
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predicting point defect equilibria across oxide hetero-interfaces: model system of ZrO2/Cr2O3†
Jing
Yang
 ab,
Mostafa
Youssef
abc and
Bilge
Yildiz
*abc
ab,
Mostafa
Youssef
abc and
Bilge
Yildiz
*abc
aLaboratory for Electrochemical Interfaces, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
cDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: byildiz@mit.edu
First published on 14th December 2016
Abstract
We present a multi-scale approach to predict equilibrium defect concentrations across oxide/oxide hetero-interfaces. There are three factors that need to be taken into account simultaneously for computing defect redistribution around the hetero-interfaces: the variation of local bonding environment at the interface as epitomized in defect segregation energies, the band offset at the interface, and the equilibration of the chemical potentials of species and electrons via ionic and electronic drift-diffusion fluxes. By including these three factors from the level of first principles calculation, we build a continuum model for defect redistribution by concurrent solution of Poisson's equation for the electrostatic potential and the steady-state equilibrium drift-diffusion equation for each defect. This model solves for and preserves the continuity of the electric displacement field throughout the interfacial core zone and the extended space charge zones. We implement this computational framework to a model hetero-interface between the monoclinic zirconium oxide, m-ZrO2, and the chromium oxide Cr2O3. This interface forms upon the oxidation of zirconium alloys containing chromium secondary phase particles. The model explains the beneficial effect of the oxidized Cr particles on the corrosion and hydrogen resistance of Zr alloys. Under oxygen rich conditions, the ZrO2/Cr2O3 heterojunction depletes the oxygen vacancies and the sum of electrons and holes in the extended space charge zone in ZrO2. This reduces the transport of oxygen and electrons thorough ZrO2 and slows down the metal oxidation rate. The enrichment of free electrons in the space charge zone is expected to decrease the hydrogen uptake through ZrO2. Moreover, our analysis provides a clear anatomy of the components of interfacial electric properties; a zero-Kelvin defect-free contribution and a finite temperature defect contribution. The thorough analytical and numerical treatment presented here quantifies the rich coupling between defect chemistry, thermodynamics and electrostatics which can be used to design and control oxide hetero-interfaces.
I. Introduction
Novel phenomena at oxide hetero-interfaces have been of great interest for both fundamental studies1 and device applications.2 In particular, ionic defect redistributions at the interface are known to influence the magnetic, electronic and transport properties of metal oxides.3–6 Understanding the redistribution of defects across such interfaces serves as the foundation for studying reaction and diffusion processes in related systems. However, addressing the electronic and ionic structures of the interfacial zone is not a trivial task. The nature of bonds between ions and chemical composition in the oxide crystal could be significantly altered near interfaces.7Three factors, the band offset, chemical potential difference and segregation energy, rendered in Fig. 1 as (a), (b) and (c), need to be addressed comprehensively and at the same level of theory in order to self-consistently describe the redistribution of charged defects at a hetero-interface. General theory of these three factors have been described in previous works, including defect chemistry at interfaces of ionic materials8–11 and semiconductors,12–14 band offset,15 bulk defect chemistry,16 and electric field discontinuity.17 Here we provide a summary of the involved theory. First (Fig. 1(a)), before considering any electronic or ionic defect, the discontinuities of valence band ΔEv and conduction band ΔEc immediately at the interface need to be calculated. Theoretically, this band discontinuity corresponds to the large electrostatic potential gradient at ideally abrupt defect-free interfaces. This discontinuity is localized in the few atomic layers in the immediate vicinity of the interface and is independent of external perturbations such as dilute doping and applied electric bias. Second (Fig. 1(b)), charged species have different electrochemical potential in equilibrium bulk materials. When two materials are put into contact, electronic and ionic defects start to diffuse, driven by this chemical potential difference. This leads to build-up of local electric field and in turn drives drift flux in the opposite direction. When the system equilibrates, drift and diffusion fluxes cancel out, leading to a reduced form of drift-diffusion equation with zero flux,12,13 which is equivalent to the Poisson–Boltzmann formalism used by the solid state ionics field.18–20 Third (Fig. 1(c)), atoms near an interface are in a distinct bonding environment compared to atoms in bulk. Defect formation energy will change due to the interfacial lattice relaxation and chemical composition variation. This adds the segregation energy, 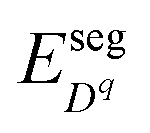 , to defect formation energy of ions in the vicinity of the interface. This region where the segregation energy is non-negligible is termed as the core zone.
, to defect formation energy of ions in the vicinity of the interface. This region where the segregation energy is non-negligible is termed as the core zone.
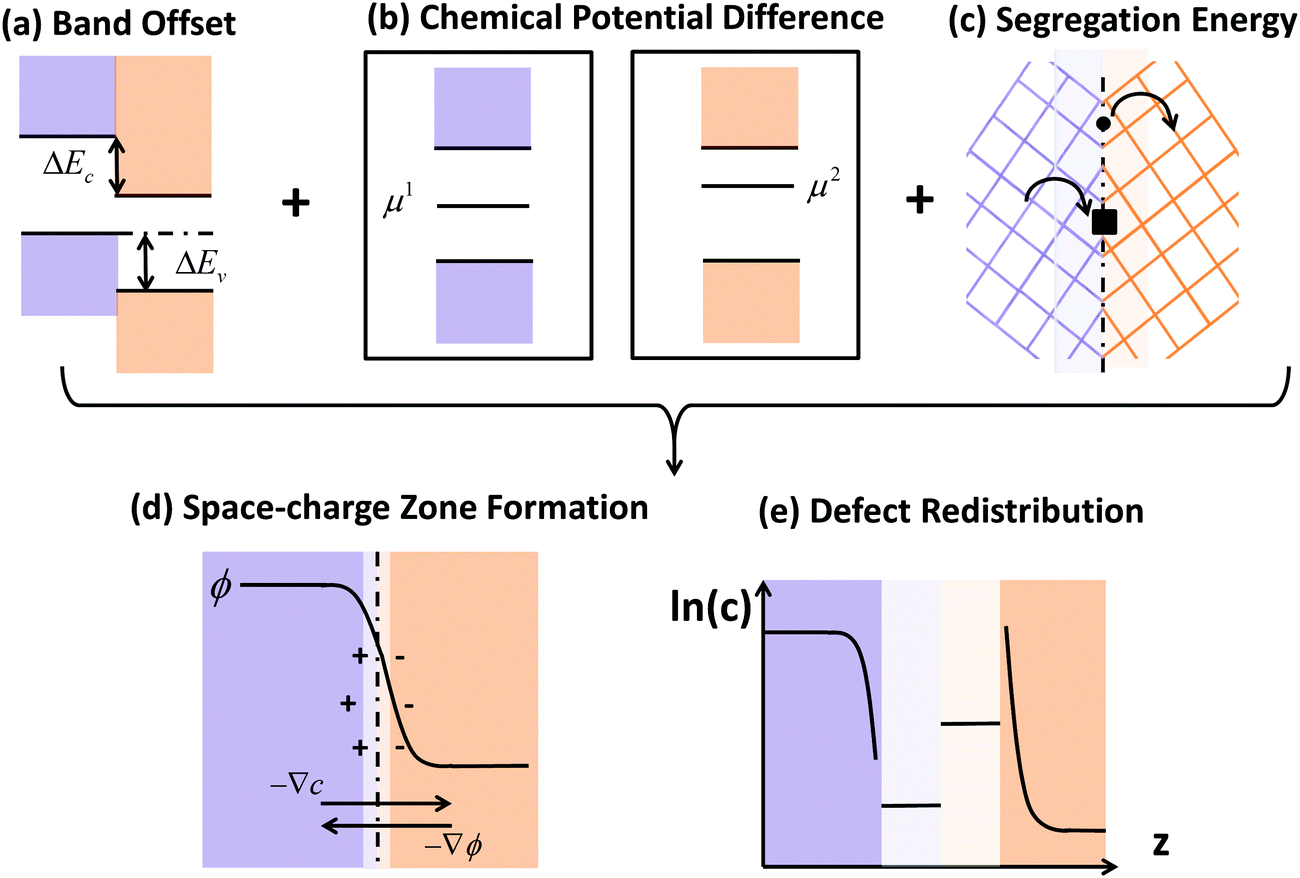 | ||
| Fig. 1 Schematic illustration of the comprehensive framework adopted in this work for predicting point defect equilibria across hetero-interfaces (ref. 8–17 in the Introduction section). (a) Band offset of the ideal interface. (b) Any species has different chemical potentials, μ1 and μ2, when bulk defect equilibria is reached on each side of the interface. This difference drives diffusion flux as the two materials are put into contact and lead to the formation of a space-charge zone. Upon equilibrium, Fermi level and the chemical potential of each element should be constant throughout the heterojunction, and the electrochemical potential of each type of ionic defect should be constant on the oxide side where it exists. (c) Defect segregation or depletion due to interfacial structural and compositional change. After assessing these three factors at the atomistic level by density functional theory (DFT), these are provided as input into the steady state drift-diffusion model. In this work, the steady state drift diffusion model is solved in a self-consistent way to generate (d) the electrostatic profiles in the space-charge and core zones, and (e) the defect redistribution profiles across the interface with explicit consideration for the interfacial core zone. When equilibrium is reached, the diffusion flux driven by concentration gradient ∇c and drift flux driven by electrostatic potential gradient ∇ϕ cancel out. Core regions extend for only several atomic layers in the immediate vicinity of the interface and are drawn with enlarged scale and light colors in (d) and (e). | ||
Each of these three factors represented in Fig. 1 have been recognized in literature, but were usually treated separately with different modeling methods. For example, numerical implementations considered either only the band offset,21–23 or only the drift-diffusion,24–26 or only the segregation energy,27–31 or the band offset and segregation energy at a semiconductor heterojunction,32,33 or the chemical potential difference and segregation energy at a grain boundary.18–20,34–36 In the rest of this section, we provide more details on these numerical implementations from literature. Predicting band offsets (factor (a)) from density functional theory (DFT) calculations has been of interest since decades ago and a number of theories have been developed.15 One commonly accepted scheme was first proposed by Van de Walle and Martin21 and applied in a number of studies,22,23 which aligns the band positions to the electrostatic potential across the heterojunction. These studies consider defect-free interfaces and only evaluate the band offsets. The electrostatic potential, ϕ, built by the drift-diffusion mechanism (factor (b)) can extend in space for several hundreds to thousands of nanometers, and is usually studied with finite-element methods in the context of semiconductor engineering. The drift-diffusion model is well-developed for semiconductor heterojunctions,12–14,32,33 where only electrons and holes are considered as the mobile species. In that case, there is no need to consider segregation energies since free holes and electrons are assumed to be unaffected by the changes in the bonding at the interface. In such model, only the space-charge potential causes changes in holes and electrons at the interface.
When it comes to ionic or mixed electronic-ionic conductors that are used in electrochemical devices, the ionic defect contribution cannot be neglected any more. Ionic and electronic drift-diffusion models based on the chemical potential difference (factor (b)) have been built for mixed electronic-ionic pn homojunctions made of p-type and n-type doped SrTiO3.24 Similar continuum level studies have also been implemented for a metal/oxide interface29 and a gas/oxide interface26 based on first-principle predictions. However, in these two systems the segregation energy term (factor (c)) is still missing from the consideration of the charged point defect redistribution. In fact in the case of the gas/oxide interface, there is the segregation of defects at the interface within the oxide side of the interface, and there is also the adsorption of charged species from the gas side. While ref. 26 chose to treat the adsorption of charged gas species only, ref. 18 chose to treat segregation of defects within the oxide only. A complete treatment of segregation/adsorption phenomena at gas/oxide interfaces starting from first-principles all the way to continuum level treatment is still missing as well, and the recent work on hydrated BaZrO3 surface27 presents a very encouraging attempt in that direction.
The defect segregation energy 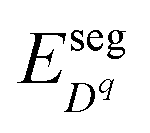 , which have discrete values in each atomic layer close to the interface, can be evaluated using atomistic simulation methods25,27,28,30,31 and are explicitly accounted for in this work. In fact these segregation energies are responsible for the formation of the core zone at the interface which exhibit very different defect concentrations in comparison with the extended space charge zone.
, which have discrete values in each atomic layer close to the interface, can be evaluated using atomistic simulation methods25,27,28,30,31 and are explicitly accounted for in this work. In fact these segregation energies are responsible for the formation of the core zone at the interface which exhibit very different defect concentrations in comparison with the extended space charge zone.
Attempts have been made to establish interfacial defect chemistry theoretically.9,11,37 These theories consider equilibrium defect reactions at the interface as boundary condition. Counting defect reactions corresponds to a canonical formalism in which defects and in particular electronic defects are tracked particle by particle. Such a canonical formalism, while tractable if one knows a priori the important defect reactions, is not in the spirit of the predictive power of first-principle modeling.16 Alternatively, a grand canonical formalism that treats individual point defects and electronic defects such that each of them is in equilibrium with a reservoir is much more compatible with atomistic simulations.16 So far, first-principles-based models have only been developed for symmetric grain boundaries in last few years,34–36 in which the segregation energy is included self-consistently into the drift-diffusion model. A symmetric grain boundary is a simplified scenario where there is no band offset on the two sides of the interface and the electrostatic profiles are symmetric across the boundary. Moreover, the electric field in the core zone was not explicitly included into the numerical treatment of grain boundaries.
