
Dehydro-oxazole, thiazole and imidazole radicals: insights into the electronic structure, stability and reactivity aspects†
Anamika
Mukhopadhyay
,
Lilit
Jacob
 and
Sugumar
Venkataramani
*
and
Sugumar
Venkataramani
*
Department of Chemical Sciences, Indian Institute of Science Education and Research, IISER Mohali, Sector 81, Knowledge City, SAS Nagar, Mohali, Punjab 140306, India. E-mail: sugumarv@iisermohali.ac.in
First published on 1st December 2016
Abstract
DFT (U)B3LYP/cc-pVTZ and (U)M06-2X/cc-pVTZ and multireference CASSCF/cc-pVTZ level of theories have been used to investigate the electronic structure of isomeric dehydrooxazole (1b–d, 3 isomers), dehydrothiazole (2b–d, 3 isomers) and dehydroimidazole (3a–d, 4 isomers) radicals. The ground state electronic structure of each radical isomer has been confirmed by predicting their doublet excited state structure and calculating the adiabatic energy difference. The stability order of the individual isomeric radicals has been estimated through the comparison of absolute energies. A hypothetical isodesmic reaction has been utilized to calculate radical stabilization energies (RSEs). The results show N-dehydro imidazole 3a as the thermodynamically most stable radical. In order to understand the structural and stability aspects of the radicals and interactions between the radical electron and the electron lone pairs, we have analysed spin densities and hybridisation changes and also performed MCSCF calculations. The reason for the higher stability of 3a has been attributed to the attainment of a π-character and subsequent delocalization, whereas, all other carbon centred radicals are found to be localized σ-radicals. Furthermore, the kinetic stability of the radicals has been investigated through unimolecular decomposition channels. All the studies showed a weak to strong coupling between the N3 and the radical centre depending on the location of the radical centre. NBO analysis suggests that through space coupling between N3 and σ* of the radical centre leads to a stabilising effect when the radical centre is adjacent to N3, whereas such interactions are absent when the radical centre is away from it. However, only a weak coupling is observed between the radical centre and X1 (O/S/NH). Particularly the interaction strength has the following trend: S < O < N–H. Indeed S, O and N–H show a stabilising effect through bond interactions with the antibonding orbitals of the alternate bonds on either side.
1. Introduction
Radicals are a well-known class of reactive intermediates that contain an unpaired electron.1–3 Generally these open-shell species are highly transient, due to their high reactivity through which they attain a closed-shell electronic configuration. The generation and reactivity of radicals play a crucial role in the combustion of petrochemicals,4 biological processes such as ageing, metabolism, and in diseases like cancer through the accumulation of reactive oxygen species (ROS),5 polymer chemistry6 and also in atmospheric chemistry.7 Moreover, radical reactions are extensively utilized in organic synthesis.8 The reactions can range from a simple hydrogen abstraction to complex cyclisation and a cascade of radical rearrangements. However, many attempts have been made in diminishing the reactivity and in enhancing the stability of the radicals through the design and incorporation of various effects such as resonance, steric factors, and creation of a radical centre at electronegative atoms etc.9Synthetic methods utilizing radical traps, and radical clocks and experimental techniques such as electron spin resonance (ESR), chemically induced dynamic nuclear polarization (CIDNP), matrix isolation, mass spectrometry, photoelectron spectroscopy and other spectroscopic techniques along with computational methods play a significant role in understanding the structural and reactivity aspects of radicals. Through these methods, extensive studies have been accomplished on several radicals,10 diradicals11 and polyradicals.12 However, only limited progress have been made in the understanding of their heterocyclic analogues. The presence of lone pairs in heterocyclic radicals potentially leads to a plethora of effects including electronic and orbital interactions via through-space (TS) and through-bond (TB) coupling.13 Thus, the studies on such open-shell systems will be challenging to both theoretical as well as practical experiments. Not only the parent radicals, but also many of their heteroatom rich ring opening products and fragments are equally interesting in an astrochemical point of view.14
Among the many species related to heterocyclic radicals, their higher analogues, didehydroheterocycles received special attention due to their synthetic utility.15 One of the pioneering studies in this regard was the direct observation of 3,4-didehydropyridine reported by Nam and Leroi.16 In yet another work on its 3,5-isomer (m-pyridyne), Sander and co-workers have reported that the overall impact of nitrogen atoms (four electron, three centred interaction) was weak. However, it was indeed influenced in the reduction of singlet–triplet energy gap by 3 kcal mol−1 compared to m-benzyne.17 Wenthold and coworkers experimentally predicted the presence of an interaction between the nitrogen lone pair and the radical centres of various isomeric didehydropyridines depending upon the position that varies from stabilizing to destabilizing effects.18 Except in the case of 2,6-didehydropyridine, which has almost degenerate singlet–triplet states, all other isomers have a singlet ground state, due to the dominant TS and TB orbital interactions and spin coupling between the two radical centres. Similarly, McMahon and coworkers reported the ring opening pathways of 2,6-didehydrothiophene.19 Overall a reasonable progress has been reached with respect to didehydroheterocycles. In order to understand the direct interaction and influence of heteroatoms on the radical centre, studies on monoradicals are unavoidable. The prototypical heterocyclic radicals, namely, the isomeric pyridine radicals have been investigated through matrix isolation infrared spectroscopy and computational studies recently. The studies included the generation and reactions with oxygen of 2-, 3- and 4-dehydropyridine radicals.20 Unimolecular decomposition channels of these three isomeric pyridine radicals have been theoretically investigated by Liu et al.21 Recently, Poskrebyshev reported the mechanism for ring opening of 2-furyl radical through computational studies.22 Hadad and coworkers and Feng et al. have investigated the bond dissociation energies (BDEs) of small heterocyclic hydrocarbons and radicals, respectively.23 Several other heterocyclic radicals have been subjected to ab initio calculations during the prediction of thermochemical data and ionization energies of parent and charged heterocycles. Potential intermediacy of many such heterocyclic radicals has also been reported during the thermal decomposition of their parent compounds.24
