
Conversion of biogenic aragonite into hydroxyapatite scaffolds in boiling solutions
D.
Reinares-Fisac
*ab,
S.
Veintemillas-Verdaguer
 a and
L.
Fernández-Díaz
*bc
a and
L.
Fernández-Díaz
*bc
aInstituto de Ciencia de Materiales de Madrid (ICMM, CSIC), C/Sor Juana Inés de la Cruz 3, Cantoblanco 28049, Madrid, Spain. E-mail: daniel.reinares@icmm.csic.es
bDepartamento de Cristalografía y Mineralogía, Universidad Complutense de Madrid, 28040 Madrid, Spain
cInstituto de Geociencias (UCM, CSIC), C/José Antonio Novais 2, 28040 Madrid, Spain. E-mail: lfdiaz@geo.ucm.es
First published on 30th November 2016
Abstract
The interaction of Sepia officinalis cuttlefish bone, made of aragonitic CaCO3, with (NH4)2HPO4 boiling solutions produces porous carbonate-bearing hydroxyapatite (Ca10(PO4)6(OH)2) scaffolds with extremely large specific surface area. Percentages of aragonite into hydroxyapatite conversion close to 90% are reached after 10 hours for all the conditions explored, though higher efficiency of the transformation process is achieved by increasing the concentration of the (NH4)2HPO4 solution. The conversion has a pseudomorphic character since it occurs with the accurate preservation of the external shape as well as the primary macro- and microtextural features of the biomineral. Moreover, the mineral replacement transformation is accompanied by a significant increase in surface area, which most likely relates to newly formed porosity. We proposed that this transformation occurs through an interface coupled dissolution–crystallization mechanism, which is continuously promoted by the release of CO2 to the atmosphere. The generation of newly formed porosity would be the consequence of the preservation of the biomineral's external shape, which requires the balance of both molar volume and solubility changes involved in the mineral transformation.
Introduction
Ceramic materials consisting of hydroxyapatite (HA; Ca10(PO4)6(OH)2), β-tricalcium phosphate (β-TCP; Ca3(PO4)2) and mixtures of both phases are commonly used in bone graft applications.1–3 Different studies have evidenced that rapid bone ingrowth and vascularisation are more feasible when implant materials contain an internal network of interconnected pores with average pore diameters in the 100–500 μm range.4–6 Biominerals are composites that consist of two components, an inorganic component, which often is calcium carbonate, and organic matter structured in membranes and fibres. Numerous calcium carbonate biominerals like brachiopod and mollusc shells, coral skeletons or sea urchin hard tissues typically contain an internal hierarchically arranged porosity whose pore sizes and distribution closely match those required in bone implant materials.7–9 This fact has driven the exploration of processing methods that allow the production of scaffolds with ideal properties for bone graft applications using calcium carbonate biominerals as raw materials. These methods commonly involve the conversion of the biogenic, inorganic calcium carbonate component into calcium phosphate phases through the interaction with phosphate-bearing aqueous solutions.6,10–15 In most cases, this interaction leads to the development of dissolution–crystallization reactions that take place under hydrothermal conditions. The role of dissolution–recrystallization reactions during mineral replacement processes has been the subject of much research conducted, mainly by Earth scientists, over the last two decades.14–17 It is now well understood that when the rate limiting process is the dissolution of the primary phase, the fine coupling of dissolution and crystallization reactions results in the accurate preservation of both the external shape (formation of pseudomorphs) and most textural features of the primary material.18–22 The development of porosity is a key aspect of dissolution–recrystallization reactions that occur with preservation of the external shape of the primary phase. This porosity allows the balance of volume change involved in the reaction when the molar volume and/or the solubility of the product phase are lower than those of the primary phase. This newly formed porosity guarantees the continuous contact between the aqueous solution and the primary phase–product phase interface, that is, the site where the reaction occurs. For further information on the characteristics of interface coupled dissolution–recrystallization reactions, the reader can consult a number of recent review papers.18,23–27 Different studies have confirmed the preservation of microtextural features, including porosity, of a variety of calcium carbonate biominerals, like sepia cuttlebone,14 coral skeletons and sea urchin spines,6 after their hydrothermal conversion into materials consisting of calcium phosphate phases, mainly hydroxyapatite. In this work, we study the conversion of a calcium carbonate biomineral (sepia cuttlebone) into calcium phosphate scaffolds through its interaction with phosphate-bearing boiling solutions in an open system. The set-up used in our experiments allows continuous re-equilibration