by the scaling relation given by46,47| ηr = ε−k; t → tgel | (1) |
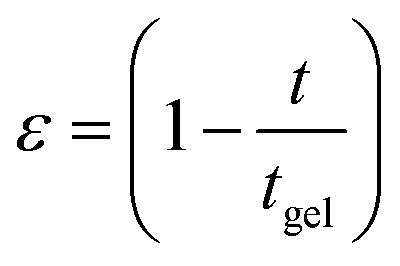 and tgel is gelation time.
and tgel is gelation time.
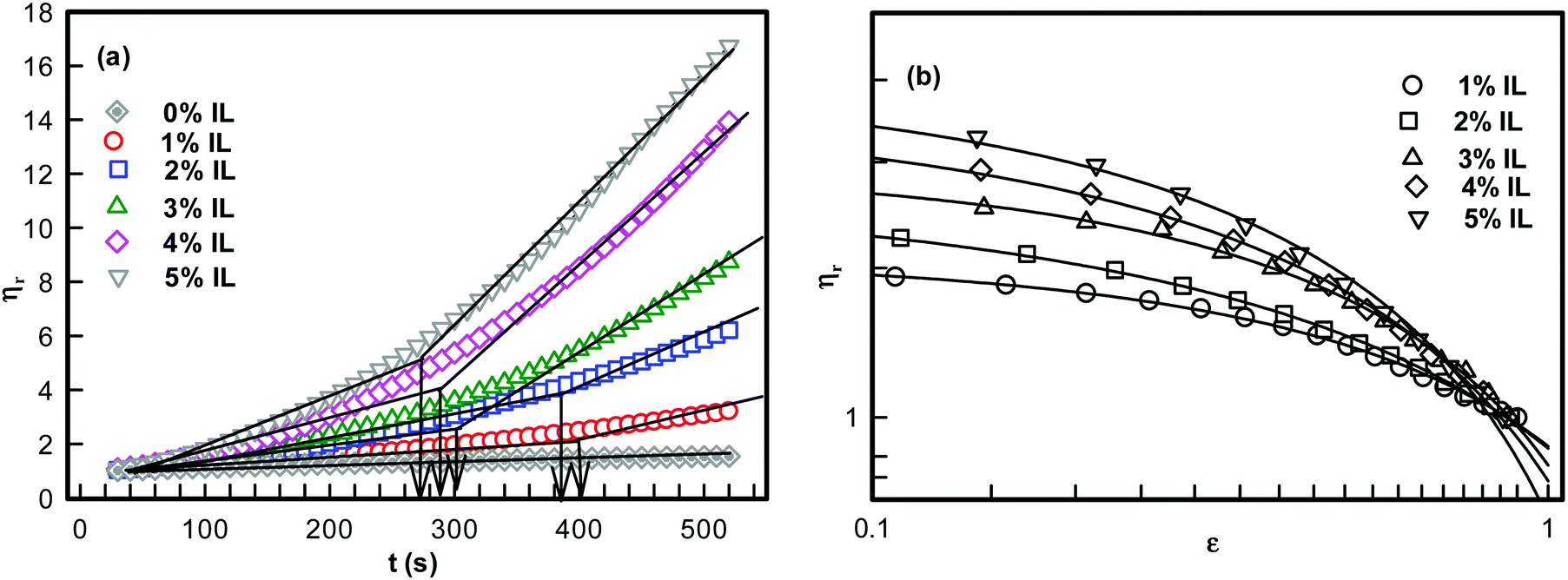 | ||
| Fig. 1 (a) Temporal evolution of relative viscosity. The arrows indicate gelation time tgel. (b) Relative viscosity as function of reduced time ε = (1 − t/tgel). Solid lines in (b) are fitting of data to eqn (1). | ||
The relative viscosity vs. time data shown in Fig. 1 yielded the characteristic time tgel deduced from the change in slope which was found to decrease with IL concentration.
It is clearly seen that, as the IL concentration was increased from 1 to 5%, the viscosity of the ionosol increased systematically. This was due to the formation of DNA–IL clusters and their interconnected networks with increase in IL concentration. The time dependent power-law growth function was used to fit the relative viscosity data to eqn (1) to determine the value of exponent k.
The exponent k described the temporal growth of the networks (Fig. 2). Two observations can be clearly made: (i) it was remarkable to note that the gelation time, tgel (1% IL) = 400 s decreased to tgel (5% IL) = 260 s (a drop of 35%) (Fig. 2(a)); and (ii) the value of k ranged between 0.6–0.9. Theoretically, Rose dynamics have shown that the exponent k = 0.7 corresponds to a conducting, and k = 1.3 to a percolating gel network.46–48 Therefore, on the basis of experimental data, it is possible to give a qualitative assessment of gelation kinetics. The exponent k decreased from 0.9 to 0.6 with increase in IL-concentration. Thus, the network which represents a conducting matrix (k ≈ 0.9) is reorganized into a percolating network when there is a propensity of IL-molecules in the dispersion. This description is nothing more than indicative, and clearly requires more experimental data to be validated.
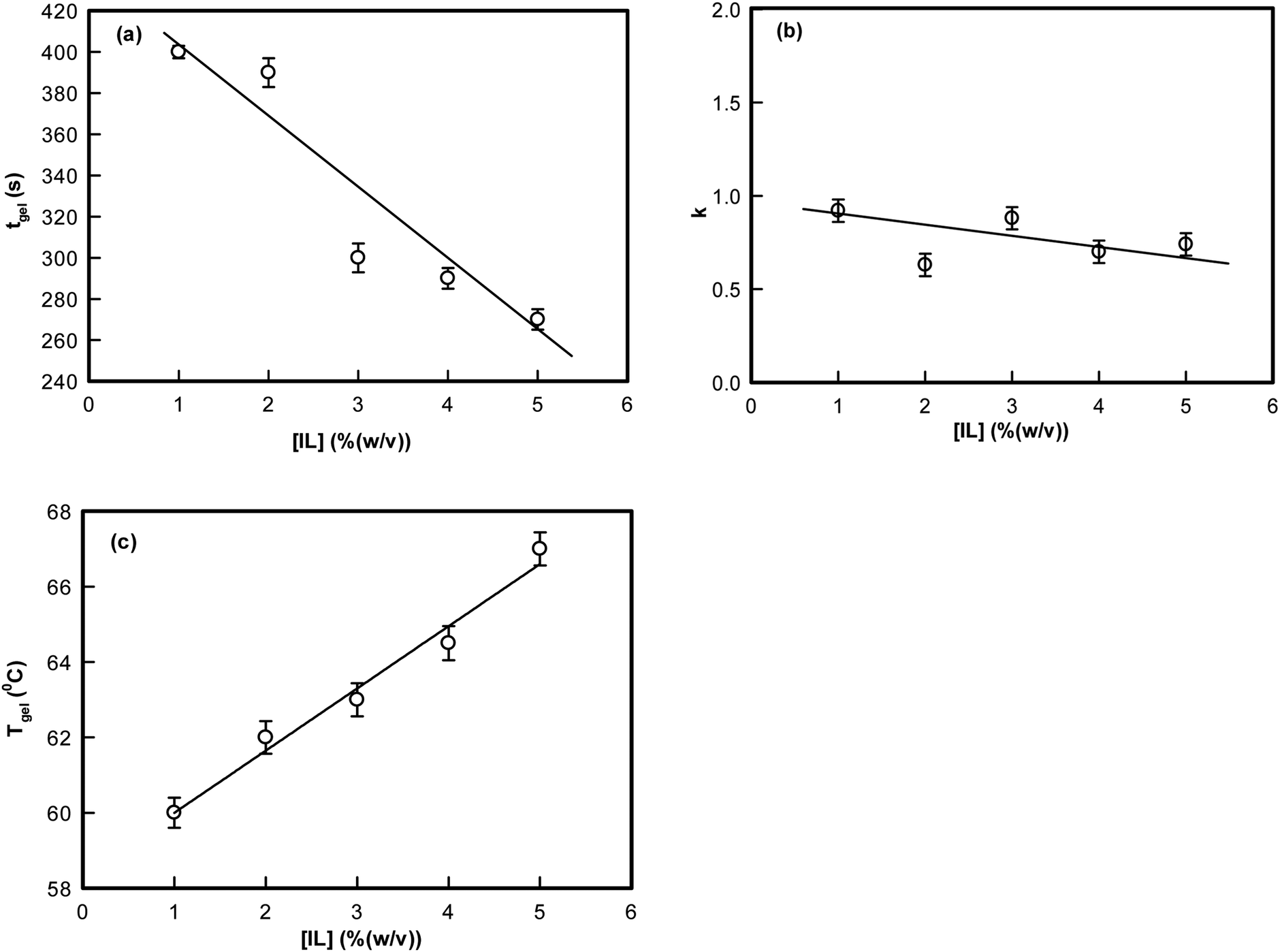 | ||
| Fig. 2 Variation of (a) gelation time tgel, (b) exponent, k, and (c) Tgel as function of ionic liquid concentration. | ||