In this work, we implement a generalized model of hetero-interfaces where no symmetry is assumed and the continuity of the displacement field17 is adhered to throughout the system by including explicitly the interface core into the solution. To our knowledge this is the first attempt to simultaneously include these three factors listed in Fig. 1 completely from ab initio calculations, without a priori assumption of dominant defect species. On the continuum modeling level, we report for the first time the solution of Poisson's equation throughout the core zone and the space-charge zone by using defect formation energy profiles of step-function behavior, while preserving the continuity of the electric displacement field throughout the entire space including the core zone. Finally, by using this ability we deconvolute the contributions to the interfacial electrostatic profiles arising from the 0 K-perfect and the finite temperature defects effects. This model and the explored concepts are applicable to any type of interface system.
Defect equilibria in bulk provide the boundary conditions needed for the interfacial defect redistribution. In previous work we assessed the defect equilibria in bulk monoclinic ZrO2.38 The key idea is to capture the defect structures and formation energies with density functional theory (DFT) calculations and feed it to the thermodynamic model where the total Gibbs free energy of the defective crystal is minimized.16 However, additional difficulty to tackle is the asymmetry of the hetero-interface. We performed DFT calculations to obtain band alignment and defect formation (segregation) energies at the interface and in bulk of both sides of the heterojunction (described below). Using these results as input, we are able to solve for the electrostatic potential generated by defect redistribution and defect concentrations across the space-charge region and in the interfacial core layer.
We selected ZrO2/Cr2O3 hetero-interface as our model system which arises in the utilization of zirconium alloys as nuclear cladding material in light water reactors.39 The oxidation of zirconium alloys by the reactor coolant (light water) could be a severe design-limiting issue under aggressive conditions inside nuclear reactors. Zirconium alloys contain additions of tin, iron, nickel and chromium which tend to precipitate in the form of intermetallics. Upon oxidation, both the host zirconium metal and the intermetallic precipitates form their respective oxides. The net outcome is multiple interfacial zones at the boundary between ZrO2 and oxides of the precipitates.40,41 Oxygen diffusion kinetics in ZrO2, which is a major consideration in designing oxidation-resistant zirconium alloys, can be significantly altered near the interface between ZrO2 and the precipitate metal oxide. In this work we focus on the interface between monoclinic ZrO2 and hexagonal Cr2O3. Our results show that throughout all temperatures and oxygen partial pressures ZrO2 exhibits a downward band bending and Cr2O3 exhibits and upward one. The core and space charge zones of ZrO2 are negatively charged, whereas those of Cr2O3 are positively charged. However, the predominant defect in each zone varies depending on temperature, oxygen partial pressure and the spatial location with respect to the junction indicating a rich coupling between defect chemistry, thermodynamics and electrostatics. In particular in the core and space charge zones of ZrO2 and under oxygen rich conditions we observe a depletion of doubly charged oxygen vacancies and a similar depletion in the sum of electron and holes. This depletion indicates a reduction in electronic and oxygen transport near Cr2O3 particles dispersed in ZrO2. This, in turn, provides an explanation for the corrosion resistance of Zr alloyed with Cr. Finally we discuss how the potential drop, interface dipole, and interface electric field are made out of two components; a 0 K defect-free contribution and a finite temperature defect contribution.
II. Methods
In this section we present a comprehensive methodology to model hetero-interfaces. First, we give a brief review of establishing point defect equilibria in the bulk of an oxide. Second, we describe how to construct a stable interface and obtain band offsets by aligning bulk band positions to a reference potential. Third, we introduce our continuum model for the equilibrium space-charge zone. Finally, we demonstrate how we explicitly treat defects in the interfacial, core zone.All density functional calculations involved in this work were performed with Vienna Ab initio Simulation Package (VASP).42–45 We used the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) functional.46,47 We employed pseudopotentials constructed using the projector augmented wave method which treat 4s24p64d25s2 electrons for Zirconium, 3s23p63d54s1 for Chromium and 2s22p4 for Oxygen as valence electrons. The plane-wave cutoff energy was set to 450 eV. A Hubbard U correction48Ueff = 5 eV was applied to the d orbitals of Chromium.49
A. Bulk defect equilibria
We adopt the framework established in our previous work50 to predict intrinsic point defect equilibria for bulk ZrO2 and Cr2O3. The results for bulk monoclinic ZrO2 were presented in ref. 38 and the detailed analysis for Cr2O3 defect chemistry is to be published elsewhere. The formation energy of a defect Dq whose charge is q is computed by: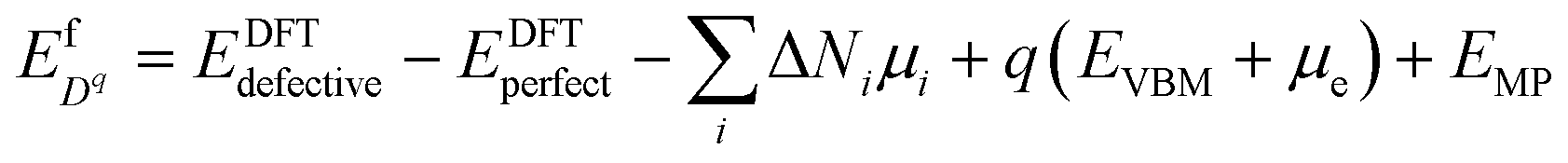 | (1) |
Here EDFTdefective is the DFT energy of the defective cell, EDFTperfect is the DFT energy of the perfect cell. μi denotes the chemical potential of each element involved in the cell, and ΔNi the relative number of each type of atoms with respect to the perfect cell (e.g. ΔNZr = −1, ΔNO = 0 for zirconium vacancies). EVBM is the valence band maximum of the perfect cell. μe is the Fermi level of electrons relative to the valence band maximum. EMP is the first-order Makov–Payne correction51 for the finite-size supercell errors in charged defect calculations.
The chemical potential of oxygen as a function of temperature T and oxygen partial pressure PO2 is calculated by:
 | (2) |
 is the DFT-calculated energy of an oxygen molecule. Eover is the correction term for O2 overbinding error created by semilocal functionals,52 which has a value of 1.22 eV for the parameters used in this work.38
is the DFT-calculated energy of an oxygen molecule. Eover is the correction term for O2 overbinding error created by semilocal functionals,52 which has a value of 1.22 eV for the parameters used in this work.38 is the tabulated reference chemical potential for O2 gas at reference pressure P0 = 0.1 MPa.53kB is the Boltzmann constant. Once μO is known, the chemical potential of cation elements are calculated by
is the tabulated reference chemical potential for O2 gas at reference pressure P0 = 0.1 MPa.53kB is the Boltzmann constant. Once μO is known, the chemical potential of cation elements are calculated by | (3) |
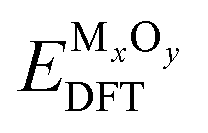 is the DFT-computed energy of the oxide material.
is the DFT-computed energy of the oxide material.
Fermi level is determined from the charge neutrality condition imposed on point defects Dq, electrons nc, and holes pv
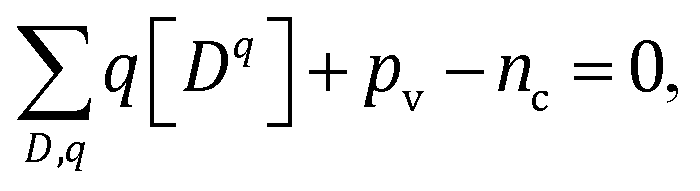 | (4) |
 | (5) |
Here nD is the number of possible sites for the defect D per unit formula. Eqn (5) is an approximate form of the expression for defect concentration sought by minimizing global Gibbs free energy.29 They are equivalent when EfD,q ≫ kBT. Similarly, the concentrations of electrons and holes, nc and pv are also functions of Fermi level and are computed by integrating the electronic density of states g(E) as follows:
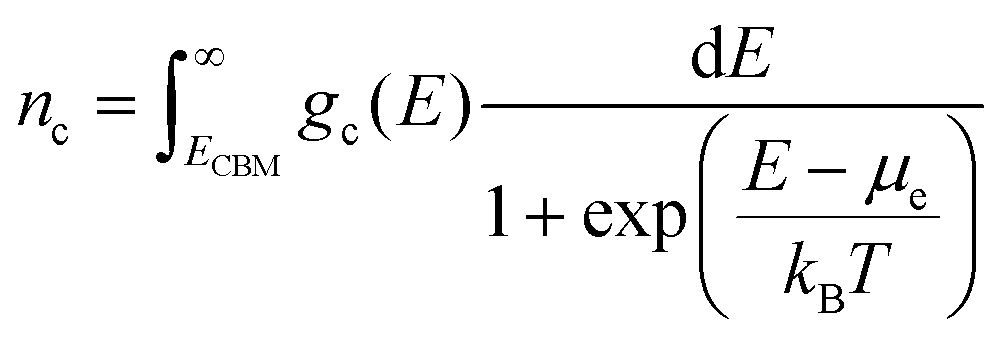 | (6) |
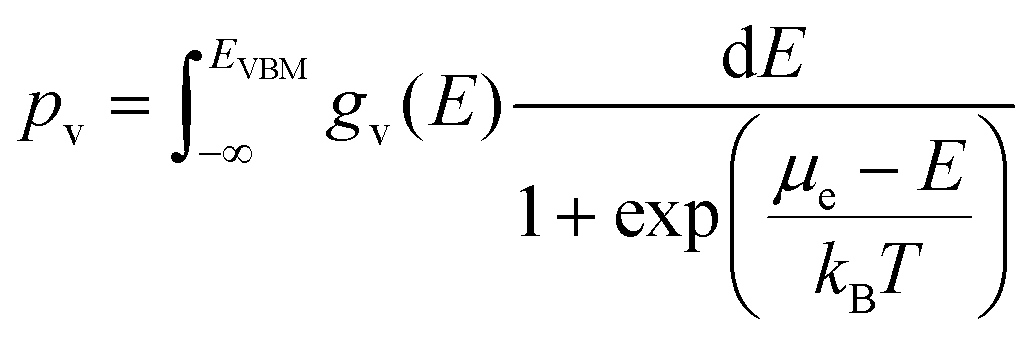 | (7) |
B. Interface band alignment
![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 2) surface55 to construct a coherent interface. Both surfaces are non-polar. This choice was made by surveying all low-energy surface combinations and minimizing the interface misfit. We built a supercell from these two surfaces with 13 atomic layers of ZrO2 and 10 atomic layers of Cr2O3, as shown in Fig. 2(c). The as-built slab model was then fully relaxed with a 4 × 4 × 1 k-points grid until the forces on ions were less than 0.02 eV Å−1, allowing cell shape and volume to change. The lattice parameters before and after relaxation are provided in Table 1.
2) surface55 to construct a coherent interface. Both surfaces are non-polar. This choice was made by surveying all low-energy surface combinations and minimizing the interface misfit. We built a supercell from these two surfaces with 13 atomic layers of ZrO2 and 10 atomic layers of Cr2O3, as shown in Fig. 2(c). The as-built slab model was then fully relaxed with a 4 × 4 × 1 k-points grid until the forces on ions were less than 0.02 eV Å−1, allowing cell shape and volume to change. The lattice parameters before and after relaxation are provided in Table 1.
 | ||
| Fig. 2 (a) m-ZrO2(001) and (b) Cr2O3(012) surface cross sections and (c) relaxed interface. Oxygen ions are drawn small and red, Cr ions are medium-sized and blue, Zr ions are largest and green. This figure is generated with visualization software VESTA.56 | ||
| a (Å) | b (Å) | ε a (%) | ε b (%) | |
|---|---|---|---|---|
| ZrO2(001) | 5.169 | 5.232 | −0.41 | 1.68 |
| Cr2O3(012) |
4.957 | 5.359 | 3.85 | −2.37 |
| After relaxation | 5.148 | 5.320 |
We comment on the method of constructing hetero-interfaces with the following: a stable interface should be built from two low-energy surfaces which have matching lattice parameters. It should be noted that the lowest-energy interface doesn't necessarily arise from two lowest-energy surfaces.57 The most rigorous method is to test all surface combinations and search for the lowest interface energy. Some of these combinations might require an extremely large supercell if the two surfaces have different periodicity. In this work m-ZrO2(001)/Cr2O3(012) is a good combination because both surfaces have rectangular cell and the mismatch is small. Additionally, neither surface is polar, so it avoids electrostatic complications with the periodic slab model.58 To confirm the stability of the constructed interface, we also performed molecular dynamics simulation on the slab model (details in ESI,† Section S1), which shows that the interface retains its phases and structure.