with the atmosphere. The boiling solution crystallization method was originally designed to produce highly perfect monocrystals for optical applications.28,29 To our knowledge, this method has only been previously used in the conversion into calcium phosphate of a calcitic biomineral, the skeleton of an echinoderm (Mellita eduardobarrosoi),30 but never before in the conversion of any aragonitic material. Here we apply the boiling solution method to the conversion of the aragonitic cuttlebone of Sepia officinalis. This biomineral is structured with chambers internally subdivided by a complex arrangement of calcified pillars and organic membranes and shows interesting properties like high compressive strength, high porosity and high permeability.31 Our aims are (i) to study the degree of preservation of both the external shape and internal architecture of the biogenic material after its transformation into calcium phosphate, (ii) to determine the reaction pathway and (iii) to evaluate the efficiency of the boiling solution method compared to traditional hydrothermal methods of inducing this mineral transformation.Experimental
Cuttlebone of Sepia officinalis collected from the coast of Northwest Spain was selected as the precursor biogenic material. Conversion experiments were performed using a 2000 mL bottom-rounded flask filled with 1400 mL of (NH4)2HPO4 solution and covered with a glass lid (Fig. 1).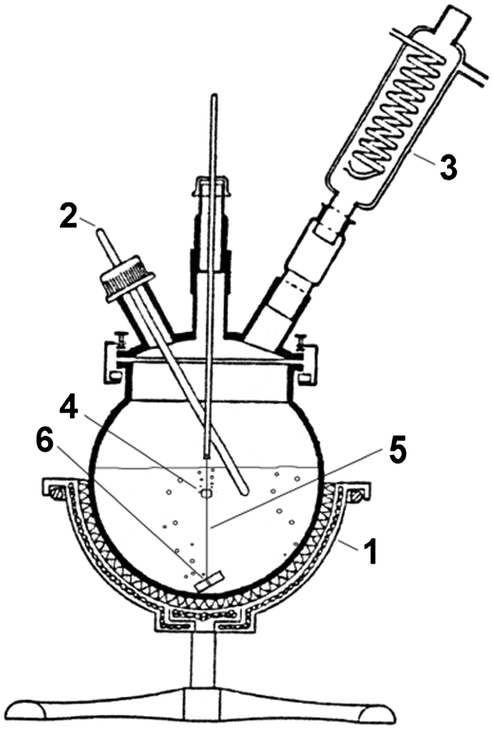 | ||
| Fig. 1 Experimental set-up. (1) Heating mantle, (2) thermocouple, (3) reflux condenser, (4) cuttlebone block, (5) nylon thread, (6) sinker. | ||
The (NH4)2HPO4 solution was prepared using high purity deionized water (Milli-Q, 18.2 MΩ) and a reagent quality chemical (Sigma-Aldrich). Different runs were conducted using three (NH4)2HPO4 solution concentrations, 0.5, 1 and 1.5 M. Measurements of the initial pH of the solution yielded a mean value of 8.4 ± 0.2, which dropped to 7.2 ± 0.2 after 24 hours of interaction with the cuttlebone samples. The system was heated by means of a heating mantle to maintain the solution under constant boiling conditions. The solution temperature was kept approximately constant at 98 °C. In order to minimize water evaporation during the experiment, a reflux condenser was inserted in the central opening of the lid that covered the flask. A constant flow of cold water (10 °C) through the condenser ensured the condensation of water vapour, which reflowed back to the flask, thereby minimizing water loss. The reflux condenser did not prevent the release of other, non-condensable, gases, namely CO2 and NH3, which were released to the atmosphere during the experiment. Once the boiling regime was stabilized, four cuttlebone 1 cm-sided cubes were immersed in the solution. To ensure that they remained completely submerged during the duration of the experiment, each cube was tied to a Teflon-coated sinker. Samples were recovered from the solution after specific reaction times (1, 3, 6, 10 and 24 hours), thoroughly rinsed, first with deionised water and then with ethanol, and dried in air at room temperature for 24 hours.
The phase composition of the samples was analyzed by powder X-ray diffraction (XRD) using a PANalytical X'Pert PRO MRD microprocessor-controlled X-ray diffractometer, Cu Kα radiation and a scan recording in the 2θ angle range between 2° and 90° with intervals of 0.017°. Standard mineral files of the ICDD-PDF database (2013 version) and diffraction diagrams were correlated to determine solid phases. A semi-quantitative percentage estimation of each mineral was then achieved using the Reference Intensity Ratio (RIR) method,32 considering the RIR values included in the PDF cards (01-076-0606 for CaCO3, 04-016-1185 for Ca5(PO4)3(OH)2) and using X'Pert High Score Plus software from PANalytical. The average crystallite size and the lattice strain of hydroxyapatite were calculated by Scherrer's equations using the half width of the (002) and (202) X-ray diffraction peaks, using the utilities of the automatic powder diffraction computer program (APD) from Phillips. The samples were further analyzed by Fourier Transform Infrared (FTIR) spectroscopy and micro-Raman spectroscopy. Infrared (IR) spectra were obtained using a BRUKER IFS 66v/S spectrophotometer equipped with a TGS detector. Each spectrum was recorded by the co-addition of 64 scans. Under these conditions, the precision was 0.2 cm−1. Micro-Raman spectra of the samples were recorded on crosscut sections of the samples using a confocal Raman microscope (WITec alpha 300 R) equipped with a SHG Nd:YAG laser (532 nm, max. power 22.5 mW) and a lens-based spectrometer that affords a lateral resolution of 200 nm. Textural characteristics of the samples were imaged using a JEOL JSM 6400 scanning electron microscope operated at 20 kV. Finally, specific surface area was determined by N2 adsorption in a Micromeritics ASAP 2000 instrument, using the BET method. Prior to the measurements, the samples were degassed under vacuum at 150 °C for 2 hours. Sorption isotherms were obtained by standard procedures.