The temperature dependent viscosity data are presented in Fig. S1 (ESI†) pertaining to the sols (ionosols) spontaneously cooling (below gelation temperature) to their gel states. It is clearly seen that as the IL concentration was increased from 1 to 5%, the temperature of gelation increased systematically from 60 to 67 °C. The respective gelation time and temperature could also be correlated from Fig. S2 (ESI†); the data are from different gelling samples. This large window of gelation temperature is remarkable, which could be achieved by the addition of a minute quantity of ionic liquid to the dispersion medium.
3.2 Dynamic arrest and ergodicity breaking
All the gel samples were rigid except for the one without IL which was in the sol phase throughout, i.e. it did not flow on inversion of the sample cell. The samples were subjected to DLS experiments in a bid to study the relaxation dynamics of the concentration fluctuations. The time dependent dynamic structure factors, g1(q,τ) obtained from different samples are depicted in Fig. S3 (ESI†). The dynamic structure factor determined from the treated intensity correlation function g2(q,t) data could be described by,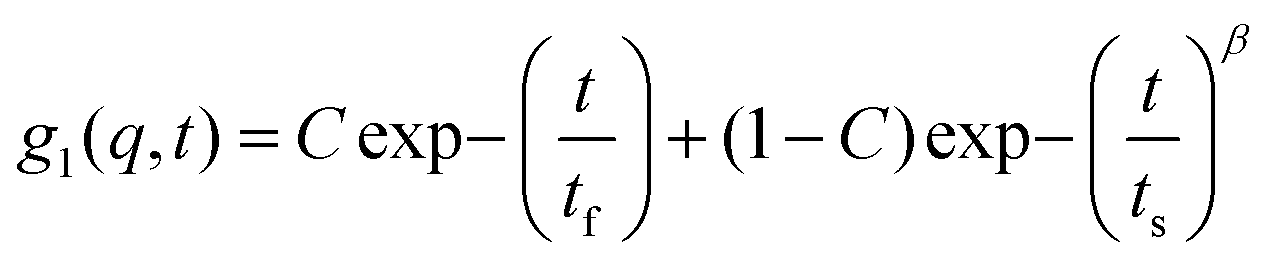 | (2) |
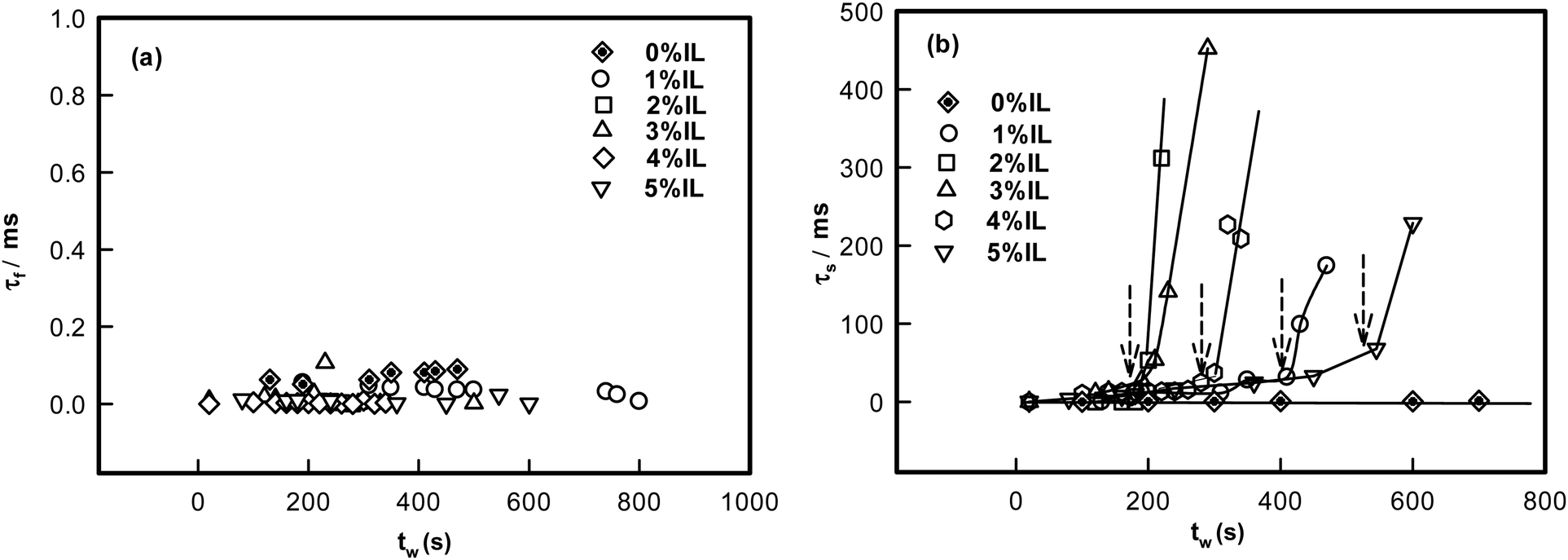 | ||
| Fig. 3 Plot of different modes of relaxation frequency with waiting time, tw shown for samples with different IL concentrations. (a) Note the invariance of the fast mode relaxation time with tw or IL concentration. (b) The slow mode relaxation time showed sharp up-turns at the time of dynamic arrest (arrows). | ||
The analysis of the DLS data also yielded the values for the fast and slow mode relaxation times τf and τs, respectively. These data are plotted as function of waiting time tw pertaining to each gel sample (Fig. 3). The fast mode relaxation time did not change with tw regardless of the increase in IL concentration. However, the slow mode exhibited a sharp increase at a specific waiting time dependent on IL content. This increase can be attributed to dynamic arrest of the networks. It was remarkable to note that the time of arrest was almost same as the ergodicity breaking time, τEB (to be discussed). Therefore, the transition to non-ergodicity was a dynamic process that was predominantly manifested in the dynamic structure factor data.