The electrostatic potential obtained from DFT calculation of the interface under consideration is shown in Fig. 3(a). To get the macroscopic average, electrostatic potential is first averaged over the plane parallel to the interface to get V(z), then averaged over one atomic layer along the z direction.60
 | (8) |
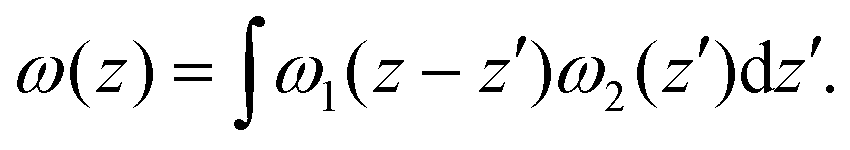 | (9) |
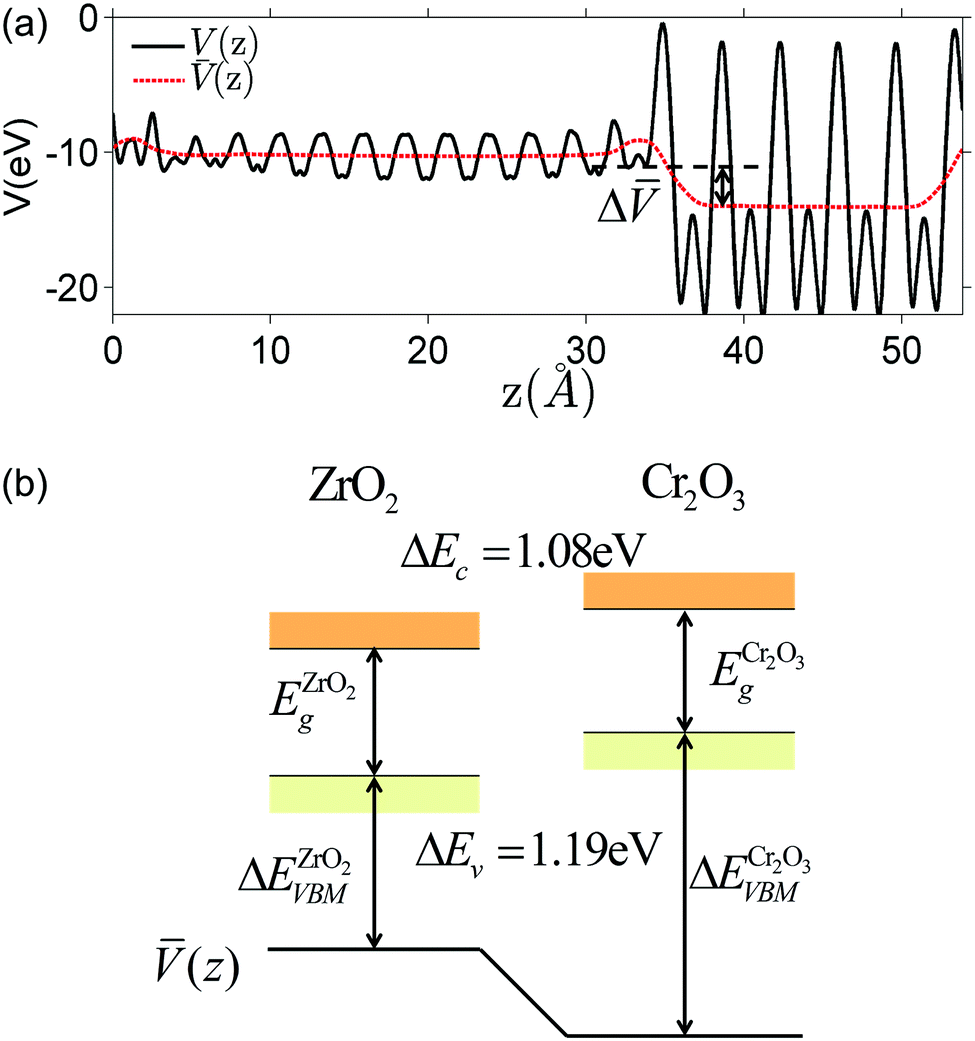 | ||
Fig. 3 (a) Planar (solid black line) and macroscopic average (dashed red line) of electrostatic potential across the ZrO2/Cr2O3 interface obtained from DFT calculation. Δ![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) ZrO2–Cr2O3 = 3.77 V is the potential difference on both sides of the interface where flat potentials are reached. (b) Aligned band positions. Band gaps of ZrO2 and Cr2O3 from DFT calculations are 3.49 eV and 3.38 eV, respectively. ZrO2–Cr2O3 = 3.77 V is the potential difference on both sides of the interface where flat potentials are reached. (b) Aligned band positions. Band gaps of ZrO2 and Cr2O3 from DFT calculations are 3.49 eV and 3.38 eV, respectively. | ||
The valence-band offset is determined by
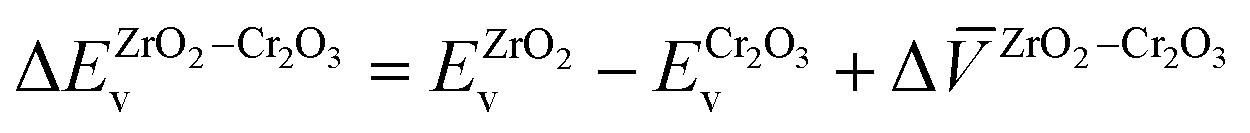 | (10) |
 and
and  are the valence band energies with respect to the local electrostatic potential obtained from two separate bulk calculations. ΔZrO2–Cr2O3 is the potential difference of the bulk-like regions of the interface, where flat potentials are reached. The aligned band positions are shown in Fig. 3(b).
are the valence band energies with respect to the local electrostatic potential obtained from two separate bulk calculations. ΔZrO2–Cr2O3 is the potential difference of the bulk-like regions of the interface, where flat potentials are reached. The aligned band positions are shown in Fig. 3(b).
We would like to clarify here the concept of band offset. As shown in Fig. 3(a), the potential in the perfect slab calculation becomes constant within two atomic layers away from the interface. This potential should not be confused with the space-charge layer potential. The equilibrium space-charge zone forms upon redistribution of various electronic and ionic charge carriers. This redistribution is a finite temperature excitation which cannot be captured directly by a zero-Kelvin DFT calculation. The band offset computed here resulted solely from the localized charge density redistribution due to the abrupt change in bonding environment at the interface. In the next section we will superimpose the defect redistribution on the defect-free heterojunction results and solve for the space-charge zone with a continuum-level model. The band discontinuities will serve as an input. The relationship between defect-free model and defective model will be further discussed at the end of this paper.
C. Equilibrium of space-charge zone
From the above calculation, we obtained the electrostatic profile of the perfect interface. Now we use these results for the perfect interface as the neutral background and superimpose the defect equilibria on it. The redistribution of defects will lead to the formation of an extended space-charge layer around the interface and a localized core layer right at the interface. The core layer is discussed in Section D below. In the space-charge layer, defect formation energies are different from bulk by a term of electrostatic potential ϕscl. Adopting a one-dimensional continuum model, ϕscl is governed by Poisson's equation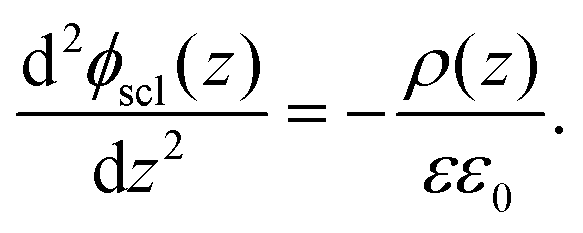 | (11) |
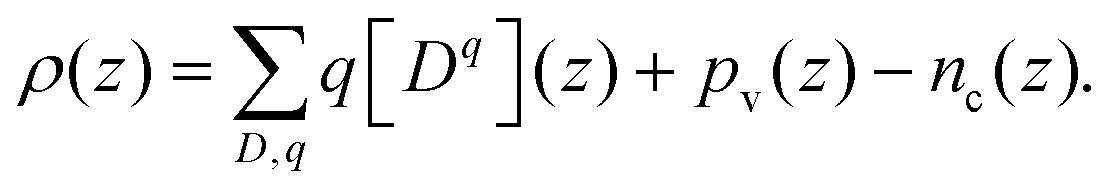 | (12) |
Defect concentrations in equilibrated heterogeneous system change with the electrostatic potential. In dilute limit where Boltzmann-like distribution is satisfied, they can be expressed with respect to their bulk value as
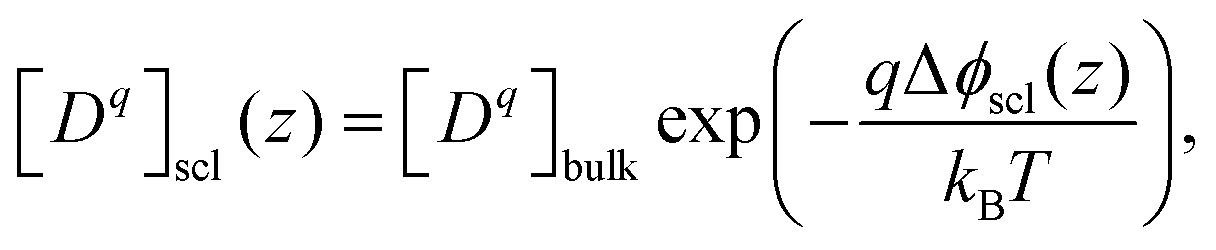 | (13) |
Eqn (11) is solved with the following boundary conditions
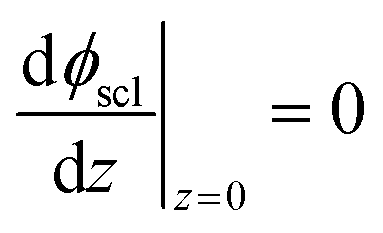 | (14) |
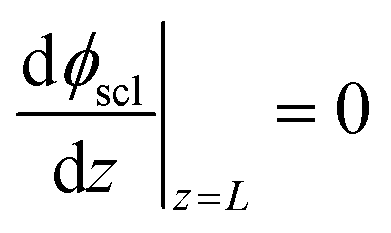 | (15) |
 | (16) |
The zero point of ϕscl is arbitrary. Here we take the convention of setting ϕscl|z=0 to zero. Built-in voltage  will be determined upon completing the solution of Poisson equation throughout the space charge zones and the core zones. A detailed description of the boundary conditions used when core zones are added is provided in ESI,† Section S2.
will be determined upon completing the solution of Poisson equation throughout the space charge zones and the core zones. A detailed description of the boundary conditions used when core zones are added is provided in ESI,† Section S2.
We point out here that using eqn (13) saves the trouble of solving defect concentrations from drift-diffusion equation coupled with Poisson equation for the electrostatic potential. The simplified form in eqn (13) is valid when  for all defects. When the formation energies start to approach kBT, their concentrations need to be described by Fermi–Dirac distribution.29 In the latter case no analytical solution for the drift-diffusion equation exists and it must be solved numerically coupled with Poisson's equation. A detailed derivation is provided in ESI,† Section S3. It is worth recalling that Poisson equation in which the charge density stems from defects described by Boltzmann-like expression under equilibrium13,61 as in this work is sometimes also termed as the Poisson–Boltzmann equation.8,18,20
for all defects. When the formation energies start to approach kBT, their concentrations need to be described by Fermi–Dirac distribution.29 In the latter case no analytical solution for the drift-diffusion equation exists and it must be solved numerically coupled with Poisson's equation. A detailed derivation is provided in ESI,† Section S3. It is worth recalling that Poisson equation in which the charge density stems from defects described by Boltzmann-like expression under equilibrium13,61 as in this work is sometimes also termed as the Poisson–Boltzmann equation.8,18,20
D. Core layer
The method presented above is already sufficient to obtain meaningful information of the space-charge region, such as the built-in potential and space-charge length. However, there still exists an important term we have ignored thus far. Defects close to interface are influenced not only by the electrostatic potential, but also by the varying chemical bonding environment in the interfacial layer. This effect is rather localized in the first few atomic layers next to the interface, compared to the space-charge zone which could extend to hundreds of nanometers spatially. To evaluate this term, we define the atomic layer in the vicinity of the interface as the core layer and treat defects in the core layer explicitly. This definition is conventional in studies of grain boundary34 and dislocations.62 However, in these existing models the core layer is not solved consistently with the space-charge layer, but treated as a homogeneous zero-electric-field zone. As a consequence of this inconsistency the electric displacement field is not continuous across the interface although it should be.17 In this section we discuss how defects in the core layer are simulated and included in the continuum model.The width of the core layer is taken to be equal to the lattice parameter in the z direction. We assume that outside the core layer, and except for the electrostatic effect, defect formation energies are not altered by the non-uniformities of the interface and remain in their bulk value. Defect formation energies in the core layer are obtained from DFT calculation to accurately evaluate the effect of structural change near interface. The cell used in Section B is not sufficient to simulate interfacial defects. A larger cell is required to model isolated, non-interacting point defects. We performed DFT calculations for defects in the core layer with a supercell doubled in the x and y direction and 2 × 2 × 1 k-points grid. Expression for defect formation energy is the same as eqn (1) except now all values are obtained from the interface calculations.