Upon introduction of a second heteroatom into the existing heterocycle, the structural and reactivity aspects of the resulting radicals are expected to be even more complex. Various electronic structural and dissociation properties of heterocycles like thiazole and oxazole molecules and their charged radicals have been studied using different methods, such as flash photolysis,25 photoelectron spectroscopy,26 MATI spectroscopy,27 VUV absorption spectroscopy,28 VUV photodissociation29 and using a strong fs laser field.30 Despite the availability of many detailed experimental and theoretical studies on charged radicals, reports on neutral radicals are scarce. Recently, Sanov and co-workers reported that the C–H bond dissociation energies in five member heterocyclic radicals are higher than that of benzene, due to a change in hybridization after the formation of σ-radicals.26
In spite of the tremendous progress in the field of radical chemistry, a few questions remain unanswered: (a) What will be the effect of heteroatoms on the overall geometry, electronic structure, stability and reactivity of the radicals? (b) When two heteroatoms are present, will there be a disparity in the influence? If yes, how and to what extent this will be? (c) What will be the modus operandi of the interaction between the radical centre and the lone pairs of the heteroatoms? To answer such questions, currently we are focusing on a series of heterocyclic radical species. In our current contribution, we target the influence of heteroatoms such as O, N, and S on the structural, stability and reactivity aspects of the five member heterocyclic radicals. The isomeric oxazole, thiazole and imidazole radicals are considered in this regard (Scheme 1).
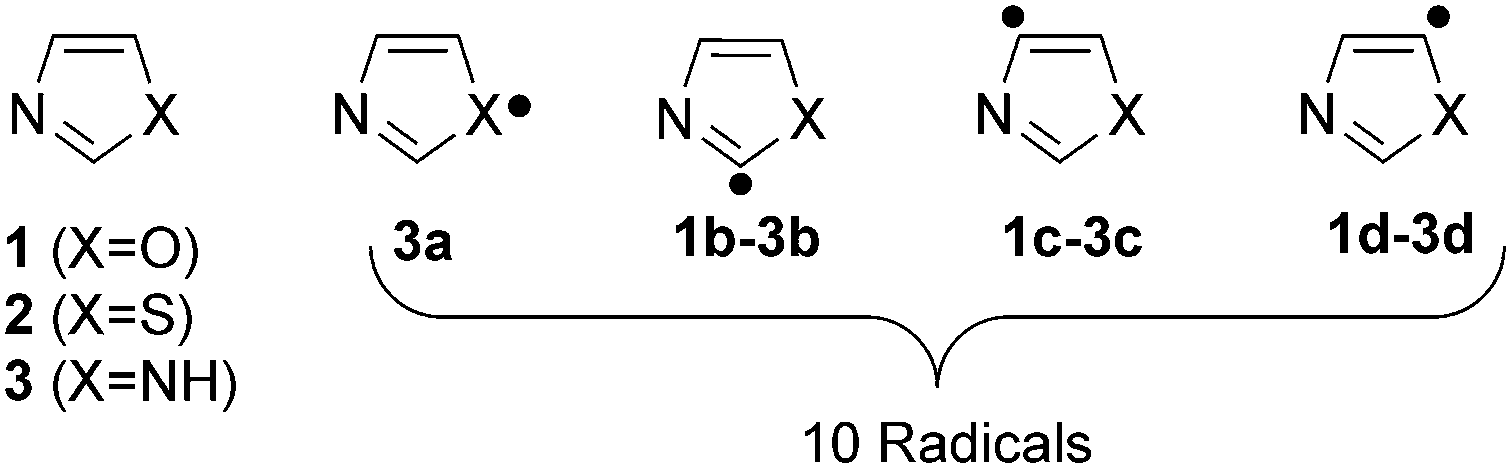 | ||
| Scheme 1 Parent heterocycles and the corresponding dehydro-oxazole, thiazole and imidazole radicals. | ||
2. Methodologies
2.1. Computational details
Full geometry optimization without any constraints of all the reactants (R), products (P), intermediates (I) and transition states (TS) were performed at the (U)B3LYP31/cc-pVTZ32 and (U)M06-2X33/cc-pVTZ levels. Single point energy calculations have been calculated at the CCSD(T)34/cc-pVTZ level using appropriate optimized geometries. Unrestricted option was used for all open shell systems. Transition states were obtained using the Synchronous Transit Guided Quasi-Newton (STQN)35 method using a QST3 approach, where the molecular specification for guess TS was provided along with the reactant and product geometries. All the transition states were confirmed by the presence of an imaginary frequency along the dissociation coordinate. IRC calculations36 were performed to verify all the TS. Natural Bond Orbital (NBO) calculations were performed to obtain the hybridization details.37 All these calculations and thermochemistry were performed using the G09 package program.38The multireference calculations were performed at the CASSCF39/cc-pVTZ level for (U)B3LYP/cc-pVTZ structures using the MOLPRO program package.40 The active space was constructed from 11 electrons in 8 orbitals of oxazole and thiazole radicals and 9 electrons in 7 orbitals for imidazole radicals. The orbital symmetry and occupancies were obtained from a prior calculation at the ROHF41/cc-pVTZ level. In all the cases, 2 of the orbitals are virtual orbitals. In the case of oxazole and thiazole radicals, the eight active space orbitals are constructed from five orbitals with A′′ symmetry and three orbitals with A′ symmetry, whereas, five out of the seven active space orbitals of imidazole radicals have A′′ symmetry and two orbitals have A′ symmetry. On the other hand, two A1, two A2 and three B1 symmetry orbitals have been considered for the C2v symmetric nitrogen centred imidazole radical (π-radical). Molecular orbitals are plotted using the MOLDEN software package.42
2.2. Unimolecular rate calculation
The rate constant for each reaction at T = 298.15 K was calculated usingwhere kB is the Boltzmann constant, h is the Planck constant and R is the gas constant.43 The concentration term co is considered to be one in all the calculations. The ΔG‡ is the free energy difference between the transition state and the reactant in every reaction.44
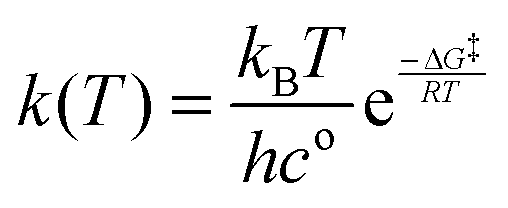
3. Results and discussion
Here, we mainly discuss the results of the (U)B3LYP/cc-pVTZ level of theory and the results from (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ have been included, wherever necessary. The results of the (U)M06-2X/cc-pVTZ level of theory almost follow the same trend as the (U)B3LYP/cc-pVTZ level (both are zero-point energy corrected values) and will be discussed only in the cases with larger deviation.453.1. Stability order
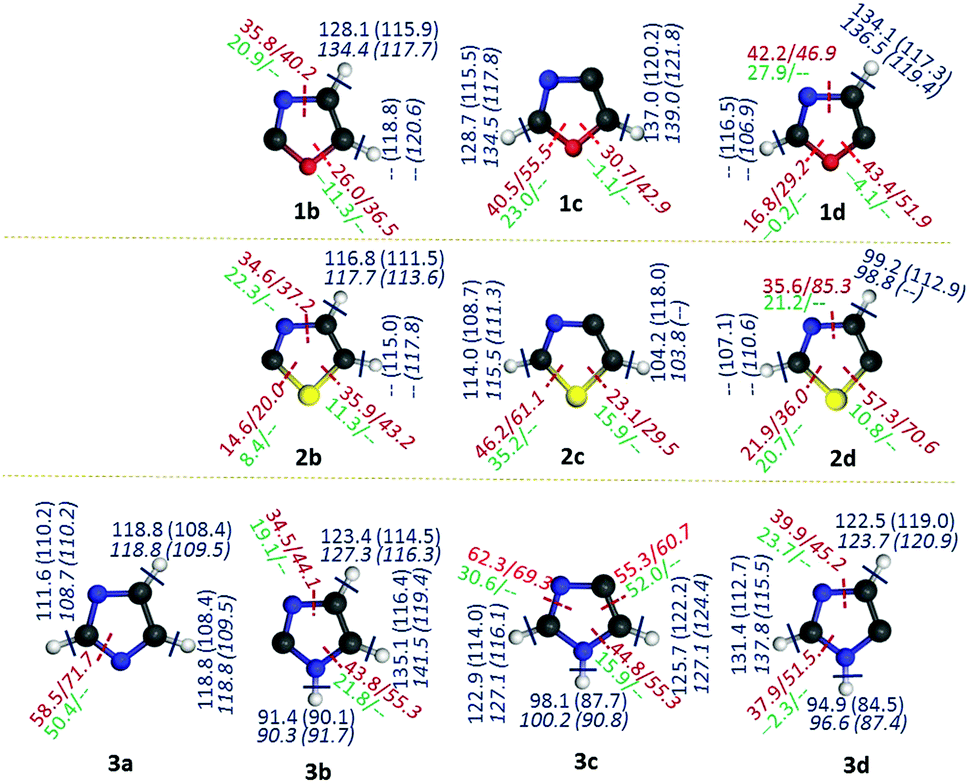
Three isomers each for both oxazole (1b–d) and thiazole (2b–d) radicals and four isomers of imidazole radicals (3a–d) were optimized at the (U)B3LYP/cc-pVTZ level and are shown in Fig. 1.46 The relative energies of three isomers using different levels of theories are listed in Table 1. The isomers 1c, 2b and 3a are found to be the most stable isomers on the respective potential energy surface (PES) for the oxazole, thiazole and imidazole radicals. The isomers 1b and 1c of the oxazole radicals lie energetically very close to each other, which varies with different levels of theory. At (U)B3LYP/cc-pVTZ the difference is 0.5 kcal mol−1, which lowers to a value of 0.1 kcal mol−1 at the (U)M06-2X/cc-pVTZ level, however the order changed and favours 1b as the minimum energy isomer when a single point energy calculation has been performed at the (U)CCSD(T)/cc-pVTZ level. In contrast, the imidazole radicals 3b and 3c are very close in energy in all the levels of theory and showed a slight preference for the 3b isomer. Unlike oxazole and imidazole radicals, there is no discrepancy in the order for thiazole radicals at all the levels of theory. The relative stability order of the corresponding thiazole radicals has been found to be 2b > 2c > 2d. Among the imidazole radicals, the nitrogen centred radical was found to be 20.1 kcal mol−1 more stable compared with the isomeric carbon centred radicals.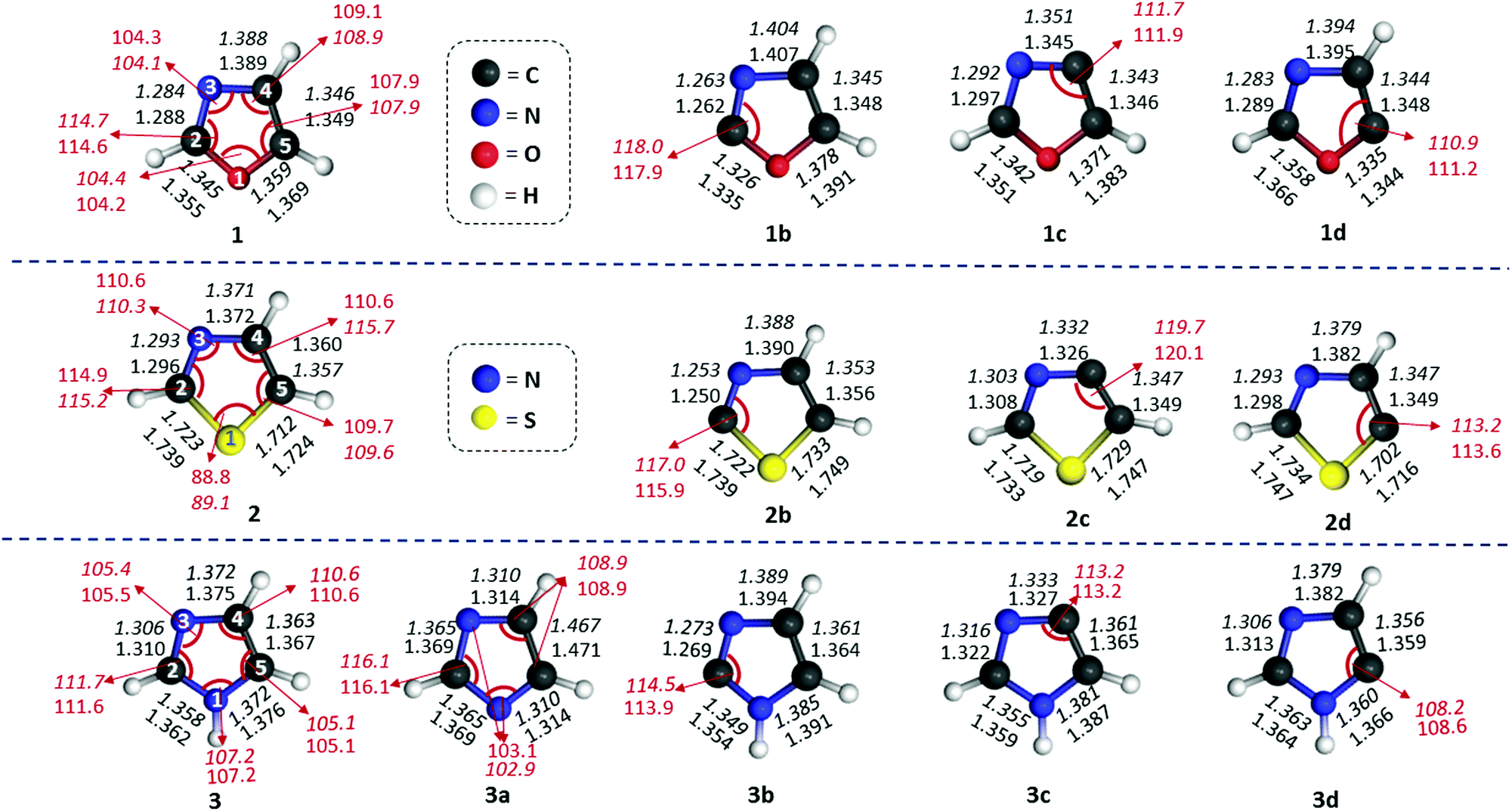 | ||
| Fig. 1 Optimized structures of oxazole, thiazole and imidazole, and their isomeric radicals. (All the radicals shown correspond to their respective ground electronic states. Selected geometrical parameters, namely, bond distances in Å and bond angles in degrees are included. The values indicated in normal font are derived from (U)B3LYP/cc-pVTZ, whereas the values mentioned in italics are derived from the (U)M06-2X/cc-pVTZ level of theory.) | ||
| Level of theory | Relative energy (kcal mol−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dehydrooxazole radicals | Dehydrothiazole radicals | Dehydroimidazole radicals | ||||||||