For an ergodic state, the dynamic structure factor g1(q,t) relaxes to the value 0 (at t → ∞) from its initial value of 1 (t = 0). For an ergodic state, the ergodicity parameter χ or the signal to noise ratio i.e. signal modulation for g1(q,t) is given by45,49
| χ = g1(q,t)∣t→0 − g1(q,t)∣t→∞ = 1 | (3) |
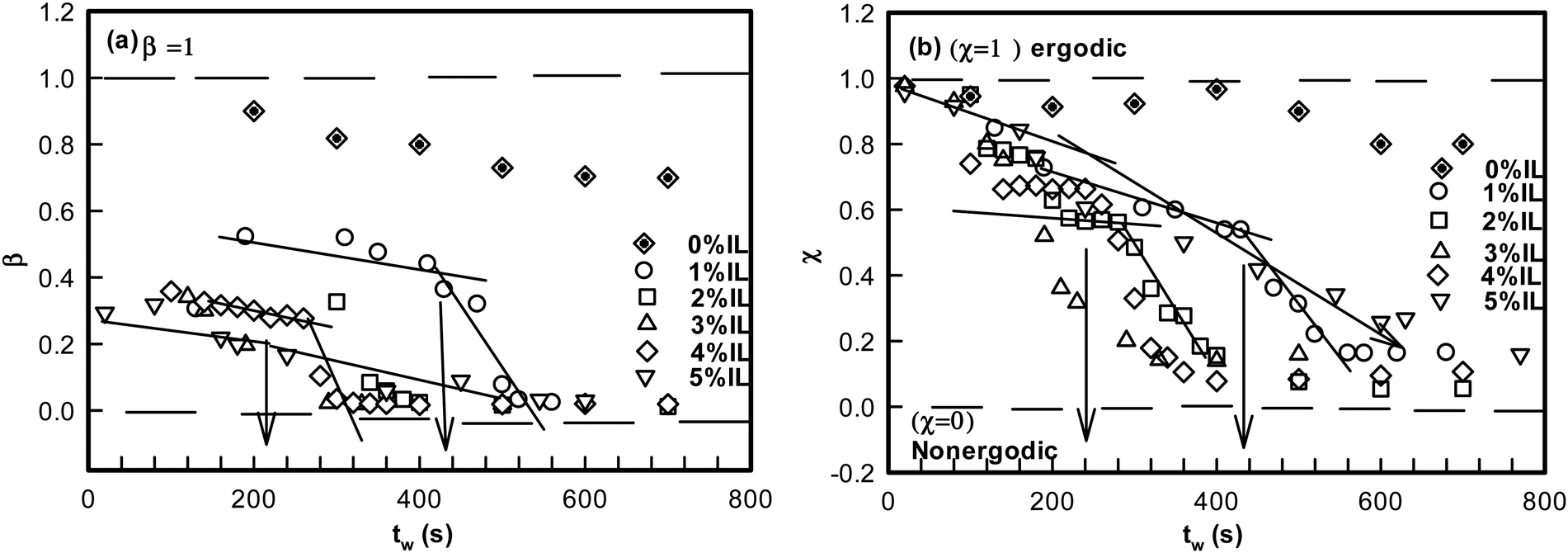 | ||
| Fig. 4 (a) Variation of stretch parameter β with waiting time tw for different IL concentrations. (b) Variation of χ as a function of waiting time, tw. The point where there is a slope change in the value of χ is defined as a ergodicity breaking time τEB shown for different IL concentrations. | ||
It is remarkable to note that both β and χ reveal an identical ergodicity breaking time τEB, which is shown in Fig. 5(a). When τEB and tgel were correlated (Fig. 5(b)), we clearly noted a linear dependence, implying the ergodicity breaking time was the same as the gelation time. Therefore, gelation referred to the dynamic arrest of the IL-cladded DNA strands that organize to yield an interconnected gel network.
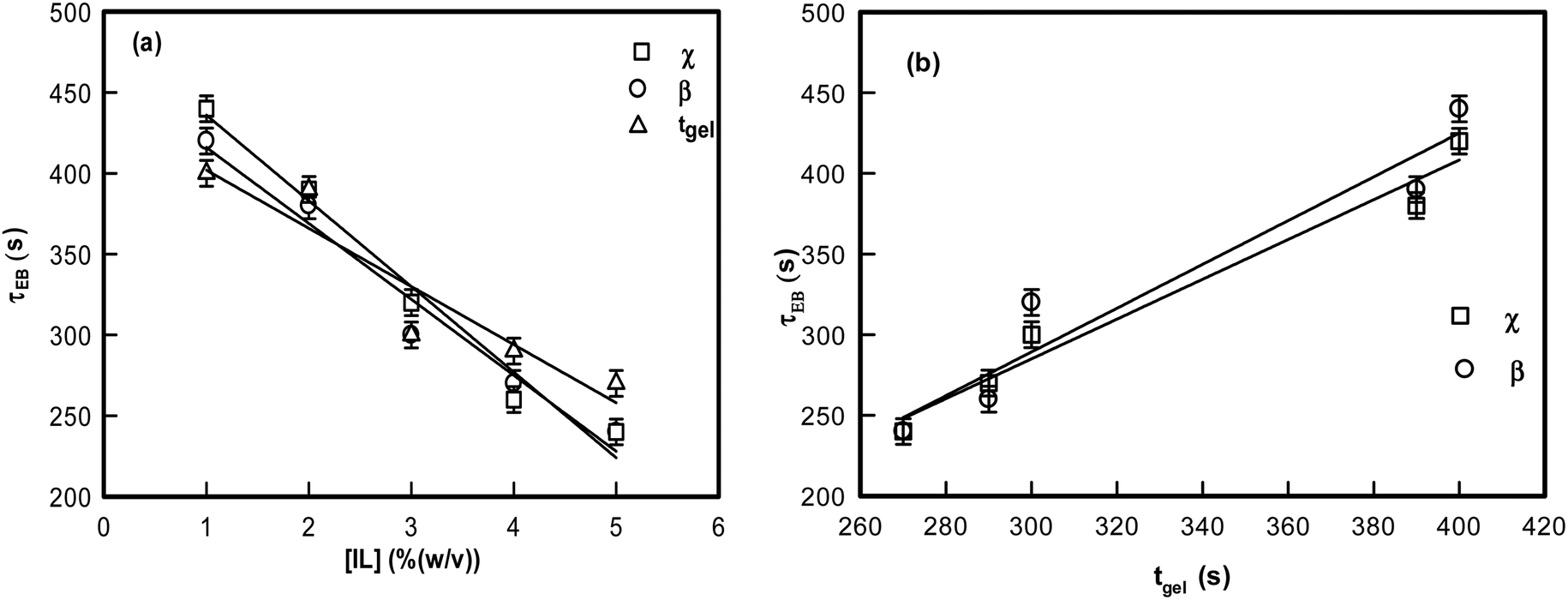 | ||
| Fig. 5 (a) Variation of ergodicity breaking time (from χ and β) and time of gelation with different IL concentrations. (b) Variation of ergodicity breaking time (from χ and β) with time of gelation tgel. | ||
3.3 Static structure factor
