
Coordination polymers constructed from tetrahedral-shaped adamantane tectons†
Teodora
Mocanu
a,
Lidia
Pop
b,
Niculina D.
Hădade
b,
Sergiu
Shova
c,
Ion
Grosu
*b and
Marius
Andruh
*a
aInorganic Chemistry Laboratory, Faculty of Chemistry, University of Bucharest, Str. Dumbrava Rosie no. 23, 020464, Bucharest, Romania. E-mail: marius.andruh@dnt.ro
bCentre of Supramolecular Organic and Organometallic Chemistry (CCSOOM), Department of Chemistry, Faculty of Chemistry and Chemical Engineering, Babeş-Bolyai University, 11 Arany Janos, 400028, Cluj-Napoca, Romania
c“Petru Poni” Institute of Macromolecular Chemistry, Aleea Grigore Ghica Voda, 41A, 700487 Iasi, Romania
First published on 30th November 2016
Abstract
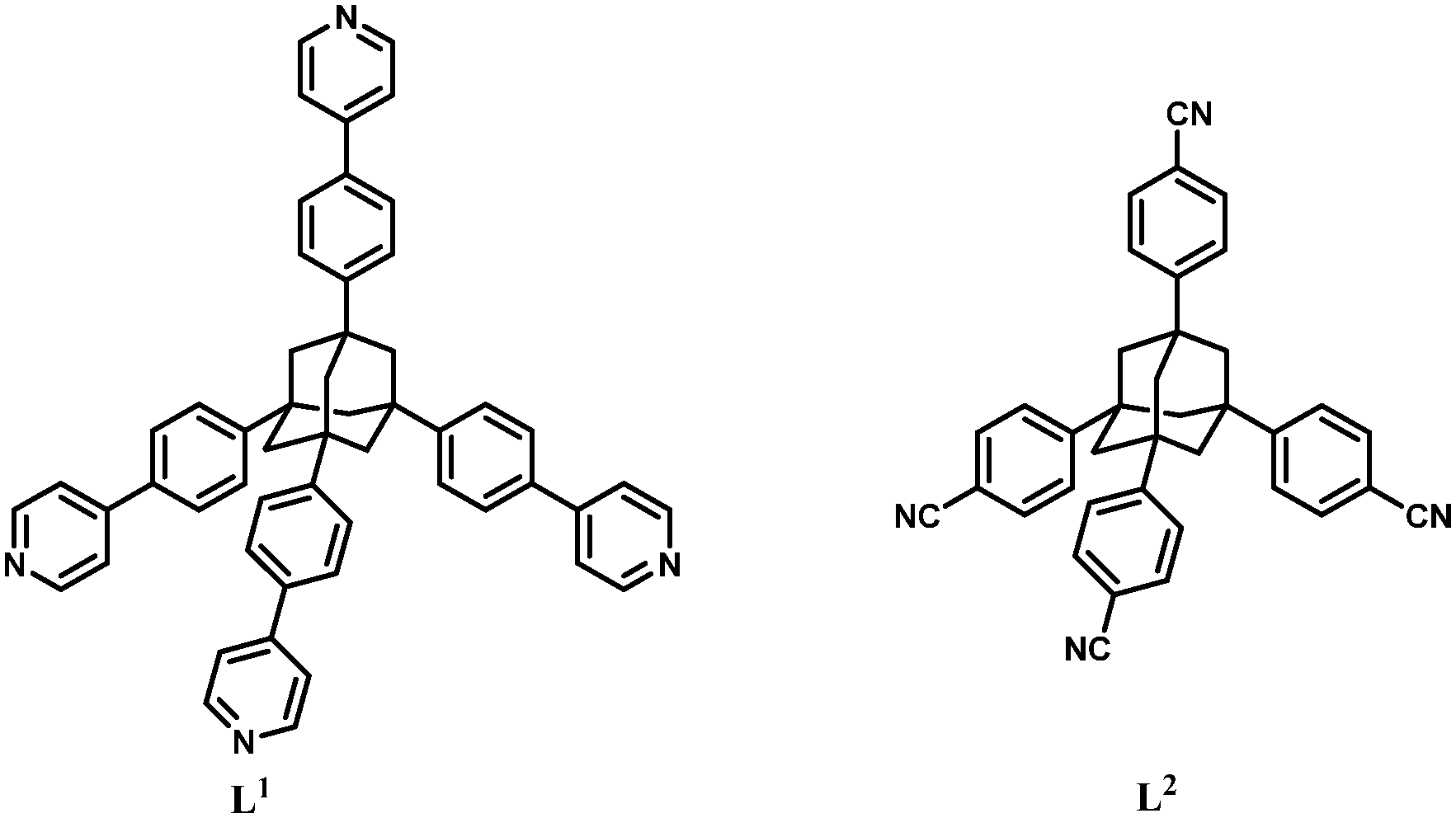
Two rigid tetrahedral organic linkers derived from adamantane have been employed in constructing a 3-D, 4-fold interpenetrated framework featuring a PtS topology, [CuL1(H2O)2](BF4)2·8H2O (1) (L1 = 1,3,5,7-tetrakis{4-(4-pyridyl)phenyl}adamantane), and a 2-fold interpenetrated grid-like coordination polymer, [Mn(hfac)2(L2)0.5] (2) (L2 = 1,3,5,7-tetrakis(4-cyanophenyl)adamantane).
Tetrahedral divergent ligands play an important role in crystal engineering.1 By developing the “node-and-spacer” approach, Robson has shown that coordination polymers featuring a diamond-like topology can be rationally obtained by combining two tectons: a metal ion with a preference for the Td coordination geometry, and an organic molecule with tetrahedrally-disposed coordination groups.2 Several other basic inorganic structures are constructed from tetrahedral tectons (mainly metal ions): lonsdaleite (lon), quartz (qtz), SrAl2 (sra) (tetrahedral nodes – uni- or binodal nets), PtS (pts) (a combination of tetrahedral and square planar nodes), Ge3N4 (tetrahedral and trigonal nodes in a 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio), fluorite (flu) (tetrahedral and cubic centres), etc.3 Even though a rich diversity of topologies can be envisioned, the number of coordination polymers described in the literature relying upon tetrahedral spacers is quite low.4 The network topologies are different from the diamondoid one when the metal ions have other stereochemical preferences.
:4 ratio), fluorite (flu) (tetrahedral and cubic centres), etc.3 Even though a rich diversity of topologies can be envisioned, the number of coordination polymers described in the literature relying upon tetrahedral spacers is quite low.4 The network topologies are different from the diamondoid one when the metal ions have other stereochemical preferences.
The coordinating-prone units attached to tetrahedral skeletons (methane, silane or adamantane derivatives) can be anionic (carboxylate,5 sulfonate,6 or phosphonate7–9 groups), or neutral (pyridine,10 triazole,11 and tetrazole8,12,13 fragments, or cyano groups2,14). Tetra-substituted adamantane derivatives are useful tectons in generating robust porous structures showing high connectivity topologies.6–8,12,14c In spite of the fact that rigid organic tetrahedral tectons are encoded to orient the donor atoms towards the vertices of a regular tetrahedron, thus extending the molecular assembling into three dimensions, organic ligands with a Td symmetry forming a 2-D layer have also been described.6a,14
Herein, we report the crystal structures of two new coordination polymers constructed from copper(II) ions and {MnII(hfac)2} entities acting as nodes: [CuL1(H2O)2](BF4)2·8H2O (1) and [Mn(hfac)2(L2)0.5] (2), where L1 and L2 are tetrahedral spacers having four phenyl–pyridyl (L1) or 4-cyanophenyl moieties (L2) attached to an adamantane backbone (Scheme 1). The two assembling species have been chosen from the following reasons: CuII adopts various coordination numbers and geometries, but, only rarely tetrahedral; {MnII(hfac)2} has two positions available for the interaction with the spacer (either cis or trans). In the first case, CuII, the topology of the resulting coordination polymer cannot be easily predicted. In the second case, a 3-D coordination polymer is expected, if the donor atoms arising from the spacer occupy the trans positions at the manganese ion, and a 2-D coordination polymer, when the donor atoms from the spacer occupy the cis positions.
 | ||
| Scheme 1 | ||
The two adamantane derivatives, L1 and L2, have been already described in the literature.8,15 In the case of tetrapyridyladamantane, L1 (Scheme S1†), relevant improvements of the synthesis, compared to the method reported by Müller,15 were elaborated. The employment of PdCl2(dppf) instead of Pd(PPh3)4 leads to an increase in the yield from 27 to 41%, a reduction of the reaction time from 72 to 12 h, and an easier chromatographic separation of the target compound.‡
The reaction of L1 with copper(II) tetrafluoroborate, in the presence of monoethanolamine, affords blue-green single crystals of [CuL1(H2O)2](BF4)2·8H2O 1.‡ The investigation of the crystal structure§ of 1 reveals that each L1 molecule connects four copper(II) ions (Fig. 1) in a 3-D network (Fig. 2). The copper ions display a distorted octahedral coordination geometry with four nitrogen atoms in the equatorial plane arising from four organic linker molecules (Cu1–N1 = 2.026(8) Å) and two semi-coordinated water molecules in trans positions (Cu1–O1W = 2.626(16) Å). The Cu⋯Cu distances are 18.130 Å (Cu1⋯Cu1a, a = 1 + x, 1 + y, z), and 20.744 Å (Cu1⋯Cu1b, b = 0.5 + x, 2.5 − y, 0.5 + z). The 3-D cationic framework can be described as a binodal (4,4)-connected network with a platinum sulfide topology, where L1 molecules play the role of tetrahedral sulfide nodes while the equatorial CuN4 units replace platinum square planar centres. The analysis of the packing diagram shows the interpenetration of four independent 3-D nets of 1 (Fig. 3). The structural changes of L1, in comparison with the homolog derived from methane,10b are reflected in an increase of the interpenetration degree. To the best of our knowledge, this is the highest interpenetration degree encountered for molecular assemblies built from a rigid organic linker with Td symmetry bearing pyridine moieties. The void space resulting upon interpenetration is filled with BF4− anions and solvent molecules. Since the number of the water molecules could not be directly determined by the crystallographic measurements, we estimated the presence of eight water molecules per formula (ESI†) from the data obtained by elemental and thermogravimetric analyses. The diffuse reflectance electronic spectrum of 1 (Fig. S3†) shows a band at 604 nm, due to the hexacoordinated CuII ions.
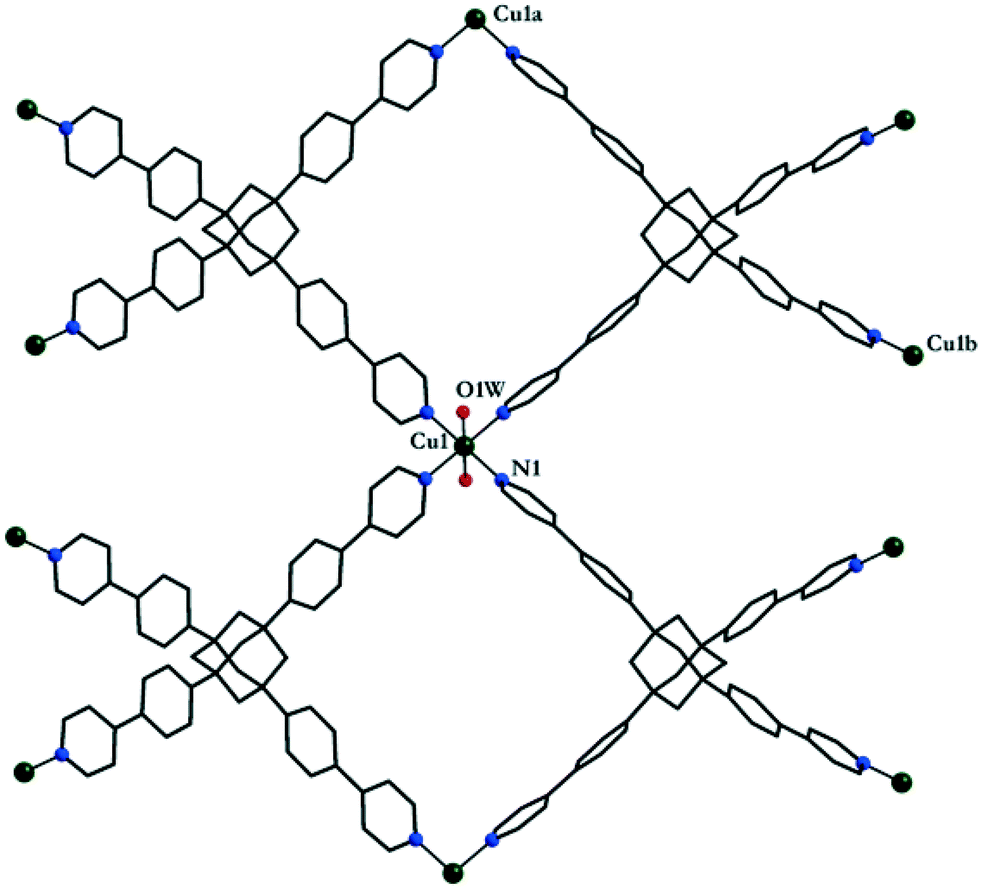 | ||
| Fig. 1 Perspective view of the coordination polymer in crystal 1 with the atom numbering scheme. | ||
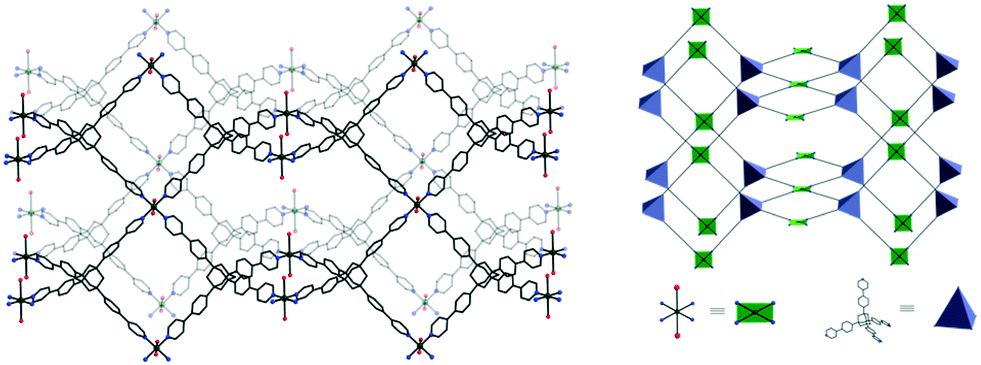 | ||
| Fig. 2 Perspective view of the 3-D coordination framework of 1 and the corresponding representation of the platinum sulfide topology. | ||
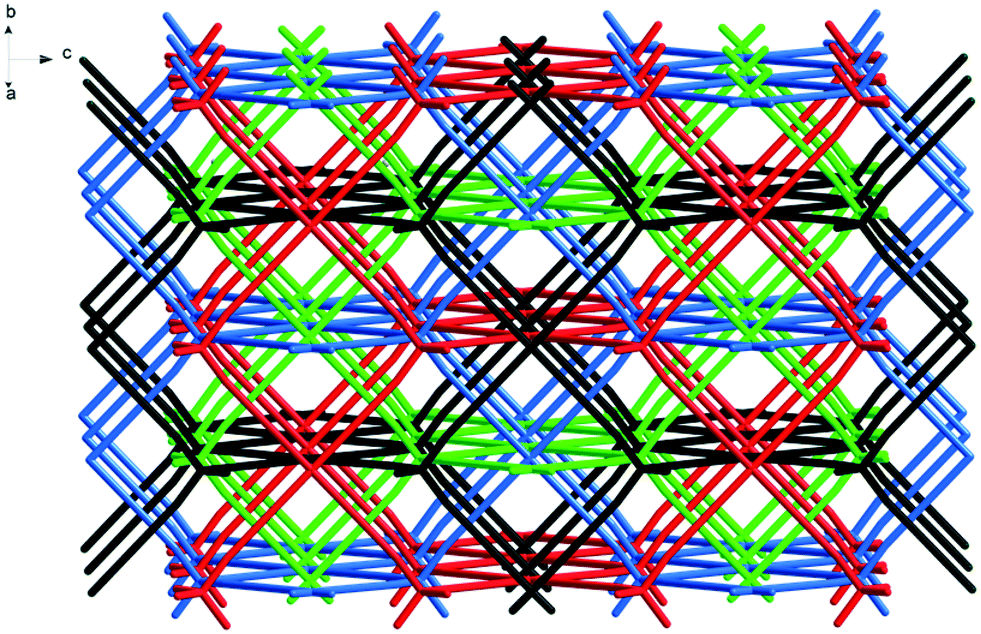 | ||
| Fig. 3 Crystal packing diagram in crystal 1, showing the four-fold interpenetration. | ||
Compound [Mn(hfac)2(L2)0.5] 2 crystallizes in the tetragonal centrosymmetric I41/a space group.§ Taking into consideration the weak coordinating ability of nitrile-containing molecules, we have employed a hexafluoroacetylacetonate metal complex, as the electron-withdrawing F atoms increase the Lewis acidity of the metal ion, thus favouring the coordination of the nitrile groups to the manganese ion, by replacing the weakly bonded aqua ligands. A neutral coordination structure is assembled by reacting [Mn(hfac)2(H2O)2] with L2 (Hhfac = 1,1,1,5,5,5-hexafluoro-2,4-pentanedione).‡
The {Mn(hfac)2} nodes are linked by L2 molecules resulting in neutral (4,4)-connected grid-like layers. The MnII ions adopt a slightly distorted octahedral geometry with two chelating hfac− ligands (Mn1–O1 = 2.128(4) Å, Mn1–O2 = 2.154(5) Å) and two nitrogen ligand atoms disposed in cis positions (Mn1–N1 = 2.239(7) Å) (Fig. 4). Since L2 acts as a 4-connecting knot, the low dimensionality of 2 can be ascribed exclusively to the cis-arrangement of nitrogen donor atoms in {Mn(hfac)2N2} units, while a trans coordination of the nitrogen L2 atoms to the metal centre would afford a 3-D net. In the case of honeycomb type layers14b,c where the tetrahedral-shaped ligand acts as a 3-connecting node, the promotion into the third dimension is disrupted by the presence of solvent molecules or coordinating anions. The inherent chirality of the cis-isomer of the metal building blocks enables the formation of chains where two L2 arms bridge metal centres of the same octahedral delta (Δ) conformation and the other two arms bridge metal centres of lambda (Λ) conformation. Viewed along the c axis, the structure of 2 can be regarded as sets of zig-zag chains constructed, each one, by MnII nodes with the same chirality, and running perpendicular to each other. Layers with meshes of square cross section are thus formed (Fig. 5). Two such layers interpenetrate (Fig. 6a). Channels within the ab plane are formed (Fig. 6b).
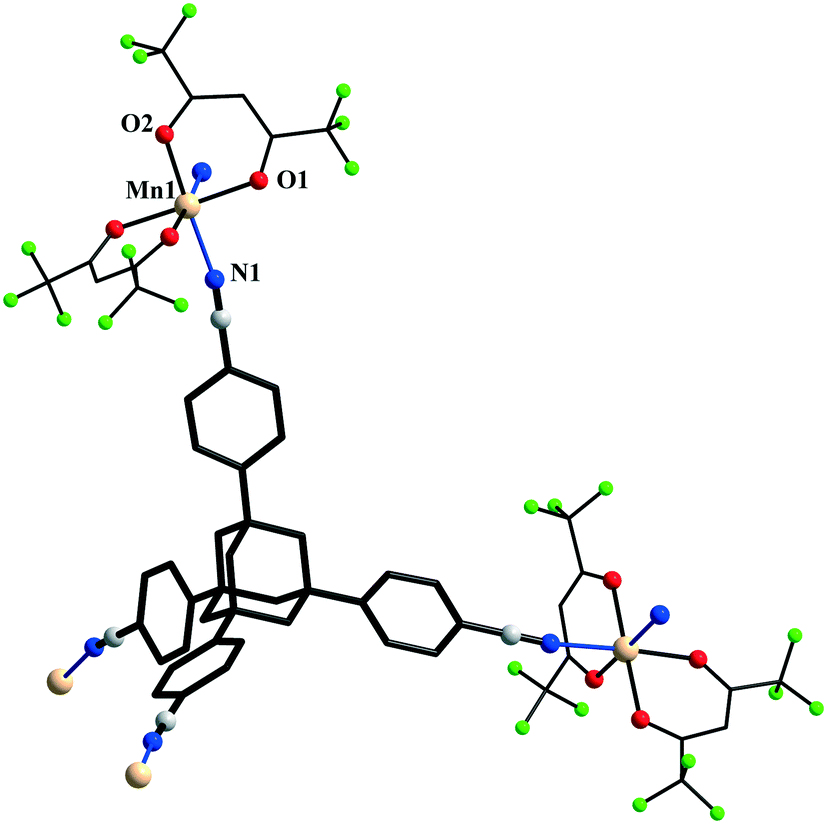 | ||
| Fig. 4 Fragment of the crystal structure of 2 showing the interaction of the tetrahedral ligand L2 with two {Mn(hfac)2} units of opposite chirality. | ||
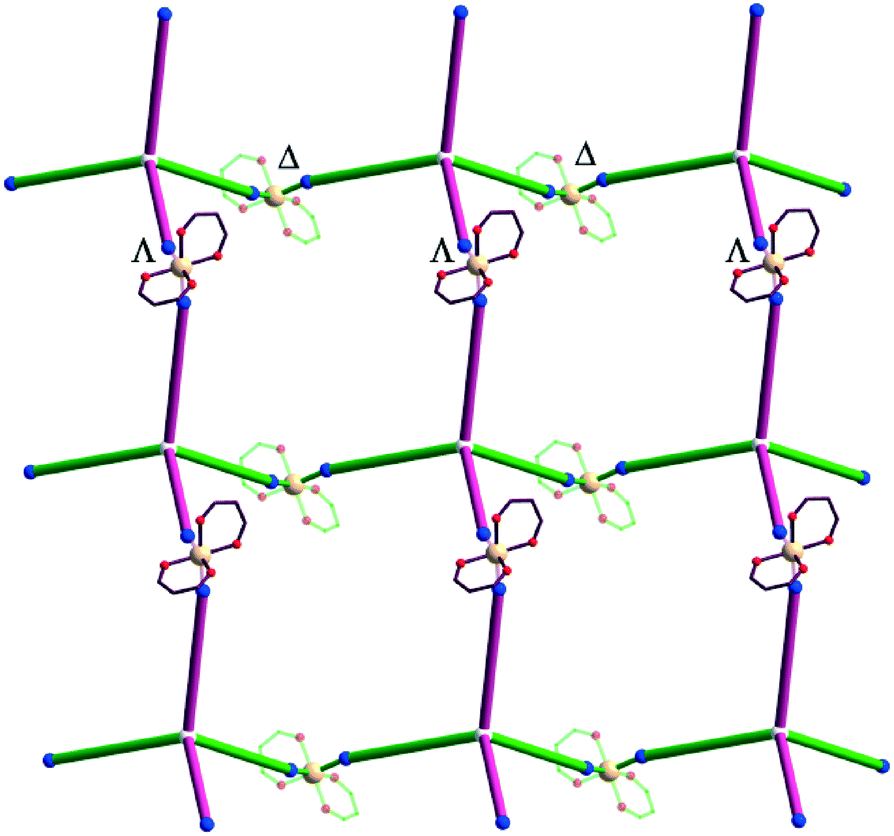 | ||
| Fig. 5 View of a layer in crystal 2 (see text: green – delta, violet – lambda configuration) (CF3 groups were omitted for clarity). | ||
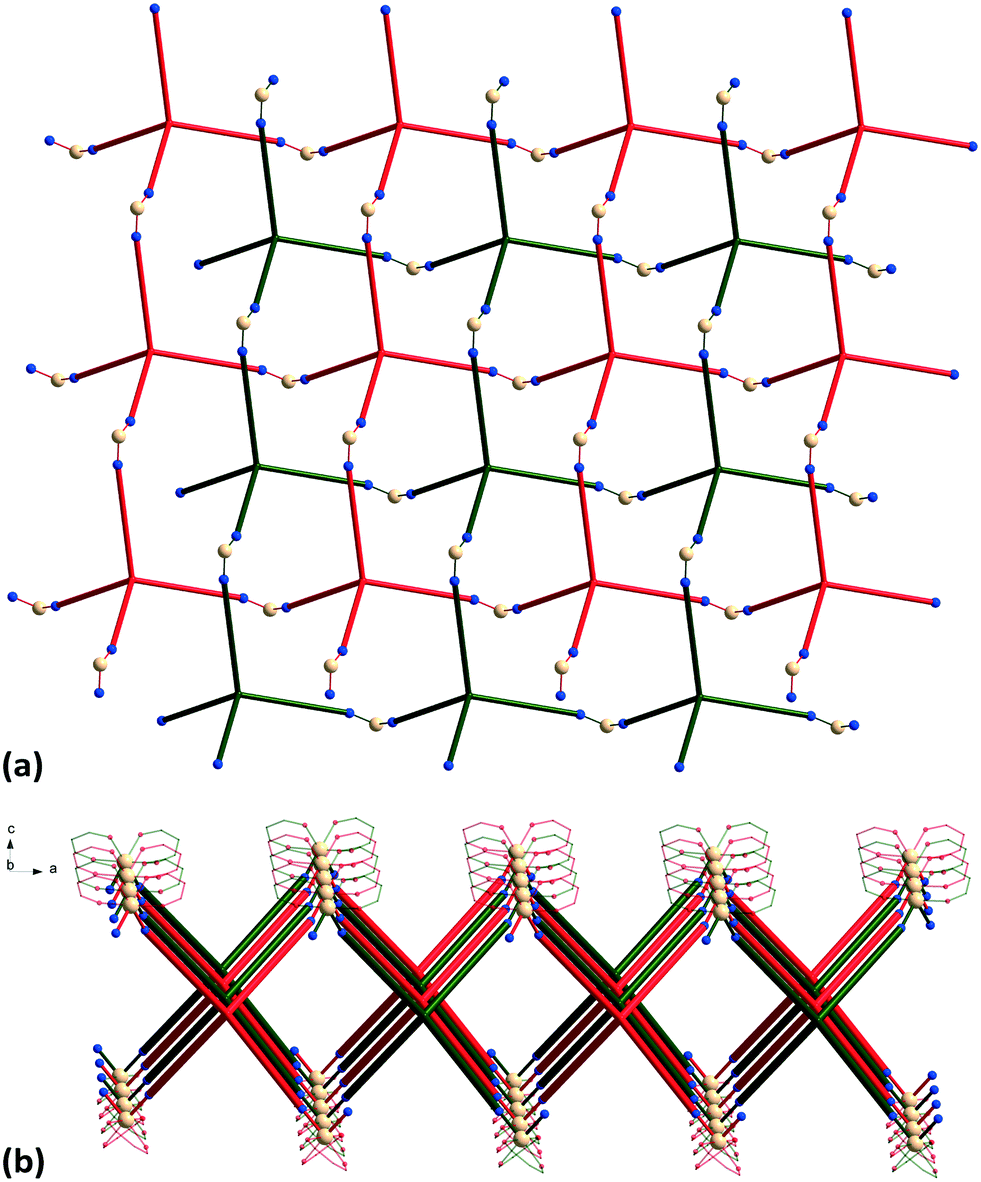 | ||
| Fig. 6 (a) Packing diagram of 2 showing the two-fold interpenetration of 2-D nets; (b) perspective view of channels. | ||
In conclusion, two coordination polymers with different dimensionalities have been synthesized from rigid tetrahedral-shaped tectons with adamantane scaffolds. We have emphasized the role played by the coordination preference of the metal centre, along with the organic linker's symmetry, in dictating the network topology. Compound 1 features a PtS-type topology of network, with four interpenetrating nets. The low dimensionality of compound 2 arises from the employment of a neutral building block displaying a cis-conformation that limits the propagation into the third dimension. Studies to check the ability of the ligands to afford high connectivity networks with other metal ions are in progress.
Acknowledgements
This work was financially supported by UEFISCDI (Grant PNII-ID-PCCE-2011-2-0050) and by a grant of the Romanian National Authority for Scientific Research and Innovation, POCPOLIG, project nr. 67/08.09.Notes and references
- (a) R. Robson, J. Chem. Soc., Dalton Trans., 2000, 3735 RSC; (b) R. Robson, Dalton Trans., 2008, 5113 RSC; (c) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546 CrossRef CAS.
- S. R. Batten, S. M. Neville and D. R. Turner, Coordination polymers: Design, analysis and applications, Royal Society of Chemistry, Cambridge, 2008 Search PubMed.
- See, for example T. Muller and S. Bräse, RSC Adv., 2014, 4, 6886 RSC and references therein.
- (a) B. Chen, M. Eddaoudi, T. M. Reineke, J. W. Kampf, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 11559 CrossRef CAS; (b) J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef CAS PubMed; (c) M. Zhang, Y.-P. Chen, M. Bosch, T. Gentle, K. Wang, D. Feng, Z. U. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 815 CrossRef CAS PubMed; (d) H. Chun, D. Kim, D. N. Dybtsev and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 971 CrossRef CAS PubMed; (e) L. Wen, P. Cheng and W. Lin, Chem. Commun., 2012, 48, 2846 RSC; (f) L. Wen, P. Cheng and W. Lin, Chem. Sci., 2012, 3, 2288 RSC; (g) C. Tan, S. Yang, N. R. Champness, X. Lin, A. J. Blake, W. Lewis and M. Schröder, Chem. Commun., 2011, 47, 4487 RSC; (h) Y. E. Cheon and M. P. Suh, Chem. Commun., 2009, 2296 RSC; (i) L. Ma, A. Jin, Z. Xie and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 9905 CrossRef CAS PubMed; (j) M. Zhang, Y.-P. Chen and H.-C. Zhou, CrystEngComm, 2013, 15, 9544 RSC; (k) R. P. Davies, R. Less, P. D. Lickiss, K. Robertson and A. J. P. White, Cryst. Growth Des., 2010, 10, 4571 CrossRef CAS; (l) J. M. Gotthardt, K. F. White, B. F. Abrahams, C. Ritchie and C. Boskovic, Cryst. Growth Des., 2012, 12, 4425 CrossRef CAS; (m) D. Liu, Z. Xie, L. Ma and W. Lin, Inorg. Chem., 2010, 49, 9107 CrossRef CAS PubMed; (n) G. A. Senchyk, A. B. Lysenko, H. Krautscheid, E. B. Rusanov, A. N. Chernega, K. W. Krämer, S.-X. Liu, S. Decurtins and K. V. Domasevitch, Inorg. Chem., 2013, 52, 863 CrossRef CAS PubMed.
- (a) D. J. Hoffart, A. P. Côté and G. K. H. Shimizu, Inorg. Chem., 2003, 42, 8603 CrossRef CAS PubMed; (b) D. J. Hoffart, S. A. Dalrymple and G. K. H. Shimizu, Inorg. Chem., 2005, 44, 8868 CrossRef CAS PubMed.
- (a) J. M. Taylor, A. H. Mahmoudkhani and G. K. H. Shimizu, Angew. Chem., Int. Ed., 2007, 46, 795 CrossRef CAS PubMed; (b) M. V. Vasylyev, E. J. Wachtel, R. Popovitz-Biro and R. Neumann, Chem. – Eur. J., 2006, 12, 3507 CrossRef CAS PubMed; (c) M. Vasylyev and R. Neumann, Chem. Mater., 2006, 18, 2781 CrossRef CAS.
- I. Boldog, K. V. Domasevitch, I. A. Baburin, H. Ott, B. Gil-Hernández, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 1235 RSC.
- A. Bulut, Y. Zorlu, E. Kirpi, A. Çetinkaya, M. Wörle, J. Beckmann and G. Yücesan, Cryst. Growth Des., 2015, 15, 5665 CAS.
- (a) K. Matsumoto, M. Kannami, D. Inokuchi, H. Kurata, T. Kawase and M. Oda, Org. Lett., 2007, 9, 2903 CrossRef CAS PubMed; (b) C. B. Caputo, V. N. Vukotic, N. M. Sirizzotti and S. J. Loeb, Chem. Commun., 2011, 47, 8545 RSC; (c) F. L. Geyer, F. Rominger, M. Vogtland and U. H. F. Bunz, Cryst. Growth Des., 2015, 15, 3539 CrossRef CAS; (d) S. Shankar, R. Balgley, M. Lahav, S. R. Cohen, R. Popovitz-Biro and M. E. van der Boom, J. Am. Chem. Soc., 2015, 137, 226 CrossRef CAS PubMed; (e) X.-Y. Chen, H.-Y. Shi, R.-B. Huang, L.-S. Zheng and J. Tao, Chem. Commun., 2013, 49, 10977 RSC; (f) X.-Y. Chen, R.-B. Huang, L.-S. Zheng and J. Tao, Inorg. Chem., 2014, 53, 5246 CrossRef CAS PubMed.
- (a) G. A. Senchyk, A. B. Lysenko, I. Boldog, E. B. Rusanov, A. N. Chernega, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2012, 41, 8675 RSC; (b) G. A. Senchyk, A. B. Lysenko, E. B. Rusanov, A. N. Chernega, J. Jezierska, K. V. Domasevitch and A. Ozarowski, Eur. J. Inorg. Chem., 2012, 5802 CrossRef CAS; (c) A. B. Lysenko, G. A. Senchyk, J. Lincke, D. Lässig, A. A. Fokin, E. D. Butova, P. R. Schreiner, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2010, 39, 4223 RSC.
- (a) I. Boldog, K. V. Domasevitch, J. Sanchiz, P. Mayerd and C. Janiak, Dalton Trans., 2014, 43, 12590 RSC; (b) I. Boldog, K. Domasevitch, J. K. Maclaren, C. Heering, G. Makhloufi and C. Janiak, CrystEngComm, 2014, 16, 148 RSC.
- M. Dincă, A. Dailly and J. R. Long, Chem. – Eur. J., 2008, 14, 10280 CrossRef PubMed.
- (a) D. Venkataraman, S. Lee, J. S. Moore, P. Zhang, K. A. Hirsch, G. B. Gardner, A. C. Covey and C. L. Prentice, Chem. Mater., 1996, 8, 2030 CrossRef CAS; (b) F.-Q. Liu and T. D. Tilley, Inorg. Chem., 1997, 36, 5090 CrossRef CAS; (c) K. M. Patil, M. E. Dickinson, T. Tremlett, S. C. Moratti and L. R. Hanton, Cryst. Growth Des., 2016, 16, 1038 CrossRef CAS.
- C. I. Schilling, O. Plietzsch, M. Nieger, T. Muller and S. Bräse, Eur. J. Org. Chem., 2011, 1743 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, crystallographic data, figures of asymmetric units, spectral data, TGA curve and experimental and calculated powder diffraction patterns. CCDC 1508551 and 1508552. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce02146h |
| ‡ Synthesis of L1: to a 100 mL three-necked round-bottom flask equipped with a reflux condenser were added 4-pyridylboronic acid (0.258 g, 2.11 mmol), Pd(dppf)Cl2 (0.043 g, 0.05 mmol), and Na2CO3 (0.452 g, 4.27 mmol) in dioxane/water (2.5:1 v/v, 35 mL) and the solution was flushed with argon for 20 min. After, 1,3,5,7-tetrakis(4-iodophenyl)adamantane (0.25 g, 0.26 mmol) was added under an argon atmosphere, and the reaction mixture was stirred at 80 °C for 12 h. The solvent was evaporated, and the crude product was dissolved in chloroform and washed with water (2 × 50 mL) and a satd. NH4Cl solution (2 × 50 mL). The separated organic layer was dried over MgSO4, filtered and concentrated under vacuum. After purification by column chromatography (silica gel, pentane/DCM/MeOH: 1.5:1:0.2, Rf = 0.45), the product was isolated as a white solid (0.082 g, 41% yield). Tetrakis(4-cyanophenyl)adamantane (L2) was obtained (Scheme S2†) in good yield (80%) starting from 1,3,5,7-tetrakis(4-iodophenyl)adamantane applying the procedure described in the literature.8 Synthesis of 1: a methanol solution (4 mL) of Cu(BF4)2·6H2O (0.0024 g, 0.007 mmol) was reacted with 0.0018 g (0.029 mmol) of monoethanolamine in 4 mL of methanol and the resulting mixture was left to slowly diffuse through an ethanol layer (5 mL) into a 5 mL CHCl3 solution of L1 (0.0019 g, 0.0025 mmol). Blue green single crystals suitable for X-ray measurements appeared in two–three weeks. Yield (based on L1): 0.0013 g, 46%. Selected IR data (KBr): 3315 (m), 3068 (w), 3033 (w), 2929 (w), 2897 (w), 2849 (w), 1664 (s), 1614 (vs), 1542 (m), 1493 (s), 1431(m), 1401 (w), 1356 (w), 1295 (w), 1236 (w), 1123 (s), 1073 (s), 1028 (s), 1007 (s), 1025 (m), 825 (w), 792 (s), 739 (w), 667 (w), 637 (w). Synthesis of 2: [Mn(hfac)2(H2O)2]·H2O (0.0038 g, 0.0072 mmol) was solubilized in 4 mL of boiling n-heptane. Upon cooling to room temperature, 4 mL of CH2Cl2 was added and the solution was carefully layered on top of a 2 mL CH2Cl2 solution of L2 (0.002 g, 0.0037 mmol) in a test tube. Yellow single crystals were formed in a few days. Yield (based on L2): 0.0015 g, 28%. Selected IR data (KBr): 3401 (m), 2939 (w), 2858 (w), 2262 (m), 2224 (m), 1647 (s), 1606 (w), 1523 (w), 1493 (s), 1405 (w), 1369 (w), 1252 (s), 1196 (s), 1149 (vs), 1096 (w), 836 (w), 794 (w), 764 (w), 665 (w). |
| § Crystal data for [CuL1(H2O)2](BF4)2·8H2O (1): C54H64CuB2F8N4O10, M = 1166.26, T = 293(2) K, tetragonal, space group I422, a = 12.8199(9), c = 32.617(4) Å, V = 5360.5(1) Å3, Dc = 1.310 g cm−3, μ = 1.218 mm−1, Z = 4, Flack parameter = 0.42(7), reflections collected = 6049, independent reflection = 1932 [Rint = 0.0566]. Before SQUEEZE: R indices [I > 2σ(I)]: R1 = 0.1033, wR2 = 0.2671, R indices (all data): R1 = 0.1507, wR2 = 0.3199, GoF = 1.086 with Δρmin/Δρmax = −0.244/1.258 e Å−3. After SQUEEZE: R indices [I > 2σ(I)]: R1 = 0.0750, wR2 = 0.1876, R indices (all data): R1 = 0.1138, wR2 = 0.2237, GoF = 0.938 with Δρmin/Δρmax = −0.216/0.939 e Å−3. Crystal data for [Mn(hfac)2(L2)0.5] (2): C29H16MnF12N2O4, M = 739.38, T = 293(2) K, tetragonal, space group I41/a, a = 11.645(5), c = 52.700(5) Å, V = 7146(6) Å3, Dc = 1.374 g cm−3, μ = 0.465 mm−1, Z = 8, reflections collected = 32 |
| This journal is © The Royal Society of Chemistry 2017 |