| 1b | 1c | 1d | 2b | 2c | 2d | 3a | 3b | 3c | 3d | |
| (U)B3LYP/6311++G(d,p) | 0.7 | 0 | 2.9 | 0 | 3.3 | 7.8 | 0 | 20.5 | 21.0 | 24.1 |
| (U)B3LYP/cc-pVTZ | 0.5 | 0 | 2.9 | 0 | 2.9 | 7.9 | 0 | 20.1 | 20.7 | 23.8 |
| (U)B3LYP/aug-cc-pVTZ | 0.5 | 0 | 2.9 | 0 | 2.8 | 7.8 | 0 | 20.2 | 20.7 | 23.9 |
| (U)M06-2X/cc-pVTZ | 0.1 | 0 | 2.5 | 0 | 2.3 | 7.3 | 0 | 18.4 | 18.7 | 22.1 |
| CCSD(T)/cc-pVTZ//(U)B3LYP/cc-pVTZ | 0 | 0.2 | 2.3 | 0 | 3.4 | 7.1 | 0 | 21.5 | 22.2 | 25.0 |
| CCSD(T)/cc-pVTZ//(U)M06-2X/cc-pVTZ | 0 | 0.1 | 2.3 | 0 | 3.4 | 7.2 | 0 | 21.4 | 22.0 | 24.9 |
3.2. Isodesmic reactions
To further understand the stability order and to estimate the radical stabilisation energy (RSE), we have performed a hypothetical isodesmic reaction of the radicals with benzene at 298.15 K.47 In this reaction, the radical centre will be transferred from the heterocyclic radicals to the benzene such that the parent heterocycle and phenyl radical will form in all the cases. The accompanying enthalpy change (ΔH298) in these reactions will provide the RSE. The isodesmic reactions are performed at (U)B3LYP/cc-pVTZ and (U)M06-2X/cc-pVTZ level of theories and the results are presented in Table 2. In general, the computed ΔH298 with more positive or less negative values indicate that the corresponding radical will be more stable (vs. phenyl radical), whereas those with more negative ΔH values will be less stable and are expected to be highly reactive. Based on these isodesmic reactions, we found out that the radical stabilization energies of all the heterocyclic radicals are in good agreement with the relative stability order obtained by comparing the absolute energies of the radicals. Upon comparison of the RSEs, we observed that the N-centred imidazole radical (3a) is found to be the most stable, followed by 2-dehydrothiazole (2b). All the heterocyclic radicals with a radical centre close to O, S and NH (1d, 2d & 3d) are found to be the least stable radicals. As mentioned earlier in the relative stability order, 1b and 1c are found to be close in energy and reflect the same trend. Interestingly, the stability order and RSEs are in good agreement with the theoretically calculated BDEs reported by Feng et al.23b|
|
||||
|---|---|---|---|---|
| Radical species | Absolute enthalpya,b | Enthalpy of parent moleculea,b | ΔH298a,c | |
| a All the values indicated are calculated from (U)B3LYP/cc-pVTZ and those mentioned in parentheses are calculated at (U)M06-2X/cc-pVTZ. (The corresponding energy valuesb for benzene = −232.22766 (−232.11593) and phenyl radical = −231.55157 (−231.44307).) b All the energy values are indicated in Hartrees. c ΔH is calculated by taking the difference between the sum of enthalpy of the products and the reactants and are expressed in kcal mol−1. | ||||
| Oxazole | 1b | −245.41175 (−245.32030) | −246.09914 (−246.00346) | −6.9 (−6.3) |
| 1c | −245.41260 (−245.32048) | −5.9 (−6.1) | ||
| 1d | −245.40800 (−245.31659) | −9.1 (−8.6) | ||
| Thiazole | 2b | −568.41163 (−568.31007) | −569.08615 (−568.98265) | 1.7 (0.4) |
| 2c | −568.40711 (−568.30643) | −1.3 (−1.9) | ||
| 2d | −568.39903 (−568.29848) | −6.2 (−6.7) | ||
| Imidazole | 3a | −225.57475 (−225.48145) | −226.22558 (−226.13208) | 14.3 (13.1) |
| 3b | −225.54255 (−225.45206) | −4.0 (−4.3) | ||
| 3c | −225.54177 (−225.45165) | −4.2 (−4.5) | ||
| 3d | −225.53669 (−225.44617) | −7.6 (−7.9) | ||

3.3. Geometry
To gain further insights into the stability and structural aspects, a closer look at the geometries is inevitable. In this regard, we compared the geometrical parameters of the optimized structures of isomeric oxazole, thiazole and imidazole radicals with their respective parent heterocycles (Fig. 1). A closer inspection at the most important parameters such as interatomic distances and the bond angles at the radical centres provided the following generalizations: (a) generation of a carbon centred radical leads to shortening of at least one of the immediate bonds. The second bond length either decreases or remains almost the same. (b) If the neighbouring position to the radical centre has a heteroatom, the corresponding bond gets shortened. If the radical centre is located between two heteroatoms, both the bond distances get shortened, except in the case of sulphur. (c) In all the radicals, the alternate bond distances from either side of the radical centre increases to a larger extent. (d) The bond distance between the atoms opposite to the radical centres remains almost the same or slightly decreases. (e) In the nitrogen centred imidazole radical 3a, there is an interesting situation, where two C–N bond lengths (C2–N1 and C2–N3) increase and two C–N bond distances (C4–N3 and C5–N1) decrease. (f) Unlike all the carbon-centred radicals, where the bond angles at the radical centres always increase the nitrogen-centred radical shows a decrease. Furthermore, the carbon flanked between the two nitrogen atoms in 3a increases its bond angle to 116.1° and decreases at another two carbons to an angle of 108.9°. With respect to bond angle variation at the radical centres, 2c showed a maximum value of 120.1°.Based on these understandings, we infer that the creation of a radical centre significantly imparts changes to the overall geometry of all the five-membered heterocycles mentioned so far.48 Furthermore, we explored the changes in hybridization at all the bonds through NBO analysis for the radicals in comparison with their respective parent compound (Table 2S in the ESI†). Only in the case of the nitrogen-centred radical 3a, the hybridization at both N–C bonds with a common carbon decrease in their π-character up to a maximum of 28%. In all other carbon-centred radicals, the π-character of the hybridized bonds at either side of the C-centres increase. However, if the radical centre is close to oxygen or sulphur (as in 1b, 2b, 1c and 2c), the corresponding π-character of the hybridized bonds also decrease. The trend indicates that the bond connecting radical centres and the oxygen or sulphur atoms attain an sp3 character, which may be due to the influence of the two lone pairs in those two atoms. Since non-bonding electrons of nitrogen exist as a σ-lone pair, their interaction with the σ-radical electron led to a change in hybridization towards an sp character. This might be the reason for the contraction of bond lengths between the C-centred radical and the adjacent nitrogen atom. However, if the radical centre is away from the nitrogen, the radical electron tends to interact with O-1 and S-1 rather than the N3 through space interaction. This can be envisaged by the change in hybridization in those corresponding bonds, which increases in p-character by 7% and 12% in 1d and 2d, respectively. On the other hand, there is a 39% decrease in p-character for 3d, due to the lack of lone pairs. The NBO analysis of the second order perturbation energies between the radical centre and the heteroatoms has been included (Table 3S in the ESI†). The results suggest that the lone pair of the nitrogen atom strongly interacts with the radical electron through space, whereas the other heteroatoms either show a weaker TS interaction or no interaction at all. However, the lone pairs of NH, O and S atoms show a stronger through bond interactions with the anti-bonding orbitals of the alternate bonds in either direction.