To map the microscopic internal structure of the ionogel samples, small angle neutron scattering (SANS) was used as a probe. The static structure factor profile for samples made with different IL concentrations is shown in Fig. 6(a). The I(q) vs. q data was first analyzed using the following protocols: (i)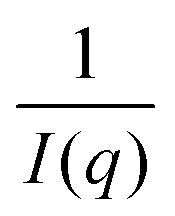 vs. q2 and (ii)
vs. q2 and (ii) 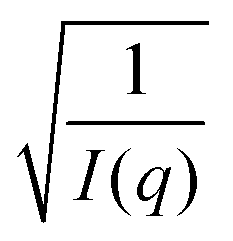 vs. q2 plots which did not yield clear straight lines or a set of two straight lines with different slopes. The best fit was obtained when the I(q) data was fitted to the following empirical functional form
vs. q2 plots which did not yield clear straight lines or a set of two straight lines with different slopes. The best fit was obtained when the I(q) data was fitted to the following empirical functional form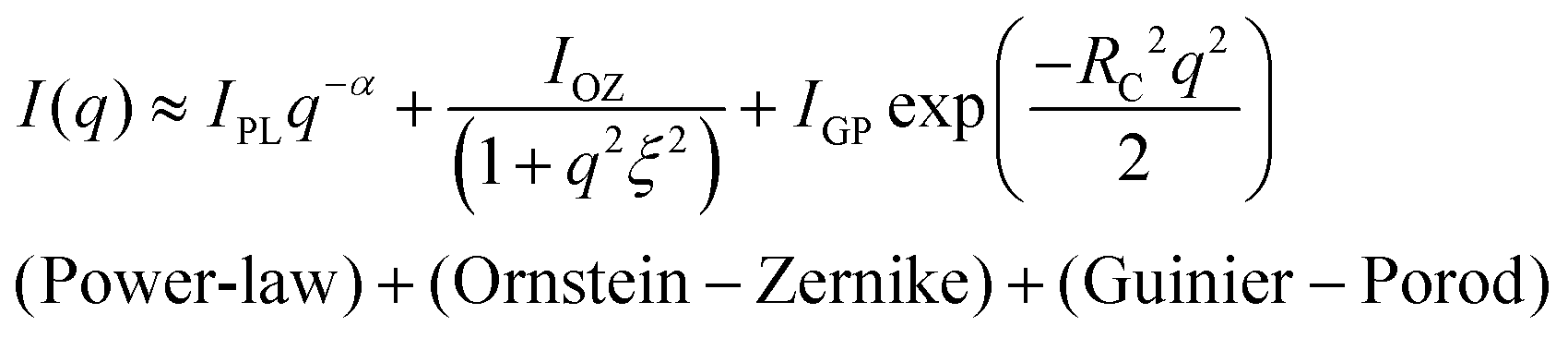 | (4) |
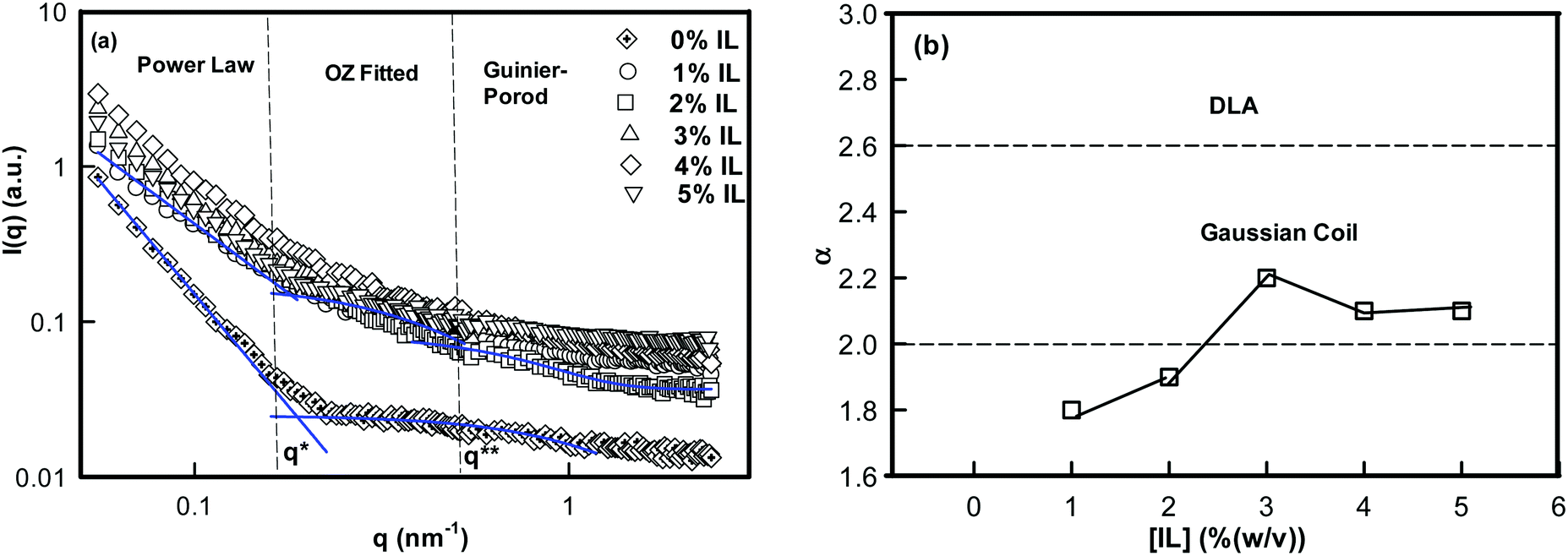 | ||
| Fig. 6 (a) Static structure factor I(q) measured by SANS for IL% (w/v) samples. Curves were fitted with the power law, the Ornstein–Zernike (OZ) function and the Guinier–Porod function in different q-regions. (b) Variation of geometrical scattering factor α with IL concentration. | ||
The low-q region (0.06 nm−1 ≤ q ≤ 0.19 nm−1) was fitted to a power-law function, the intermediate-q region (0.19 nm−1 ≤ q ≤ 0.53 nm−1) was fitted to the Ornstein–Zernike function, and the asymptotic range at q → ∞ (q ≥ 0.53 nm−1) was fitted to the Guinier–Porod function. Here, α is related to the geometry of the scattering moiety.