Results and discussion
XRD patterns of the calcium carbonate biogenic precursor and samples partially converted into calcium phosphate after different times of interaction with boiling (NH4)2HPO4 solutions are depicted in Fig. 2. The mineral component of the precursor was confirmed as the calcium carbonate polymorph aragonite (Fig. 2, top diffraction pattern). The diffraction patterns of all samples recovered after one hour of interaction showed well developed sharp peaks that matched well those expected for a mixture of hydroxyapatite and aragonite, regardless of the initial concentration of the aqueous solution (Fig. 2). Upon interaction with the boiling solution, the intensity of the hydroxyapatite peaks progressively increased paralleling the decrease of the intensity of those peaks corresponding to aragonite. The percentage of transformation of aragonite into hydroxyapatite (ft) as estimated by XRD semiquantitative analyses for each experiment is shown in Table 1.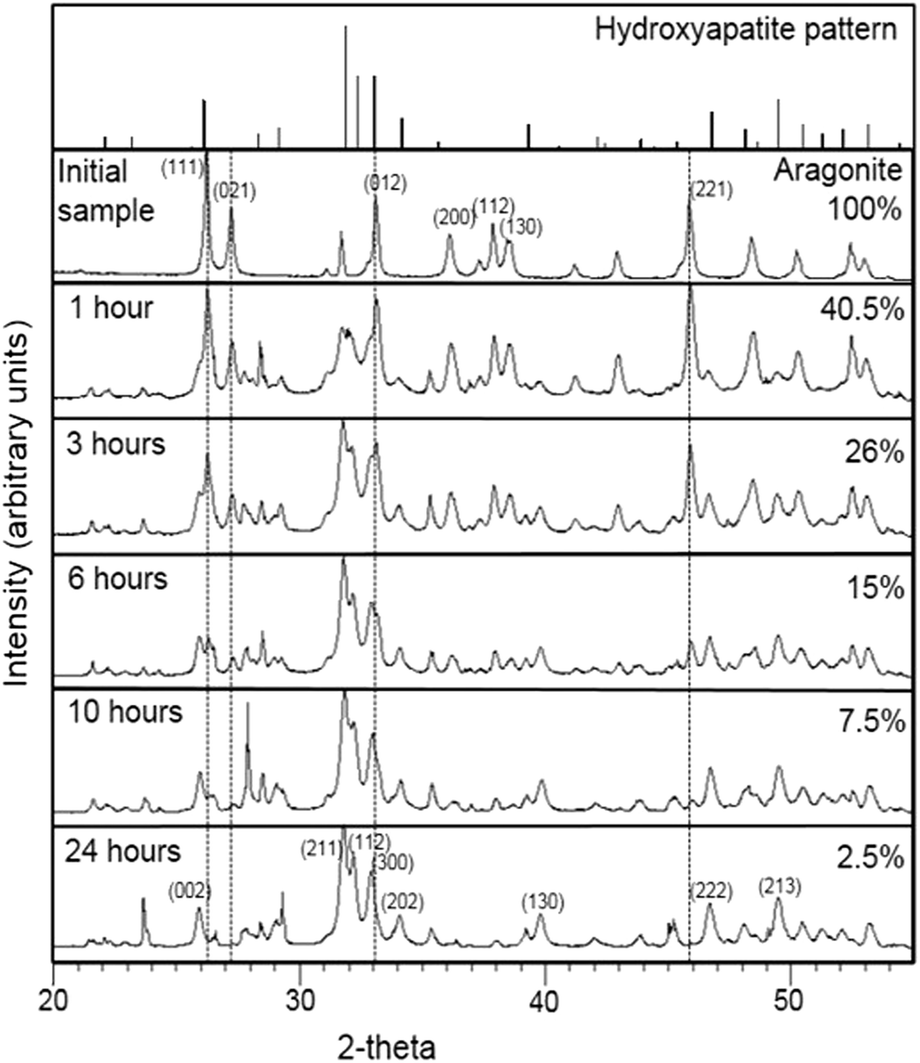 | ||
| Fig. 2 XRD patterns of samples interacted for various periods of time with 1 M boiling solution. Small peaks at 2θ = 27.8, 28.5, 29.1 and 36.9, which do not match the aragonite nor hydroxyapatite diffraction patterns, can be assigned to (NH4)2HPO4 (PDF card 00-029-0111). | ||
| t (hours) | Percentage (%) of hydroxyapatite | ||
|---|---|---|---|
| 0.5 M | 1 M | 1.5 M | |
| 1 | 56.5 ± 2.5 | 59.5 ± 0.5 | 59 ± 1.5 |
| 3 | 66.5 ± 1.5 | 74 ± 1 | 80 ± 1.8 |
| 6 | 82 ± 3 | 85 ± 1 | 90 ± 2 |
| 10 | 85.5 ± 4.5 | 92.5 ± 2.5 | 94 ± 2.5 |
| 24 | 93 ± 2 | 97.5 ± 2.5 | 98 ± 2 |
Hydroxyapatite crystallinity values were found below one for all samples irrespective of the interaction time, indicating a low crystallinity product (Table 2).