3.4. Spin density and electrostatic potential
Spin densities and electrostatic potentials of all the 10 radicals have been calculated at the (U)B3LYP/cc-pVTZ and (U)M06-2X/cc-pVTZ levels of theory. The spin density values are listed in Fig. 2. The spin density values exhibit the extent of delocalization of the spin centres. In the nitrogen-centred radical 3a, the spin densities are negative at both the nitrogen centres, whereas all its other carbons have positive values. This clearly indicates the delocalization of spin towards the carbon centres. This also explains the drastic changes in the geometrical parameters in 3a compared to the parent compound. On the other hand, the majority of the carbon-centred radicals have spin density values close to 1, which indicates that the unpaired electron is localized on that atom. The spin densities of all the carbon-centred radicals are localized, and the values also reflect the stability order except in the case of oxazole radicals. Similarly the electrostatic potentials have also been derived for all the radicals (Fig. 3S in the ESI†). They showed an appearance of a weak negative potential close to the radical centres. Interestingly, the nitrogen-centred radical of imidazole showed strong negative potentials at both nitrogen atoms. This clearly indicates that the nitrogen centres have sigma lone pairs, which further envisage the delocalization of a radical electron towards the carbon atoms. Except in the case of sulphur, a weak negative potential in all the radicals combines with the strong neighbouring negative potential of the heteroatoms.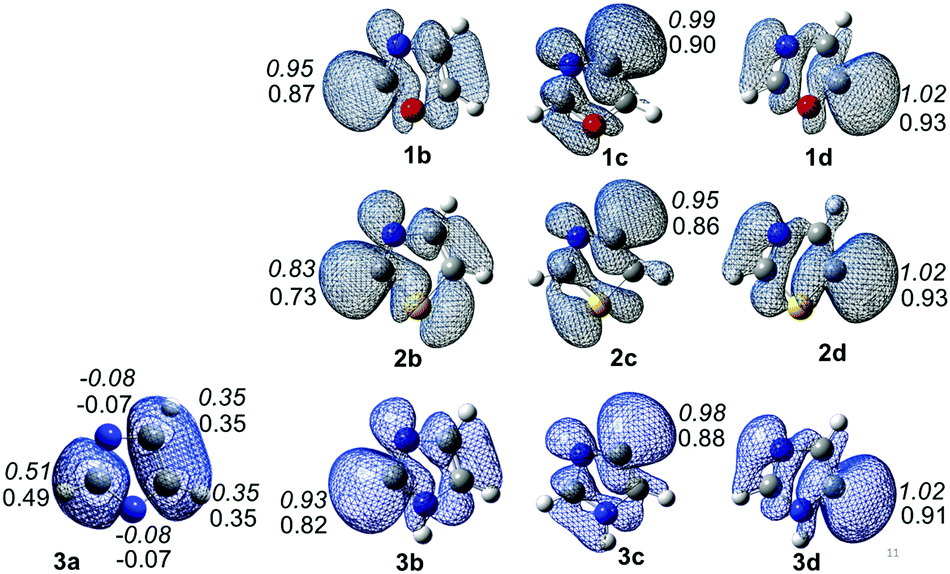 | ||
| Fig. 2 Spin densities of dehydro-oxazole, thiazole and imidazole radicals. (Each radical showing only positive spin densities are rendered at an isovalue of 0.002. The positive spin density values next to the radical centres are indicated. Normal font: (U)B3LYP/cc-pVTZ; italics: (U)M06-2X/cc-pVTZ. In the case of 3a, the nitrogen centres show a negative spin density, whereas the carbon centres show positive spin density values.) | ||
3.5. Multireference calculations
The geometrical parameters, NBO analysis and electrostatic potentials showed weak to strong interactions between the radical centre and the electron lone pairs in all the radicals. In order to understand further, we performed single point energy calculations at the multireference electronic structure mode using CASSCF. In these calculations, the five aromatic π-orbitals, σ-radical centred orbital and the orbitals corresponding to the non-bonded electrons of the heteroatoms have been chosen in the active space. The six electrons corresponding to the aromatic sextet, radical electron and the lone pairs corresponding to the heteroatoms are considered in this regard (Fig. 3). Except in the case of the nitrogen-centred radical, all the carbon-centred radicals showed a singly occupied molecular orbital (SOMO) corresponding to σ-radicals. In the former case, it was found to be an aromatic π-orbital, which had an occupancy close to 1, an indication of strong delocalization of the radical electron. In all the cases, the orbitals corresponding to the lone pair are more stabilized and are lying well below the filled aromatic π-orbitals. Interestingly, all the carbon-centred radicals acquire a 2A′ ground state electronic structure. In order to understand the interaction between the radical electron and the lone pair from the heteroatoms, we inspected the singly occupied molecular orbital (SOMO) of all the radical species at different isovalues (Fig. 5S in the ESI†). Through this, we found out that in all the C-centred radicals there was a very weak anti-bonding interaction between the radical centre and the lone pair of the electrons irrespective of their position.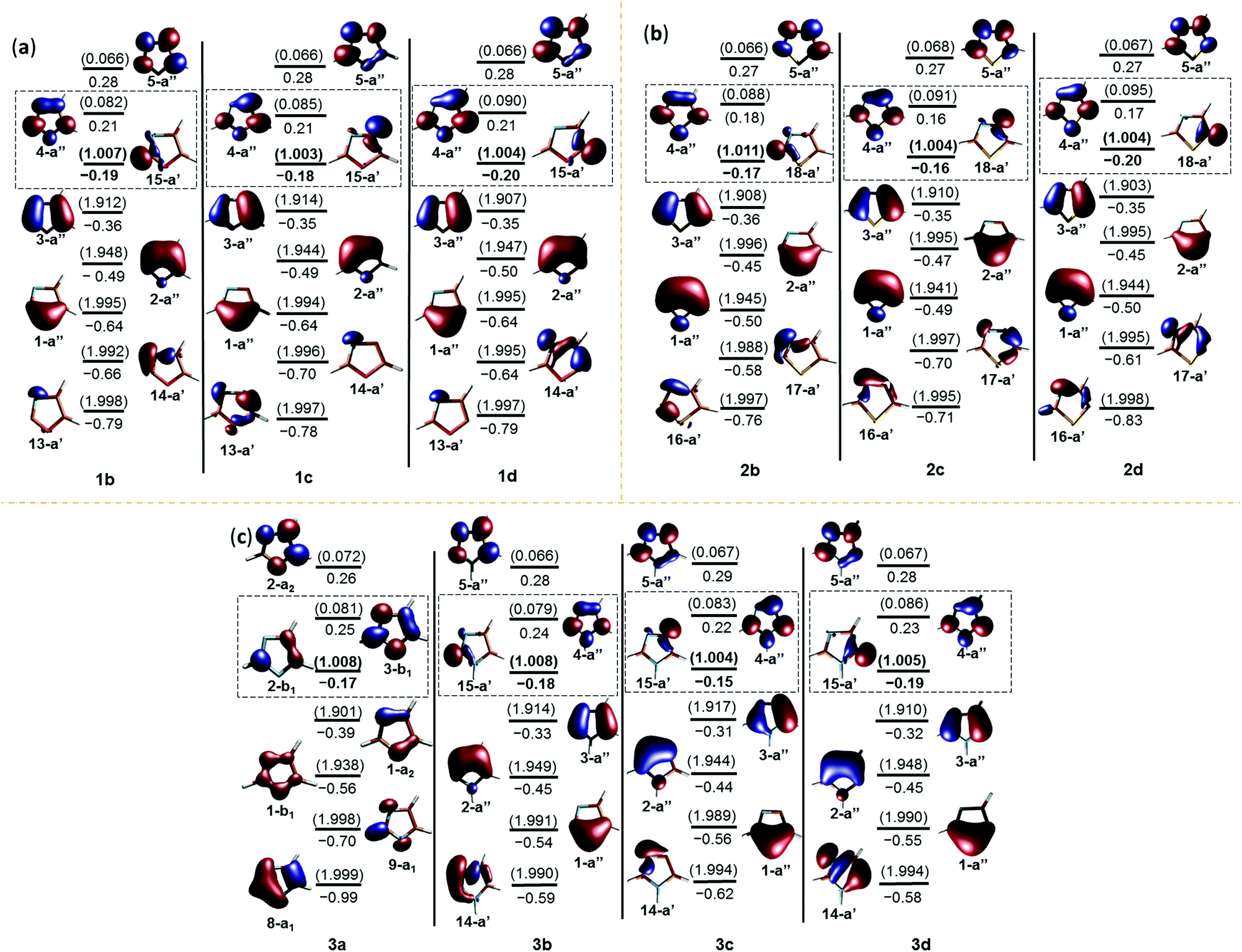 | ||
| Fig. 3 CASSCF/cc-pVTZ//(U)B3LYP/cc-pVTZ orbitals of the dehydro- (a) oxazole, (b) thiazole and (c) imidazole radicals (the active space orbitals are indicated along with their energies in Hartrees and natural orbital occupancy numbers (NOON) in parentheses; their respective orbital symmetries are indicated below. The singly occupied molecular orbital (SOMO) and the lowest unoccupied molecular orbital (LUMO) have been highlighted by a box. The parameters corresponding to the SOMO are indicated in bold. All the orbitals have been rendered at an isovalue of 0.05). | ||
In contrast, the nitrogen-centred radical 3a possesses a 2B1 ground state with C2v geometry. Since all the C-centred radicals have Cs symmetry, we tried to optimize 3a at Cs symmetry as well.49 Both Cs and C2v symmetric structures were found to be almost degenerate.50 In both the structures, the orbitals corresponding to the nitrogen centres are doubly occupied and low-lying, due to a through space three-centred interaction, where the common carbon is also participating in the interactions. Hence, one of the aromatic π-orbital turns out to be the SOMO that accounted for the enhanced stability and negative spin density at the nitrogen centres in the C2v and Cs structures. This unique behaviour of 3a is comparable to the 3-centred 5-electron boryl radicals, such as the borole radical, 1,3,2-diazaborole radical and 2,5-dihydro-1,3,2-diazaborinine radical, which were reported as π-radicals instead of the expected σ-radicals.51 The striking aspect in both the cases is the flipping of electrons from the orthogonal σ-orbital (A′ structure) to the π-orbital (A′′ structure), through which they attain delocalization. However, the major difference is the interaction between the σ-radical electron in both cases with the fully filled nitrogen π-orbital in 3a and empty boron π-orbital in the boryl radicals, respectively. An alternative way of inspecting the electronic structure of 3a is to relate it to a cyclopentadienyl (C5H5, Cp) radical, where homolysis of the formal C–H σ-bond of cyclopentadiene leads to a π-radical. The resulting radical can attain either a highly symmetric D5h or C2v structure, which makes this molecule a target for experimental and theoretical investigations.52 Although ESR spectroscopic studies confirmed the highly symmetric structure, below certain temperatures (transition temperatures) anisotropic behaviour with non-uniform spin distributions was observed that was believed to be due to pseudo rotations arising from Jahn–Teller distortions. However, recent vibrational spectroscopic studies on the Cp radical in a helium nanodroplet experiment ruled out this possibility and favoured the highly symmetric structure.53 The interaction with the host accounted for the anisotropic behaviour at low temperatures. In this regard, both boryl radicals and cyclopentadienyl radicals support the possibility of a spin flip from σ- to π-orbitals, which is the case in 3a. To emulate this flipping and to understand the delocalization of spin, a model has been proposed in Fig. 4, which is analogous to the borole radical reported by Lu et al.51 Through this, we can explain the accumulation of spin densities at the three carbon centres instead of nitrogen.