At equilibrium, for polymers in a good solvent, the mean-field theory describes that the structure factor of concentration fluctuation is defined by the Ornstein–Zernike (O–Z) function.51 Here, ξ is the correlation length or mesh size of the network. It was possible to probe the intermediate-q domain because of the high resolution of the spectrometer available in PSI. In the far-q domain, called the asymptotic regime, the chain cross-section makes a finite contribution to measured structure factor. In the Guinier–Porod formalism, at q → ∞ the structure factor is described by
| I(q) ∼ exp(−Rc2q2/2) | (5) |
The analysis of SANS data yielded α value in between 1.8 ± 0.1 and 2.1 ± 0.1 (Fig. 6(b)) for 1–5% (w/v) ionogels, and 2.8 for DNA sols. The value of α = 2 corresponds to a Gaussian coil type structure while at α = 2.6 the conformation is closer to diffusion limited aggregation clusters in 3D-space.
Furthermore, in the intermediate-q region data were analyzed by the Ornstein–Zernike relation that emerged from density fluctuations existing in the condensed phase.51 The average mesh size or correlation length of these fluctuation was given by ξ = 3 ± 0.7 nm for different ionogel samples, and a smaller mesh size for sols, ξ = 0.73 nm. The correlation length of only DNA sol (0% IL) was 76.7% less than that of DNA ionogels. The asymptotic regime was analyzed using eqn (5). The cross-section of DNA strands determined was, Rc = 1.7 ± 0.1 nm independent of IL concentration.
The variation of mesh size ξ and cross sectional radius Rc for different IL concentrations are depicted in Fig. S4(a) (ESI†). The characteristic cross-over lengths referred to the boundaries between the various q-regions mentioned earlier, are designated as q* and q** in Fig. 6(a). Therefore, the length scales 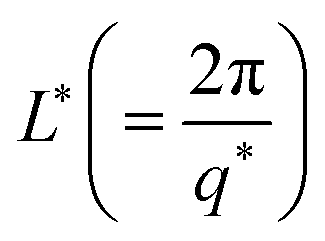 and
and 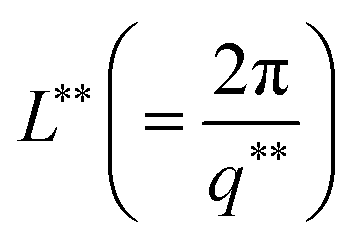 were determined to be equal to 33 ± 5 nm and 12 ± 1 nm, respectively. This revealed that inter particle distance decreased as the mesh-like structures appeared within the ionogels. The characteristic cross-over lengths L* and L** for different IL concentrations are depicted in Fig. S4(b) (ESI†).
were determined to be equal to 33 ± 5 nm and 12 ± 1 nm, respectively. This revealed that inter particle distance decreased as the mesh-like structures appeared within the ionogels. The characteristic cross-over lengths L* and L** for different IL concentrations are depicted in Fig. S4(b) (ESI†).
It was pertinent to note that the rod-like DNA strand, when cladded with IL-molecules loses its rigidity and ability to assume the Gaussian coil conformation due to localized surface charge neutralization. Thus, the binding of positively charged imidazolium head groups to DNA resulted in a softening of the stiffness of the nucleic acid. DNA–protein binding has shown similar results earlier.7–9
3.4 Rheology
The ionogel samples were subjected to dynamic rheology studies to evaluate their viscoelastic properties. Linear viscoelastic theory predicts that the storage modulus G′ exhibits a frequency dependent dispersion behaviour given by45| G′(ω) = G0ωn | (6) |
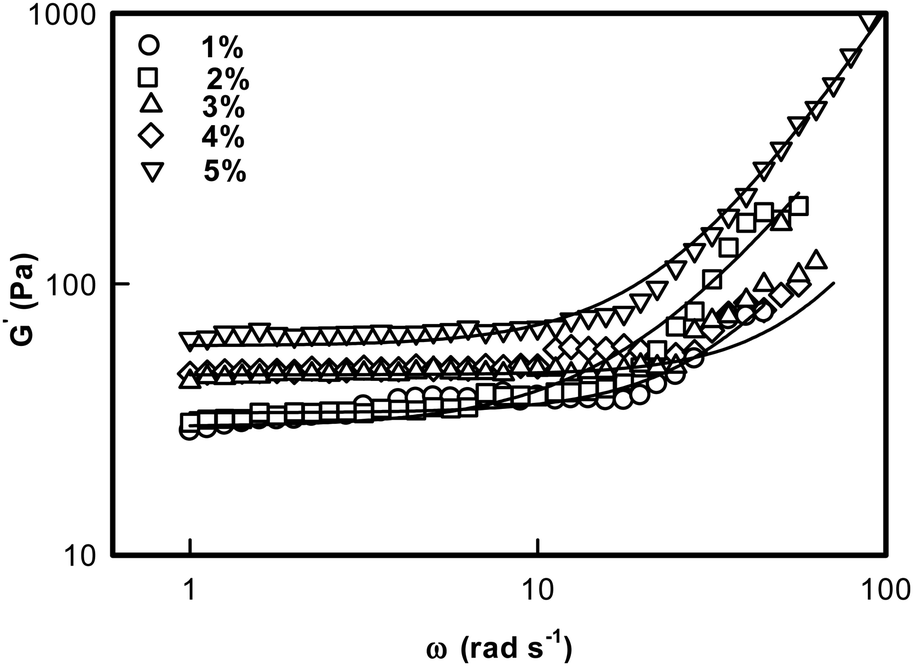 | ||
| Fig. 7 Storage G′(ω) modulus of DNA ionogel plotted as a function of frequency. Solid line is a fit to eqn (6). | ||
Lodge et al. have explained the self assembly of ion gels of tri-block copolymers and have extensively discussed the viscoelastic behavior of these chemically crosslinked gels.52Fig. 7 is a log–log plot depiction of eqn (6). Thus, the exponent ‘n’ could be determined from the slope of the line: log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) G′(ω) = G0 + nlogω.
G′(ω) = G0 + nlogω.
Two observations are clearly made from these data: (i) the low frequency storage modulus, G0 given by value of G′(ω) at lowest frequency used in the experiment, showed a characteristic dependence with G0 assuming a value ∼27 Pa for 1% and it sharply increased to ∼70 Pa for the [IL] = 5% sample; and (ii) we found that the value of n = 1.8 ± 0.2 was universal. These results clearly indicated that these ionogels were viscoelastic in nature irrespective of their actual [IL] concentration (Fig. 8, n-data) which constrained the self assembly of DNA to be hierarchical (Fig. 8, G0 data).