| t (hours) | Line width (202), FWHMa (°) | Average crystallite size, L (nm) by Scherrer's equation | Crystallinity (Xc) |
|---|---|---|---|
| 24 | 0.239 | 49 | 0.326 |
| 10 | 0.228 | 53 | 0.309 |
| 6 | 0.293 | 37 | 0.405 |
| 3 | 0.344 | 30 | 0.480 |
| 1 | 0.394 | 26 | 0.553 |
The study of the precursor and partially transformed samples by FTIR confirms the conclusions of the XRD analysis. FTIR spectra corresponding to samples interacted with a 1 M (NH4)2HPO4 boiling solution for 1 hour (a) and 10 hours (b) are depicted in Fig. 3. The spectrum of the 1 hour-interacted sample shows sharp bands that can be assigned to aragonite at 700 and 712 cm−1 (ν4), 855 cm−1 (ν2) and 1080 (ν1) cm−1 as well as a broad band around 1475 cm−1 (ν3).12–14 Though very weak, these bands can also be detected in the spectrum of the sample interacted for 10 hours. Both spectra show well defined bands at 470 cm−1 (ν2), 564 and 602 cm−1 (ν4), 957 cm−1 (ν1), and 1043 cm−1 (ν3) that can be assigned to phosphate groups in hydroxyapatite.12–14 Additional bands that can be attributed to carbonate incorporated in a-type (877 and 1546 cm−1) and b-type (1405 and 1460 cm−1) sites in the structure of hydroxyapatite33 are also found in both spectra. A small band at 633 cm−1 in the 10 hours sample can be assigned to OH−,34 whereas a broad band visible in both spectra between 3000 and 3750 cm−1 can be attributed to both absorbed water and OH− vibrations.35 A small shoulder at 3572 cm−1, present in both spectra but more intense in that corresponding to 10 hours, can be attributed to the OH− stretching mode. The band at 1638 cm−1 can also be indicative of absorbed water.34
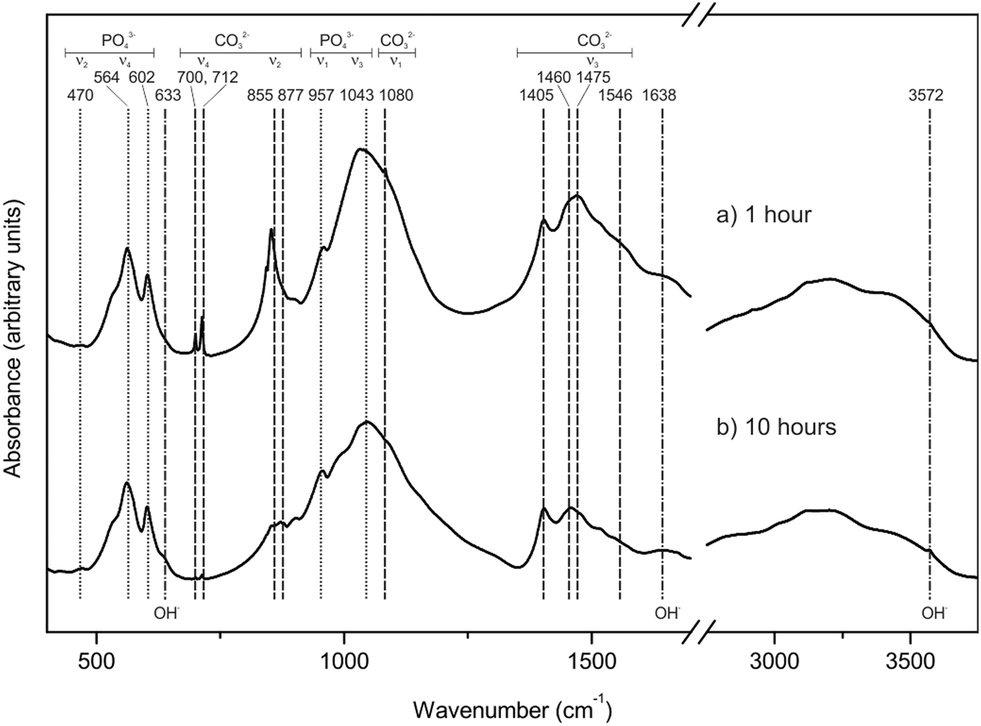 | ||
| Fig. 3 IR spectra of samples interacted for 1 and 10 hours with a 1 M (NH4)2HPO4 boiling solution. | ||
SEM micrographs of Sepia officinalis cuttlebone prior to and after the conversion of the primary aragonitic component into hydroxyapatite are shown in Fig. 4. The original inner structure of Sepia officinalis cuttlebone consists of chambers which appear compartmentalized by an arrangement of vertical pillars and horizontal layers and further subdivided by thin horizontal membranes (Fig. 4a). Distances between the horizontal layers are in the range ∼300–400 micrometers, whereas distances between the pillars are ∼80–140 micrometers. Both pillars and horizontal layers are fairly well preserved during the aragonite–hydroxyapatite conversion as can be seen in Fig. 4b, which shows a cuttlebone sample after 10 hours of reaction with a 1 M (NH4)2HPO4 boiling solution. The preservation of the horizontal thin membranes is significantly worse, most likely due to their mainly organic nature. Moreover, the observation under higher magnification of the surfaces of layers and pillars in the transformed samples evidences that the smaller, delicate textural details of these surfaces disappear during the conversion to be substituted by micron-sized newly formed spherulitic aggregates. Differences in the concentration of the phosphate-bearing aqueous solution result in small differences in the characteristics of these aggregates, which are built up of fibres when a 0.5 M (NH4)2HPO4 solution is used (Fig. 4c and d), but consist of plate-shaped crystals in a radial arrangement when more concentrated phosphate-bearing solutions are used (Fig. 4e and f).