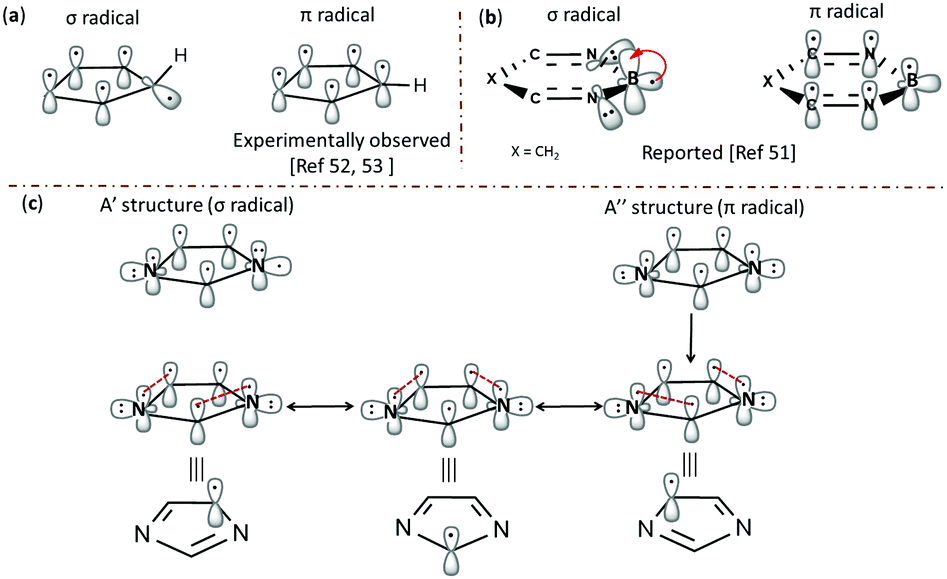 | ||
| Fig. 4 Delocalization of spin in (a) C5H5 radical, (b) 2,5-dihydro-1,3,2-diazaborinine radical and (c) the 1-dehydroimidazole radical 3a. | ||
3.6. Reactivity studies
The reactivity studies of dehydro-oxazole, thiazole and imidazole radicals are very important in understanding the complex reaction channels and the ring opening products. Equally, this will be helpful in estimating the low energy pathways and in comparison of their kinetic stability. In this regard, we considered four different approaches, namely, isomerization of the radicals through a 1,2-H shift, unimolecular decomposition to ring opening products, C–H bond cleavage to form biradicals followed by ring opening and concerted ring opening channels. Although the first two approaches were successful, the remaining approaches did not lead to the desired results. Indeed earlier studies of unimolecular decomposition on pyridine radicals also discussed the failure in capturing the transition states for such C–H bond cleavage reactions.21,54 In the same line, we were not able to obtain the transition states corresponding to the concerted ring opening pathways. A few transition state structures corresponding to concerted pathways have been optimized at the HF level, which were found to be either unstable at DFT or leading to a transition state that resembles stepwise cleavage. The rate constants for all the channels have also been calculated.55 The computational results on the unimolecular decomposition channels and most important properties of all the reactants, intermediates, products and transition states are tabulated. (Table 7S comprising the results from the (U)B3LYP/cc-pVTZ level and Table 8S comprising the results from the (U)M06-2X/cc-pVTZ level of theory in the ESI†). However, only the energy values from the (U)B3LYP/cc-pVTZ level and the single point energies calculated based on these geometries at the (U)CCSDT/cc-pVTZ level of theory are indicated in all the figures related to the unimolecular dissociation channels. Also, the energy barriers for each TS relative to the immediate reactants are described in the text, whereas the relative energy with respect to the parent radical is reported in all the figures.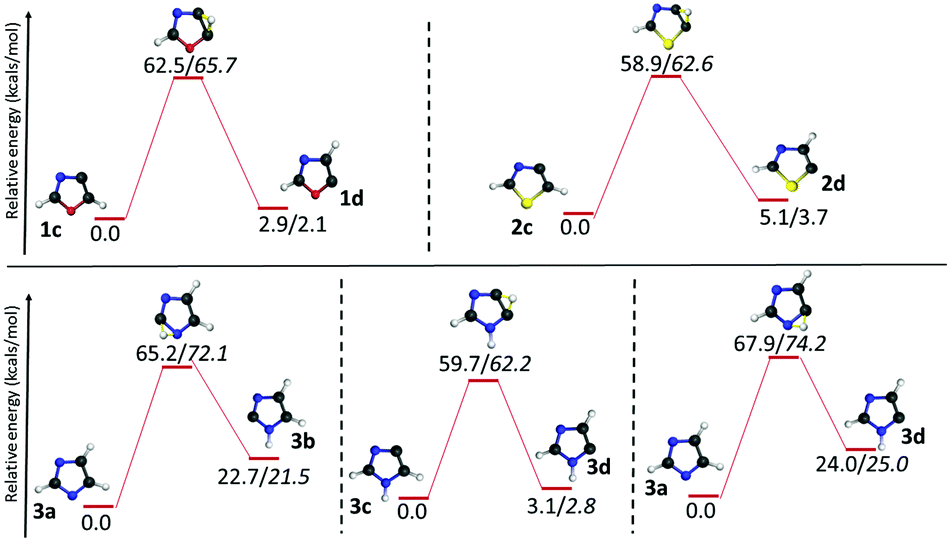 | ||
| Fig. 5 Isomerization channels of dehydro-oxazole, thiazole and imidazole radicals through 1,2-H shift (normal font = (U)B3LYP/cc-pVTZ; Italics = (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ). | ||
3.7. Unimolecular decomposition pathways
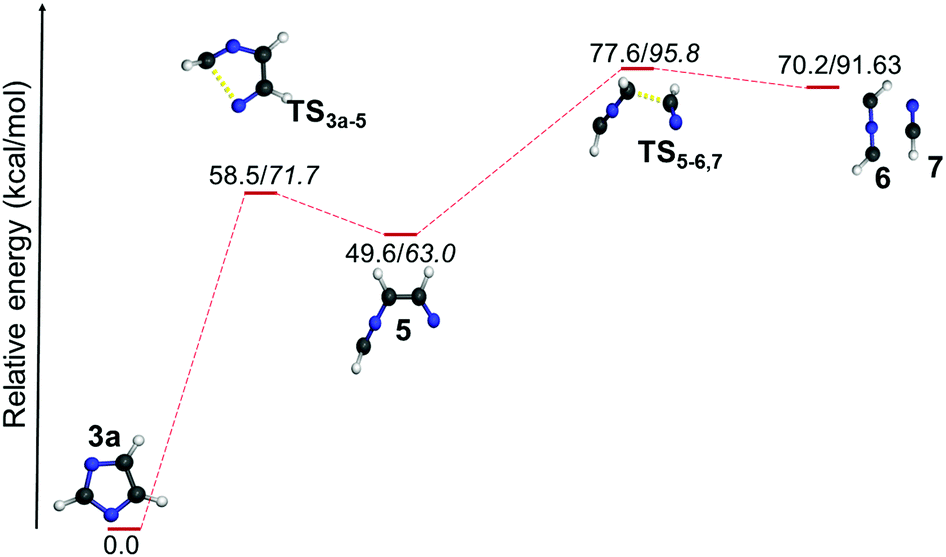 | ||
| Fig. 6 Ring-opening pathway of the N-dehydroimidazole radical 3a (normal font = (U)B3LYP/cc-pVTZ; italics = (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ). | ||
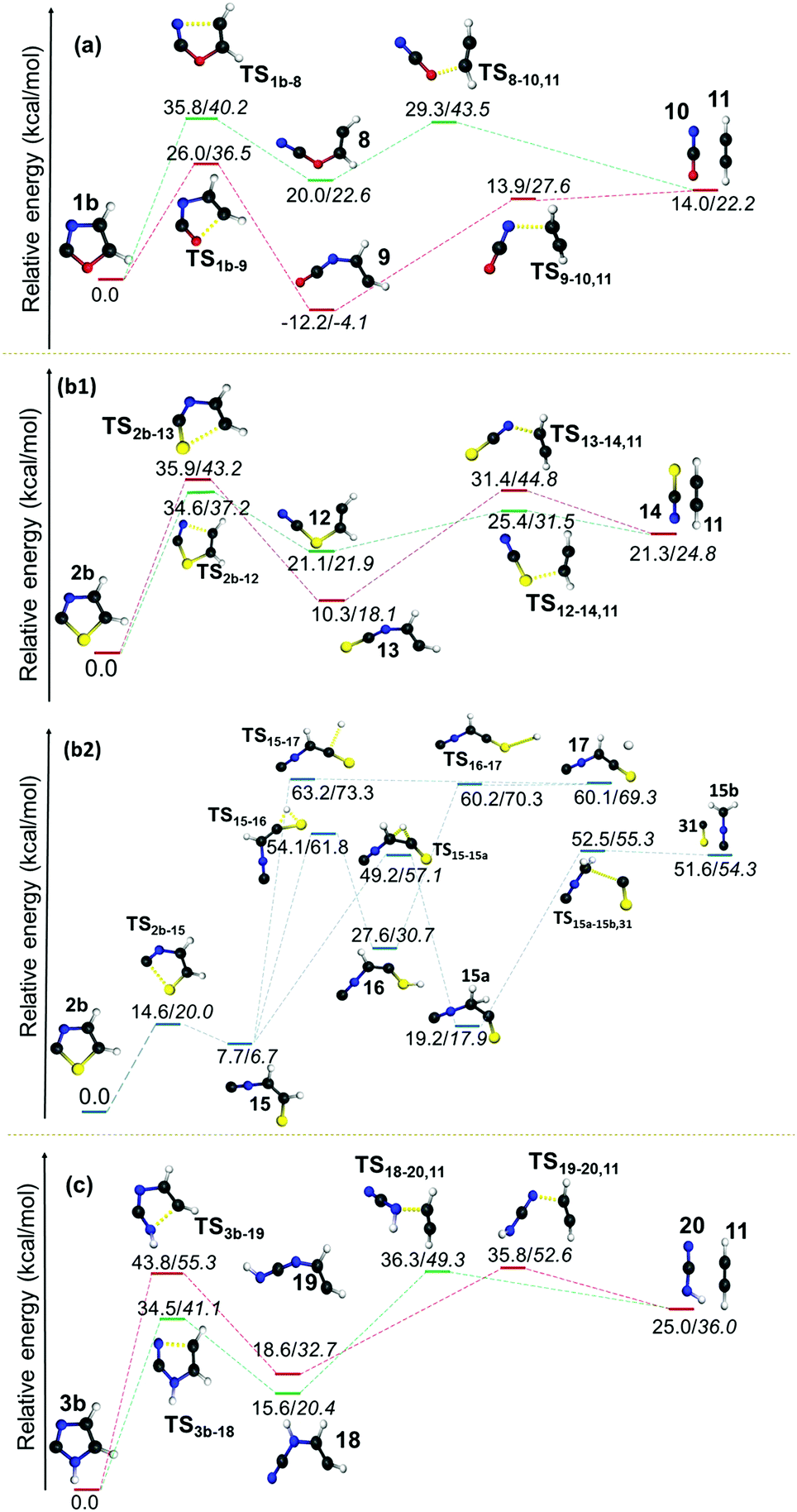 | ||
| Fig. 7 Unimolecular decomposition pathways of 1b, 2b and 3b (normal font = (U)B3LYP/cc-pVTZ; italics = (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ). For clarity, the unimolecular decomposition channels in 2b are split into two separate parts b1 and b2. | ||
However, the resulting product 15 was found to be more stable compared to the products from other channels, and it bifurcated into three further decomposition channels with very high barriers at 46.4, 55.5 and 41.5 kcal mol−1, respectively. All three channels involved a H-atom shift within the intermediate 15. Among these the lowest barrier (41.5 kcal mol−1) channel is 1,2-H shift from C5 to C4 in 15, which leads to the formation of intermediate 15a. The intermediate 15a further decomposes into an isocyanomethyl radical and CS, which is located 51.6 kcal mol−1 higher in energy, in comparison with the precursor 2b radical as obtained at the (U)B3LYP/cc-pVTZ level. Except in the case of the above mentioned channel in the thiazole radical (2b to 15), the resulting ring opened radicals from all three isomeric radicals generally led to the dissociative end products in the second and final steps. One of the common final products in all the cases is acetylene, whereas the other product is found to be the OCN radical5810 in the case of 1b, the NCS radical59 in 2b and the HNCN60 radical in 3b. The barriers for acetylene formation are between 4.3 and 25.6 kcal mol−1, which makes this reaction kinetically as well as entropically more favourable. However, in all these cases, the resulting end products are thermodynamically destabilized compared to the ring-opened analogues of the heterocyclic radicals. The barrier was the least in the case of thiazole (12 to 11/14). On the other hand, the corresponding ring-opening channel in the oxazole radical 1b led to the thermodynamically stable radical 9 that may dissociate further to 10 and 11. At (U)B3LYP/cc-pVTZ, we have found out that the TS is 0.1 kcal mol−1 lower in energy compared to the products. However, the energy of the products drops to 5.4 kcal mol−1 below the TS, when the same channel has been calculated at the (U)CCSDT/cc-pVTZ level of theory.
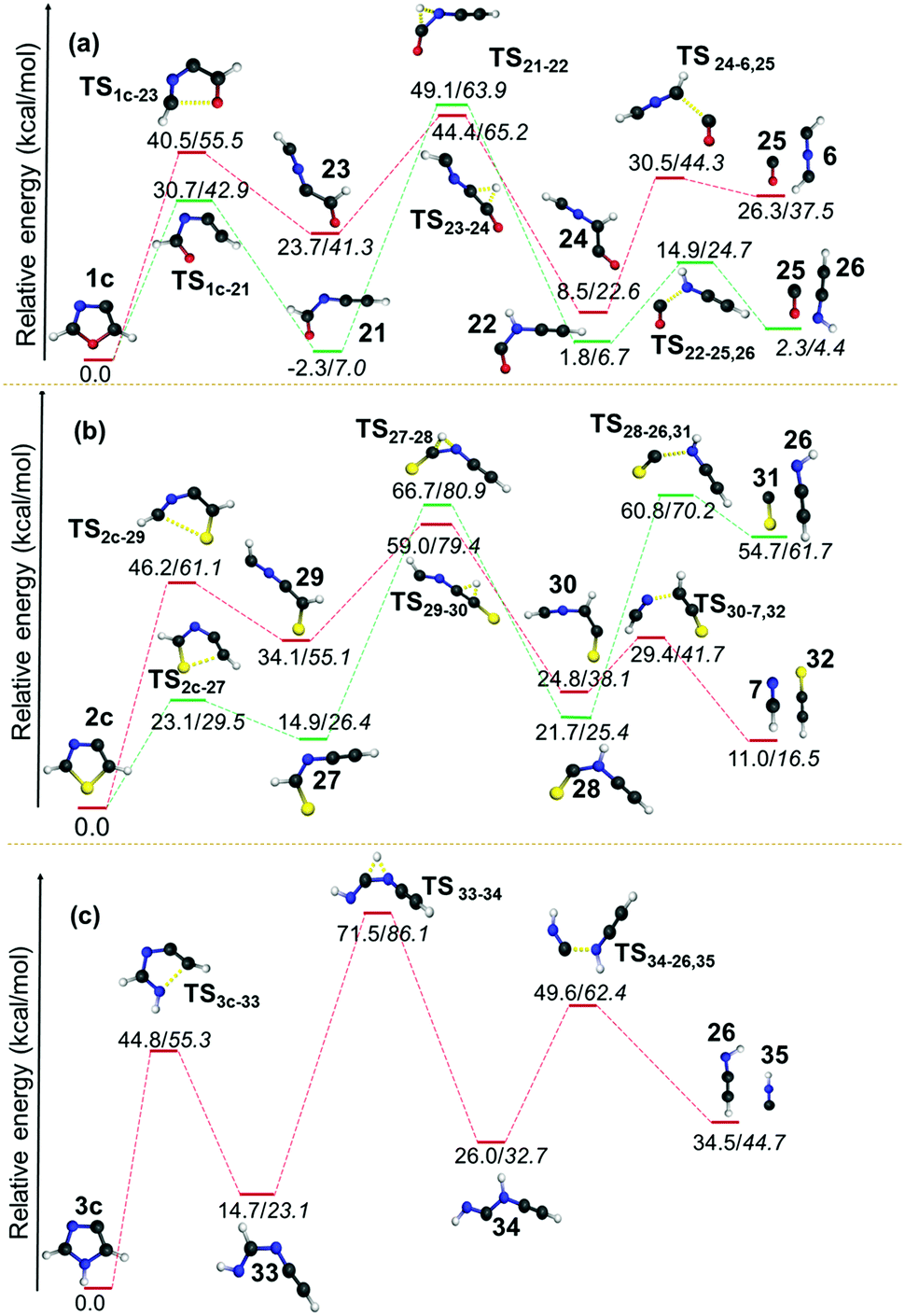 | ||
| Fig. 8 Unimolecular decomposition pathways of 1c, 2c and 3c (normal font = (U)B3LYP/cc-pVTZ; italics = (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ). | ||
The same barrier was found to be lower and comparable for the second pathway i.e. for the O–C2 and S–C2 bond cleavage pathways with values 25.4 and 24.9 kcal mol−1, respectively. In both the cases, early TSs are observed, which indicates that the reactions are found to be thermodynamically favourable. However, the isomerization of kinetically favoured products from the first step involves a TS corresponding to hydrogen shifts that are almost symmetrical. The fact that both the reactants and products are of comparable energy means that the barriers lie in the higher side for those two channels (21 to 2257 and 27 to 28) for 1c and 2c. After the isomerization step, the dissociation of neutral molecules such as CO, HCN and HNC are favoured end products along with HCNCH,56 HCCNH56 and HCCS62 in all these channels. Since this step is entropically favourable, the barriers are also found to be in the lower side, which range between 4.6 and 23.6 kcal mol−1. The dissociation of CS is estimated to have a barrier of 39.1 kcal mol−1 in the case of 2c.