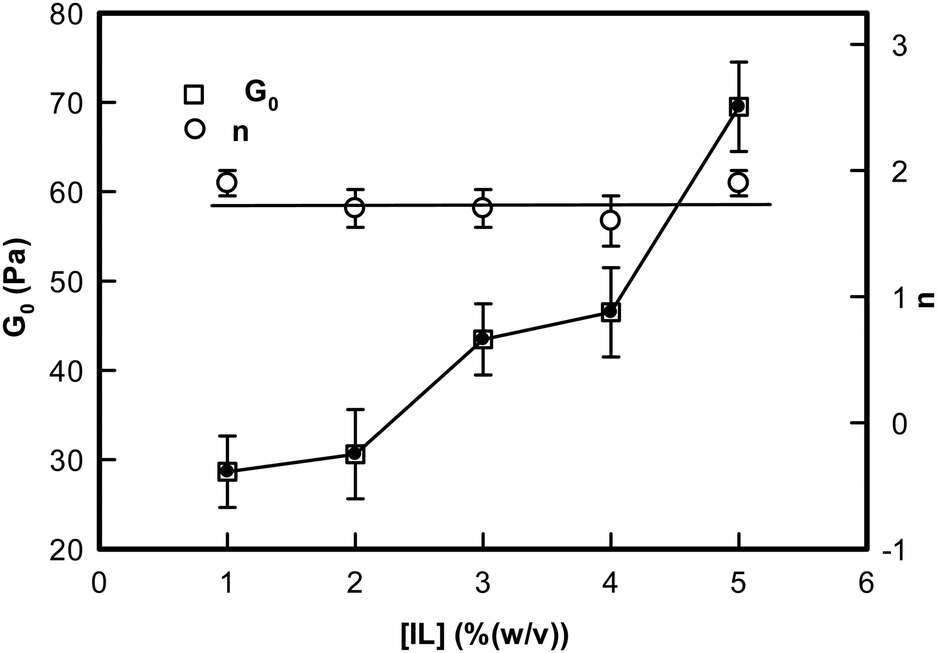 | ||
| Fig. 8 Variation of the rigidity modulus G0 and the frequency exponent n of ionogels shown as a function of IL concentration. Solid lines are guide to the eye. | ||
The gel phase can be visualized as a transient polymer network of viscoelastic length, ζel through the relation given by53
| G0 ∼ kBT/ζel3 | (7) |
The data in hand yielded ζel ≈ 45 ± 3 nm for [IL] = 1–3%; 38 nm for [IL] = 4% and 35 nm for [IL] = 5% ionogels (Fig. S5, ESI†). Thus, the networks become more compact with the increase in IL concentration which imparted higher values to its storage modulus.
In the next step, isochronal temperature sweep experiments were carried out to characterize the melting profiles of these gels (Fig. S6, ESI†). It was remarkable to observe that sharp transitions were found at two distinct temperatures. We shall refer to these as Tmelt and Tgel, respectively. These Tgel values were consistent with the same determined from viscosity experiments. This is illustrated in Fig. 9. It was remarkable to note that Tgel could be raised (shifted) by ≈7 °C because of the presence of a tiny volume fraction of ionic liquid. Therefore, the ionogels are associated with a gelation zone that spans from 60 to 67 °C, which will enable design of more flexible DNA based biomaterials in the future.
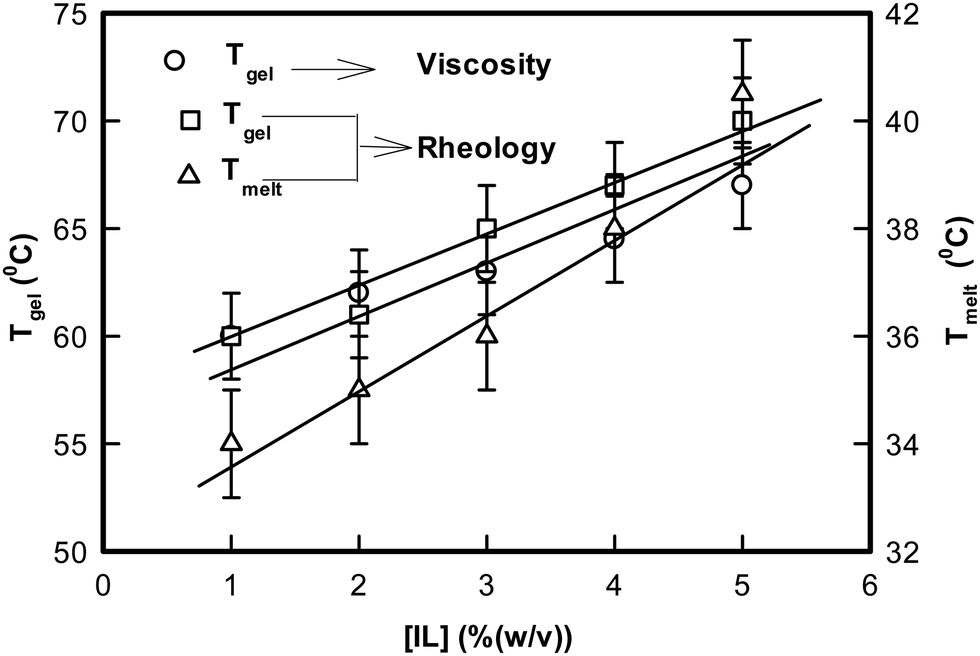 | ||
| Fig. 9 Variation of the temperature of melting, Tmelt, and gelation, Tgel, of DNA ionogels as a function of IL concentration obtained from rheology and viscosity. | ||
Let us discuss the significance of Tmelt. We did not observe an equivalent of Tmelt from the viscosity data, which may be due to the poor resolution of the viscometer apparatus. However, it can be argued that initial self-organization of IL-cladded DNA strands formed primary transient networks with a characteristic enthalpy defined by Tmelt. This temperature increased from 34 °C for 1% to 40 °C for 5% ionogel. Further, the G0 value noted a decrease of typically 40% as this temperature was approached. Compared to this, the drop in G0 value was almost 100% as Tgel approached. Clearly, the gelation transition that occurred at Tgel was associated with a larger enthalpy compared to the melting transition encountered at Tmelt.