 | ||
| Fig. 4 SEM micrographs of untransformed Sepia officinalis cuttlebone (a), cuttlebone after 24 hours of interaction with 1.5 M (NH4)2HPO4 boiling solution (98% conversion of aragonite into hydroxyapatite) (b), hydroxyapatite crystal aggregates grown on the surface of cuttlebone after interaction with a boiling (NH4)2HPO4 solution for specific times (t): 0.5 M, t = 24 hours (c and d); 1.5 M, t = 1 hour (e); 1.5 M, t = 24 hours (f). For micrographs a and b, scale = 500 microns; for micrographs c, d, e and f, scale = 5 microns. | ||
The interaction of the biogenic material with a boiling (NH4)2HPO4 solution leads to a percentage of transformation well above 50 after 1 hour irrespective of the initial concentration of the solution. After 3 hours of interaction, the percentage of transformation varies between 65 (concentration of the solution = 0.5 M) and 80 (concentration of the solution = 1.5 M). The percentage of transformation reaches values ≥90% after 24 hours, 10 hours and 6 hours in contact with 0.5, 1 and 1.5 M solutions, respectively, which indicates that increasing the solution concentration results in the conversion significantly speeding up. The transformation approaches completion after 24 hours in contact with 1 and 1.5 M solutions.
We interpret that the conversion is the result of interface coupled dissolution–recrystallization reactions. When the experiments start, the phosphate-bearing solutions are undersaturated with respect to aragonite. This drives the dissolution of Sepia officinalis cuttlebone samples, which results in the release of Ca2+ and CO32− ions to the fluid. Immediately, phosphate-bearing solutions become highly supersaturated with respect to hydroxyapatite due to the extremely low solubility of this phase (SPHydroxyapatite = 6.3 × 10−59 at 25 °C).36 It is worthwhile to note that the supersaturation with respect to hydroxyapatite of a 0.5 M (NH4)2HPO4 aqueous solution saturated with respect to aragonite (SParagonite = 10−8.336 at 25 °C)37 is 1015.47 (calculation conducted with the PHREEQC code38). It is to be expected that hydroxyapatite crystals form on the surface of the Sepia officinalis cuttlebone samples long before the solution reaches equilibrium with respect to aragonite. The formation of hydroxyapatite removes Ca2+ and, though to a lesser extent, also CO32− ions from the solution, further undersaturating it with respect to aragonite and, thereby, promoting the dissolution of this phase. An interface coupling between both reactions, aragonite dissolution and hydroxyapatite crystallization, explains the good preservation of both the external shape and the original inner structure of Sepia officinalis cuttlebone during the conversion.18,23–27
The percentage of transformation values determined in this work are strikingly higher than those reported for the conversion into hydroxyapatite of the calcitic Mellita eduardobarrosoi by the boiling solution method using similar conditions.30 The faster conversion of aragonitic Sepia officinalis cuttlebone most likely is the consequence of the higher porosity and permeability of the latter material, which results in a larger solid–fluid interface where the coupled dissolution–crystallization reaction occurs.