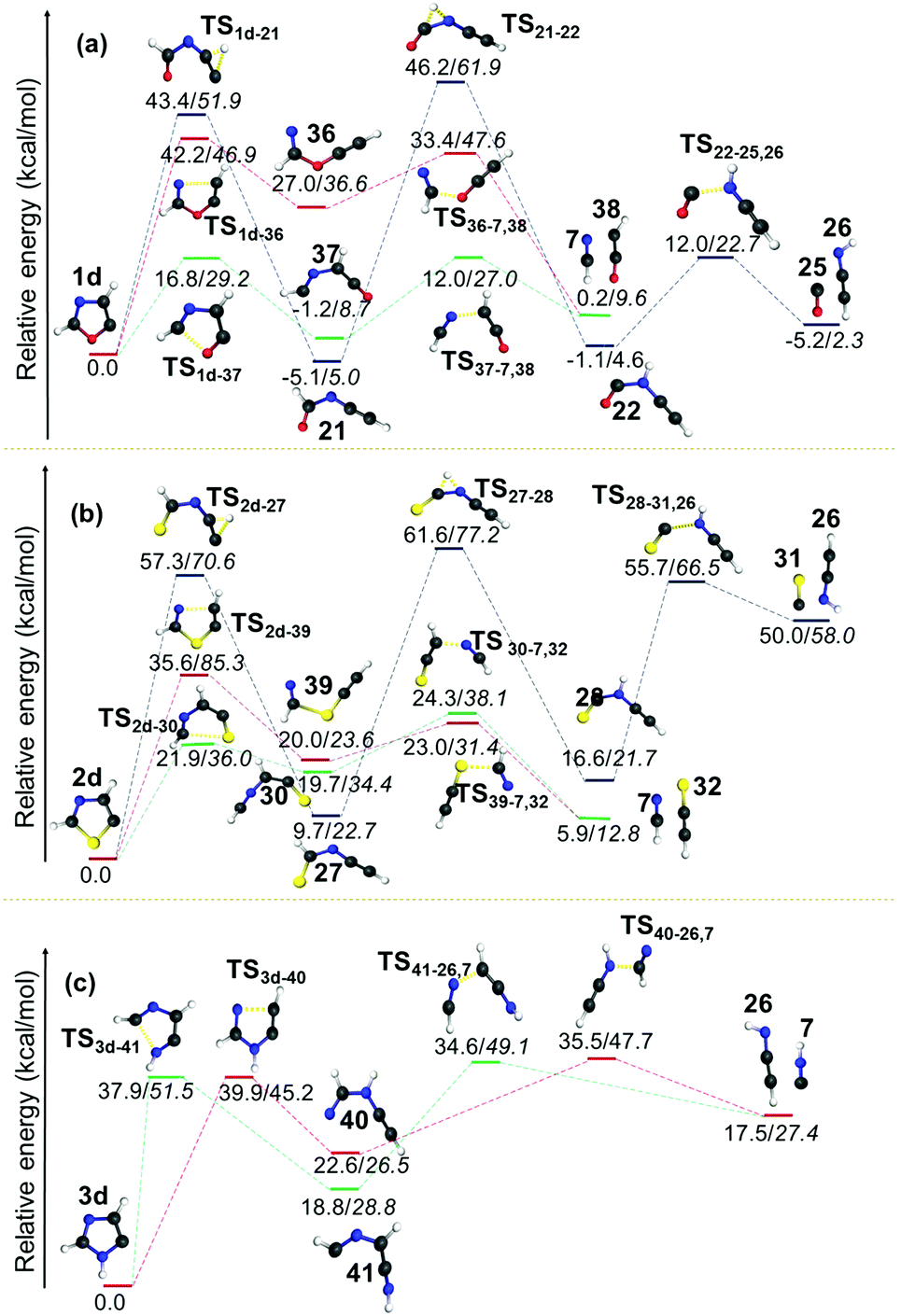 | ||
| Fig. 9 Unimolecular decomposition pathways of 1d, 2d and 3d (normal font = (U)B3LYP/cc-pVTZ; italics = (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ). | ||
In all the pathways the ring-opening products were destabilized with respect to their parent radical. An additional pathway was also calculated in the case of both 1d and 2d radicals, which involves the cleavage of the C5–X bond. These pathways were found to be higher in energy as compared to the C2–X and C4–N bond cleavage pathways, and this can be attributed to the reduction in C5–X bond lengths in the radicals with respect to their parent heterocycles. The most striking aspect in this channel is the formation of the thermodynamically more stable ring-opening products 2157 and 27 from 1d and 2d, respectively. Indeed, the product 21 is found to be −5.1 kcal mol−1 more stable compared to the parent radical 1d. In both cases, this ring-opening product is further progressed by an isomerisation step through a 1,2-hydrogen shift. Both the transition states corresponding to this isomerisation in 1d and 2d are nearly symmetrical with respect to a hydrogen shift with larger energy barriers of 51.3 and 51.9 kcal mol−1, respectively. The first intermediate 27 formed in the case of 2d was also found to be relatively stable compared to the isomers 22 (from 21) and 28 (from 27) that are destabilized with respect to their starting material by 4.0 and 6.9 kcal mol−1, respectively. The resultant products from C5–O/S scission undergo a hydrogen shift to form ring-opened products from another two channels. Furthermore, in all three cases, the HCCNH56 radical 26 and HCN 7 were obtained as two common end products. On the other hand, the C2–X bond breaking pathway led to the formation of HCN as the end product along with HCCO,61 HCCS62 and HCCNH56 radicals in 1d, 2d and 3d, respectively.
In general, energetically favourable dissociation occurs at the bond, which is alternate to the radical centre. Exceptions can be observed when sulphur or N–H is at the alternate position from the radical centre. Here, C–S cleavage is more facile, whereas C–NH cleavage is energetically less probable. In all the thiazole radicals, the C–S bond scission can happen at both sides, however, either cleavage between the carbon closest to the radical centre or the radical carbon itself will be the most favourable channel. The C2–X bond weakens when a radical centre is created at the C5 position. As a result, the ring opening at C2–X is more favourable. These clearly indicate that a destabilizing interaction between the radical centre and the S, O and N–H atoms (at 1-position) leads to weak coupling in the order S < O < N–H. More destabilising effects of sulphur and oxygen can be attributed to the presence of two lone pairs, which may not lie in the same direction as the radical electron. On the other hand, the N3 exhibits a reasonably stronger stabilizing interaction with the radical centre either TS or TB. Due to delocalization, the C–N bond cleavage is the only pathway in the case of the N-centred imidazole radical 3a, which is the most stable species among the radicals investigated in this contribution.
In summary, the kinetic stability of all the isomeric oxazole, thiazole and imidazole radicals have been estimated using unimolecular ring opening followed by fragmentation channels and isomerisation pathways through 1,2-H shifts. In majority of the ring opening, the first step has been found to be the rate-determining step. The second C–H and N–H bond dissociation of the radicals leading to the formation of a biradical has also been calculated.54 However, the barriers for the concerted ring opening channels could not be estimated. The most important results comprising the barriers and enthalpy changes corresponding to the first ring opening step and the BDEs with respect to the C–H and N–H bonds are included in Fig. 10. Based on the various possible channels, we found that all the ring opening channels are lower in energy compared to the C–H and N–H bond dissociation energies. Hence, the barriers for the ring opening channels are important for estimating the kinetic stability of all the heterocyclic radicals discussed thus far. The barriers also suggest that the thiazole and oxazole radicals are more susceptible to ring opening rather than the imidazole radicals. The weakening of the C–O and C–S bonds in the radicals results in the lower barrier and in turn lowers the kinetic stability of those radicals.
 | ||
| Fig. 10 Summary of the unimolecular ring-opening pathways (energy values indicated in kcal mol−1; (normal font = (U)B3LYP/cc-pVTZ; italics = (U)CCSDT/cc-pVTZ//(U)B3LYP/cc-pVTZ)). The values in red colour indicate the barriers corresponding to the first step of the unimolecular decomposition pathways, relative to their corresponding parent radical. The corresponding enthalpy changes in the (U)B3LYP/cc-pVTZ level are indicated in green. The second bond dissociation energy54 (BDE) values for all possible biradicals are indicated in a blue colour ((U)B3LYP/cc-pVTZ – normal font and (U)M06-2X/cc-pVTZ – Italics). Of the two values mentioned as the second BDE, the first one corresponds to the singlet biradical formation, whereas the values indicated in the parentheses correspond to the triplet biradical formation. | ||
In the case of all the isomeric oxazole radicals, ring opening through C–O bond cleavage is both kinetically as well as thermodynamically favoured in all the cases, due to the lower barrier and negative enthalpy changes. In spite of the positive enthalpy changes associated with the ring opening channels, thiazole radicals prefer C–S bond cleavage channels due to lower barriers. Similar to the other radicals, imidazole radicals also preferentially ring open through CH–N1H rather than CH–N3 bond cleavage. The only exception is 3b, where C4–N3 is getting cleaved. The stronger interaction between the radical centre and the N3, and the consequent weakening of the C4–N3 bond can potentially lead to such ring opening pathways. All the imidazole radicals other than 3d show ring-opening channels with positive enthalpy changes. All these clearly indicate that the radical centre exerts a stronger interaction with N3 than with N1–H/S/O in all the heterocyclic radicals.
4. Conclusions
Throughout this investigation, computational studies have been carried out on the electronic structure, stability and reactivity aspects of dehydro- oxazole, thiazole and imidazole radicals. The ground electronic structure of each radical has been confirmed by predicting their doublet excited state structure and also by estimating the adiabatic energy difference. The electronic structure calculations reveal that nitrogen-centred imidazole radical 3a was found to be the most stable radical due to attainment of its π-radical character and a consequent delocalization of the formal σ-radical electron into the π-system. On the other hand, the carbon centred radicals 1c and 2b are the minimal energy structures in their respective PES of dehydro- oxazole and thiazole radicals. Unlike 3a all the C-centred radicals are found to be highly localized σ-radicals.The relative position of the radical centre with respect to the nitrogen and other heteroatoms strongly influences the structural and stability aspects within the isomers. Among the radicals, the maximum structural alteration was observed for the N-dehydroimidazole radical 3a. In other carbon-centred radicals, the radicals flanked between nitrogen (N3) and heteroatoms (N1–H/O1/S1) and the radicals close to N3 were found to be closer in energy except in the case of thiazole. With the change in the level of theory, the order interchanged in the case of oxazole radicals. Among the carbon centred radicals those with a radical centre at C5 i.e. adjacent to X1 (N–H/O/S) were found to be the least stable isomers. Larger spin density values indicate a higher reactivity for most of the C-centred radicals. To understand further about the interactions between the heteroatoms and the radical centres, NBO analysis and multireference CASSCF calculations were performed. Overall, the results suggest a strong through space coupling between the radical centre and the N3 lone pair, when the radical centre is adjacent to it. The heteroatoms at 1-position, on the other hand, show a stronger through bond coupling with alternate bonds rather than a through space interaction with the radical centre. In none of the cases, is any clear evidence for the existence of or absence of cooperative interactions between the radical centre and both the heteroatoms observed. However, the direct interaction between N3 with the radical centre and the indirect stabilizing effects through interaction between the lone pair of electrons of X1 (NH/O/S) with the anti-bonding orbitals of the alternate bonds with respect to X1 are operating simultaneously. The non-participation of the lone pair of electrons of X1 towards the interaction with the radical centre can be attributed to the orientation of the lone pair with respect to the radical orbital.