4. Conclusion
The microscopic structure of the sol and gel state of the DNA–IL system was studied exhaustively as function of ionic liquid concentration. DNA (1% w/v) was in the sol phase (it does not gel) which exhibited random Brownian motion and ergodicity. On addition of IL, there was a gradual transition from a random to an arrested state because of the cessation of Brownian dynamics owing to the formation of IL-mediated network structures, which is a first order phase transition from the ergodic to non-ergodic state. Inter-particle distance reduced, and mesh-like structures appeared in the presence of IL. Self-organization of DNA in IL solution occurred systematically, driven mostly by electrostatic interaction. Note that presence of IL molecules, introduces an enhanced possibility of hydrogen bonding, Coulombic interactions and hydrophobic forces. The self-assembly leading to gelation observed in this work may be viewed as arising from an assortment of aforesaid forces. This has been conceptualized in a schematic representation shown in Scheme 1.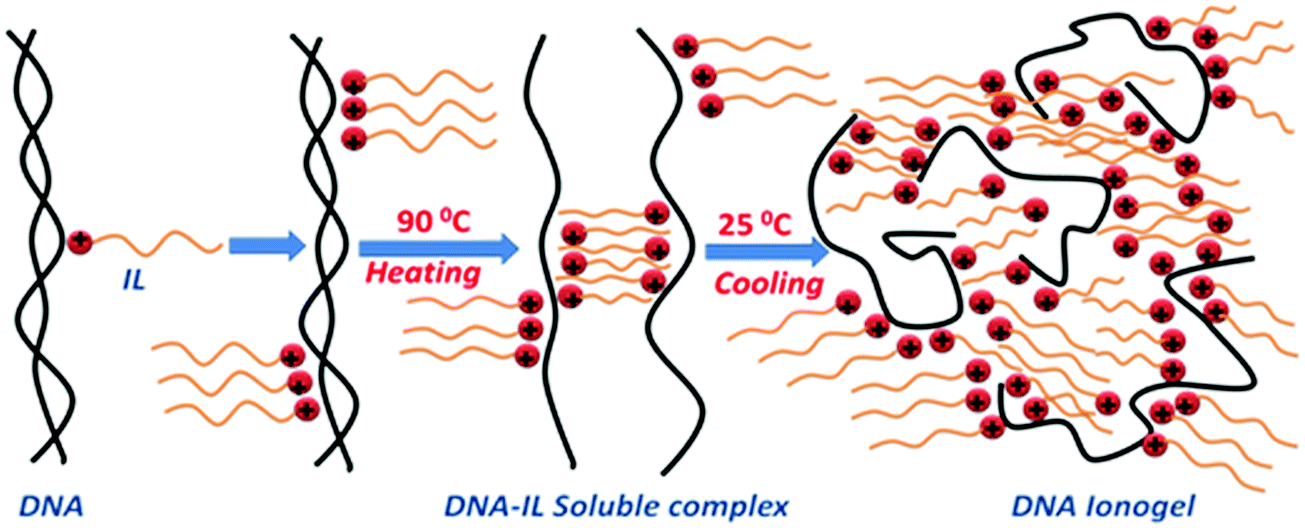 | ||
| Scheme 1 Schematic representation of the formation of DNA ionogels (not to scale). | ||
It is known that ssDNA can be prepared by heating the dsDNA stock solutions to 80 °C for 15 min followed by quenching to ice temperature, which prevents its renaturation.10 We followed the same procedure, but used charge quenching to keep the strands separated. The positively charged imidazolium head groups of IL molecules present in the solution were attracted towards the negatively charged phosphate groups on DNA due to electrostatic forces to form a monolayer of IL on the single strands. However, it should also be realized that the hydrophobic interaction between the alkyl groups (ethyl-moieties) of the ionic liquid molecules and the hydrophobic nitrogenous bases in DNA also played a role (marginal). This prevented the re-naturation of the DNA strand. What followed was a self-assembly process, where the hydrophobic tails of IL were bound to DNA with similar units present in their neighborhood. This resulted in the formation of an amorphous network of IL–DNA clusters. Since the system was undergoing a sol–gel transition, the conclusion of the formation of the DNA–IL networks shown in Scheme 1 was logical. The low-q region available to our SANS instrument did not yield a robust Guinier regime which could have yielded information about the physical size of these growing clusters. This phenomenological model is a conjecture. The relaxation dynamics were hierarchical within ionogels. These results draw our attention to the very marginally understood self-organization of DNA in an ionic liquid medium. Furthermore, the observation of an increase in the gel strength leading to an almost 10 °C increase in the gelation temperature when the IL concentration did not exceed 5% (w/v) was remarkable. Thus, we believe the results obtained will widen the potential application of DNA gels.
Acknowledgements
P. K. P. acknowledges UGC-BSR, Government of India for a Junior Research Fellowship. K. R. is thankful to the Department of Science and Technology, Government of India-Inspire Faculty Award. This study was funded by the Department of Science and Technology (DST), India.References
- K. Rawat, P. R. Solanki, K. Arora and H. B. Bohidar, Response of gelatin modified electrode towards sensing of different metabolites, Appl. Biochem. Biotechnol., 2014, 174, 1032–1042 CrossRef CAS PubMed.
- S. Chaleawlert-Umpon and N. Pimpha, Preparation of magnetic polymer microspheres with reactive epoxide functional groups for direct immobilization of antibody, Colloids Surf., A, 2014, 461, 66–75 CrossRef.
- W. Suginta, P. Khunkaewla and A. Schulte, Electrochemical biosensor applications of polysaccharides chitin and chitosan, Chem. Rev., 2013, 113, 5458–5479 CrossRef CAS PubMed.
- A. D. Dias, D. M. Kingsley and D. T. Corr, Recent advances in bioprinting and applications for biosensing, Biosensors, 2014, 4, 111–136 CrossRef CAS PubMed.
- V. V. Singh, A. K. Nigam, A. Batra, M. Boopathi, B. Singh and R. Vijayaraghavan, Applications of ionic liquids in electrochemical sensors and biosensors, Int. J. Electrochem., 2012, 165683 Search PubMed.
- R. Yan, F. Zhao, J. Li, F. Xiao, S. Fan and B. Zeng, Direct electrochemistry of horseradish peroxidase in gelatin–hydrophobic ionic liquid gel films, Electrochim. Acta, 2007, 52, 7425–7431 CrossRef CAS.
- N. Arfin and H. B. Bohidar, Condensation, complex coacervation, and overcharging during DNA–gelatin interactions in aqueous solutions, J. Phys. Chem. B, 2012, 116, 13192–13199 CrossRef CAS PubMed.
- K. Rawat, V. K. Aswal and H. B. Bohidar, DNA–gelatin complex coacervation, UCST and first-order phase transition of coacervate to anisotropic ion gel in 1-methyl-3-octylimidazolium chloride ionic liquid solutions, J. Phys. Chem. B, 2012, 116, 14805–14816 CrossRef CAS PubMed.
- K. Rawat, J. Pathak and H. B. Bohidar, Effect of persistence length on binding of DNA to polyions and overcharging of their intermolecular complexes in aqueous and in 1-methyl-3-octyl imidazolium chloride ionic liquid solutions, Phys. Chem. Chem. Phys., 2013, 15, 12262–12273 RSC.
- M. C. Moran, M. G. Miguel and B. Lindman, DNA gel particles: Particles preparations and release characteristics, Langmuir, 2007, 23, 6478–6481 CrossRef CAS PubMed.
- F. Hokey and P. J. Passer, Osmotic observation on chemically cross linked DNA gels in physiological salt solution, Biomacromolecules, 2004, 5, 232–237 CrossRef PubMed.
- D. Costa, P. Harisson, S. Scheneider, M. G. Miguel and B. Lindmann, Interaction between covalent DNA gels and cationic surfactant, Biomacromolecules, 2006, 7, 1090–1095 CrossRef CAS PubMed.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Ionic liquid (molten salt) phase organometallic catalysis, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Ionic liquids—solvents of the future?, Science, 2003, 302, 792–793 CrossRef PubMed.
- P. Wasserscheid, Chemistry: Volatile times for ionic liquids, Nature, 2006, 439, 797 CrossRef CAS PubMed.
- W. T. Leitner, Catalysis: A greener solution, Nature, 2003, 423, 930–931 CrossRef CAS PubMed.
- S. OzBas-Turan, C. Aral, L. Kabasakal, M. Keyer-Uysal and J. Akbuga, Co-encapsulation of two plasmids in chitosan microspheres as a non-viral gene delivery vehicle, J. Pharm. Pharm. Sci., 2003, 6, 27–32 CAS.
- C. Aral and J. Akbuga, Preparation and in vitro transfection efficiency of chitosan microspheres containing plasmid DNA: poly(L-lysine) complexes, J. Pharm. Pharm. Sci., 2003, 6, 321–326 CAS.
- J. Le Bideau, L. Viau and A. Vioux, Ionogels, ionic liquid based hybrid materials, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- K. Haraguchi and T. Takehisa, Nanocomposite hydrogels: A unique organic inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- A. Sharma, K. Rawat, P. R. Solanki and H. B. Bohidar, Gelatin–ionic liquid based platform for glucose detection, Curr. Top. Med. Chem., 2015, 15, 1257–1267 CrossRef CAS PubMed.
- X. Zhu, H. Zhang and J. Wu, Chemiresistive ionogel sensor array for the detection and discrimination of volatile organic vapor, Sens. Actuators, B, 2014, 202, 105–113 CrossRef CAS.
- J. B. Lee, S. Peng, D. Yang, Y. H. Roh, H. Funabashi, N. Park, E. J. Rice, L. Chen, R. Long, M. Wu and D. Luo, A mechanical metamaterial made from a DNA hydrogel, Nat. Nanotechnol., 2012, 7, 816–820 CrossRef CAS PubMed.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Enzyme-catalysed assembly of DNA hydrogel, Nat. Mater., 2006, 5, 797–801 CrossRef CAS PubMed.
- X. Xiong, C. Wu, C. Zhou, G. Zhu, Z. Chen and W. Tan, Responsive DNA-based hydrogels and their applications, Macromol. Rapid Commun., 2013, 34, 1271–1283 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Self-assembled multicompartment liquid crystalline lipid carriers for protein, peptide, and nucleic acid drug delivery, Acc. Chem. Res., 2011, 44, 147–156 CrossRef CAS PubMed.
- B. Angelov, A. Angelova, S. K. Filippov, G. Karlsson, N. Terrill, S. Lesieur and P. Stepaneka, Topology and internal structure of PEGylated lipid nanocarriers for neuronal transfection: Synchrotron radiation SAXS and cryo-TEM studies, Soft Matter, 2011, 7, 9714–9720 RSC.
- B. Angelov, A. Angelova, S. Filippov, G. Karlsson, N. Terrill, S. Lesieur and P. Štěpánek, SAXS study of sterically stabilized lipid nanocarriers functionalized by DNA, J. Phys.: Conf. Ser., 2012, 351, 012004 CrossRef.
- B. Angelov, A. Angelova, S. K. Filippov, T. Narayanan, M. Drechsler, P. Štěpánek, P. Couvreur and S. Lesieur, DNA/Fusogenic lipid nanocarrier assembly: Millisecond structural dynamics, J. Phys. Chem. Lett., 2013, 4, 1959–1964 CrossRef CAS PubMed.
- B. Angelov, A. Angelova, M. Drechsler, V. M. Garamus, R. Mutafchieva and S. Lesieur, Identification of large channels in cationic PEGylated cubosome nanoparticles by synchrotron radiation SAXS and Cryo-TEM imaging, Soft Matter, 2015, 11, 3686–3692 RSC.
- H. Rosa, D. F. S. Petri and A. M. Carmona-Ribeiro, Interactions between bacteriophage DNA and cationic biomimetic particles, J. Phys. Chem. B, 2008, 112, 16422–16430 CrossRef CAS PubMed.
- I. S. Kikuchi, W. Viviani and A. M. Carmona-Ribeiro, Nucleotide insertion in cationic bilayers, J. Phys. Chem. A, 1999, 103, 8050–8055 CrossRef CAS.
- I. S. Kikuchi and A. M. Carmona-Ribeiro, Interactions between DNA and synthetic cationic liposomes, J. Phys. Chem. B, 2000, 104, 2829–2835 CrossRef CAS.
- N. Orakdogen, B. Erman and O. Okay, Evidence of strain hardening in DNA gels, Macromolecules, 2010, 43, 1530–1538 CrossRef CAS.
- R. Thomas, Recherches sur la d'enaturation des acides desoxyribonucléiques, Biochim. Biophys. Acta, 1954, 14, 231–240 CrossRef CAS.
- S. A. Rice and P. Doty, The thermal denaturation of desoxyribose nucleic acid, J. Am. Chem. Soc., 1957, 79, 3937–3947 CrossRef CAS.
- P. Doty, H. Boedtker, J. R. Fresco, R. Haselkorn and M. Litt, Secondary structure in ribonucleic acids, Proc. Natl. Acad. Sci. U. S. A., 1959, 45, 482–499 CrossRef CAS.
- R. B. Inman and R. L. Baldwin, Helix-random coil transitions in synthetic DNAs of alternating sequence, J. Mol. Biol., 1962, 6, 172–184 CrossRef.
- J. Marmur and P. Doty, Determination of the base composition of deoxyribonucleic acid from its thermal denaturation temperature, J. Mol. Biol., 1962, 6, 109–118 CrossRef.
- W. F. Dove and N. Davidson, Cation effects on the denaturation of DNA, J. Mol. Biol., 1962, 5, 467–478 CrossRef CAS.
- P. O. Ts'o, G. Helmkamp and C. Sander, Interaction of nucleosides and related compounds with nucleic acids as indicated by the change of helix–coil transition temperature, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 686–698 CrossRef.
- C. Schildkraut and S. Lifson, Dependence of the melting temperature of DNA on salt concentration, Biopolymers, 1965, 3, 195–208 CrossRef CAS PubMed.
- J. Kohlbrecher and W. Wagner, The new SANS instrument at the Swiss spallation source SINQ, J. Appl. Crystallogr., 2000, 33, 804–806 CrossRef CAS.
- B. J. Berne and R. Pecora, Dynamic Light Scattering: With Applications to Chemistry, Biology and Physics, New York, USA, 1976 Search PubMed.
- H. A. Barnes, Handbook of Elementary Rheology, University of Wales Press, Wales, 2000 Search PubMed.
- A. Aharony and D. Stauffer, Introduction to Percolation Theory, Taylor and Francis, London, 1994 Search PubMed.
- D. Stauffer, A. Coniglio and A. Adams, Gelation and critical phenomena, Adv. Polym. Sci., 1982, 44, 103–158 CrossRef CAS.
- L. de Arcangelis, E. Del Gado and A. Coniglio, Complex dynamics in gelling systems, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 9, 277–282 CrossRef CAS PubMed.
- A. B. Rodd, D. E. Dunstan, D. V. Boger, J. Schmidt and W. Burchard, Heterodyne and nonergodic approach to dynamic light scattering of polymer gels: Aqueous xanthan in the presence of metal ions (aluminum(III)), Macromolecules, 2001, 34, 3339–3352 CrossRef CAS.
- R. K. Pujala and H. B. Bohidar, Ergodicity breaking and aging dynamics in Laponite–Montmorillonite mixed clay dispersions, Soft Matter, 2012, 8, 6120–6127 RSC.
- P. G. De Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, Ithaca, NY, 2nd edn, 1985 Search PubMed.
- Y. He, P. G. Boswell, P. Bu1hlmann and T. P. Lodge, Ion gels by self-assembly of a triblock copolymer in an ionic liquid, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef CAS PubMed.
- A. Ajji and L. Choplin, Rheology and dynamics near phase separation in a polymer blend: Model and scaling analysis, Macromolecules, 1991, 24, 5221–5223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The variation of relative viscosity with different temperature, gelation temperature Tgel as a function of gelation time tgel, time dependent dynamic structure factor, g1(τ) intensity profile, mesh size ξ, cross sectional radius Rc, characteristic cross over lengths for different DNA–IL samples at various IL concentrations undertaken in this study. See DOI: 10.1039/c6cp06229f |
| This journal is © the Owner Societies 2017 |