The important role of the original porosity of Sepia officinalis cuttlebone in facilitating the conversion is supported by micro-Raman analyses conducted on crosscut sections of partially transformed samples. Micro-Raman spectra recorded after different times of interaction with the phosphate-bearing solution evidence that the aragonite into hydroxyapatite conversion simultaneously initiates and progresses in the rim and core areas of the sample (Fig. 5). This is in clear contrast with previous results of the development of the conversion of aragonite single crystals under hydrothermal conditions.15 In this latter case, the conversion starts on the surface of the single crystals and progressively advances towards their core. This advancement is accompanied by the development of a sharp interface between the aragonite core and a polycrystalline hydroxyapatite rim which persists until the conversion is completed.15 The reasons for these differences between the conversion of aragonite single crystals and Sepia officinalis cuttlebone most likely relate to the hierarchically arranged porosity present in the latter, which provides the phosphate-bearing solution access to inner parts of the sample from the very beginning of the interaction.
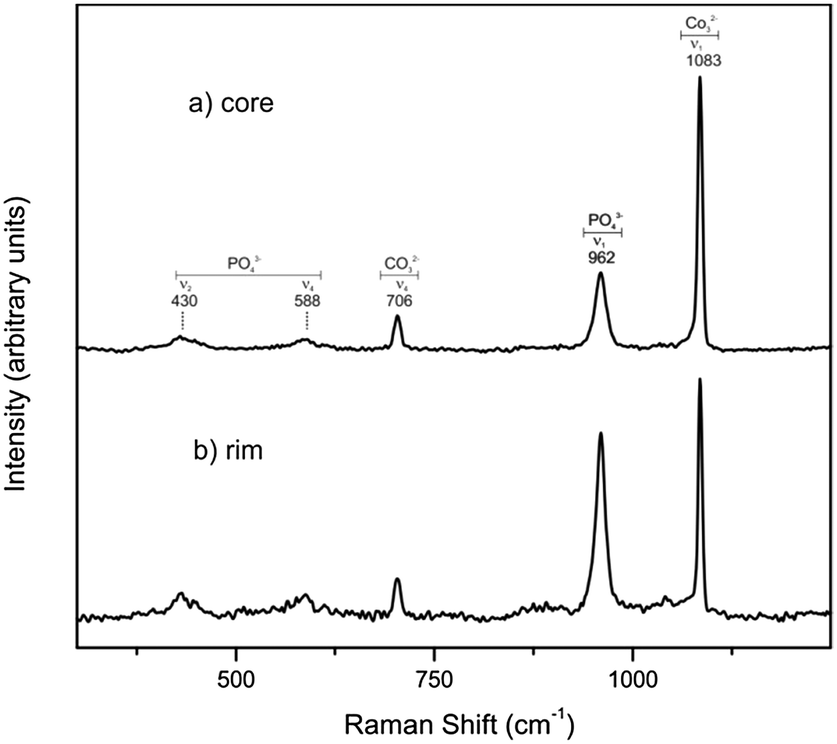 | ||
| Fig. 5 Micro-Raman spectra recorded on crosscut sections of samples interacted for 3 hours with a 1 M (NH4)2H2PO4 boiling solution. Spectrum (a) was recorded from the core of the sample, whereas spectrum (b) was collected from its rim. | ||
The efficiency of the boiling solution method for the conversion of Sepia officinalis cuttlebone is comparable to that of the hydrothermal procedure when the latter is operated at temperatures around 100 °C.14 The high efficiency of the boiling solution method can be related to a number of factors. On the one hand, it is known that the use of a boiling solution (reflux conditions) facilitates the control of temperature during chemical reactions in solution. This is, indeed, why boiling solutions are extensively used in organic and organometallic chemistry. On the other hand, it has been demonstrated that significantly high crystal growth rates are achieved in boiling solutions compared to those measured at temperatures slightly below boiling conditions.29,39 This latter effect has been related to a more efficient transfer of solute during bubbles' collision with the surface of the growing crystal.39 In the particular case of sepia's cuttlebone, the sample itself is a source of bubbles due to its high porosity. Finally, the boiling solution method deals with an open system in which the continuous elimination of NH3 and CO2 displaces the reaction towards the hydroxyapatite side.