For each radical, reactivity studies have also been done using unimolecular decomposition channels. The kinetically favourable ring opening channels are at the positions where the bonding is weaker compared to their parent compound. In particular, the alternate bonds from either side of the radical centres are weakened. This led to the possibility of more than one ring opening channel. In such situations, the interaction between the N3 and radical centre dictates the lower energy pathways. The ring opening led to several small molecules such as acetylene, HCN, HNC, CO, HCCO, HCCS, OCN radical, NCS radical and several isomeric fragment molecules of molecular formulae C2H2N, CHN2 and C3H2NO. The reaction rate constants for each step have also been calculated, which can be useful in understanding the major pathways. Experimental generation followed by reactivity studies and confirmation of these computational results on these heterocyclic radicals through matrix isolation infrared spectroscopy is currently underway.
Acknowledgements
We are grateful to the Science and Engineering Research Board (SERB), New Delhi, for the financial support (EMR/2014/000780). We are also thankful to IISER Mohali for the start-up grant and providing research facilities including computational facilities. AM thanks CSIR for the Senior Research Associateship (No. 13(8767-A)/2015-Pool). We thank Dr P. Balanarayan for his help in sharing the computational resources. The research data supporting this publication can be accessed at http://dx.doi.org/10.1039/c6cp05677f.References
- (a) M. Gomberg, Chem. Rev., 1924, 1, 91–141 Search PubMed; (b) R. A. Moss, in Reactive Intermediate Chemistry, ed. M. S. Platz and M. Jones, Wiley-Blackwell, USA, 2003 Search PubMed.
- G. Pratsch and M. R. Heindrich, in Radicals in synthesis III in Topics in current chemistry, ed. M. R. Heinrich and A. Gansäuer, Springer-Verlag Berlin, Heidelberg, 2012 Search PubMed.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed; (b) M. L. Coote, C. Y. Lin, A. L. J. Beckwitha and A. A. Zavitsasb, Phys. Chem. Chem. Phys., 2010, 12, 9597–9610 Search PubMed.
- (a) N. J. Labbe, R. Sivaramakrishnan, C. F. Goldsmith, Y. Georgievskii, J. A. Miller and S. J. Klippenstein, J. Phys. Chem. Lett., 2016, 7, 85–89 Search PubMed; (b) D. S. N. Parker, R. I. Kaiser, T. P. Troy, O. Kostko, M. Ahmed and A. M. Mebe, J. Phys. Chem. A, 2015, 119, 7145–7154 CrossRef CAS PubMed; (c) M. J. Fadden and C. M. Hadad, J. Phys. Chem. A, 2000, 104, 6324–6331 Search PubMed; (d) V. R. Morris, S. C. Bhatia, A. W. Stelson and J. H. Hall, Jr., Energy Fuels, 1991, 5, 126–133 Search PubMed.
- (a) J. Bejma, P. Ramires and L. L. Ji, Acta Physiol. Scand., 2000, 169, 343–351 Search PubMed; (b) S. B. Nimse and D. Pal, RSC Adv., 2015, 5, 27986–28006 Search PubMed; (c) A. T. Lam, E. P. Faragó, M. C. Owen, B. Fiser, B. Jójárt, S. J. K. Jensen, I. G. Csizmadiaab and B. Viskolcz, Phys. Chem. Chem. Phys., 2014, 16, 9602–9609 Search PubMed; (d) P. Cysewski, D. Bednarek and K. Kozłowska, Phys. Chem. Chem. Phys., 2003, 5, 4899–4904 Search PubMed.
- (a) B. D. Gervasoni, G. N. Khairallah, R. A. J. O’. Hair and U. Wille, Phys. Chem. Chem. Phys., 2015, 17, 9212–9221 Search PubMed; (b) G. Wang and J. Huang, Polym. Chem., 2014, 5, 277–308 Search PubMed.
- (a) J. S. Francisco, J. T. Muckerman and H.-G. Yu, Acc. Chem. Res., 2010, 43, 1519–1526 Search PubMed; (b) J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 Search PubMed.
- (a) Z.-M. Chen, X.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2015, 44, 5220–5245 Search PubMed; (b) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 Search PubMed; (c) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 Search PubMed; (d) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 Search PubMed; (e) L. J. Sebren, J. J. Devery, III and C. R. J. Stephenson, ACS Catal., 2014, 4, 703–716 Search PubMed.
- (a) F. G. Bordwell, M. J. Bausch, J.-P. Cheng, T. H. Cripe, T.-Y. Lynch and M. E. Mueller, J. Org. Chem., 1990, 55, 58–63 Search PubMed; (b) X. Creary, M. E. M. Mohammadi and S. McDonald, J. Org. Chem., 1989, 54, 2904–2910 Search PubMed; (c) X. Creary, Acc. Chem. Res., 2006, 39, 761–771 Search PubMed; (d) R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 Search PubMed; (e) D. A. Shultz and G. T. Farmer, J. Org. Chem., 1998, 63, 6254–6257 Search PubMed.
- For example: (a) M. L. Coote, C. Y. Lina and A. A. Zavitsas, Phys. Chem. Chem. Phys., 2014, 16, 8686–8696 Search PubMed; (b) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 Search PubMed; (c) J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560–2575 Search PubMed; (d) J. Hioe, D. Šakić, V. Vrček and H. Zipse, Org. Biomol. Chem., 2015, 13, 157–169 Search PubMed; (e) L. Debien, B. Quiclet-Sire and S. Z. Zard, Acc. Chem. Res., 2015, 48, 1237–1253 Search PubMed.
- For example: (a) M. Abe, Chem. Rev., 2013, 113, 7011–7088 Search PubMed; (b) J.-L. Lin, C.-H. Ye, B.-C. Lin, S.-H. Li, Z.-X. Yang, Y.-H. Chiang and S.-W. Chen, J. Phys. Chem. C, 2015, 119, 26471–26480 Search PubMed; (c) K. Hirai, T. Itoh and H. Tomioka, Chem. Rev., 2009, 109(8), 3275–3332 Search PubMed; (d) B. K. Carpenter, Chem. Rev., 2013, 113, 7265–7286 Search PubMed.
- For example: (a) A. Rajca, Chem. Rev., 1994, 94, 871–893 Search PubMed; (b) H. F. Bettinger and W. Sander, J. Am. Chem. Soc., 2003, 125, 9726–9733 Search PubMed; (c) A. I. Krylov, J. Phys. Chem. A, 2005, 109, 10638–10645 Search PubMed; (d) P. U. Manohar, L. Koziol and A. I. Krylov, J. Phys. Chem. A, 2009, 113, 2591–2599 Search PubMed; (e) T. E. Munsch, L. V. Slipchenko, A. I. Krylov and P. G. Wenthold, J. Org. Chem., 2004, 69, 5735–5741 Search PubMed.
- (a) R. Hoffmann, Acc. Chem. Res., 1971, 4, 1–9 Search PubMed; (b) K. Yoshizawat and R. Hoffmann, J. Am. Chem. Soc., 1995, 117, 6921–6926 Search PubMed; (c) W. Adam, A. Grimison and R. Hoffmann, J. Am. Chem. Soc., 1969, 91, 2590–2599 Search PubMed; (d) H. Wei, D. A. Hrovat, Y. Mo, R. Hoffmann and W. T. Borden, J. Phys. Chem. A, 2009, 113, 10351–10358 Search PubMed; (e) R. Improta and V. Barone, Chem. Rev., 2004, 104, 1231–1253 Search PubMed.
- (a) K. I. Öberg, Chem. Rev., 2016, 116, 9631–9663 Search PubMed; (b) C. A. Cole, N. J. Demarais, Z. Yang, T. P. Snow and V. M. Bierbaum, Astrophys. J., 2013, 779, 1–9 Search PubMed; (c) F. A. Ribeiro, G. C. Almeida, W. Wolff, H. M. B. Roberty and M. L. M. Rocco, J. Phys. Chem. C, 2014, 118, 25978–25986 Search PubMed; (d) M. Lattelais, Y. Ellinger, A. Matrane and J.-C. Guillemin, Phys. Chem. Chem. Phys., 2010, 12, 4165–4171 Search PubMed; (e) Y. Osamura and S. Petrie, J. Phys. Chem. A, 2004, 108, 3615–3622 Search PubMed.
- (a) R. Levine and W. W. Leake, Science, 1955, 121, 780 Search PubMed; (b) A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 Search PubMed; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645–3651 Search PubMed.
- H.-H. Nam and G. E. Leroi, J. Am. Chem. Soc., 1988, 110, 4096–4097 Search PubMed.
- M. Winkler, B. Cakir and W. Sander, J. Am. Chem. Soc., 2004, 126(19), 6135–6149 Search PubMed.
- N. J. Rau and P. G. Wenthold, J. Phys. Chem. A, 2011, 115, 10353–10362 Search PubMed.
- Y. S. Kim and R. J. McMahon, J. Org. Chem., 2005, 70(20), 8171–8179 Search PubMed.
- A. Korte, A. Mardyukov and W. Sander, Aust. J. Chem., 2014, 67(9), 1324–1329 Search PubMed.
- R. Liu, T. T. S. Huang, J. Tittle and D. Xia, J. Phys. Chem. A, 2000, 104, 8368–8374 Search PubMed.
- G. A. Poskrebyshev, Chem. Phys., 2016, 466, 52–64 Search PubMed.
- (a) C. Barckholtz, T. A. Barckholtz and C. M. Hadad, J. Am. Chem. Soc., 1999, 121, 491–500 Search PubMed; (b) Y. Feng, J. Wang, L. Liu and Q. Guo, J. Phys. Org. Chem., 2003, 16, 883–890 Search PubMed.
- (a) X. Cheng, et al. , Chin. J. Chem., 2005, 23, 1017–1020 Search PubMed; (b) V. Vrček and H. Zipse, J. Org. Chem., 2009, 74, 2947–2957 Search PubMed; (c) R. Liu, T. T.-S. Huang, J. Tittle and D. Xia, J. Phys. Chem. A, 2000, 104, 8368–8374 Search PubMed; (d) P.-K. Lo and K.-C. Lau, J. Phys. Chem. A, 2011, 115, 932–939 Search PubMed.
- R. Venkatasubramanian and S. L. N. G. Krishnamachari, Praman, 1988, 30, 529–533 Search PubMed.
- L. M. Culberson, C. C. Blackstone, A. A. Wallace and A. Sanov, J. Phys. Chem. A, 2015, 119, 9770–9777 Search PubMed.