Following the arguments in Section C, it is straightforward to extend eqn (13) to the core layer and the defect concentration expression in that case reads
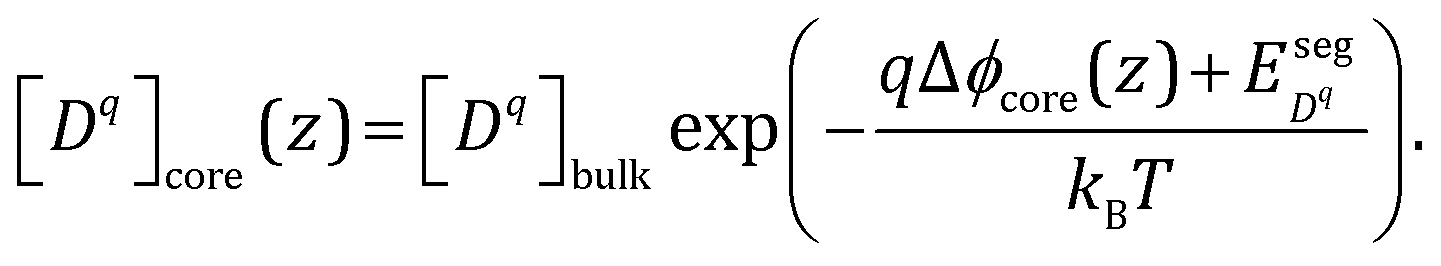 | (17) |
 is the difference in formation energy between defects in bulk and core layer, excluding the contribution of electrostatic potential. Values of
is the difference in formation energy between defects in bulk and core layer, excluding the contribution of electrostatic potential. Values of  for selected defects are listed in Table 2. This value reflects the change of formation energy due to interfacial bonding environment variation and is independent of temperature and oxygen partial pressure.
for selected defects are listed in Table 2. This value reflects the change of formation energy due to interfacial bonding environment variation and is independent of temperature and oxygen partial pressure.
| ZrO2 side |
|
Cr2O3 side |
|
|---|---|---|---|

|
3.61 |
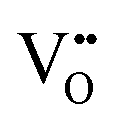
|
−2.84 |
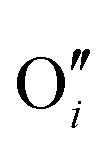
|
−1.44 |
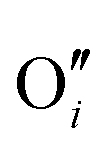
|
−7.75 |

|
−3.00 |

|
−3.98 |


Poisson equation is solved explicitly inside the core zone subject to the boundary conditions described in Section C and subject to the global charge neutrality as described in eqn (16). A detailed solution scheme is in ESI,† Section S2. It is this explicit solution of Poisson equation inside the core zone that preserves the continuity of the displacement field throughout the heterojunction. Poisson equation is discretized onto a grid with mesh spacing of 1 Å outside core layer and 0.1 Å inside core layer and numerically solved with Newton's method. More details of the numerical implementation are discussed in ESI,† Section S2.
III. Results
We divide this section into four parts. First we analyze the electrostatic profiles resulted by the redistribution of charged defects across the interface. In particular we focus on the non-uniformities of the core layer and explicitly inspect the electric field and charge concentrations in this region. In the second part we take a close look at the spatially-resolved concentration profiles of the dominant defects and discuss how the existence of such interfaces influences the corrosion of zirconium alloys. In the third part we elucidate the general implications that this work provides on studying hetero-interface by making a comparison between the contribution of the defect-free interface and defect redistribution. We present all the results obtained at 1200 K for a range of oxygen pressures. Results obtained for 800 K are in the Supporting Information, presenting the same trends as shown here. Last, we list possible future improvements for the computational method.A. Electrostatic profiles
In this section we represent electrostatic profiles under a selected thermodynamic condition (1200 K, PO2 = 10−5 atm) to demonstrate the features of defect redistribution across the interface. Fig. 4 shows the electrostatic potential, the band bending diagram, and the formation energy of across this hetero-interface.
across this hetero-interface.
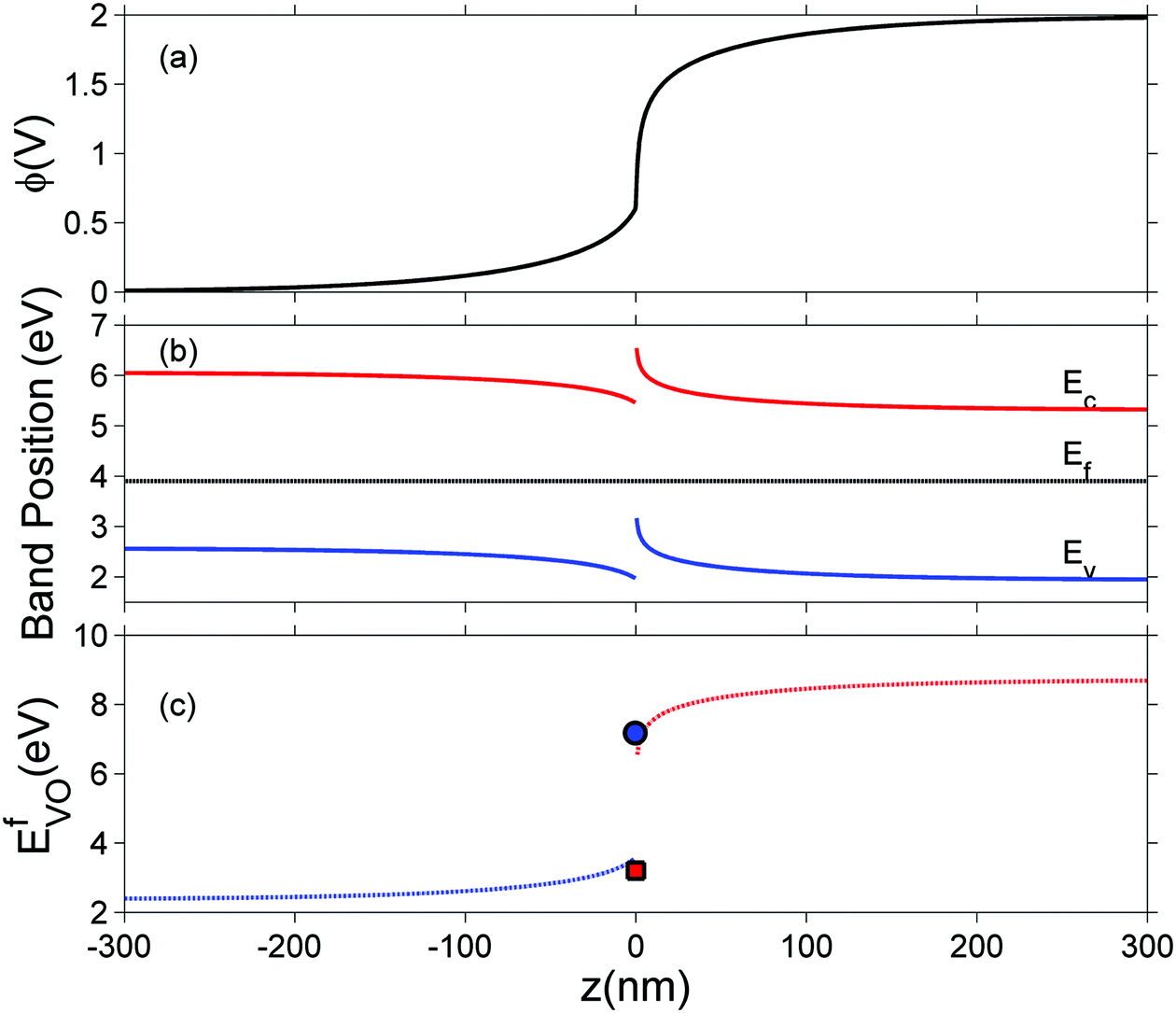 | ||
| Fig. 4 (a) Electrostatic potential, (b) band edges positions for valence band Ev and conduction band Ec, and the Fermi level Ef, (c) formation energy of doubly charged oxygen vacancy calculated at 1200 K and PO2 = 10−5 atm. The interface is placed at z = 0 with ZrO2 on the left side and Cr2O3 on the right side. In (c), the blue circle (ZrO2 side) and red square (Cr2O3 side) show explicitly the formation energy in core layers on each side. | ||
Under these conditions, the total built-in potential is ϕbuilt-in = 1.98 V. At the interface, the band discontinuities from core/space-charge boundary in ZrO2 to core/space-charge boundary in Cr2O3 are ΔEv = 1.19 eV and ΔEc = 1.08 eV, as sought from the method described in Section IIB. Bands bend down on the ZrO2 side and up on the Cr2O3 side. As such, the positively charged defect species are depleted and negatively charged defects accumulate in both the space-charge and core zones of ZrO2. The converse occurs on Cr2O3 side. The presence of the core layer adds a potential drop of 0.05 V on the ZrO2 side and 0.10 V on Cr2O3 side. Therefore, right at the interface (core/core) both band discontinuities are increased by 0.05 eV due to defect segregation in the core layer. We plot the formation energy of  in Fig. 4(c) with the circle and square explicitly showing values in the core layer on ZrO2 and Cr2O3 side, respectively. Outside the core layer (in the space charge zone),
in Fig. 4(c) with the circle and square explicitly showing values in the core layer on ZrO2 and Cr2O3 side, respectively. Outside the core layer (in the space charge zone),  differs from bulk value by twice the electrostatic potential change as it is a doubly charge defect. Going from outside the core to its interior,
differs from bulk value by twice the electrostatic potential change as it is a doubly charge defect. Going from outside the core to its interior, 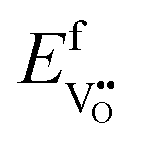 raises abruptly by 3.61 eV on the ZrO2 side and drops by 2.84 eV on the Cr2O3 side. The values here show that formation energy change due to interfacial effects is much more significant than due to the electrostatic potential variations.
raises abruptly by 3.61 eV on the ZrO2 side and drops by 2.84 eV on the Cr2O3 side. The values here show that formation energy change due to interfacial effects is much more significant than due to the electrostatic potential variations.
From Fig. 4 we can see that the electrostatic potential is not smooth at the interface, implying a discontinuity of the electric field. To further examine the interfacial electric field, we zoom on the interfacial zone in Fig. 5. Several features are captured in this plot. First, the electric displacement field D = εε0E (Fig. 5(b)) is continuous throughout, as required by Poisson's equation. At ZrO2/Cr2O3 boundary, there is a discontinuity in both the charge density and the dielectric constant, this results in a discontinuity in the electric field at z = 0. In addition, at the boundaries core/space charge within each oxide, we assume ε is constant and not affected by the electric fields. However, because of the abrupt change in charge density (Fig. 5(c)) at these boundaries, the first derivative of electric field (its divergence) is discontinuous.
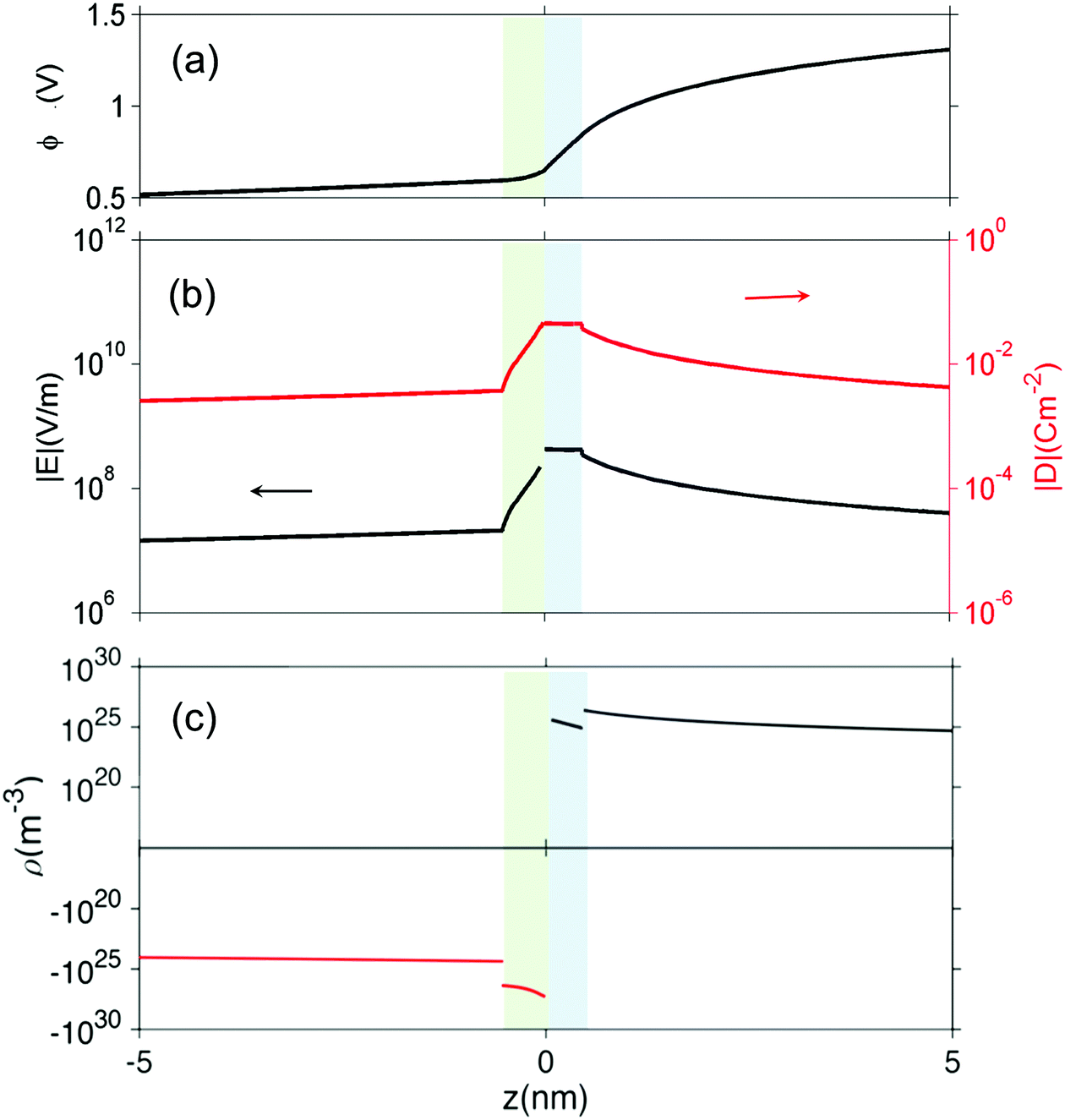 | ||
| Fig. 5 (a) Electrostatic potential ϕ, (b) the magnitude of the electric field E and electric displacement field D and (c) charge concentration ρ close to the interface. Regions with green and blue background color represent core layer on ZrO2 and Cr2O3 side, respectively. | ||
Under the above mentioned thermodynamic conditions and because of the downward band bending of ZrO2 and upward band bending of Cr2O3, ZrO2 side is negatively charged and Cr2O3 side is positively charged. The core layer of ZrO2 is more heavily charged in comparison with its space charge layer by several orders of magnitude. This is mainly caused by the segregation of 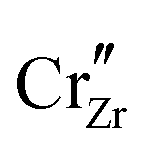 and
and 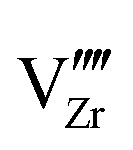 to the core layer. On the other hand, the core layer of Cr2O3 is positively charged due to the abundance of valence band holes and
to the core layer. On the other hand, the core layer of Cr2O3 is positively charged due to the abundance of valence band holes and 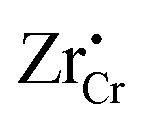 . However, contrary to ZrO2, the charge density in the core layer of Cr2O3 is less than the charge density in it space charge zone. Details of the defect chemistry will be discussed in the following section.
. However, contrary to ZrO2, the charge density in the core layer of Cr2O3 is less than the charge density in it space charge zone. Details of the defect chemistry will be discussed in the following section.
It is common in literature to treat the core layer as a homogeneous region where the electrostatic potential is flat and defect concentrations have constant values, excluding it from the continuum solution. This is a reasonable approximation because the core is a single atomic layer. Such approximate models have produced reliable results in reproducing the electric conductivity of various systems.34,36,63–68 However, this approximation did not capture the magnitude of electric field in the core region. There are some encouraging attempts of treating core layer explicitly in literature. Tschöpe et al. performed a series of studies on cerium oxide grain boundaries, where the potential profile across a tri-layer crystal is shown.68–70 However, details of how the core layer was treated were not given and the electric field in the core layer was not mentioned.68–70 Mebane et al. took another approach by treating the non-zero segregation energy in the core zone as a boundary condition. This approach bypasses the complication of solving the Poisson's equation with a step-function-like formation energy profile and does not account for the large electric field that arises in this core zone.71,72
The localized field is important in understanding localized processes at the interface. Recent studies have both measured experimentally73 and predicted theoretically74,75 the existence of such large electric fields at a water/oxide interface, an oxide surface, and oxide heterostructures, and their impact on the field-dependent properties such as local bonding interaction, dielectric constant, and 2D electron gas density. When these properties are of interest, accurate prediction of local electric field accounting for both perfect crystal contribution and defect contribution would be necessary. Here we address these two contributions for ZrO2/Cr2O3 system.
In this study, we explicitly included the variations of the potential and field inside core layer into the continuum model. Thus, Poisson's equation is adhered to and the displacement field D is continuous throughout the core layer and the space-charge layer. Based on our results, the potential drop in the core layer is on the order of 10−3–10−1 V, as such defects concentrations do not deviate much from the constant-potential approximation. The advantage of our method, however, is that we can now quantitatively evaluate the electric field at the interface caused by defect redistribution.
B. Defect concentrations
In this section we look at the calculated defect concentrations across ZrO2/Cr2O3 interface and deduce how Cr2O3 grown on the Cr secondary phase particles may affect the degradation processes in ZrO2 passive film formed on zirconium alloys which are used in nuclear reactors. The alloying of Fe and Cr to the zirconium alloys significantly enhances their corrosion resistance.41 Zircaloy-4, for example, contains 0.05–0.15% Cr and 0.12–0.18% Fe. These alloying elements have very low solubility in zirconium metal and precipitate as intermetallic secondary phase particles (SPPs) Zr(Cr,Fe)2. These intermetallic precipitates are incorporated in the oxide layer as it starts to grow, and later, these incorporated precipitates get oxidized. It has been observed that Cr2O3 will grow as an outer shell as Zr(Cr,Fe)2 precipitates are oxidized and thus, form an interface with ZrO2 host.40 The existence of these interfaces will impact the transport of oxygen, hydrogen, and electrons inside the ZrO2 layer and thus, affect the corrosion rate of the underlying zirconium alloy. To quantify this interfacial effect, defect concentrations in the space-charge and core layers are needed.In Fig. 6(a) we show the defect concentrations of undoped bulk m-ZrO2 at 1200 K and different oxygen partial pressures. ZrO2 exhibits oxygen deficiency at low PO2 with positively-charged oxygen vacancies compensating electrons. The dominant type of oxygen vacancy is  at PO2 < 10−15 atm and then switches to
at PO2 < 10−15 atm and then switches to 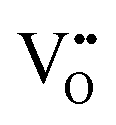 at higher oxygen partial pressures. ZrO2 becomes an intrinsic semiconductor at PO2 > 10−7 atm where electronic defects (holes and electrons) predominate. The results here differ from our previously reported result for bulk m-ZrO238 because the vibrational free energy is excluded here in order to be consistent with the results for the interface, where vibrational free energy calculations are prohibitively too expensive.
at higher oxygen partial pressures. ZrO2 becomes an intrinsic semiconductor at PO2 > 10−7 atm where electronic defects (holes and electrons) predominate. The results here differ from our previously reported result for bulk m-ZrO238 because the vibrational free energy is excluded here in order to be consistent with the results for the interface, where vibrational free energy calculations are prohibitively too expensive.
 | ||
| Fig. 6 Defect concentrations at 1200 K and different oxygen partial pressures in ZrO2 (a) in bulk, (b) far from the space-charge zone but within the reach of Cr defects, (c) outside the core layer within the space-charge zone, and (d) in the core layer. | ||
When ZrO2 is brought into contact with Cr2O3, chromium ions diffuse into ZrO2 and introduce chromium substitutional defects. We assume the diffusion length LD of defects are longer than the space-charge zone width Lscl, thus complete equilibrium is reached. Far enough from the interface (Lscl < L < LD), local charge neutrality is maintained with addition of CrZr defects, as shown in Fig. 6(b). Chromium ions tend to stay in their +3 oxidation state, acting as acceptors in ZrO2. 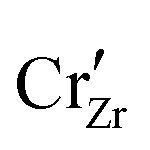 is compensated by oxygen vacancy at low PO2 (dominant charge states for the vacancy changes from +1 to +2 as PO2 gets higher). At much higher PO2
is compensated by oxygen vacancy at low PO2 (dominant charge states for the vacancy changes from +1 to +2 as PO2 gets higher). At much higher PO2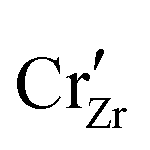 defects are compensated by holes. Dissolution of chromium into ZrO2 extends the regime of PO2 over which the oxygen vacancy predominates. Approaching to the interface in the space-charge layer in Fig. 6(c), positively-charged defects are depleted and negatively-charged defects are accumulated.
defects are compensated by holes. Dissolution of chromium into ZrO2 extends the regime of PO2 over which the oxygen vacancy predominates. Approaching to the interface in the space-charge layer in Fig. 6(c), positively-charged defects are depleted and negatively-charged defects are accumulated.  and electrons predominate in this zone, leading to a negatively-charged space-charge layer. In the core zone, CrZr defects are enriched significantly, as a sign of intermixing between ZrO2 and Cr2O3. Multiple oxidation states (0 to +3) of chromium exist in the core zone with concentrations much appreciable than the rest of ZrO2 thanks to their segregation energy in the core zone. Moreover
and electrons predominate in this zone, leading to a negatively-charged space-charge layer. In the core zone, CrZr defects are enriched significantly, as a sign of intermixing between ZrO2 and Cr2O3. Multiple oxidation states (0 to +3) of chromium exist in the core zone with concentrations much appreciable than the rest of ZrO2 thanks to their segregation energy in the core zone. Moreover 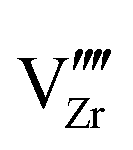 and
and 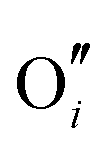 start to populate the core zone of ZrO2 at high PO2, showing that the segregation energies in the core zone not only affect extrinsic defects, but intrinsic ones, as well. By tracing the dominant defect in the core zone as a function of PO2 we observe a switch from doubly charged
start to populate the core zone of ZrO2 at high PO2, showing that the segregation energies in the core zone not only affect extrinsic defects, but intrinsic ones, as well. By tracing the dominant defect in the core zone as a function of PO2 we observe a switch from doubly charged  when PO2 < 10−7 atm to quadruply charged
when PO2 < 10−7 atm to quadruply charged 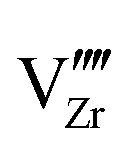 when PO2 > 10−7 atm. This shows the strong coupling between thermodynamic conditions, defect chemistry and the electrostatics of oxide heterojunctions with implications for the possibility of experimentally controlling these junctions via controlling growth conditions.
when PO2 > 10−7 atm. This shows the strong coupling between thermodynamic conditions, defect chemistry and the electrostatics of oxide heterojunctions with implications for the possibility of experimentally controlling these junctions via controlling growth conditions.
Similar analysis can be done on the Cr2O3 side, as shown in Fig. 7. In undoped bulk Cr2O3, there is no significant concentration of ionic defects (Fig. 7(a)) and only electronic defects predominate. This is consistent with the conclusions of the conductivity experiments conducted on undoped Cr2O3.76 Far away from the interface such that local charge neutrality is maintained but within the diffusion length of Zr defects, 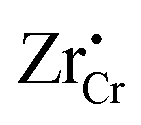 and
and 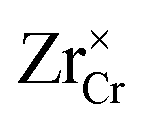 (Zr in +4 and +3 oxidation states, respectively) start to show up with appreciable concentrations. Getting closer to the interface, the bands of Cr2O3 bend up, and the concentration of the positively-charged
(Zr in +4 and +3 oxidation states, respectively) start to show up with appreciable concentrations. Getting closer to the interface, the bands of Cr2O3 bend up, and the concentration of the positively-charged 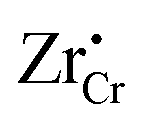 is increased. Inside the core layer, oxygen vacancies tend to segregate but the predomination in this zone remains for
is increased. Inside the core layer, oxygen vacancies tend to segregate but the predomination in this zone remains for  and holes. The core zone of Cr2O3 maintains the predomination of singly positively charged defect throughout all oxygen partial pressures considered here at 1200 K but with a switching from
and holes. The core zone of Cr2O3 maintains the predomination of singly positively charged defect throughout all oxygen partial pressures considered here at 1200 K but with a switching from  to holes at PO2 = 10−18 atm. This again brings up the rich coupling between thermodynamics, chemistry and electrostatics in oxide hetero-interfaces.
to holes at PO2 = 10−18 atm. This again brings up the rich coupling between thermodynamics, chemistry and electrostatics in oxide hetero-interfaces.
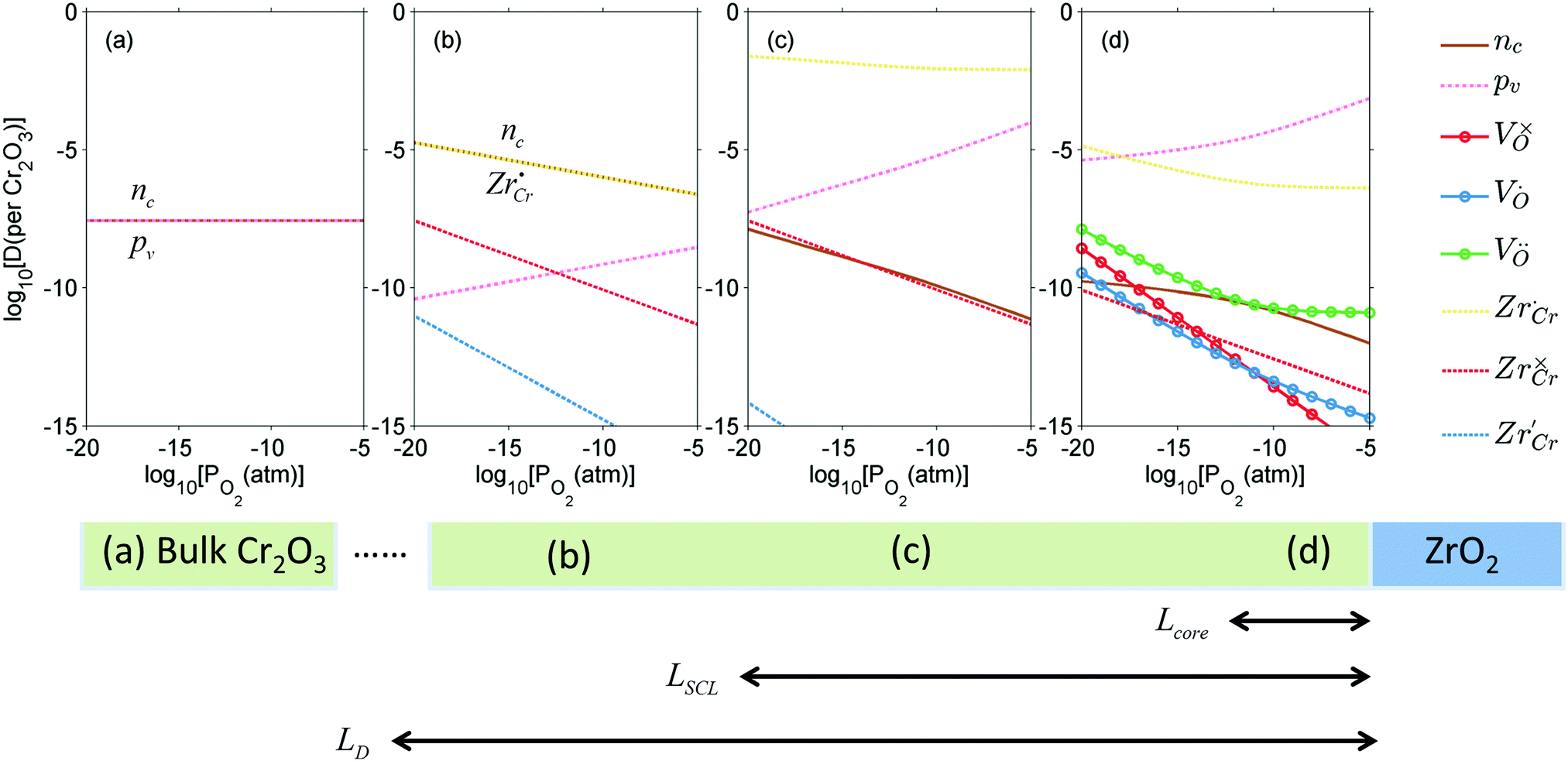 | ||
| Fig. 7 Defect concentrations in Cr2O3 at 1200 K and different oxygen partial pressures (a) in bulk, (b) far from the space-charge zone but within the reach of Zr defects, (c) outside the core layer within the space-charge zone, and (d) in the core layer. | ||
For symmetric grain boundaries in oxides it is customary to postulate that the core zone has a charge opposing and compensating that of the space charge zone.34,36 The symmetry of the grain boundary justifies this postulate. At a hetero-interface, however, the symmetry is broken and in the case of the ZrO2/Cr2O3 system studied here we found that the core and the space charge have similar charge (both are negative in the case of ZrO2 and both are positive in the case of Cr2O3). When the space charge and the core zone possess similar charges, the electric compensation occurs on the other side of the junction (the other oxide). In fact this charge compensation scenario in heterojunctions suggests the possibility of the presence of a thermodynamic point of zero charge in analogue with what happens when an oxide is immersed in an aqueous solution.77 That is a point in which the two sides of the junction are uncharged. We systematically varied temperature (between 400 K to 1400 K) and oxygen partial pressures (between 10−30 atm to 1 atm) to search for such a neutral point in the ZrO2/Cr2O3 system but did not observe it. Its absence in this system does not preclude its existence in other oxide hetero-interfaces.
We turn now to the implications of this analysis on zirconium alloys in nuclear reactors. The growth rate of zirconium oxide layer is thought to be limited by either electron transport or oxygen diffusion in ZrO2.78Fig. 8 shows the concentrations of free electrons, holes and 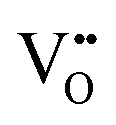 throughout the heterojunction at various oxygen partial pressures at 1200 K. By looking at the concentration distribution of oxygen-related defects and electronic defects, we can analyze how electron transport and oxygen diffusion are changed in the space-charge layer of ZrO2 compared to its bulk due to the change in the concentration of these defects in the space-charge layer. In Fig. 8(a), because the bands bend down on the ZrO2 side, the concentration of electrons is increased in the space charge layer compared to bulk, and subsequently holes are decreased as well in this layer. This enhancement of electron concentration has a favorable effect on preventing hydrogen, generated by the corrosion reaction, from getting into ZrO2.38,79 In addition, the sum of electron and hole concentrations is plotted in Fig. 8(b). If we assume that the mobilities of electrons and holes are on the same order of magnitude, this quantity should give us an estimation of the change in total electronic conductivity in the space-charge layer. Fig. 8(b) shows that when PO2 < 10−12 atm, the space charge layer of ZrO2 is in its n-type regime. In this case, the total electronic conductivity in the space-charge zone is increased. On the other hand, when ZrO2 becomes p-type at higher PO2, the total electronic conductivity is decreased in the space-charge layer in comparison to bulk due the downward band bending. If electron transport is the rate limiting step of the corrosion reaction, then the oxidized Cr precipitates lying in the oxygen rich, and hence the p-type part of ZrO2, closer to the interface with water coolant in the nuclear reactor, have a desirable of effect of slowing down the electronic conductivity and consequently reducing the corrosion rate. In fact, the ZrO2 oxide layer grown is exposed to a gradient in oxygen chemical potential between an oxygen poor zone at the interface with the underlying Zr alloy and an oxygen rich zone at the interface with water. As such, even if the electronic conductivity is enhanced around the precipitates in the oxygen-poor part of ZrO2 (n-type), it is inevitable that this conductivity is reduced around the precipitates in the oxygen-rich part (p-type).
throughout the heterojunction at various oxygen partial pressures at 1200 K. By looking at the concentration distribution of oxygen-related defects and electronic defects, we can analyze how electron transport and oxygen diffusion are changed in the space-charge layer of ZrO2 compared to its bulk due to the change in the concentration of these defects in the space-charge layer. In Fig. 8(a), because the bands bend down on the ZrO2 side, the concentration of electrons is increased in the space charge layer compared to bulk, and subsequently holes are decreased as well in this layer. This enhancement of electron concentration has a favorable effect on preventing hydrogen, generated by the corrosion reaction, from getting into ZrO2.38,79 In addition, the sum of electron and hole concentrations is plotted in Fig. 8(b). If we assume that the mobilities of electrons and holes are on the same order of magnitude, this quantity should give us an estimation of the change in total electronic conductivity in the space-charge layer. Fig. 8(b) shows that when PO2 < 10−12 atm, the space charge layer of ZrO2 is in its n-type regime. In this case, the total electronic conductivity in the space-charge zone is increased. On the other hand, when ZrO2 becomes p-type at higher PO2, the total electronic conductivity is decreased in the space-charge layer in comparison to bulk due the downward band bending. If electron transport is the rate limiting step of the corrosion reaction, then the oxidized Cr precipitates lying in the oxygen rich, and hence the p-type part of ZrO2, closer to the interface with water coolant in the nuclear reactor, have a desirable of effect of slowing down the electronic conductivity and consequently reducing the corrosion rate. In fact, the ZrO2 oxide layer grown is exposed to a gradient in oxygen chemical potential between an oxygen poor zone at the interface with the underlying Zr alloy and an oxygen rich zone at the interface with water. As such, even if the electronic conductivity is enhanced around the precipitates in the oxygen-poor part of ZrO2 (n-type), it is inevitable that this conductivity is reduced around the precipitates in the oxygen-rich part (p-type).
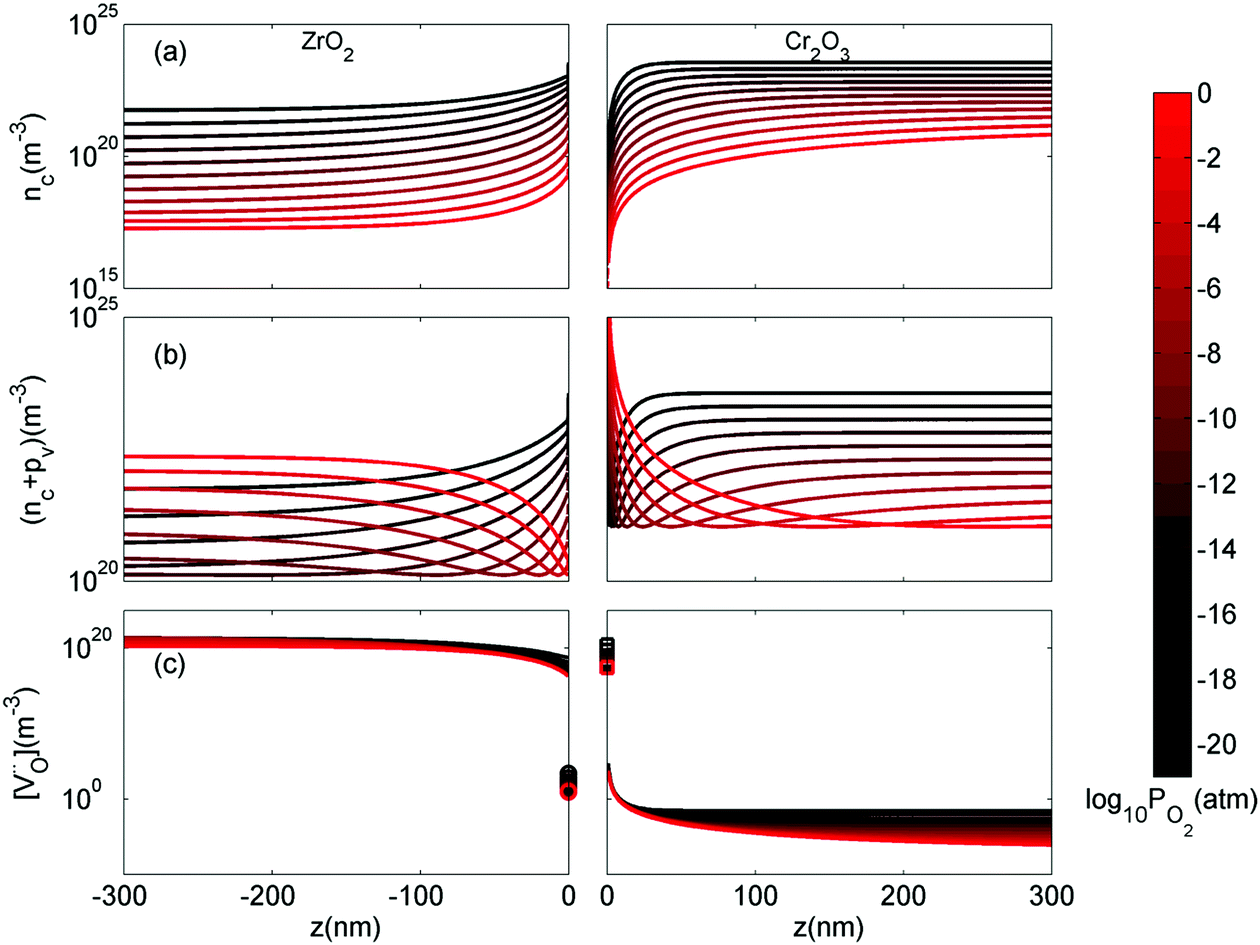 | ||
Fig. 8 Spatially-resolved concentrations of (a) electrons, (b) sum of electrons and holes and (c) 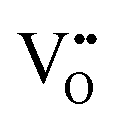 as a function of oxygen partial pressure at T = 1200 K. Circles and squares in panel (c) represent the concentrations in the core layer on the ZrO2 and Cr2O3 sides, respectively. as a function of oxygen partial pressure at T = 1200 K. Circles and squares in panel (c) represent the concentrations in the core layer on the ZrO2 and Cr2O3 sides, respectively. | ||
Cr2O3 has low oxygen diffusivity (∼10−16 cm2 s−1 at 1200 K, 0.1 atm80) compared to monoclinic ZrO2 (∼10−13 cm2 s−1 at 1200 K, 0.4 atm81), which indicate that the in-precipitate oxygen diffusion should be negligible, especially that the concentration of 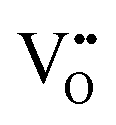 in Cr2O3 is several orders of magnitude lower than in ZrO2 as shown in Fig. 8(c). Since the precipitates are discrete particles that do not form a continuous path through which oxygen ions can diffuse, we focus here on oxygen diffusion in the matrix, ZrO2. The extended space charge zones around the oxidized Cr precipitates can form a significant fraction of the matrix with possibilities for space charge percolation. Fig. 8(c) shows that oxygen vacancies are strongly depleted throughout the space-charge layer and in particular in the core zone on ZrO2 side, which indicates that oxygen diffusion in ZrO2 is suppressed around the Cr oxidized precipitates. This observation suggests that if oxygen diffusion is the rate limiting step of the corrosion reaction, then the presence of oxidized Cr precipitates has also a desirable effect of slowing down oxygen diffusion in the extended space charge zones around them. While the conclusions here are based on the defect profiles evaluated at 1200 K, they are still applicable to lower temperatures closer to the actual reactor operation temperature (see ESI,† Section S4 for calculated profiles at 800 K). Modeling the space charge zone at lower temperatures becomes more challenging in the dilute limit because the difference in the spatial extent of the core layer and the space charge layer becomes increasingly large.
in Cr2O3 is several orders of magnitude lower than in ZrO2 as shown in Fig. 8(c). Since the precipitates are discrete particles that do not form a continuous path through which oxygen ions can diffuse, we focus here on oxygen diffusion in the matrix, ZrO2. The extended space charge zones around the oxidized Cr precipitates can form a significant fraction of the matrix with possibilities for space charge percolation. Fig. 8(c) shows that oxygen vacancies are strongly depleted throughout the space-charge layer and in particular in the core zone on ZrO2 side, which indicates that oxygen diffusion in ZrO2 is suppressed around the Cr oxidized precipitates. This observation suggests that if oxygen diffusion is the rate limiting step of the corrosion reaction, then the presence of oxidized Cr precipitates has also a desirable effect of slowing down oxygen diffusion in the extended space charge zones around them. While the conclusions here are based on the defect profiles evaluated at 1200 K, they are still applicable to lower temperatures closer to the actual reactor operation temperature (see ESI,† Section S4 for calculated profiles at 800 K). Modeling the space charge zone at lower temperatures becomes more challenging in the dilute limit because the difference in the spatial extent of the core layer and the space charge layer becomes increasingly large.
It is worth noting that the above analysis considers chromium as the only alloying element in ZrO2, and Zr as the only alloying element in Cr2O3. In actual zirconium alloys used by the nuclear industry, other alloying elements dissolving into ZrO2 can also impact its defect chemistry and corrosion resistance.38,82 Fe alone (zircaloy-4) or Ni and Fe (zircaloy-2) are key alloying elements in Zr alloys that contain Cr as well. In addition, hydrogen from the aqueous environment has a non-negligible contribution to the defect chemistry. Recently we have established the defect equilibria in M-ZrO2 when it is co-doped with hydrogen and 3d transition metals.38 Future work will build on the analysis presented here and our previous work, to quantify the combined effect of these doping elements on the interfacial defect distributions and electrical properties in oxide hetero-interfaces including ZrO2.
C. Potential drop, electric field and interface dipole: finite temperature vs. 0 K
In this study, we have obtained two sets of solutions of Poisson's equation: (1) from first-principle calculations on the defect-free interface at 0 K, and (2) from continuum level modeling of defect equilibria, where ionic and electronic defects redistribute due to chemical and electrical forces and reach thermal equilibrium at finite-temperature. Therefore, our multi-scale method allows us to separate the contributions of the perfect crystal (0 K) and the defect formation and segregation (finite T) on the potential drop, the electric field, and the interface dipole obtained from solving Poisson's equation. These three quantities are all experimentally measurable83 and are important for understanding the physical and chemical properties of hetero-structures.84,85 In this section we compare the origins and consequences of these two contributions. The summary of the comparison is shown in Table 3.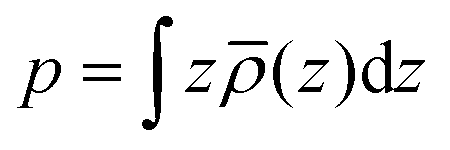
| Potential drop (V) | Maximum electric field (V m−1) | Dipole density (e m−1) | |
|---|---|---|---|
| Perfect interface | −3.8 | 4.9 × 109 | 3.0 × 1011 |
| Defect contribution | 2.0 | −4.3 × 108 | 1.2 × 109 |
Fig. 9 shows a comparison between the macroscopic averages of the solution of Poisson's equation of the defect-free interface and the solution of Poisson's equation for the charged defects at T = 1200 K and PO2 = 10−5 atm. The potential drop across perfect interface ΔV, as shown in Fig. 9(a), is −3.8 V. This potential drop results from the forming of polar bonds at the interface. Electrons distribute unevenly on the two sides of the interface and a localized interface dipole is built. Notice from Fig. 9 that the electrostatic potential becomes flat, and electric field and charge concentration of the defect-free interface become zero already at one atomic layer away from the interface. Because of the highly localized nature of this potential drop, electric field and charge concentrations have very high values right at the defect-free interface. In the system under consideration, the maximum electric field is 4.9 × 109 V m−1 and the interface dipole density is 3.0 × 1011 e m−1.
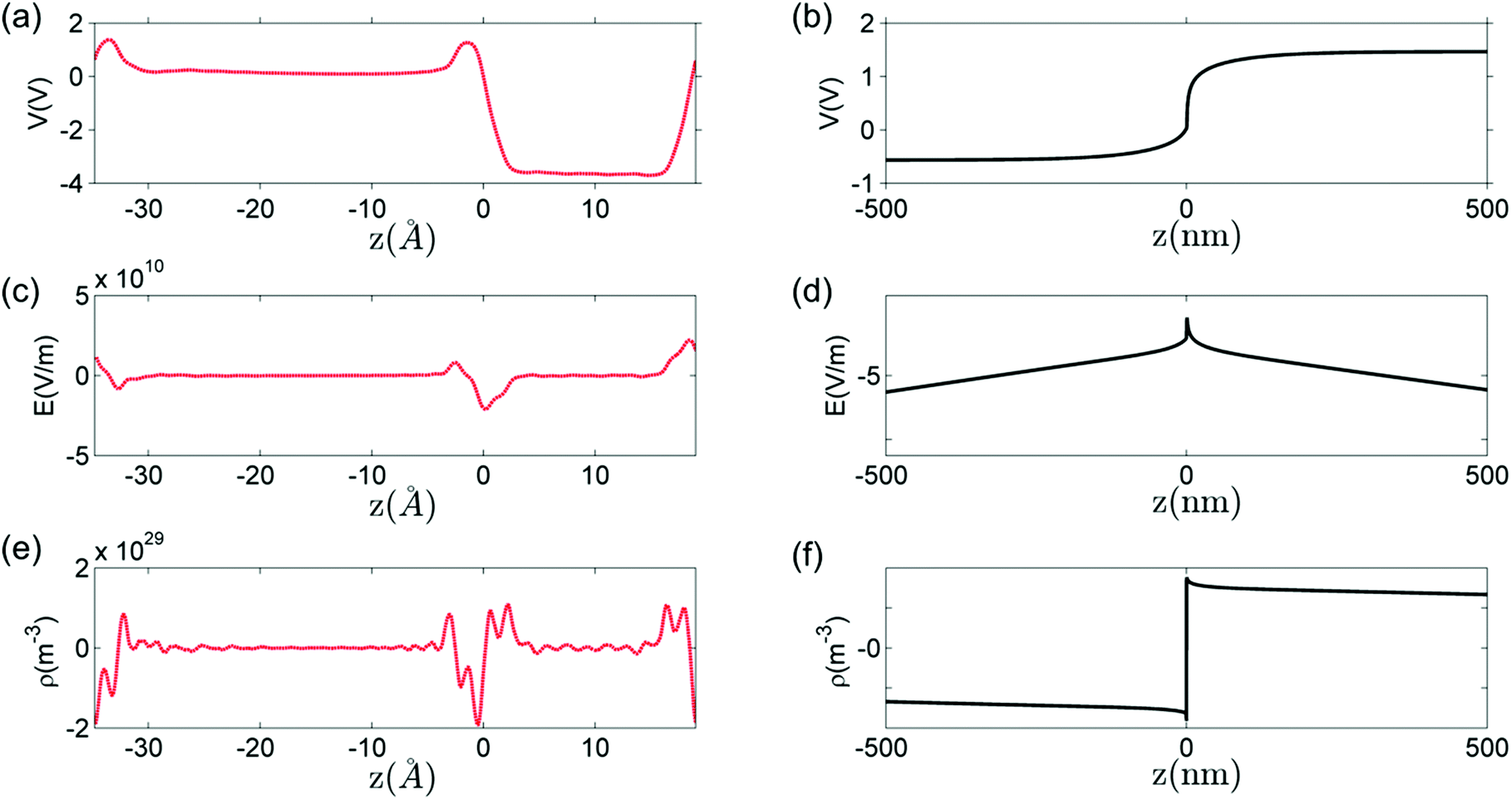 | ||
| Fig. 9 Macroscopic average of (a) electrostatic potential V, (c) electric field E and (e) charge density ρ from the perfect interface calculated by DFT at 0 K, compared with the same quantities (b), (d) and (f) contributed by the redistribution of defects at T = 1200 K and PO2 = 10−5 atm. | ||
In contrast, the effect of the defects is extended in space (hence the denotation space charge). As shown in Fig. 9, the length of space-charge layer is several hundred nanometers. The potential drop is 2.0 V across the space-charge layer, with only 0.2 V drop in the core zone. The maximum electric field is −4.3 × 108 V m−1 and the dipole density is 1.2 × 109 e m−1, which are order-of-magnitude lower than the perfect interface contribution. The two contributions are, however, mathematically additive as required by the superposition principle.86
These two effects coexist in every interface between two semiconductors (or insulators) and can both influence the interface properties. However, because of the difference in spatial scales of these two effects, either could be dominating at different spatial positions around the interface. When electrostatic profiles are discussed in literature, these two effects are seldom distinguished or contrasted. We believe that the highly localized nature of the field and the dipole arising from the defect-free interface gives rise to the catalytic activity of exposed or barely hidden interfaces.87 It can also influence the transport processes right at the interface (parallel or across). On the other hand, the field and dipole generated by defects at finite temperatures influence the transport processes over an extended region around the interface.
D. Possible future improvements for the computational method
There are several improvements that could be made to further refine our predictions. First, Fermi–Dirac distribution could be used instead of Boltzmann distribution in calculating defect concentrations. This requires including explicit solution of the drift-diffusion equations for all defects. This is particularly important when the formation energies start approaching kBT. Second, we did not consider finite temperature effects in this work. To accurately evaluate defect concentrations at various temperatures, formation energy should be replaced by formation free energy, by adding the contribution of phonons and the electronic entropy.50 Third, it has been reported in literature that the formation energy of a point defect with respect to its distance to the interface should be an oscillating function, which gradually approaches its bulk value after a few atomic layers.25,30,88 In this work we only treat the first atomic layer as the core layer, which is the computationally affordable approximation for evaluating segregation energies for all types of defects in the system at this time. In principle we could obtain spatially-resolved defect formation energy profiles from first-principles calculations by inserting defects in each atomic layer and add as many core layers as needed into the continuum model. Fourth, we treated electrons and holes as free carriers in bulk, space-charge layer and the core zone and thus their concentrations were calculated by applying Fermi–Dirac distribution to the electronic density of states. However, it is known that these electronic defects can self-trap in polarizable oxides forming small polarons which is indeed the case for holes in monoclinic ZrO2.89 Small polarons should be treated as point defects when calculating their concentrations. The self-trapping energy for holes in monoclinic ZrO2 is only 0.13 eV89 indicating that the results presented here remain qualitatively correct even if self-trapping is accounted for. However, it is of interest to explore the differences in self-trapping energies between bulk and the core region in future work. Finally, more accurate electronic structure methods, such as hybrid functionals22 and G0W0,90 might be needed to correctly predict band alignment at the interface and electronic structures of the defects. Additional modeling difficulties could arise while going to more complicated oxide systems, but the framework provided here addresses defect chemistry at hetero-interface in a generalized way and could potentially be applied to other oxide interfaces and surfaces.Lastly, results for temperatures lower than 800 K are not presented because the space charge layer length becomes too large (∼100 microns at 600 K), requiring to use a very coarse mesh to do the calculations, and also because these really large space charge zones are beyond the thickness of the corrosion passive films. In reality, the space charge length could be significantly reduced by other alloying elements that exist in the system, which is not considered in this work.
IV. Conclusion
In this paper, we implemented a thorough framework to predict the equilibrium defect concentrations across a hetero-interface starting from first principles-based calculations. Our method takes into account both long-range Coulomb interaction in the space charge layer and the localized effects of band discontinuity and bonding environment variation, allowing for a comprehensive description of defect redistribution across hetero-interfaces.The addition of core layers explicitly into the solution provides a closer view on the electric field and defect chemistry in the vicinity of the interface. Our model features a consistent treatment of Poisson's equation throughout the core and space charge layers preserving the continuity of the displacement field, whereas prior studies treat the core layer as a field-free homogeneous zone. Our results suggest that the variation in the formation energy of defects can be up to several eVs in the core region, causing a localized change in the electrostatic potential on the order of 0.1 V inside the core zone. Consequently, a giant electric field arises in this core zone and hence it deserves explicit modeling when studying oxide interfaces.
Our selected model interface, ZrO2/Cr2O3, is of interest to understand and improve the performance of zirconium alloys as nuclear cladding materials. This interface arises from the oxidation of the host Zr and the Cr secondary phase particles embedded inside the host. Here we clarified the beneficial effect of this interface in improving the corrosion and hydrogen resistance of Zr alloys. In particular, in the extended space charge layer around these interfaces and under oxygen rich conditions, we predict a decrease in the total electronic defect concentrations and in the oxygen vacancy concentration. Both of these results should decrease the metal oxidation kinetics. In spite of the reduction in the sum of electronic charge carriers under oxygen rich conditions, we found an increase of free electron concentration in the ZrO2 space-charge zone compared to its bulk for all oxygen partial pressures. This effect should promote proton reduction and hydrogen evolution at the surface, and thus, suppress hydrogen pickup into the alloy. Both effects are beneficial towards protecting the integrity of zirconium alloys. The model and the analyses presented here provide a utility to understand the effect of alloying elements on the corrosion resistance of zirconium alloys when these alloying elements tend to precipitate as secondary phase particles and oxidize within the ZrO2 passive layer.
This systematic study also facilitates a comparison of two contributions to the interfacial electrostatic properties; a 0 K contribution originating from the formation of a perfectly abrupt interface, and a finite temperature excitation due to the equilibration of electronic and ionic defects. Finally, we discussed several improvements to enhance the predictive power of the herein presented model. Although here we presented the application of the model to the problem of zirconium alloy corrosion resistance, we believe that it is broadly applicable to oxide interfaces arising in other important technologies such as lithium batteries, fuel cells, and computer memories.
Acknowledgements
This work is supported by the Consortium for Advanced Simulation of Light Water Reactors (CASL), an Energy Innovation Hub for Modeling and Simulation of Nuclear Reactors under U.S. Department of Energy Contract No. DE-AC05-00OR22725. We acknowledge the Extreme Science and Engineering Discovery Environment (XSEDE) program for calculations performed under allocation No. TG-DMR120025.References
- H. Y. Hwang, Y. Iwasa, M. Kawasaki, B. Keimer, N. Nagaosa and Y. Tokura, Nat. Mater., 2012, 11, 103–113 CrossRef CAS PubMed.
- J. Mannhart and D. G. Schlom, Science, 2010, 327, 1607–1611 CrossRef CAS PubMed.
- S. V. Kalinin, A. Borisevich and D. Fong, ACS Nano, 2012, 6, 10423–10437 CrossRef CAS PubMed.
- S. V. Kalinin and N. A. Spaldin, Science, 2013, 341, 858–859 CrossRef CAS PubMed.
- F. Baiutti, G. Logvenov, G. Gregori, G. Cristiani, Y. Wang, W. Sigle, P. A. van Aken and J. Maier, Nat. Commun., 2015, 6, 8586 CrossRef CAS PubMed.
- Q. Cao, Y.-F. Cheng, H. Bi, X. Zhao, K. Yuan, Q. Liu, Q. Li, M. Wang and R. Che, J. Mater. Chem. A, 2015, 3, 20051–20055 CAS.
- D. Colleoni, G. Miceli and A. Pasquarello, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 125304 CrossRef.
- J. Maier, Physical chemistry of ionic materials: ions and electrons in solids, John Wiley & Sons, 2004 Search PubMed.
- J. Maier, Solid State Ionics, 1994, 70–71(Pt 1), 43–51 CrossRef CAS.
- J. Maier, Prog. Solid State Chem., 1995, 23, 171–263 CrossRef CAS.
- J. Maier, Solid State Ionics, 2001, 143, 17–23 CrossRef CAS.
- B. Van Zeghbroeck, Principles of semiconductor devices, 2004 Search PubMed.
- P. A. Markowich, C. A. C. A. Ringhofer and C. C. Schmeiser, Semiconductor equations, Springer-Verlag, 1990 Search PubMed.
- R. F. Pierret, Semiconductor device fundamentals, Addison Wesley, 1996 Search PubMed.
- A. Franciosi and C. G. Van de Walle, Surf. Sci. Rep., 1996, 25, 1–140 CrossRef CAS.
- C. Freysoldt, B. Grabowski, T. Hickel, J. Neugebauer, G. Kresse, A. Janotti and C. G. Van de Walle, Rev. Mod. Phys., 2014, 86, 253–305 CrossRef.
- M. Stengel and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 241103 CrossRef.
- R. A. De Souza, V. Metlenko, D. Park and T. E. Weirich, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 174109 CrossRef.
- J. Maier, Nat. Mater., 2005, 4, 805–815 CrossRef CAS PubMed.
- S. Kim, J. Fleig and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 2268–2273 RSC.
- C. G. Van de Walle and R. M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 8154–8165 CrossRef CAS.
- A. Alkauskas, P. Broqvist, F. Devynck and A. Pasquarello, Phys. Rev. Lett., 2008, 101, 106802 CrossRef PubMed.
- J. C. Conesa, J. Phys. Chem. C, 2012, 116, 18884–18890 CAS.
- S. Saraf, M. Markovich and A. Rothschild, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245208 CrossRef.
- S. Kasamatsu, T. Tada and S. Watanabe, e-J. Surf. Sci. Nanotechnol., 2010, 8, 93–100 CrossRef CAS.
- A. R. Hassan, A. El-Azab, C. Yablinsky and T. Allen, J. Solid State Chem., 2013, 204, 136–145 CrossRef CAS.
- J. M. Polfus, T. S. Bjorheim, T. Norby and R. Bredesen, J. Mater. Chem. A, 2016, 4, 7437–7444 CAS.
- D. M. Duffy, J. H. Harding and A. M. Stoneham, J. Appl. Phys., 1994, 76, 2791–2798 CrossRef CAS.
- S. Kasamatsu, T. Tada and S. Watanabe, Solid State Ionics, 2011, 183, 20–25 CrossRef CAS.
- Y. Kuru, D. Marrocchelli, S. R. Bishop, D. Chen, B. Yildiz and H. L. Tuller, J. Electrochem. Soc., 2012, 159, F799–F803 CrossRef CAS.
- T. Tauer, R. O'Hayre and J. W. Medlin, J. Mater. Chem. A, 2013, 1, 2840–2846 CAS.
- K. Yang, J. R. East and G. I. Haddad, Solid-State Electron., 1993, 36, 321–330 CrossRef CAS.
- K. Horio and H. Yanai, IEEE Trans. Electron Devices, 1990, 37, 1093–1098 CrossRef.
- R. A. De Souza, Phys. Chem. Chem. Phys., 2009, 11, 9939–9969 RSC.
- J. M. Polfus, K. Toyoura, F. Oba, I. Tanaka and R. Haugsrud, Phys. Chem. Chem. Phys., 2012, 14, 12339–12346 RSC.
- M. C. Gobel, G. Gregori and J. Maier, Phys. Chem. Chem. Phys., 2014, 16, 10214–10231 RSC.
- J. Jamnik, J. Maier and S. Pejovnik, Solid State Ionics, 1995, 75, 51–58 CrossRef CAS.
- M. Youssef, M. Yang and B. Yildiz, Phys. Rev. Appl., 2016, 5, 014008 CrossRef.
- A. Yilmazbayhan, E. Breval, A. T. Motta and R. J. Comstock, J. Nucl. Mater., 2006, 349, 265–281 CrossRef CAS.
- Y. Hatano and M. Sugisaki, J. Nucl. Sci. Technol., 1997, 34, 264–268 CrossRef CAS.
- D. Pêcheur, F. Lefebvre, A. T. Motta, C. Lemaignan and J. F. Wadier, J. Nucl. Mater., 1992, 189, 318–332 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- F. Lebreau, M. M. Islam, B. Diawara and P. Marcus, J. Phys. Chem. C, 2014, 118, 18133–18145 CAS.
- M. Youssef and B. Yildiz, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 144109 CrossRef.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- M. W. Chase, S. National Institute of and Technology, NIST-JANAF thermochemical tables, American Chemical Society; American Institute of Physics for the National Institute of Standards and Technology, Washington, D.C., Woodbury, N.Y., 1998 Search PubMed.
- A. Christensen and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 8050–8064 CrossRef CAS.
- N. J. Mosey and E. A. Carter, J. Mech. Phys. Solids, 2009, 57, 287–304 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. A. J. Fisher and H. Matsubara, Philos. Mag., 2005, 85, 1067–1088 CrossRef CAS.
- B. E. Gaddy, E. A. Paisley, J.-P. Maria and D. L. Irving, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 125403 CrossRef.
- K. Steiner, W. Chen and A. Pasquarello, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 205309 CrossRef.
- S. Baroni, R. Resta, A. Baldereschi and M. Peressi, in Spectroscopy of Semiconductor Microstructures, ed. G. Fasol, A. Fasolino and P. Lugli, Springer, US, 1989, ch. 16, vol. 206, pp. 251–271 Search PubMed.
- B. Van Zeghbroeck, Principles of semiconductor devices, Colarado University, 2004 Search PubMed.
- S. P. Waldow and R. A. De Souza, ACS Appl. Mater. Interfaces, 2016, 8, 12246–12256 CAS.
- S. Kim and J. Maier, J. Electrochem. Soc., 2002, 149, J73–J83 CrossRef CAS.
- R. A. De Souza, F. Gunkel, S. Hoffmann-Eifert and R. Dittmann, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 241401 CrossRef.
- E. E. Helgee, A. Lindman and G. Wahnström, Fuel Cells, 2013, 13, 19–28 CrossRef CAS.
- B. Joakim Nyman, E. E. Helgee and G. Wahnström, Appl. Phys. Lett., 2012, 100, 061903 CrossRef.
- X. Guo and J. Maier, Adv. Funct. Mater., 2009, 19, 96–101 CrossRef CAS.
- A. Tschöpe, S. Kilassonia and R. Birringer, Solid State Ionics, 2004, 173, 57–61 CrossRef.
- A. Tschöpe, MRS Online Proc. Libr., 2002, 756, EE4.2.1 Search PubMed.
- A. Tschöpe, C. Bäuerle and R. Birringer, J. Appl. Phys., 2004, 95, 1203–1210 CrossRef.
- D. S. Mebane, Comput. Mater. Sci., 2015, 103, 231–236 CrossRef CAS.
- D. S. Mebane and R. A. De Souza, Energy Environ. Sci., 2015, 8, 2935–2940 CAS.
- T. Siebert, B. Guchhait, Y. Liu, B. P. Fingerhut and T. Elsaesser, J. Phys. Chem. Lett., 2016, 3131–3136, DOI:10.1021/acs.jpclett.6b01369.
- K. Krishnaswamy, C. E. Dreyer, A. Janotti and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 085420 CrossRef.
- H. Peelaers, K. Krishnaswamy, L. Gordon, D. Steiauf, A. Sarwe, A. Janotti and C. G. Van de Walle, Appl. Phys. Lett., 2015, 107, 183505 CrossRef.
- A. Holt and P. Kofstad, Solid State Ionics, 1994, 69, 127–136 CrossRef CAS.
- J. Cheng and M. Sprik, Phys. Chem. Chem. Phys., 2012, 14, 11245–11267 RSC.
- B. Cox, J. Nucl. Mater., 2005, 336, 331–368 CrossRef CAS.
- A. Couet, A. T. Motta and R. J. Comstock, J. Nucl. Mater., 2014, 451, 1–13 CrossRef CAS.
- S. C. Tsai, A. M. Huntz and C. Dolin, J. Mater. Sci. Eng. A, 1996, 212, 6–13 CrossRef.
- A. Madeyski and W. W. Smeltzer, Mater. Res. Bull., 1968, 3, 369–375 CrossRef CAS.
- A. Couet, A. T. Motta, B. de Gabory and Z. Cai, J. Nucl. Mater., 2014, 452, 614–627 CrossRef CAS.
- S. Balaz, Z. Zeng and L. J. Brillson, J. Appl. Phys., 2013, 114, 183701 CrossRef.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 CAS.
- Y. Hikita, K. Nishio, L. C. Seitz, P. Chakthranont, T. Tachikawa, T. F. Jaramillo and H. Y. Hwang, Adv. Energy Mater., 2016, 6, 1502154 CrossRef.
- D. J. Griffiths, Introduction to electrodynamics, Prentice Hall, Upper Saddle River, NJ, 1999 Search PubMed.
- J. H. Lee and A. Selloni, Phys. Rev. Lett., 2014, 112, 196102 CrossRef PubMed.
- A. Lindman, E. E. Helgee and G. Wahnström, Solid State Ionics, 2013, 252, 121–125 CrossRef CAS.
- K. P. McKenna, M. J. Wolf, A. L. Shluger, S. Lany and A. Zunger, Phys. Rev. Lett., 2012, 108, 116403 CrossRef PubMed.
- R. Shaltaf, G. M. Rignanese, X. Gonze, F. Giustino and A. Pasquarello, Phys. Rev. Lett., 2008, 100, 186401 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp04997d |
| This journal is © the Owner Societies 2017 |