BET measurements indicate that the aragonite–hydroxyapatite conversion is accompanied by a marked increase in specific surface area, which evolves from an initial value of 7.5 m2 g−1 to reach 26.3 and 29.5 m2 g−1 in samples obtained after 1 and 10 hours of reaction with a 1 M (NH4)2HPO4 boiling solution, respectively. This dramatic increase in surface area can be interpreted as the result of two factors. The first factor could relate to a decrease in the size of the particles that constitute the hydroxyapatite-after-aragonite-cuttlebone pseudomorph. However, the evolution of the average crystallite size of hydroxyapatite calculated by Scherrer's equation does not support this interpretation, since this parameter slightly increases as the interaction progresses (see Table 2). The second factor could relate to the generation of porosity associated with the aragonite–hydroxyapatite conversion. It has been demonstrated that preservation of the external shape during dissolution–crystallization reactions involves porosity generation when the molar volume decreases and/or the secondary phase is less soluble than the primary one.15–18,23–27 This is the case in the aragonite–hydroxyapatite conversion which involves a molar volume reduction of ∼6%.14,15 Moreover, the solubility of hydroxyapatite is more than thirty orders of magnitude lower than the solubility of aragonite. This very large difference can also contribute to the development of new porosity.14 Most likely a combination of factors is responsible for the increase in specific surface area. It is worthwhile mentioning that longer interaction times do not result in a further increase of the specific surface area but in its decrease. Thus, after 24 hours of reaction, the specific surface area has a value of 17.6 m2 g−1. This decrease most likely is the consequence of hydroxyapatite crystal growth. This interpretation is in good agreement with the variation of hydroxyapatite average crystallite size calculated by Scherrer's equation (Table 2).
Conclusions
We have successfully transformed an aragonitic biomineral, Sepia officinalis cuttlebone, into hydroxyapatite after interaction with (NH4)2HPO4 boiling solutions. The obtained materials accurately reproduce most of the original morphological and textural characteristics of the biogenic precursor. Moreover, they consist of hydroxyapatite with carbonate substitutions in both A- and B-sites and show large specific surface areas and interconnected porosity. These are desirable properties in hydroxyapatite-based scaffold materials for bone grafting applications that guarantee good compatibility for organic matrices and high reactivity.40–42 For the aragonite–hydroxyapatite mineral replacement, we proposed a reaction mechanism that involves coupled dissolution–crystallization reactions continually promoted by CO2 release to the atmosphere. Mineral replacement in boiling solution can potentially be applied to the production of porous scaffolds using calcium carbonate biominerals as raw material.Acknowledgements
Financial support from projects CGL2013-47988-C2-1-P and MAT2014-52069-R (Spanish Ministry of Economy and Competitiveness) is gratefully acknowledged. The authors thank the staff of ICTS Centro Nacional de Microscopía Electrónica for kindly providing technical support. The authors are indebted to Dr. Emilio Matesanz from the X-ray Diffraction Central Service of the Complutense University (UCM) for assistance with XRD measurements and interpretation. D. R.-F. thanks the Spanish Ministry of Education, Culture and Sport for funding through a research initiation scholarship.Notes and references
- M. Jarcho, Clin. Orthop. Relat. Res., 1981, 157, 259 CAS.
- R. Z. LeGeros, Chem. Rev., 2008, 108, 4742 CrossRef PubMed.
- K. Zhou, Y. Zhang, D. Zhang, X. Zhang, Z. Li, G. Liu and T. W. Button, Scr. Mater., 2011, 64, 426 CrossRef CAS.
- K. A. Hing, S. M. Best, K. E. Tanner, W. Bonfield and P. A. Revell, J. Mater. Sci.: Mater. Med., 1999, 10, 663 CrossRef CAS PubMed.
- M. Okamoto, Y. Dohi, H. Ohgushi, H. Shimaoka, M. Ikeuchi, A. Matsushima, K. Yonemasu and H. Hosoi, J. Mater. Sci.: Mater. Med., 2006, 17, 327 CrossRef CAS PubMed.
- M. Schlosser, S. Fröls, U. Hauf, I. Sethmann, S. Schultheiss, F. Pfeifer and H. J. Kleebe, J. Am. Ceram. Soc., 2013, 96, 412 CAS.
- I. Sethmann and G. Worheide, Micron, 2008, 39, 209 CrossRef CAS PubMed.
- D. Vielzeuf, J. Garrabou, A. Baronnet, O. Grauby and C. Marschal, Am. Mineral., 2008, 93, 1799 CrossRef CAS.
- E. Griesshaber, A. J. Goetz, L. Howard, A. Ball, S. Ruff and W. W. Schmahl, Bioinspired, Biomimetic Nanobiomater., 2012, 1, 133 CrossRef.
- B. Ben-Nissan, Curr. Opin. Solid State Mater. Sci., 2003, 7, 283 CrossRef CAS.
- J. H. G. Rocha, A. F. Lemos, S. Agathopoulos, P. Valerio, S. Kannan and F. N. Oktar, et al. , Bone, 2005, 37, 850 CrossRef CAS PubMed.
- S. Kannan, J. H. G. Rocha, S. Agathopoulos and J. M. F. Ferreira, Acta Biomater., 2007, 3, 243 CrossRef CAS PubMed.