- S. Han, T. Y. Kang, S. Choi, K. W. Choi, S. J. Baek, S. Leeb and S. K. Kim, Phys. Chem. Chem. Phys., 2008, 10, 3883–3887 Search PubMed.
- M. H. Palmer, Chem. Phys., 2008, 344, 21–34 Search PubMed.
- A. F. Lago, R. D. Januário, M. Simonb and J. Z. Dávalosd, J. Mass Spectrom., 2014, 49, 1163–1170 Search PubMed.
- P. Tzallas, C. Kosmidis, J. G. Philis, K. W. D. Ledingham, T. McCanny, R. P. Singhal, S. M. Hankin, P. F. Taday and A. J. Langley, Chem. Phys. Lett., 2001, 343, 91–98 Search PubMed.
- M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499–505 Search PubMed.
- (a) T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 Search PubMed; (b) D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys., 1993, 98, 1358–1371 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) G. D. Purvis, III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 Search PubMed; (b) G. E. Scuseria, C. L. Janssen and H. F. Schaefer, III, J. Chem. Phys., 1988, 89, 7382–7387 Search PubMed; (c) J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968–5975 Search PubMed.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449 Search PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 Search PubMed.
- (a) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO version 3.1; (b) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 Search PubMed.
- M. J. Frisch, et al., GAUSSIAN 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) R. H. A. Eade and M. A. Robb, Chem. Phys. Lett., 1981, 83, 362–368 Search PubMed; (b) M. J. Frisch, I. N. Ragazos, M. A. Robb and H. B. Schlegel, Chem. Phys. Lett., 1992, 189, 524–528 Search PubMed.
- (a) H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 Search PubMed; (b) H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, MOLPRO, version 2012.1, a package of ab initio programs, Cardiff, UK, 2012 Search PubMed; (c) H. J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053 Search PubMed.
- We used the restricted open shell (RO) method for all open shell radical species. This method uses doubly occupied spatial part for α and β spins for the closed shell part and singly occupied spatial part for the unpaired electron. The RO method was used over the unrestricted method, as the orbital analysis is simpler in the former one. The latter one uses singly occupied spatial part for α and β spins. See (a) C. C. J. Roothaan, Rev. Mod. Phys., 1960, 32(2), 179–185 Search PubMed; (b) R. Carbó and J. M. Riera, A General SCF Theory, Lecture Notes in Chemistry, Springer, 1978, vol. 5, pp. 1–4 Search PubMed; (c) R. McWeeny, Methods of Molecular Quantum Mechanics, Academic Press, 2nd edn, 1992 Search PubMed.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 Search PubMed.
- D. A. McQuarrie and J. D. Simon, Physical Chemistry, A Molecular Approach, University Science Books, California, 1997, vol. 28, pp. 1137–1180 Search PubMed.
- The free energy change (ΔG‡) has been calculated from the data available in thermochemistry part of the frequency calculations using Gaussian 09 suite of programme. See http://www.gaussian.com/g_whitepap/thermo.htm.
- Many cited reports in this contribution dealing with open shell systems have included the results using the (U)B3LYP/cc-pVTZ level of theory. Although all the calculations for our systems have also been performed at the (U)M06-2X/cc-pVTZ level of theory and indicated at appropriate places and in the ESI,† for consistency only the former values have been generally used.
- Out of 10 heterocyclic radicals in our current investigation, 9 radicals are C-centred and are found to have a 2A′ electronic structure, whereas the lone N-centred radical 3a has a 2A′′ electronic state. To understand whether these are the ground electronic state structures, for all the 10 radical isomers, we calculated the 2A′′ electronic structures (for all the C-centred radicals) and 2A′ electronic structure (for the N-centred radical). All the optimized structure corresponding to nine 2A′′ geometries of C-centred radicals and 2A′ electronic structure for the N-centred radical have been given as Fig. 1S, ESI†. The results show that the energy gaps (ΔE = E2A′′ − E2A′) are positive for all except in the case of N-centred radical 3a (Table 1S in the ESI†). This confirms that the 2A′ and 2A′′ are indeed the ground state electronic structures for the C-centred and N-centred radicals, respectively.
- (a) D. A. Ponomarev and V. V. Takhistov, J. Chem. Educ., 1997, 74(2), 201 Search PubMed; (b) A. Colson, D. Becker, I. Eliezer and M. D. Sevilla, J. Phys. Chem. A, 1997, 101(47), 8935–8941 Search PubMed.
- The interaction between the electron lone pairs and radical electron has been examined based on the internuclear distances between the corresponding atoms (Fig. 2S in the ESI†). In many cases the distances are found to be reduced compared to the respective distances in the parent compound. Despite the fact that the changes can be a crude indicative of bonding interaction, the behaviour has been corroborated to the overall changes in the molecular structure due to the creation of the radical centre rather than the interaction. This was further confirmed through the change in hybridisation upon radical centre formation.
- The Cs structure and C2v structures of 3a are found to have identical geometrical parameters. The geometries, important parameters and CASSCF results for both structures are compared in Fig. 6S available in the ESI†.
- The C2v and Cs structures that have been computed at different levels of theory showed a difference only at the fourth decimal. Indeed the (U)M06-2X level of theory showed zero energy difference. The energies at different levels of theory for both the structures are indicated in Table 4S of the ESI.† Since the calculations are performed using tight convergence criteria, both structures are found to be almost identical with respect to the geometries (too negligible deviations with respect to the geometrical parameters) as well as the energies (Fig. 6S in the ESI†).
- D. Lu, C. Wu and P. Li, Org. Lett., 2014, 16, 1486–1489 Search PubMed.
- (a) G. R. Liebling and H. M. McConnell, J. Chem. Phys., 1965, 42, 3931 Search PubMed; (b) E. Hedaya, Acc. Chem. Res., 1969, 2, 367–373 Search PubMed; (c) J. C. R. Kiracofe, D. E. Graham and H. F. Schaefer, III, Mol. Phys., 1998, 94(5), 767–787 Search PubMed; (d) M. J. Bearpark, M. A. Robb and N. Yamamoto, Spectrochim. Acta, Part A, 1999, 55, 639–646 Search PubMed.
- D. Leicht, M. Kaufmann, G. Schwaab and M. Havenith, J. Chem. Phys., 2016, 145, 74304 Search PubMed.
- The second C–H and N–H bond dissociation energies have been estimated by considering a reaction, where the radical leads to a biradical and H atom as the products. The computed enthalpy of the corresponding reaction provides the second C–H and N–H BDE of the isomeric radicals (Table 5S in the ESI†).
- The rate constants for all the channels have been estimated at the (U)B3LYP/cc-pVTZ level of theory and the results are tabulated (Table 6S in the ESI†).
- C2H2N isomers: Different structural isomers of the C2H2N radical have been obtained as the end product of unimolecular dissociation channels. The isomeric HCNCH 6 has been observed as the end product in the case of 3a and 1c in the present study. This isomer has not been previously reported. The other isomer ketene iminyl radical (HNCCH, 26) has been observed as the end product of the unimolecular dissociation of 1c, 2c, 3c, 1d, 2d and 3d radicals. On the other hand, an isocyanomethyl radical (CH2NC, 15b) has been obtained as the end product in one of the channels of thiazole radical (2b). Both ketene iminyl and isocyanomethyl radicals have been studied. (See (a) K. Sendt, E. Ikeda, G. B. Bacskay and J. C. Mackie, J. Phys. Chem. A, 1999, 103, 1054–1072 Search PubMed; (b) P. M. Mayer, M. S. Taylor, M. W. Wong and L. Radom, J. Phys. Chem. A, 1998, 102, 7074 Search PubMed ) This isomer has been detected from acetonitrile in matrix IR upon laser ablation of metals. (See ; (c) H.-G. Cho and L. Andrews, J. Phys. Chem. A, 2011, 115(31), 8638–8642 Search PubMed ).
- C3H2NO isomers: The structural isomers of C3H2NO such as species 8, 9, 22, 23, 3 have been obtained as intermediates in unimolecular pathways of oxazole radicals. Except for isomer 9, other species are reported for the first time. A theoretical study has already been reported for 9, where this radical was identified as an intermediate in the [3+2] cycloaddition reaction of NCO + HCCH. (See H. Chen and J. Ho, J. Phys. Chem. A, 2003, 107, 7643–7649 Search PubMed ).
- OCN radical: Y. S. Kim, F. Zhanga and R. I. Kaiser, Phys. Chem. Chem. Phys., 2011, 13, 15766–15773 Search PubMed.
- NCS radical: (a) R. Tarroni, J. Chem. Phys., 2011, 135, 164310 Search PubMed; (b) M. Ouazbir, G. Chambaud, P. Rosmus and P. J. Knowles, Phys. Chem. Chem. Phys., 1999, 1, 2649–2655 Search PubMed; (c) R. E. Baren and J. F. Hershberger, J. Phys. Chem. A, 1999, 103(51), 11340–11344 Search PubMed.
- CHN2 isomers: The cyanoamino radical (HNCN, 20) has been reported here as the end product in the unimolecular dissociation of the 3b radical. The absorption spectrum of this radical was produced in the flash photolysis of diazomethane. (See (a) G. Herzberg and P. A. Warsop, Can. J. Phys., 1963, 41, 286 Search PubMed ) Jet cooled Laser Induced Fluorescence (LIF) spectrum of the HNCN radical was reported. (See ; (b) M. Wu, G. Hall and T. J. Sears, J. Chem. Soc., Faraday Trans., 1993, 89, 615 Search PubMed ). Ab initio calculation on this radical has also been performed by several groups using different level of theories (See ; (c) F. M. Tao, W. Klemperer, M. C. McCarthy, C. A. Gottlieb and P. Thaddeus, J. Chem. Phys., 1994, 100, 3691 Search PubMed; (d) E. P. Clifford, P. G. Wenthold, W. C. Lineberger, G. A. Petersson and G. B. Ellison, J. Phys. Chem. A, 1997, 101, 4338–4345 Search PubMed; (e) C. Puzzarini and A. Gambi, J. Chem. Phys., 2005, 122, 064316 Search PubMed ). Microwave spectrum for the HNCN radical has also been reported. (See ; (f) S. Yamamoto and S. Saito, J. Chem. Phys., 1994, 101, 10350 Search PubMed ).
- HCCO radical: See S. L. N. G. Krishnamachari and R. Venkata-subramanian, Pramana, 1984, 23, 321 Search PubMed.
- HCCS radical: See (a) S. L. N. G. Krishnamachari and T. V. Venkitachalam, Chem. Phys. Lett., 1978, 55, 116 Search PubMed; (b) J. R. Flores, J. Phys. Chem. B, 2003, 107, 9711 Search PubMed; (c) J. R. Dunlop, J. Karolczak and D. J. Clouthier, Chem. Phys. Lett., 1988, 151, 362 Search PubMed; (d) H. Kohguchi, Y. Ohshima and Y. Endo, Chem. Phys. Lett., 1996, 254, 397 Search PubMed; (e) E. Kim, H. Habara and S. Yamamoto, J. Mol. Spectrosc., 2002, 212, 83 Search PubMed; (f) P. G. Szalay, J. Chem. Phys., 1996, 105, 2735 Search PubMed; (g) P. G. Szalay and J. P. Blandeau, J. Chem. Phys., 1997, 106, 436 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05677f |
| This journal is © the Owner Societies 2017 |