- K. S. Vecchio, X. Zhang, J. M. Massie, M. Wang and C. W. Kim, Acta Biomater., 2007, 3, 785 CrossRef CAS PubMed.
- A. Kasioptas, T. Geisler, C. V. Putnis, C. Perdokouri and A. Punis, J. Cryst. Growth, 2010, 312, 2431 CrossRef CAS.
- A. Kasioptas, T. Geisler, C. V. Putnis, C. Perdokouri, C. Trepmann, N. Gussone and A. Punis, Geochim. Cosmochim. Acta, 2011, 75, 3486 CrossRef CAS.
- P. Pöml, M. Menneken, T. Stephan, D. R. D. Niedermeier, T. Geisler and A. Putnis, Geochim. Cosmochim. Acta, 2007, 71, 3311 CrossRef.
- K. Pollok, C. V. Putnis and A. Putnis, Am. J. Sci., 2011, 311, 211 CrossRef CAS.
- A. Putnis, Rev. Mineral. Geochem., 2009, 30, 87 CrossRef.
- L. Fernández-Díaz, C. M. Pina, J. M. Astilleros and N. Sánchez-Pastor, Am. Mineral., 2009, 94, 1223 CrossRef.
- F. Xia, J. Brugger, G. Chen, Y. Ngothai, B. O'Neill, A. Putnis and A. Pring, Geochim. Cosmochim. Acta, 2009, 73, 1945 CrossRef CAS.
- F. Xia, J. Brugger, Y. Ngothai, B. O'Neill, G. Chen and A. Pring, Cryst. Growth Des., 2009, 9, 4902 CAS.
- A. Putnis and C. V. Putnis, J. Solid State Chem., 2007, 180, 1783 CrossRef CAS.
- A. Putnis, Mineral. Mag., 2002, 66, 689 CrossRef CAS.
- C. V. Putnis and L. Fernández-Díaz, EMU Notes, 2010, 10, 189 Search PubMed.
- E. Ruiz-Agudo, C. V. Putnis and A. Putnis, Chem. Geol., 2014, 383, 132 CrossRef CAS.
- A. Putnis, Rev. Mineral. Geochem., 2015, 80, 1 CrossRef.
- A. Altree-Williams, A. Pring, Y. Ngothai and J. Brugger, Earth-Sci. Rev., 2015, 150, 628 CrossRef CAS.
- K. Nassau, J. Cryst. Growth, 1972, 15, 171 CrossRef CAS.
- S. Veintemillas-Verdaguer, R. Rodriguez-Clemente and K. Sangwald, J. Cryst. Growth, 1987, 83, 367 CrossRef CAS.
- M. A. Araiza, J. Gómez, R. Rodríguez-Clemente and V. M. Castaño, J. Mater. Synth. Process., 1999, 7, 211 CrossRef CAS.
- A. G. Checa, J. H. E. Cartwright, I. Sánchez-Almazo, J. P. Andrade and F. Ruiz-Raya, Sci. Rep., 2015, 5, 11513, DOI:10.1038/srep11513.
- R. L. Snyder, Powder Diffr., 1992, 7, 186 CrossRef CAS.
- J. C. Elliott, D. W. Holcomb and R. A. Young, Calcif. Tissue Int., 1985, 37, 372 CrossRef CAS PubMed.
- S. Raynaud, E. Champion, D. Bernache-Assollant and P. Thomas, Biomaterials, 2002, 23, 1065 CrossRef CAS PubMed.
- N. S. Chickerur, M. S. Tung and W. E. Brown, Calcif. Tissue Int., 1980, 32, 55 CrossRef CAS PubMed.
- Y. Avnimelech, E. C. Moreno and W. E. Brown, J. Res. Natl. Bur. Stand., Sect. A, 1973, 77, 149 CrossRef CAS.
- L. N. Plummer and E. Busenberg, Geochim. Cosmochim. Acta, 1982, 46, 1011 CrossRef CAS.
- D. L. Parkhurst and C. A. J. Appelo, User's guide to PHREEQC (Version 2)-A computer program for speciation, batch-reaction, one-dimensional transport and inverse geochemical calculations, USGS, Denver, 1999 Search PubMed.
- R. Rodriguez-Clemente, S. Veintemillas Verdaguer and F. Rull-Perez, in Morphology and Growth Units of Crystals, ed. I. Sunagawa, Terra Scientific Publishing Company, Tokyo, 1989, vol. 7, pp. 479–512 Search PubMed.
- C. Rey, C. Combes, C. Drouet, H. Sfihi and A. Barrougm, Mater. Sci. Eng., C, 2007, 27, 198 CrossRef CAS.
- J. A. F. Gamelasand and A. G. Martins, Colloids Surf., A, 2015, 478, 62 CrossRef.
- D. S. H. Lee, Y. Pai, S. Chang and D. H. Kim, Mater. Sci. Eng., C, 2016, 58, 97 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |