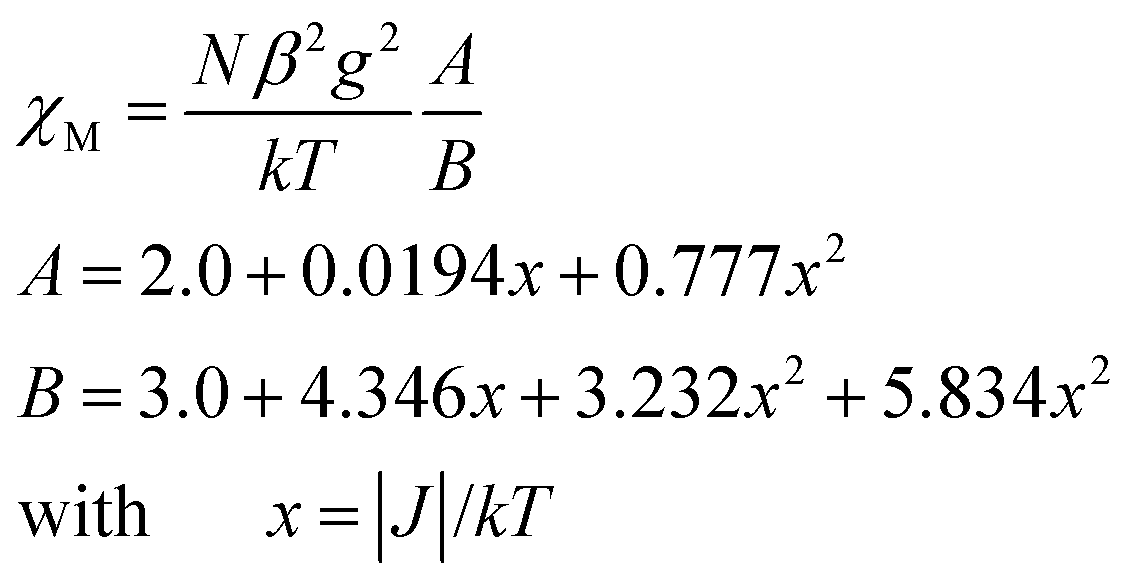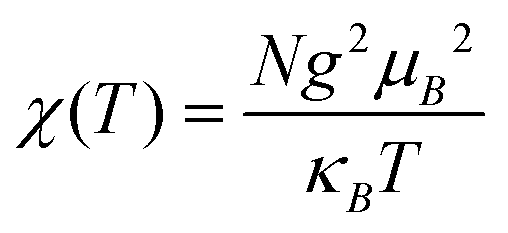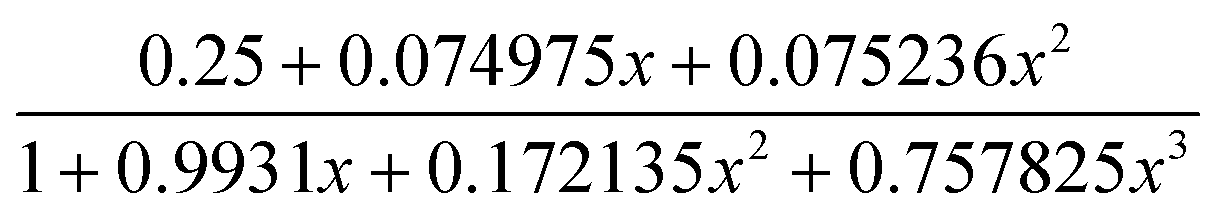DOI:
10.1039/C6CE02115H
(Paper)
CrystEngComm, 2017,
19, 117-128
Exploring 4-(3-carboxyphenyl)picolinic acid as a semirigid building block for the hydrothermal self-assembly of diverse metal–organic and supramolecular networks†
Received
3rd October 2016
, Accepted 21st November 2016
First published on 21st November 2016
Abstract
4-(3-Carboxyphenyl)picolinic acid (H2cppa) was applied as a new and virtually unexplored building block for the synthesis of eleven new coordination compounds, namely [M(Hcppa)2(H2O)2]·2H2O {M = Ni (1) and Zn (2)}, [M(μ3-cppa)(H2O)2]n {M = Ni (3) and Co (4)}, {[Co(μ-cppa)(2,2′-bipy)(H2O)]·H2O}n (5), [Zn(μ-cppa)(2,2′-bipy)]n (6), [M(μ-cppa)(phen)(H2O)]n {M = Co (7), Zn (8), Mn (9), and Cu (10)}, and {[Cd3(μ3-cppa)3(phen)2]·4H2O}n (11). All these compounds were generated by a hydrothermal self-assembly method using the corresponding metal(II) chlorides, H2cppa, and two types of N-donor ancillary ligands, selected from 2,2′-bipyridine (2,2′-bipy) or 1,10-phenanthroline (phen). The obtained products 1–11 were characterized by standard methods, including IR spectroscopy, elemental and thermogravimetric analyses, and powder and single-crystal X-ray diffraction. They represent the first structurally characterized compounds derived from H2cppa. The structures of 1–11 vary from a complex 3D metal–organic framework (MOF) 11 to 2D coordination polymers 3 and 4, 1D coordination polymers 5–10, and discrete 0D monomers 1 and 2. Such a structural diversity is guided by the nature of the metal(II) node, level of deprotonation of 4-(3-carboxyphenyl)picolinic acid, and type of applied ancillary ligand. In addition, the structures of 1, 2, 5, and 7–10 are further extended [0D → 3D (1 and 2), 1D → 2D (5), and 1D → 3D (7–10)] into various H-bonded supramolecular networks. Both the supramolecular (in 1, 2, 5, and 7–10) and metal–organic (in 3, 4, 6, and 11) underlying networks were analyzed and classified from the topological viewpoint, revealing the distinct pcu (in 1 and 2), fes (in 3 and 4), gek1 (in 5), 2C1 (in 6), and srs (in 7–10) topological nets, whereas a topologically unique framework was identified in MOF 11. The magnetic behavior of 3–5, 7, 9, and 10 and luminescence properties of 2, 6, 8, and 11 were also investigated and discussed.
Introduction
Research on coordination polymers that feature one-, two-, or three-dimensional (1D, 2D, or 3D) structures has received significant attention in recent years due to their intricate structural architectures and topologies, as well as notable functional properties and applications in different fields.1–6 However, there is still a significant challenge to construct coordination polymers having predictable structural types, since their assembly process can depend on a variety of important factors, such as the geometry of metal nodes, interconnectivity of organic spacers, stoichiometry and reaction conditions, type of solvent, and presence of ancillary ligands.7–9 The search for novel types of organic ligands toward the synthesis of coordination polymers with predictable architectures represents an important research direction. It is particularly interesting to explore various flexible organic building blocks with adjustable backbones as spacers for the generation of coordination polymers that are based on metal nodes with distinct coordination preferences. Within a large family of organic spacers applied in crystal engineering of coordination polymers, aromatic polycarboxylic acids have been extensively used due to their high stability, coordination versatility and flexibility, as well as the presence of different binding sites or functional groups.1c,4a,d,6d,8a,10 On the other hand, the N-donor ligands also have a significant influence on the assembly process of coordination polymers.8a,10e,11
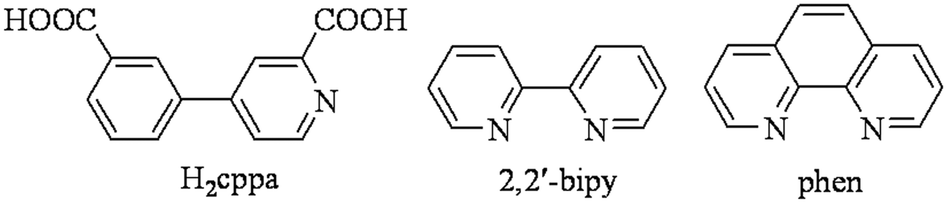
With the objective of exploring new multifunctional polycarboxylate building blocks for the preparation of coordination polymers, we have focused our attention on the relatively simple but poorly explored pyridine-dicarboxylic acids, which have a considerable potential to result in complex and topologically distinct coordination or supramolecular networks. Hence, in the present study, we have selected 4-(3-carboxyphenyl)picolinic acid (H2cppa, Scheme 1) as a promising but virtually unexplored N,O-building block for the design of coordination polymers. The selection of this acid has been governed by the following considerations. (1) H2cppa possesses two COOH groups with a long separation distance between them and a possibility of being fully or partially deprotonated, depending on the pH of the reaction medium. (2) One aromatic ring of H2cppa bears a picolinate N,O-functionality, thus allowing a relatively strong chelating behavior to different metal ions. (3) H2cppa represents a considerable flexibility owing to a possible rotation of pyridine and phenyl rings around the C–C single bond. (4) H2cppa has remained unexplored in the design of coordination polymers, as attested by a search of the Cambridge Structural Database (CSD).
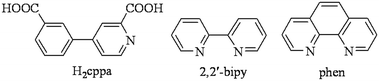 |
| | Scheme 1 Structures of H2cppa and auxiliary ligands (2,2′-bipy and phen). | |
Bearing these points in mind, we report herein the hydrothermal synthesis, X-ray crystal structures, full characterization, topological classification, thermal stability, and magnetic or luminescence properties of eleven novel coordination compounds derived from H2cppa as a principal building block, namely, [M(Hcppa)2(H2O)2]·2H2O {M = Ni (1) and Zn (2)}, [M(μ3-cppa)(H2O)2]n {M = Ni (3) and Co (4)}, {[Co(μ-cppa)(2,2′-bipy)(H2O)]·H2O}n (5), [Zn(μ-cppa)(2,2′-bipy)]n (6), [Co(μ-cppa)(phen)(H2O)]n {M = Co (7), Zn (8), Mn (9), and Cu (10)}, and {[Cd3(μ3-cppa)3(phen)2]·4H2O}n (11). To our knowledge, these compounds represent the first structurally characterized products that were assembled from 4-(3-carboxyphenyl)picolinic acid.
Experimental section
Materials and methods
All chemicals and solvents were of A.R. grade and used without further purification. H2cppa was obtained from Jinan Henghua Sci. & Tec. Co., Ltd. C, H, and N elemental analyses were performed on an Elementar Vario EL elemental analyzer. IR spectra were recorded using KBr pellets and a Bruker EQUINOX 55 spectrometer. Thermogravimetric analyses (TGA) were carried out under N2 atmosphere on a LINSEIS STA PT1600 thermal analyzer (heating rate 10 °C min−1); see the ESI† for a discussion of the TGA data. Magnetic susceptibility data were collected on a Quantum Design SQUID Magnetometer MPMS XL-7 in the 2–300 K temperature range and a field of 0.1 T. A correction was made for the diamagnetic contribution prior to data analysis. Excitation and emission spectra were recorded on an Edinburgh FLS920 fluorescence spectrometer using the solid samples at room temperature. Powder X-ray diffraction (PXRD) patterns were measured on a Rigaku-Dmax 2400 diffractometer using Cu-Kα radiation (λ = 1.54060 Å); the X-ray tube was operated at 40 kV and 40 mA. The experimental PXRD patterns of the as-synthesized bulk samples of 1–11 match well the simulated plots obtained from the single-crystal X-ray data, thus proving the purity of the bulk samples (Fig. S3, ESI†).
Synthesis of compounds
1–11
All compounds were prepared under similar hydrothermal conditions and the synthesis details are summarized in Table 1. A general synthetic procedure is described for a nickel(II) derivative 1 as a representative example. Synthesis and analytical data for 2–11 are given in the ESI.†
Table 1 Synthetic details for 1–11a
| Compound |
Reaction mixture composition (molar ratio) |
Yieldb (%) |
|
General reaction conditions: metal(II) chloride (0.3 mmol), H2ccpa (0.3 mmol), NaOH (0.3–0.6 mmol), 2,2′-bipy or phen (0.3 mmol, optional), H2O solvent (10 mL); hydrothermal synthesis in a Teflon-lined stainless steel vessel (25 mL), 160 °C, 3 d.
Yield based on H2cppa.
|
|
1
|
NiCl2·6H2O, H2cppa, NaOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) :1:1) |
60 |
|
2
|
ZnCl2, H2cppa, NaOH (1:1:1) |
55 |
|
3
|
NiCl2·6H2O, H2cppa, NaOH (1:1:2) |
60 |
|
4
|
CoCl2·6H2O, H2cppa, NaOH (1:1:2) |
55 |
|
5
|
CoCl2·6H2O, H2cppa, 2,2′-bipy, NaOH (1:1:1:2) |
55 |
|
6
|
ZnCl2, H2cppa, 2,2′-bipy, NaOH (1:1:1:2) |
55 |
|
7
|
CoCl2·6H2O, H2cppa, phen, NaOH (1:1:1:2) |
60 |
|
8
|
ZnCl2, H2cppa, phen, NaOH (1:1:1:2) |
60 |
|
9
|
MnCl2·6H2O, H2cppa, phen, NaOH (1:1:1:2) |
60 |
|
10
|
CuCl2·6H2O, H2cppa, phen, NaOH (1:1:1:2) |
35 |
|
11
|
CuCl2·H2O, H2cppa, phen, NaOH (1:1:1:2) |
60 |
[Ni(Hcppa)
2
(H
2
O)
2
]·2H
2
O
.
A mixture of NiCl2·6H2O (71.1 mg, 0.3 mmol), H2cppa (73.0 mg, 0.3 mmol), NaOH (12.0 mg, 0.3 mmol), and H2O (10 mL) was stirred at room temperature for 15 min, then sealed in a 25 mL Teflon-lined stainless steel vessel and heated at 160 °C for 3 days, followed by cooling to room temperature at a rate of 10 °C h−1. Green crystals were isolated manually, washed with distilled water, and dried to furnish compound 1. Yield: 60% (based on H2cppa). Calcd for C26H24NiN2O12: C 50.76, H 3.93, N 4.55%. Found: C 50.53, H 3.91, N 4.52%. IR (KBr, cm−1): 3469 m, 3244 m, 1713 s, 1595 s, 1544 m, 1411 s, 1376 s, 1314 w, 1227 s, 1160 w, 1115 w, 1084 w, 1059 m, 1028 w, 906 w, 865 w, 834 m, 809 m, 757 m, 706 m, 654 w, 634 w, 589 w, 538 w.
X-ray data collection and structure determination
Single-crystal X-ray data for 1–11 were collected on a Bruker Smart CCD or an Agilent SuperNova diffractometer, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Semiempirical absorption corrections were applied using the SADABS program. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using the SHELXL-2014 software.12 All the non-hydrogen atoms were refined anisotropically by full-matrix least-squares methods on F2. All the H atoms (except the those of H2O molecules) were placed in calculated positions with fixed isotropic thermal parameters, and included in structure factor calculations in the final stage of full-matrix least-squares refinement. The hydrogen atoms of water molecules were located by difference maps and constrained to ride on their parent O atoms. In the structure of compound 11, as it was not possible to see clear electron-density peaks in difference maps which would correspond with acceptable locations for the various H atoms bonded to water oxygen atoms O13, O14, O15 and O16, the refinement was completed with no allowance for these water H atoms in the model. The crystal data for 1–11 are summarized in Table 2. The selected bond lengths are listed in Table S1,† whereas H-bonding parameters are given in Table S2 (ESI†). Topological analysis of metal–organic or H-bonded networks was carried out using Topos software and following the concept of the simplified underlying net.13,14 Such nets were generated by contracting cppa2− spacers (in 3–11), [M(H2O)2]2+ (in 3, 4), [Co(2,2′-bipy)(H2O)]2+ (in 5), [Zn(bpy)]2+ (in 6), and [M(phen)(H2O)]2+ (in 7–10) blocks, or [M(Hcppa)2(H2O)2] (in 1, 2) molecular units to their centroids, maintaining connectivity via coordination bonds or hydrogen bonds. For the analysis of the nets involving H-bonding interactions, only strong D–H⋯A hydrogen bonds were considered, wherein the H⋯A < 2.50 Å, D⋯A < 3.50 Å, and ∠(D–H⋯A) > 120°; D and A stand for donor and acceptor atoms, respectively.13 CCDC 1498139 for 1, 1505548 for 2, 1498140 for 3, 1496266 for 4, 1496267 for 5, 1505549 for 6, 1496268 for 7, and 1505550–1505553 for 8–11 contain the supplementary crystallographic data for this paper.
Table 2 Crystallographic data and structure refinement summary for 1–11
| Compound |
1
|
2
|
3
|
4
|
5
|
6
|
| Chemical formula |
C26H24NiN2O12 |
C26H24ZnN2O12 |
C13H11NiNO6 |
C13H11CoNO6 |
C23H19CoN3O6 |
C23H15ZnN3O4 |
| Formula weight |
615.18 |
621.84 |
335.94 |
336.16 |
492.34 |
462.75 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
P21/c |
P21/n |
P21/n |
|
a/Å |
12.5528(7) |
12.523(4) |
12.5405(4) |
12.5902(5) |
11.4833(8) |
9.0764(14) |
|
b/Å |
14.2273(7) |
14.220(3) |
13.8008(4) |
13.8557(6) |
12.8687(6) |
16.0794(16) |
|
c/Å |
7.1047(4) |
7.0884(19) |
7.0319(3) |
7.0755(3) |
15.1022(8) |
13.913(3) |
|
α/° |
90 |
90 |
90 |
90 |
90 |
90 |
|
β/° |
93.315(5) |
93.40(4) |
93.828(3) |
93.443(4) |
97.349(6) |
106.70(2) |
|
γ/° |
90 |
90 |
90 |
90 |
90 |
90 |
|
V/Å3 |
1266.73(12) |
1260.1(6) |
1214.29(7) |
1232.07(9) |
2213.4(2) |
1944.9(6) |
|
T/K |
293(2) |
296(2) |
293(2) |
293(2) |
293(2) |
293(2) |
|
Z
|
2 |
2 |
4 |
4 |
4 |
4 |
|
D
c/g cm−3 |
1.613 |
1.639 |
1.838 |
1.812 |
1.477 |
1.580 |
|
μ/mm−1 |
0.839 |
1.048 |
1.628 |
1.422 |
0.820 |
1.300 |
|
F(000) |
636 |
640 |
688 |
684 |
1012 |
944 |
| Refl. measured |
4342 |
4028 |
4412 |
4870 |
7838 |
7322 |
| Unique refl. (Rint) |
2239 (0.0496) |
2273 (0.0539) |
2161 (0.0328) |
2193 (0.0403) |
3925 (0.0356) |
3429 (0.1007) |
| GOF on F2 |
1.071 |
0.928 |
1.065 |
1.060 |
1.004 |
1.003 |
|
R
1 [I > 2σ(I)]a |
0.0498 |
0.0561 |
0.0352 |
0.0411 |
0.0478 |
0.0684 |
| wR2 [I > 2σ(I)]b |
0.0831 |
0.1023 |
0.0753 |
0.0764 |
0.0865 |
0.0781 |
| Compound |
7
|
8
|
9
|
10
|
11
|
|
|
R
1 = ∑||Fo| − |Fc||/|Fo|.
wR2 = {∑[w(Fo2 − Fc2)]2/∑[w(Fo2)]2}1/2.
|
| Chemical formula |
C25H17CoN3O5 |
C25H17ZnN3O5 |
C25H17MnN3O5 |
C25H17CuN3O5 |
C63H45Cd3N7O16 |
|
| Formula weight |
498.35 |
504.78 |
494.35 |
502.96 |
1493.26 |
|
| Crystal system |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Orthorhombic |
Monoclinic |
|
| Space group |
P212121 |
P212121 |
P212121 |
P212121 |
P21/n |
|
|
a/Å |
10.0350(3) |
10.0211(5) |
10.1184(4) |
10.0257(3) |
10.9405(7) |
|
|
b/Å |
11.6320(3) |
11.6063(8) |
11.5420(4) |
11.6347(6) |
21.7240(17) |
|
|
c/Å |
18.6660(6) |
18.7216(11) |
18.8913(9) |
18.6389(5) |
23.8970(11) |
|
|
α/° |
90 |
90 |
90 |
90 |
90 |
|
|
β/° |
90 |
90 |
90 |
90 |
91.988(5) |
|
|
γ/° |
90 |
90 |
90 |
90 |
90 |
|
|
V/Å3 |
2178.84(11) |
2177.5(2) |
2206.23(16) |
2174.16(14) |
5676.2(6) |
|
|
T/K |
293(2) |
293(2) |
293(2) |
293(2) |
293(2) |
|
|
Z
|
4 |
4 |
4 |
4 |
4 |
|
|
D
c/g cm−3 |
1.519 |
1.540 |
1.488 |
1.537 |
1.738 |
|
|
μ/mm−1 |
0.832 |
1.172 |
0.642 |
1.049 |
1.194 |
|
|
F(000) |
1020 |
1032 |
1012 |
1028 |
2944 |
|
| Refl. measured |
7868 |
8475 |
8062 |
8242 |
23475 |
|
| Unique refl. (Rint) |
3836 (0.0381) |
3706 (0.0560) |
3890 (0.0458) |
3841 (0.0410) |
10031 (0.0627) |
|
| GOF on F2 |
1.012 |
1.074 |
1.013 |
1.137 |
1.023 |
|
|
R
1 [I > 2σ(I)]a |
0.0399 |
0.0464 |
0.0471 |
0.0522 |
0.0916 |
|
| wR2 [I > 2σ(I)]b |
0.0798 |
0.0646 |
0.0725 |
0.1218 |
0.1722 |
|
| Flack parameter |
−0.018(12) |
−0.032(13) |
0.003(18) |
0.080(12) |
|
|
Results and discussion
Crystal structures of [M(Hcppa)2(H2O)2]·2H2O {M = Ni (1) and Zn (2)}
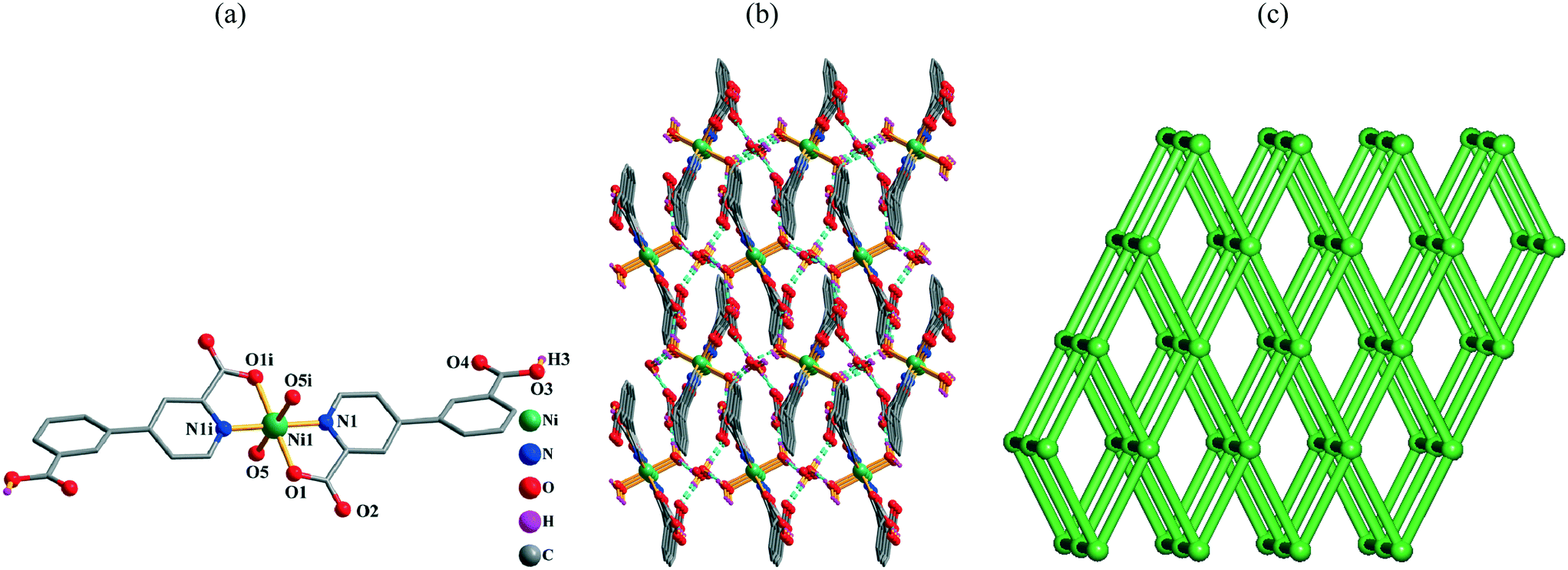
Single-crystal X-ray analyses revealed that compounds 1 and 2 are isostructural discrete 0D monomers; the structure of 1 is described in detail. Its asymmetric unit bears one crystallographically unique Ni(II) atom (half occupancy, located at an inversion center), one Hcppa− ligand, one coordinated and one lattice H2O molecule. The equatorial sites of the six-coordinate Ni1 center are occupied by two O and two N atoms (picolinate functionality) from two symmetry equivalent Hcppa− blocks (Fig. 1a), whereas the axial sites are taken by O atoms from two equivalent water ligands, thus forming an ideal octahedral {NiN2O4} geometry. The Ni–N distances are 2.014(3) Å, while the Ni–O bonds are in the 2.023(2)–2.119(2) Å range, being in agreement with related distances in other Ni(II) complexes.2c,6d,10e,11e In 1, the Hcppa− moiety acts as a terminal N,O-ligand (Scheme 2, mode I), in which the deprotonated carboxylate group adopts a monodentate mode. The dihedral angle between the two rings in Hcppa− is 31.60°. The discrete molecular units of 1 are interconnected by the O–H⋯O hydrogen bonds to form a 3D H-bonded supramolecular network (Fig. 1b and Table S2†). From the topological point of view, this network (Fig. 1c) can be classified as a uninodal 6-connected net with the pcu [alpha-Po primitive cubic] topology and the point symbol of (412·63).
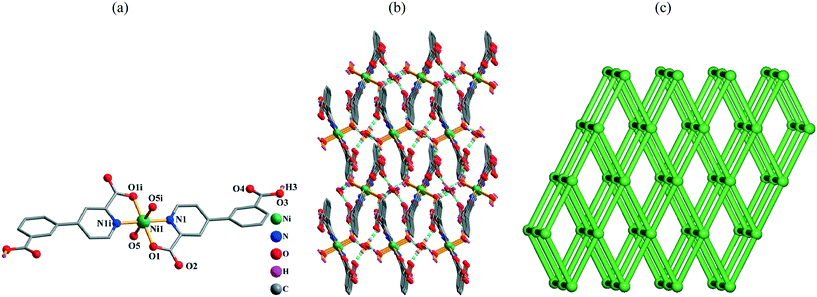 |
| | Fig. 1 Structural fragments of 1. (a) Coordination environment of the Ni1 atom (except for COOH groups, H atoms are omitted for clarity). Symmetry code: i: −x + 1, −y + 1, −z + 1. (b) 3D H-bonded supramolecular network viewed along the bc plane (H bonds are represented by blue lines). (c) Topological representation of the underlying 3D supramolecular net showing a uninodal 6-connected network with the pcu topology; rotated view along the a axis; color codes: centroids of 6-connected [Ni(Hcppa)2(H2O)2] molecular nodes (green balls). | |
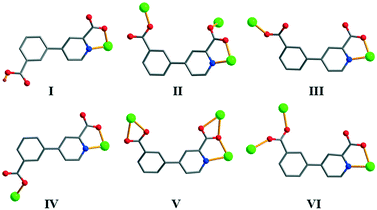 |
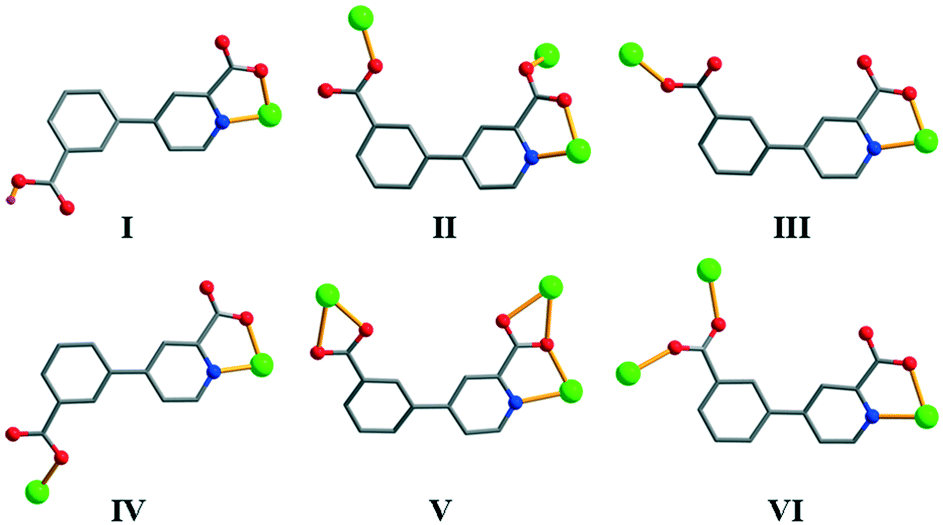
| | Scheme 2 Various coordination modes of Hcppa−/cppa2− blocks in 1–11. | |
Crystal structures of [M(μ3-cppa)(H2O)2]n {M = Ni (3) and Co (4)}
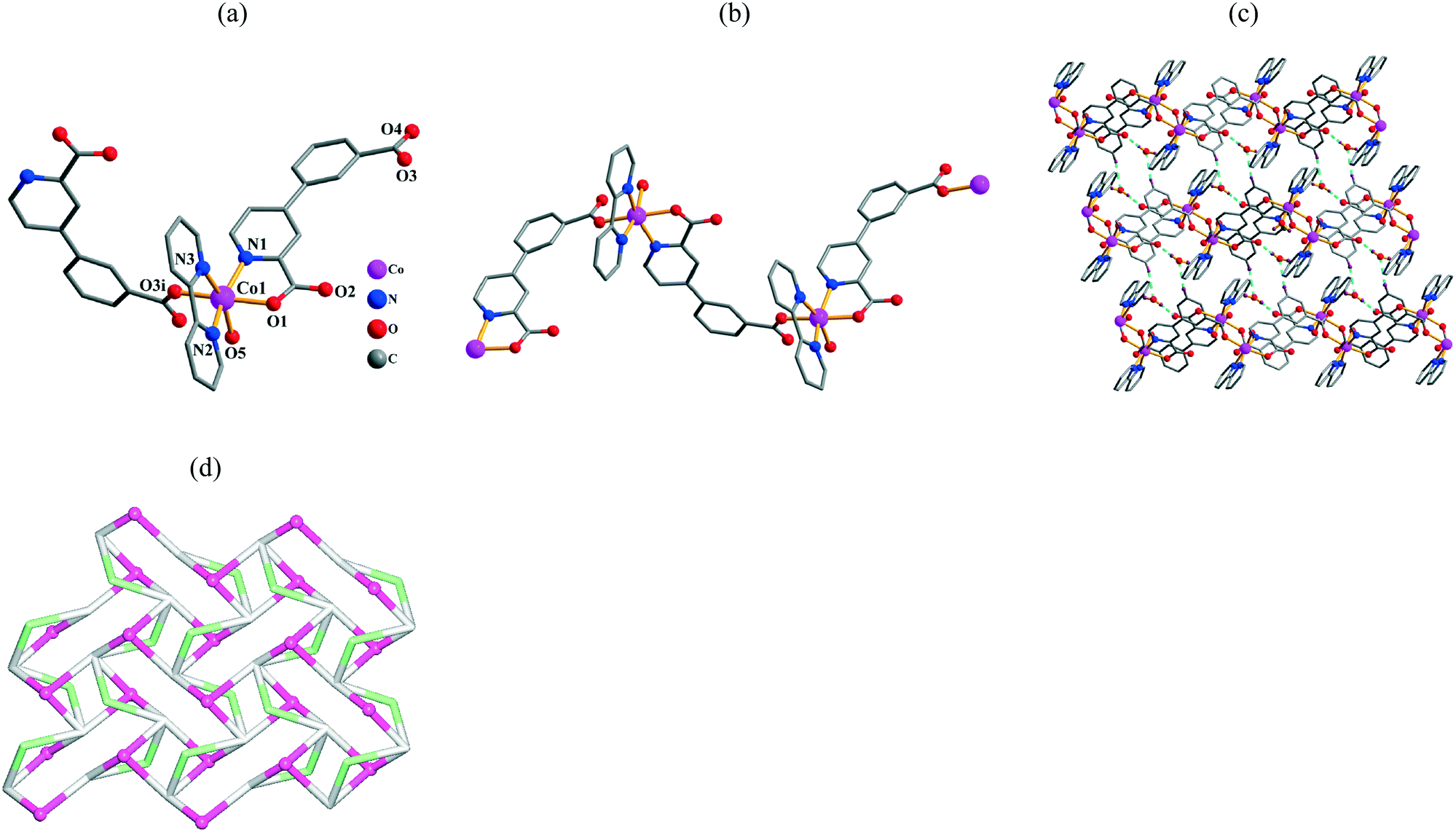
Single-crystal X-ray diffraction analyses revealed that compounds 3 and 4 are isostructural; the structure of 3 is described in detail as a representative example (Fig. 2). It is a 2D coordination polymer and its asymmetric unit consists of one Ni(II) atom, one μ3-cppa2− block, and two coordinated water molecules. Each six-coordinate Ni(II) center adopts a distorted octahedral {NiNO5} geometry (Fig. 2a), which is filled by three carboxylate O atoms and one N atom from three different cppa2− blocks and two O atoms from two H2O ligands. The Ni–O bonds are in the 1.997(2)–2.190(2) Å range, while the Ni–N bond is 2.031(3) Å; both are within typical ranges.6d,g,10f,11e In 3, the cppa2− moiety acts as a μ3-spacer (Scheme 2, mode II), wherein the picolinate carboxylate group adopts a μ-bridging bidentate mode; the second carboxylate group features a monodentate mode. The dihedral angle between the pyridyl and phenyl rings in the cppa2− spacer attains 11.27°. As represented in Fig. 2b, the μ3-cppa2− blocks connect adjacent Ni(II) atoms to form a 2D coordination polymer network. For the sake of topological classification of this layer, we generated an underlying net (Fig. 2c). It is composed of the 3-connected Ni and μ3-cppa nodes (these are topologically equivalent). The resulting net can be described as a uninodal 3-connected layer with the fes [Shubnikov plane net (4·82)] topology and point symbol of (4·82). The neighboring metal–organic layers are further assembled, via O–H⋯O hydrogen bonds (Fig. S1, Table S3†), into a 3D supramolecular net.
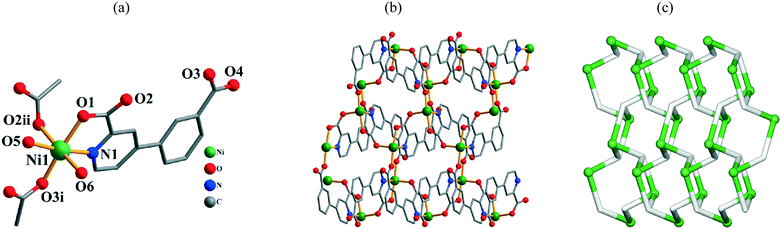 |
| | Fig. 2 Structural fragments of 3. (a) Coordination environment of the Ni1 atom (H atoms are omitted for clarity). Symmetry codes: i: −x + 2, y − 1/2, −z + 3/2; ii: x, −y + 3/2, z − 1/2. (b) 2D coordination polymer along the bc plane. (c) Topological representation of the underlying 2D network showing a uninodal 3-connected layer with the fes [Shubnikov plane net (4·82)] topology; rotated view along the a axis; color codes: 3-connected Ni nodes (green balls), centroids of 3-connected μ3-cppa2− nodes (gray). | |
Crystal structure of {[Co(μ-cppa)(2,2′-bipy)(H2O)]·H2O}n (5)
The asymmetric unit of 5 has one crystallographically unique Co(II) atom, one μ-cppa2− block, one 2,2′-bipy ligand, one coordinated and one lattice H2O molecule. Each six-coordinate Co1 center displays a distorted octahedral {CoN3O3} environment (Fig. 3a), which is filled by two carboxylate O and one N atom from two different cppa2− blocks, two N atoms from the 2,2′-bipy moiety, and one O atom from the H2O ligand. The lengths of the Co–O bonds are in the 2.087(2)–2.133(3) Å range, while the Co–N bonds are 2.109(3)–2.148(3) Å; these bonds are within normal ranges reported for related Co(II) compounds.6c,8a,10a,d In 5, the cppa2− block acts as a μ-linker (Scheme 2, mode III) with the COO groups taking the monodentate mode. The dihedral angle within the aromatic rings in the cppa2− linker is 24.83°. The μ-cppa2− ligands interlink alternately the adjacent Co1 atoms to form a zigzag 1D coordination polymer chain with a Co1⋯Co1 separation of 10.746(2) Å and a Co1⋯Co1⋯Co1 angle of 111.49° (Fig. 3b). From the topological viewpoint, these chains can be classified as 2-connected underlying chains with the 2C1 topology. In addition, the adjacent 1D chains are further interconnected by H-bonds between each other and involving water molecules of crystallization, thus resulting in the generation of a complex 2D H-bonded supramolecular network (Fig. 3c). After simplification, topological analysis of such a layer reveals a binodal 3,5-connected net with the gek1 topology (Fig. 3d). It is defined by the point symbol of (3·4·5)(32·4·5·62·74), wherein the (3·4·5) and (32·4·5·62·74) notations correspond to the 3-connected [Co(2,2′-bipy)(H2O)]2+ and the 5-connected cppa2− nodes, respectively; there are also the 2-connected H2O linkers.
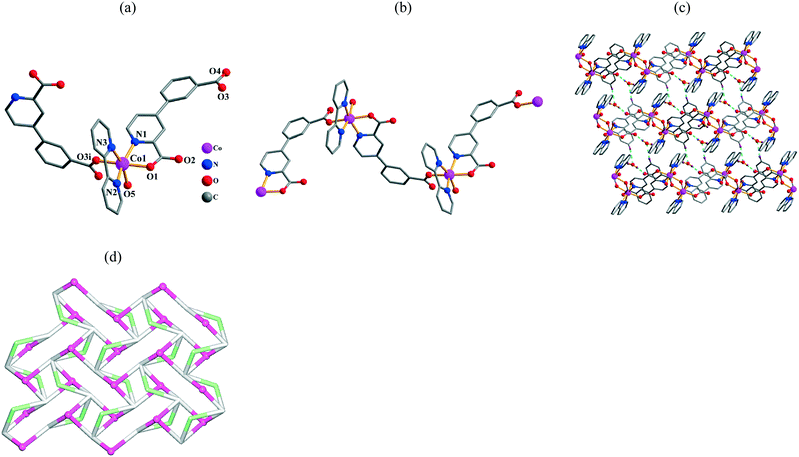 |
| | Fig. 3 Structural fragments of 5. (a) Coordination environment of the Co1 atom (H atoms are omitted for clarity). Symmetry codes: i: x − 1/2, −y + 3/2, z − 1/2. (b) Zigzag 1D coordination polymer chain along the ac plane. (c) 2D H-bonded supramolecular layer along the ac plane. (d) Topological representation of the underlying 2D H-bonded network showing a binodal 3,5-connected net with the gek1 topology; view along the a axis; color codes: centroids of 5-connected cppa2− nodes (gray), centroids of 3-connected [Co(2,2′-bipy)(H2O)]2+ nodes (pink balls), and centroids of 2-connected H2O linkers (pale green). | |
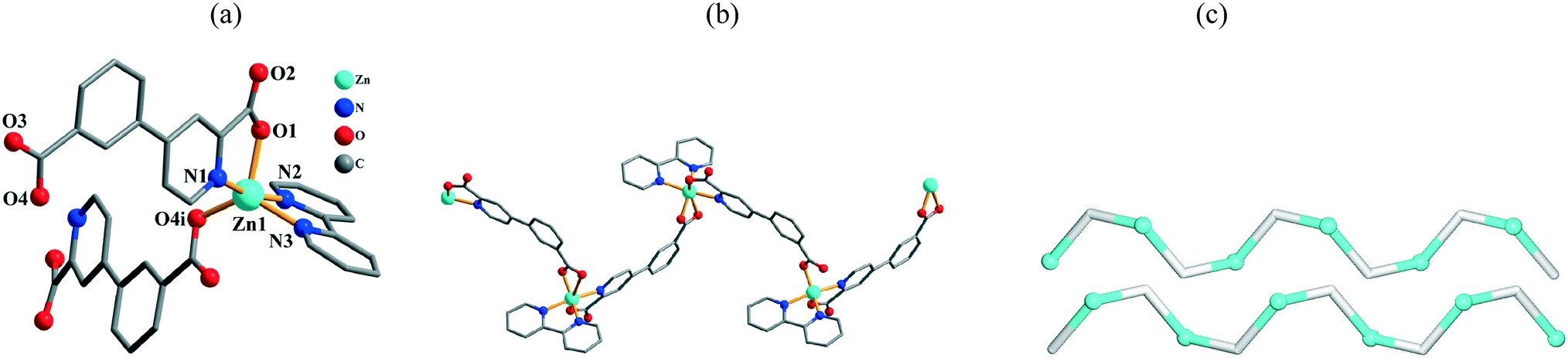
Crystal structure of [Zn(μ-cppa)(2,2′-bipy)]n (6)
The structure of 6 is also a 1D coordination polymer. The asymmetric unit comprises one crystallographically independent Zn(II) center, one μ-cppa2− moiety, and one 2,2′-bipy ligand. The five-coordinate Zn1 atom possesses a distorted trigonal bipyramidal {ZnN3O2} environment (Fig. 4a). The Zn1 center is surrounded by two O and one N atom from two distinct μ-cppa2− linkers and two N atoms from the 2,2′-bipy moiety. The Zn–O and Zn–N bond distances are in the 1.972(4)–2.028(4) and 2.069(5)–2.135(5) Å ranges, respectively; these are within the normal values observed in related coordination compounds.1c,4d,10e,11e
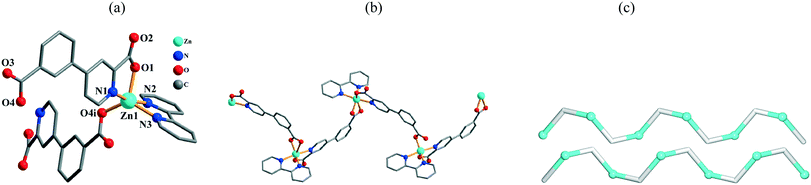 |
| | Fig. 4 Structural fragments of 6. (a) Coordination environment of the Zn1 atom (H atoms are omitted for clarity). Symmetry codes: i = −x + 1/2, y + 1/2, −z + 3/2. (b) Zigzag 1D coordination polymer chain along the ab plane. (c) Topological representation of two underlying 1D chains showing a uninodal 2-connected net with the 2C1 topology; view along the a axis; color codes: 2-connected [Zn(2,2′-bipy)]2+ nodes (cyan balls), centroids of 2-connected μ-cppa2− linkers (gray). | |
In 6, the cppa2− spacer adopts a μ-coordination mode (Scheme 2, mode IV) with both COO groups showing monodentate modes. In the cppa2−, the dihedral angle between the aromatic rings is 8.63°. The μ2-cppa2− linkers connect the neighboring Zn1 centers to generate a zigzag 1D coordination polymer chain with the Zn1⋯Zn1 separation of 10.672(2) Å and Zn1⋯Zn1⋯Zn1 angle of 97.77° (Fig. 4b). Topological analysis of the underlying 1D network (Fig. 4c) shows uninodal 2-connected chains with the 2C1 topology. In contrast to 5, no extension of the metal–organic chains is observed in 6 due to the lack of strong H-bonding interactions between the adjacent chains.
Crystal structures of [M(μ-cppa)(phen)(H2O)]n {M = Co (7), Zn (8), Mn (9), and Cu (10)}
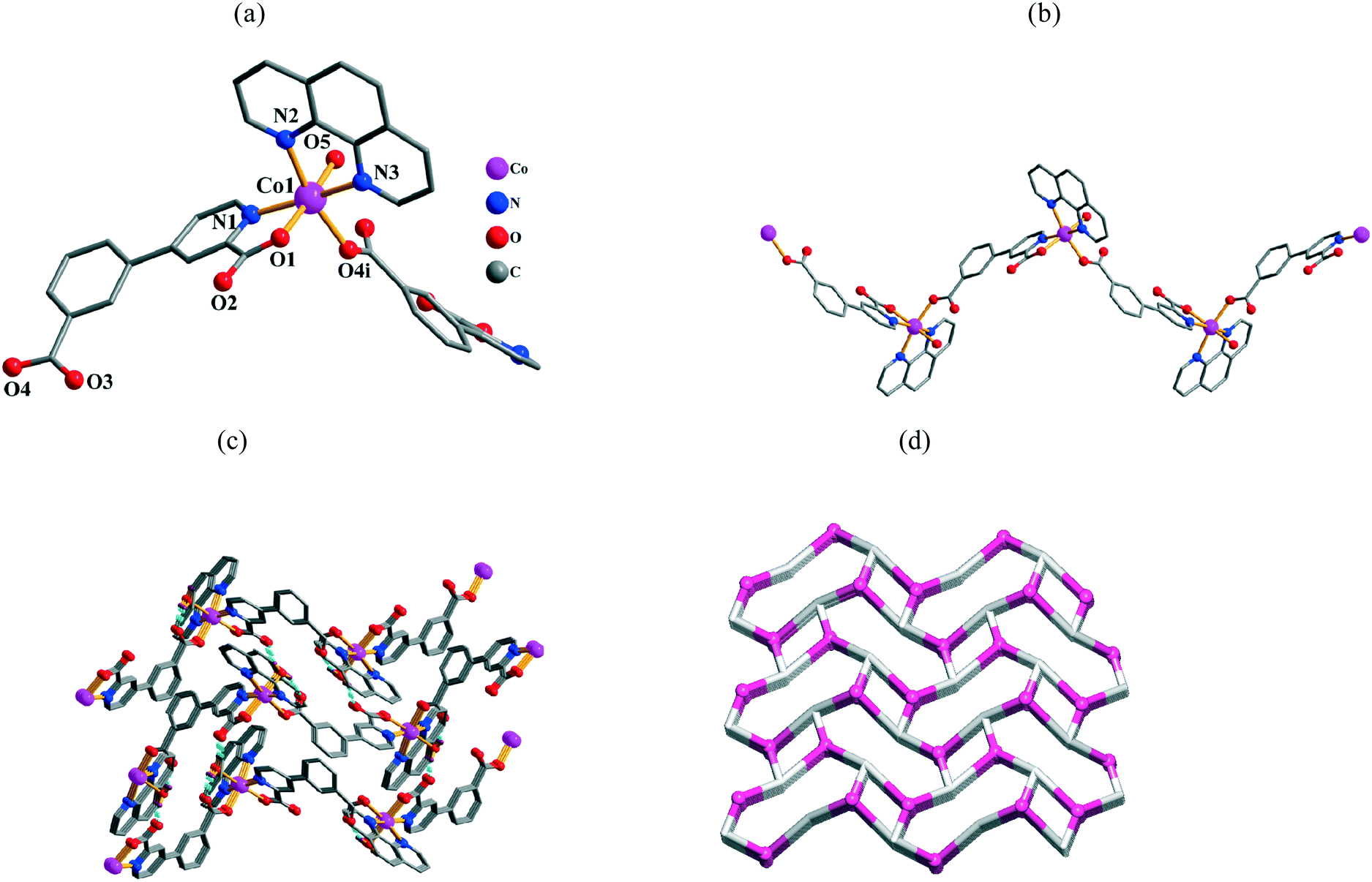
Single-crystal X-ray diffraction analyses reveal that the compounds 7–10 are isomorphous and, therefore, only 7 is described in detail as a representative example. Compound 7 is also shows a zigzag 1D coordination polymer structure. Its asymmetric unit bears one Co(II) atom, one μ-cppa2− block, one phen moiety, and one water ligand. The six-coordinate Co1 atom (Fig. 5a) is surrounded by two carboxylate O and one N atom from two different μ-cppa2− linkers, two phen N atoms, and one O atom from H2O ligand, thus forming a distorted octahedral {CoN3O3} geometry. The Co–O [2.054(3)–2.107(3) Å] and Co–N [2.125(4)–2.176(4) Å] bond distances agree with those in some related Co(II) compounds.8a,10a,f,11e In 7, the cppa2− block acts as a μ-linker (Scheme 2, mode III), in which both COO groups exhibit a monodentate mode. In the μ-cppa2− moiety, the dihedral angle between the phenyl and pyridyl is 18.92°. The μ-cppa2− linkers connect the adjacent Co(II) centers to generate a zigzag 1D coordination polymer chain with a Co1⋯Co1 separation of 12.537(2) Å and a Co1⋯Co1⋯Co1 angle of 96.22° (Fig. 5b). However, in contrast to 5, the crystal packing pattern and H-bonding interactions are essentially different in 7, resulting in the extension of the structure into a 3D H-bonded supramolecular network (Fig. 5c). From the topological viewpoint, this network is composed of the 3-connected [Co(phen)(H2O)]2+ and cppa2− nodes (topologically equivalent) and can be classified as a uninodal 3-connected net (Fig. 5d) with the srs (SrSi2) topology and the point symbol of (103).
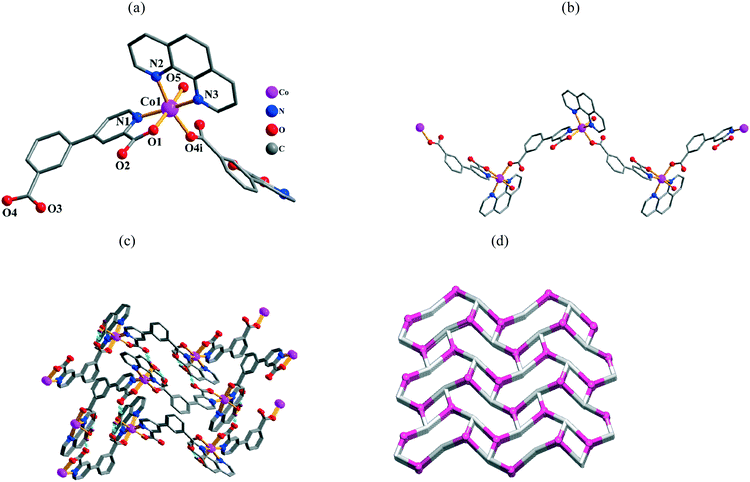 |
| | Fig. 5 Structural fragments of 7. (a) Coordination environment of the Co1 atom (H atoms are omitted for clarity). Symmetry codes: i = −x + 3/2, −y, z + 1/2. (b) Zigzag 1D coordination polymer chain along the bc plane. (c) 3D H-bonded supramolecular network along the bc plane. (d) Topological representation of the underlying 3D H-bonded network showing a uninodal 3-connected net with the srs topology; view along the a axis; color codes: centroids of 3-connected [Co(phen)(H2O)]2+ (pink balls) and cppa2− (gray) nodes. | |
Crystal structure of {[Cd3(μ3-cppa)3(phen)2]·4H2O}n (11)
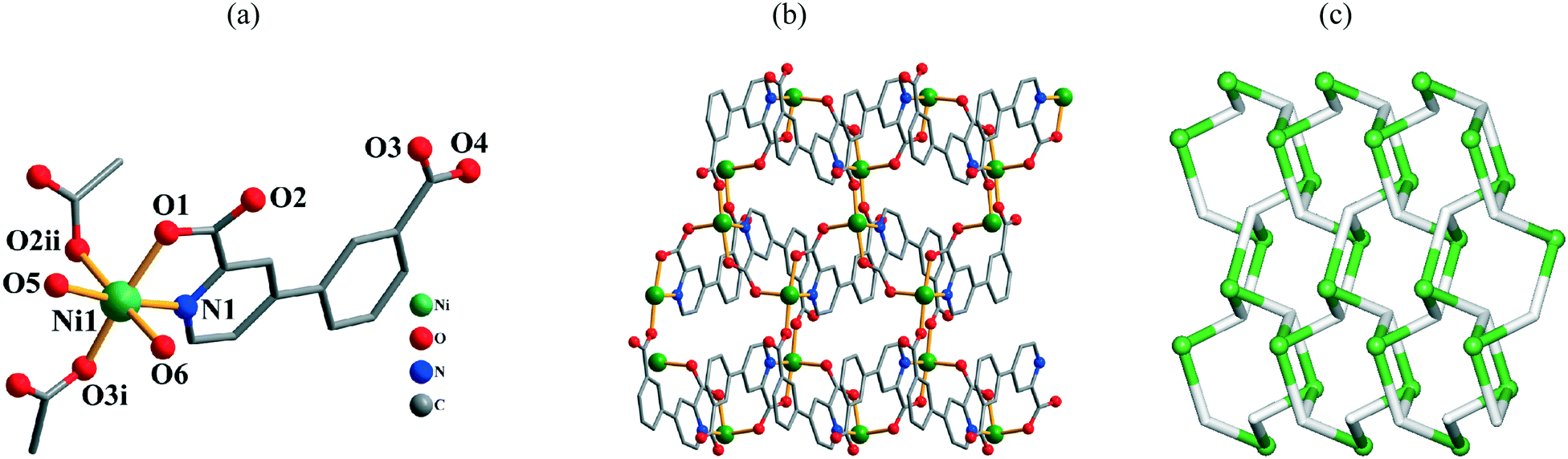
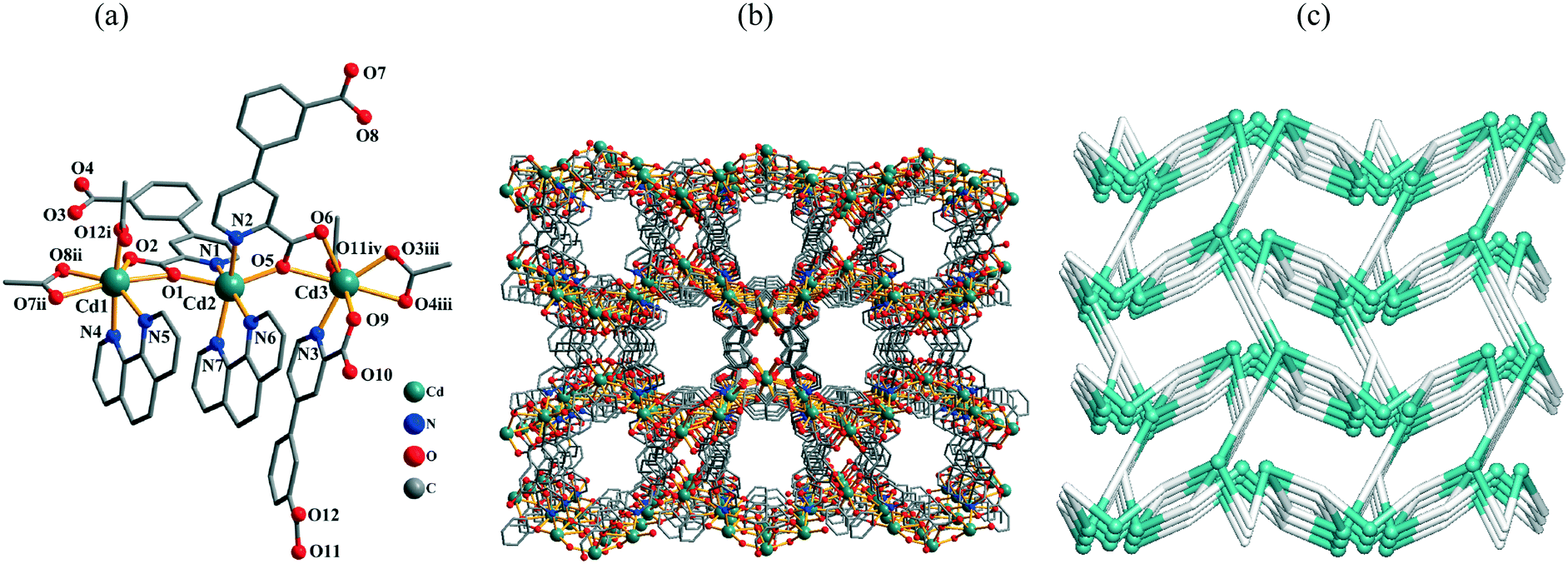
The structure of 11 demonstrates an extremely complex 3D metal–organic framework. In the asymmetric unit, there are three crystallographically unique Cd(II) atoms, three μ3-cppa2− blocks, two phen ligands, and four H2O molecules of crystallization. As shown in Fig. 6a, three Cd centers adopt distinct coordination environments. The seven-coordinate Cd1 atom is surrounded by five carboxylate O atoms from three different μ3-cppa2− blocks and two phen N atoms, forming a distorted pentagonal bipyramidal {CdN2O5} geometry. The six-coordinate Cd2 atom adopts a distorted octahedral {CdN4O2} geometry, which is filled by two O and two N atoms from two different μ3-cppa2− ligands and two phen N atoms. The Cd3 center is also seven-coordinate and is bound by six carboxylate O atoms and one N atom from the four different μ3-cppa2− blocks, thus constructing a distorted pentagonal pyramidal {CdNO6} geometry. The Cd–O bonds vary from 2.250(9) to 2.601(10) Å, whereas the Cd–N bonds range from 2.330(16) to 2.394(10) Å (Table S1, ESI†), being within normal values for Cd(II) coordination compounds.1c,4b,5d,6c In 11, the μ3-cppa2− ligands show two different coordination modes (Scheme 2, modes V and VI), in which the COO groups are monodentate, bidentate, or μ-bridging bidentate. The dihedral angles of 16.04 and 10.48° are observed between the pyridyl and phenyl rings in the μ3-cppa2− blocks. The three neighboring Cd(II) centers are interconnected by means of two COO groups from the two individual cppa2− blocks, giving rise to a tricadium(II) subunit (Fig. 6a). In this Cd3 subunit, the Cd1⋯Cd2, Cd2⋯Cd3, and Cd1⋯Cd3 distances are 4.606(2), 4.563(2), and 9.163(2) Å, respectively. The Cd1⋯Cd2⋯Cd3 angle is 175.91°. The adjacent Cd3 subunits are multiply interlinked by the cppa2− blocks into a 3D MOF (Fig. 6b). The framework possesses channels with a dimension of 7.48 × 5.16 Å, measured by atom-to-atom distances. If the coordinated and lattice water molecules are removed, the PLATON analysis reveals that the network's free volume attains 7.73% of the crystal volume.15 Given the presence of three different types of Cd atoms interlinked by the μ3-cppa2− blocks, compound 11 shows a very complicated 3D metal–organic framework. As a result, a tetranodal 3,3,3,4-connected underlying net can be generated (Fig. 6c). It is composed of the 3-connected Cd1 and 4-connected Cd3 nodes, as well as two topologically distinct types of the 3-connected μ3-cppa2− nodes; in addition, there are also the 2-connected Cd2 atoms that are considered as linkers from the topological viewpoint. This complex net features a unique topology that is defined by the point symbol of (4·102)2(4·82·102·12)(4·82)(8·102). Herein, the (4·102) and (8·102) indices correspond to different μ3-cppa2− nodes, whereas the (4·82·102·12) and (4·82) notations correspond to the Cd3 and Cd1 nodes, respectively. The novelty of this topological type was confirmed by a search of different databases.13,14
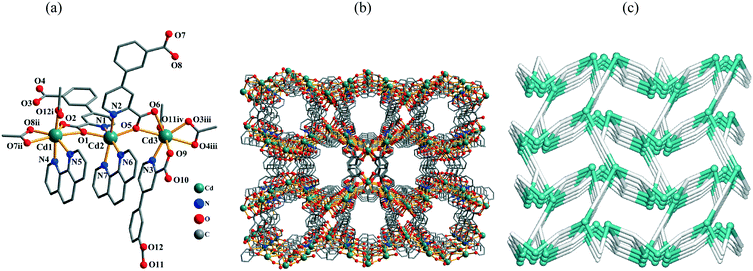 |
| | Fig. 6 Structural fragments of 11. (a) Coordination environment of the Cd(II) atoms (H atoms are omitted for clarity). Symmetry codes: i = −x + 1/2, y − 1/2, −z + 3/2; ii = x − 1/2, −y + 1/2, z + 1/2; iii = x + 1/2, −y + 1/2, z − 1/2; iv = −x + 3/2, y − 1/2, −z + 3/2. (b) 3D metal–organic framework along the ac plane. (c) Topological representation of the underlying 3D framework showing a tetranodal 3,3,3,4-connected net with the unique topology and point symbol of (4·102)2(4·82·102·12)(4·82)(8·102); view along the a axis; color codes: 2-, 3-, and 4-connected Cd atoms (turquoise balls), centroids of 3-connected μ3-cppa2− nodes (gray). | |
Coordination modes of Hcppa−/cppa2− blocks and structural comparison
In compounds 1–11, the deprotonation of 4-(3-carboxyphenyl)picolinic acid (H2cppa) resulted in the Hcppa− or cppa2− ligands that exhibit six different coordination modes (Scheme 2). Depending on the molar ratio of NaOH and H2cppa used during the synthesis of compounds, the dicarboxylate moiety adopts partially and fully deprotonated forms. For example, a 1:1 molar ratio between NaOH and H2cppa resulted in the Hcppa− form in 1 and 2. When NaOH is used in excess (2:1 molar ratio relative to H2cppa), a completely deprotonated cppa2− form is generated in 3–11. The carboxylate groups of 4-(3-carboxyphenyl)picolinate ligands adopt four typical modes, including monodentate, bidentate, μ-bridging bidentate, and μ-bridging tridentate modes (Scheme 2), whereas the pyridyl N atom behaves as an N-donor for metal(II) centers in all products. Moreover, a noteworthy property of semirigid 4-(3-carboxyphenyl)picolinate ligands consists of the rotation of the C–C single bond between the pyridyl and phenyl rings (dihedral angles are in the 8.63–31.65° range), thus facilitating the formation of a certain coordination environment around metal ions during the self-assembly process. Hence, the ligands derived from H2cppa can adopt various coordination modes and behave as either terminal (in 1 and 2) or distinct μ- (in 5–10) or μ3-building blocks (in 3, 4, and 11).
Compounds 1 and 3 were prepared under similar reaction conditions, except for a difference between the molar ratios of NaOH and H2cppa (1:1 for 1 and 2:1 for 3). Their structural differences indicate that the assembly process is dependent on the molar ratio of NaOH and the main building block. Products 4, 5, and 7 were also assembled under similar conditions, except for the introduction of the auxiliary ligands in 5 and 7 (2,2′-bipy for 5 and phen for 7). As a result, distinct structures were generated, namely a 2D metal–organic layer in 4 (in the absence of any auxiliary ligand) and zigzag 1D chains in 5 and 7 (in the presence of auxiliary ligands). The decrease in the network dimensionality from 2D to 1D can be attributed to a large steric hindrance of the auxiliary ligands. Products 7 and 11 were also prepared under essentially similar reaction conditions, except for the type of metal chloride used (CoCl2·6H2O for 7 and CdCl2·H2O for 11). Their structural differences show that the assembly process is metal ion-dependent, thus resulting in a 1D coordination polymer 7 and a 3D metal–organic framework 11. All the above-mentioned features indicate that the molar ratio of reagents, the presence of auxiliary ligands, and the type of the metal ions significantly influence the assembly process and the resulting structures of complexes or coordination polymers.
Apart from the distinct dimensionality of 1–11 that ranges from 0D (in 1 and 2) to 1D (in 5–10), 2D (in 3 and 4), and 3D (in 11), all the obtained products also display various types of topological networks, including an undocumented topology of MOF 11. In fact, the topological analysis and classification of the H-bonded or metal–organic networks in the obtained structures revealed the following underlying nets: pcu in 1 and 2, fes in 3 and 4, 2C1 in 5–10, and a topologically unique 3D framework in 11.
Luminescence properties
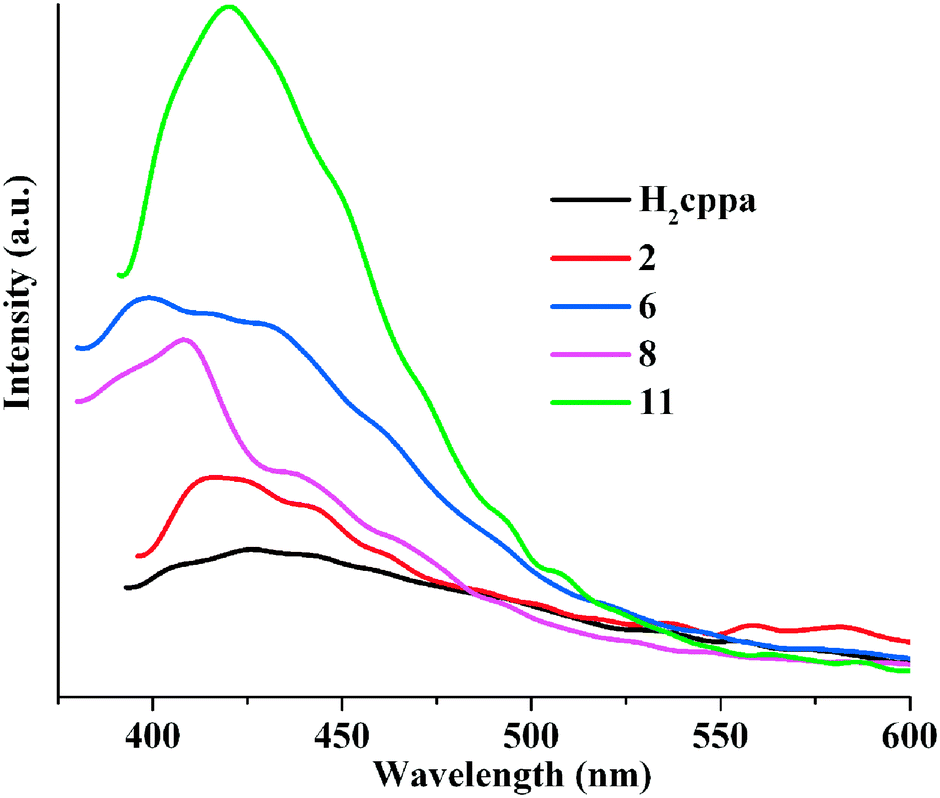
The emission spectra of free H2cppa and Zn and Cd containing products 2, 6, 8, and 11 were measured at room temperature in the solid state (Fig. 7). H2cppa displays a weak photoluminescence with an emission maximum at 421 nm if excited at 341 nm. However, compounds 2, 6, 8, and 11 exhibit much more intense emission bands with the maximum at 415 nm for 2, 405 nm for 6, 411 nm for 8, and 418 nm for 11 (λex was 341 nm in all cases). These bands can be assigned to an intraligand (π* → n or π* → π) emission.8a,16,17 The more intense luminescence of the metal-containing compounds can be explained by a binding of ligands to metal centers, which increases the ligand's rigidity and may diminish an energy loss due to radiationless decay.10f,18,19 It is interesting to highlight that the MOF 11 exhibits a stronger emission intensity in comparison with 2, 6, and 8 (Zn(II)-containing products), which can potentially be explained by their structural differences and the presence of Cd(II) atoms. In 2, the Hcppa− blocks act as simple terminal ligands, while in 6, 8, and 11 the cppa2− moieties adopt a μ- and μ3-coordination fashion, respectively. The latter may turn the ligands more rigid, thus reducing a vibration-induced deactivation.11e,18,19 An intense photoluminescence of 11 suggests that this compound can potentially be considered as a candidate for use in photoactive materials.
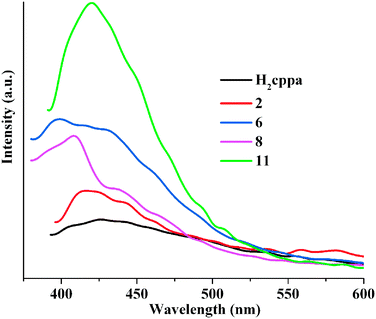 |
| | Fig. 7 Solid state emission spectra of H2cppa, 2, 6, 8, and 11 [λex was 341 nm]. | |
Magnetic properties
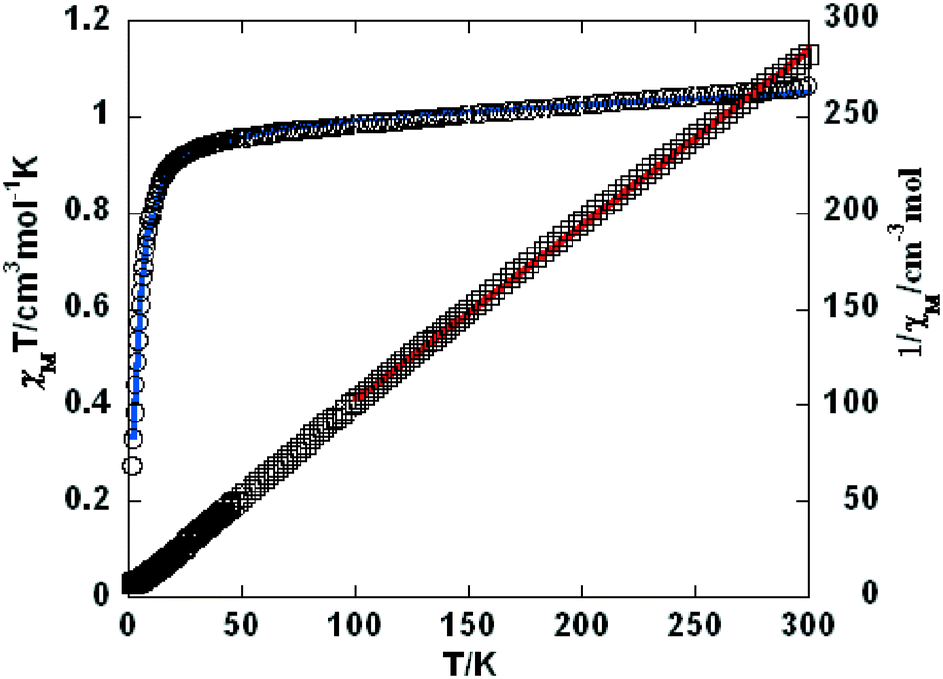
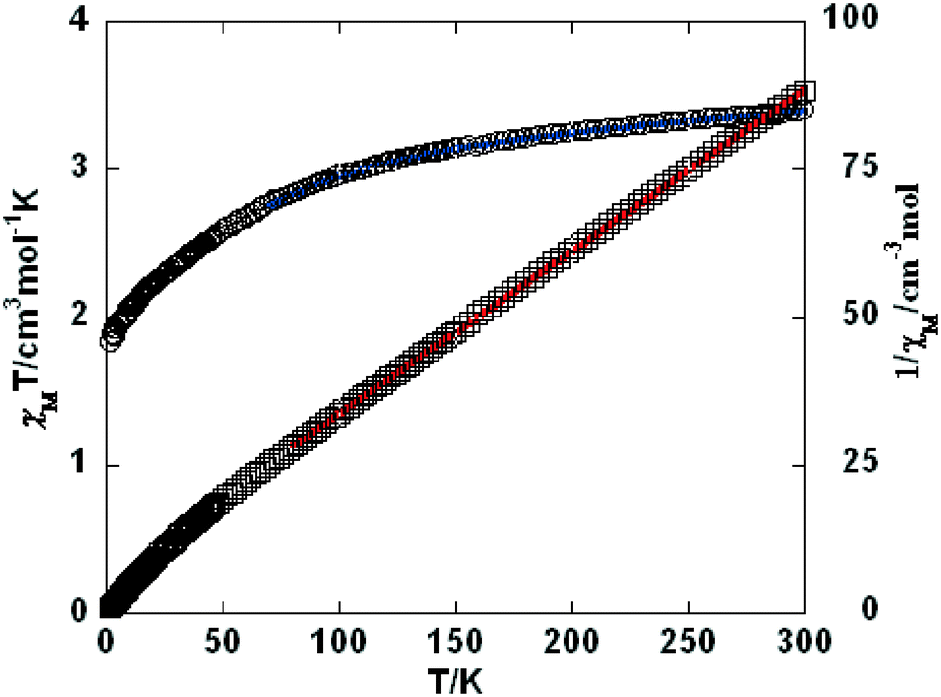
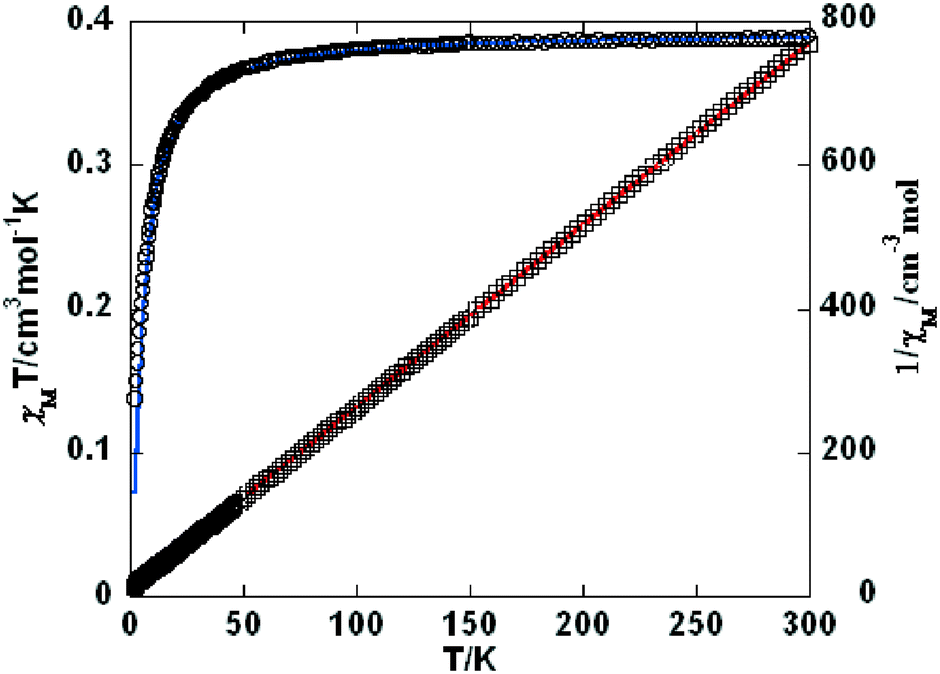
Variable-temperature magnetic susceptibility investigation was carried out on powder samples of 3–5, 7, 9, and 10 in the 2–300 K temperature range. For 3 (Fig. 8), the χMT value of 1.06 cm3 mol−1 K at room temperature is close to the expected one (1.00 cm3 mol−1 K) for one magnetically isolated Ni(II) ion (S = 1, g = 2.0). Upon cooling, the χMT value decreases very slowly from 1.06 cm3 mol−1 K at 300 K to 0.956 cm3 mol−1 K at 50 K, and then decreases steeply to 0.274 cm3 mol−1 K at 2 K. In the 100–300 K interval, the χM−1vs. T plot for 3 obeys the Curie–Weiss law with a Weiss constant θ of −25.7 K and a Curie constant C of 1.15 cm3 mol−1 K. An empirical (Weng's) formula can be applied to analyze the 1D systems with S = 1, using numerical procedures:20,21
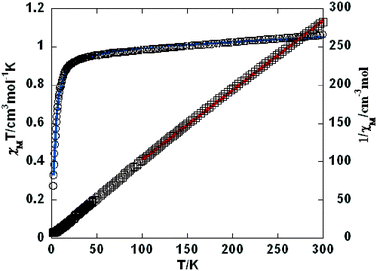 |
| | Fig. 8 Temperature dependence of χMT (○) and 1/χM (□) vs. T for compound 3. The blue line represents the best fit to the equations in the text. The red line shows the Curie–Weiss fitting. | |
Using this method, the best-fit parameters for 3 were obtained: g = 2.04, J = −9.65 cm−1, and R = 6.1 × 10−5, where R = ∑(χobsT − χcalcT)2/∑(χobsT)2. The J value of −9.65 cm−1 indicates that the coupling between the Ni(II) centers is antiferromagnetic.
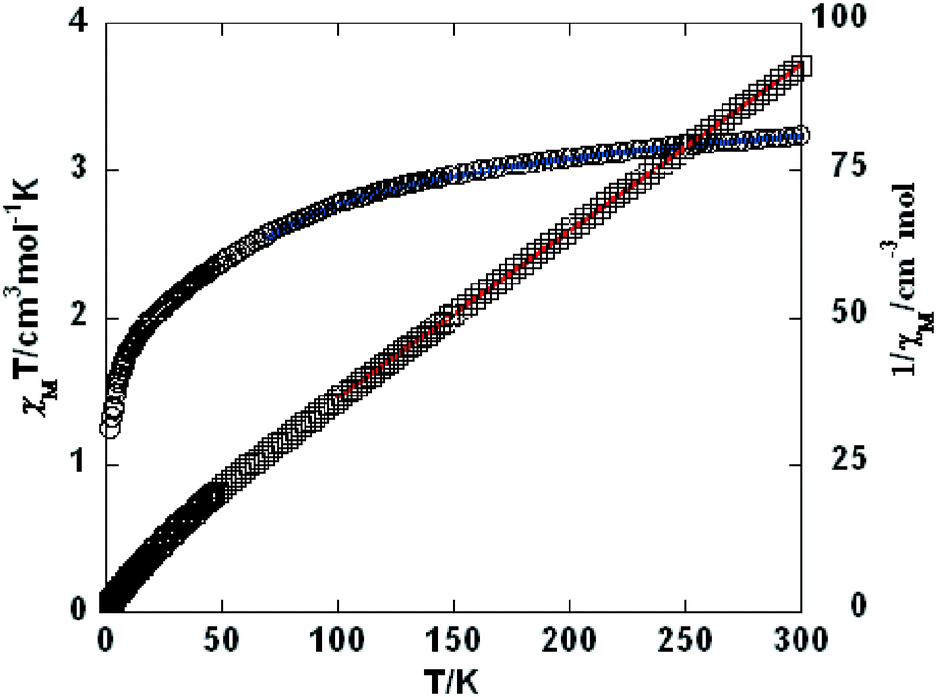
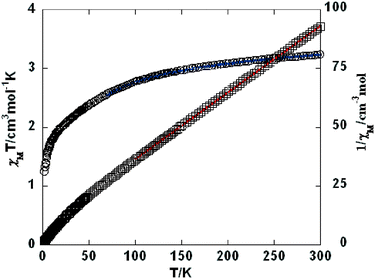
As shown in Fig. 9–11, the temperature dependence of magnetic susceptibility for compounds 4, 5, and 7 is rather similar. The room temperature values of χMT, 3.24 cm3 mol−1 K for 4, 3.40 cm3 mol−1 K for 5, and 3.36 cm3 mol−1 K for 7, are superior to the value (1.83 cm3 mol−1 K) expected for a magnetically isolated high-spin Co(II) ion (S = 3/2, g = 2.0). This is a common phenomenon for Co(II) ions due to their strong spin–orbital coupling interactions.10a,d,f,11e On lowering the temperature, the χMT values decrease slowly until about 120 K, then decrease quickly to 1.25, 1.84, and 1.33 cm3 mol−1 K at 2.0 K for 4, 5, and 7, respectively. Between 90 and 300 K, the magnetic susceptibility can be fitted to the Curie–Weiss law with C = 3.53 cm3 mol−1 K and θ = −28.4 K for 4, C = 3.50 cm3 mol−1 K and θ = −11.4 K for 5, and C = 3.45 cm3 mol−1 K and θ = −10.3 K for 7. These results indicate an antiferromagnetic interaction between the adjacent Co(II) centers in these three compounds. We attempted to fit the data for 4, 5, and 7 by applying the following expression for a 1D Co(II) chain:20
| χchain = (Ng2β2/kT)[2.0 + 0.0194x + 0.77x2][3.0 + 4.346x + 3.232x2 + 5.834x3]−1 x = ∣J∣/kT. |
 |
| | Fig. 9 Temperature dependence of χMT (○) and 1/χM (□) vs. T for compound 4. The blue line represents the best fit to the equations in the text. The red line shows the Curie–Weiss fitting. | |
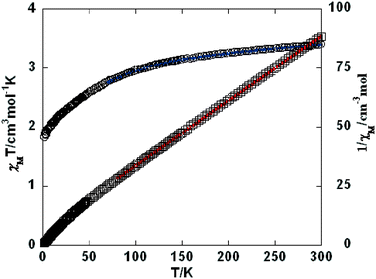 |
| | Fig. 10 Temperature dependence of χMT (○) and 1/χM (□) vs. T for compound 5. The blue line represents the best fit to the equations in the text. The red line shows the Curie–Weiss fitting. | |
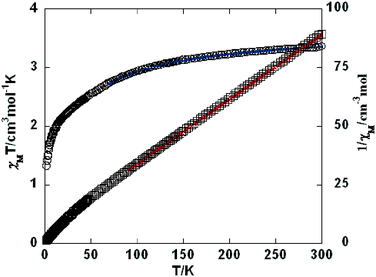 |
| | Fig. 11 Temperature dependence of χMT (○) and 1/χM (□) vs. T for compound 7. The blue line represents the best fit to the equations in the text. The red line shows the Curie–Weiss fitting. | |
By applying this rough model, the susceptibility for 4, 5, and 7 above 70 K was simulated, leading to J = −9.87 cm−1, g = 2.43 for 4, J = −3.52 cm−1, g = 2.45 for 5, and J = −2.87 cm−1, g = 2.46 for 7. The negative J parameters indicate a weak antiferromagnetic exchange coupling between the adjacent Co(II) centers in 4, 5, and 7, which is in agreement with negative θ values. The structures of 3 and 4 show that there is only one magnetic exchange pathway within the chain (via a syn–anti carboxylate bridge), which is responsible for the observed antiferromagnetic exchange.
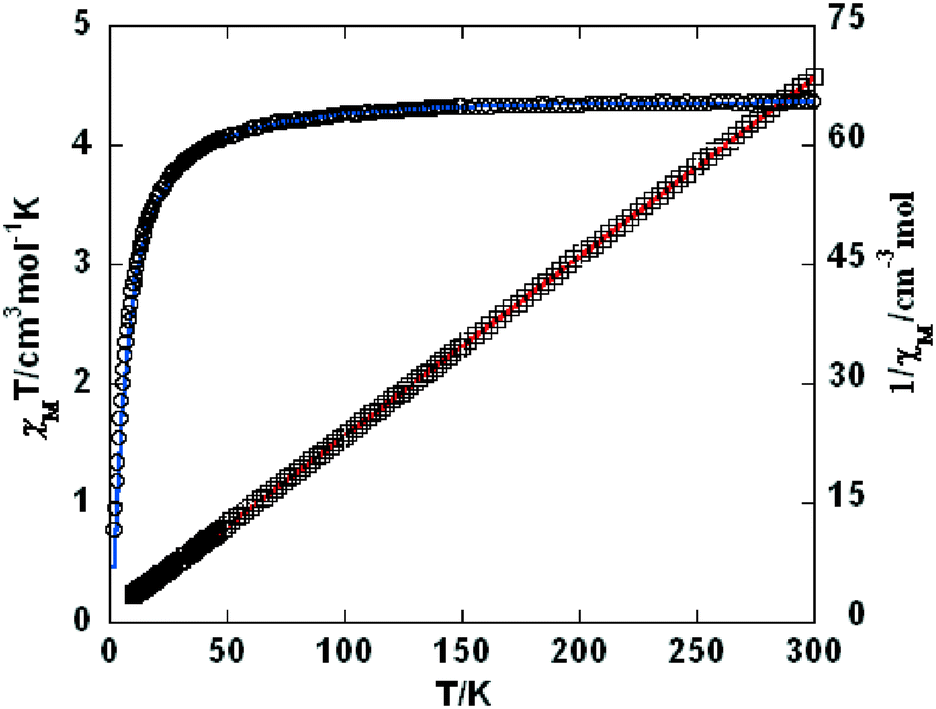
For 9, the χMT value at 300 K is 4.37 cm3 mol−1 K, which is equal to the spin only value of 4.38 cm3 mol−1 K for a magnetically isolated Mn(II) center (SMn = 5/2, g = 2.0). Upon cooling, the χMT value decreases very slowly from 4.44 cm3 mol−1 K at 300 K to 4.20 cm3 mol−1 K at 72 K, and then decreases steeply to 0.78 cm3 mol−1 K at 2 K (Fig. 12). In the 2–300 K interval, the χM−1vs. T plot for 9 obeys the Curie–Weiss law with a Weiss constant θ of −3.17 K and a Curie constant C of 4.41 cm3 mol−1 K. We fitted the magnetic data of 9 using the following expression22 for a 1D Mn(II) chain:
| χchain = (Ng2β2/kT)[A + Bx2][1 + Cx + Dx3]−1 with A = 2.9167, B = 208.04, C = 15.543, D = 2707.2, and x = ∣J∣/kT |
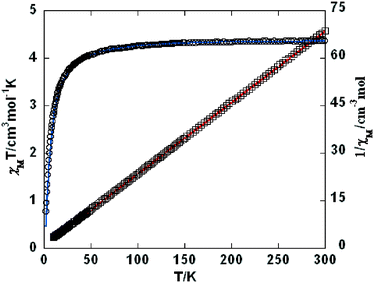 |
| | Fig. 12 Temperature dependence of χMT (○) and 1/χM (□) vs. T for compound 9. The red line represents the best fit to the equations in the text. The blue line shows the Curie–Weiss fitting. | |
By applying this model, the susceptibility for 9 was simulated, leading to J = −2.25 cm−1, g = 2.06, and the agreement factor R = 5.69 × 10−6 (R = ∑(χobsT − χcalcT)2/∑(χobsT)2). A low J parameter confirms that an extremely weak antiferromagnetic exchange coupling exists between the adjacent Mn(II) centers, which also agrees with a negative θ value.
For 10, the χMT value of 0.39 cm3 mol−1 K at 300 K is very close to the value (0.38 cm3 mol−1 K) expected for a magnetically isolated Cu(II) ion (S = 1/2, g = 2.0). Upon cooling, χMT decreases continuously to a minimum of 0.14 cm3 mol−1 K at 2 K (Fig. 13). Between 2 and 300 K, the magnetic susceptibilities can be fitted to the Curie–Weiss law with C = 0.39 cm3 mol−1 K and θ = −1.50 K. The negative θ value points out the presence of dominant antiferromagnetic interactions between the Cu(II) ions. χ(T) was also analyzed as an isotropic Heisenberg spin chain using the Bonner–Fisher model for a 1D, S = (1/2), spin chain system with the nearest neighbor spin AF coupling J as:23,24
where
x = |
J|/
kBT. The
χ(
T) is fitted satisfactorily in the temperature range of 2–300 K. The fitted parameters are
J/
kB = −2.37 K and
g = 2.02. A low
J parameter confirms an extremely weak antiferromagnetic exchange coupling between the adjacent Cu(
II) centers, which is in agreement with a negative
θ value.
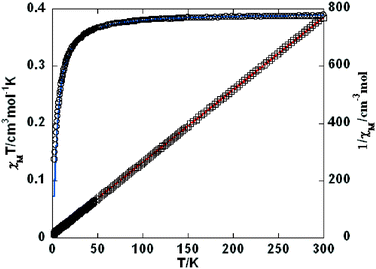 |
| | Fig. 13 Temperature dependence of χMT (○) and 1/χM (□) vs. T for compound 10. The red line shows the Curie–Weiss fitting. | |
In 5, 7, 9, and 10, a weak antiferromagnetic coupling can be attributed to the large M⋯M separations [10.746(3) Å for 5, 12.537(3) Å for 7, 12.620(3) Å for 9, and 12.522(4) Å for 10].
Conclusions
In the present work, we showed that 4-(3-carboxyphenyl)picolinic acid can act as a versatile N,O-building block for the generation, via a hydrothermal self-assembly pathway, of a series of novel metal(II) (Ni, Zn, Co, Mn, Cu, and Cd) coordination compounds. By selecting different metal(II) nodes and adjusting the molar ratio between a metal ion, H2cppa main building block, NaOH, and/or the type of the aromatic N-donor ancillary ligand (optional), we synthesized eleven new coordination compounds, also using water as a benign reaction medium. All the obtained products were fully characterized and represent the first structurally characterized coordination compounds derived from H2cppa. Their structures range from the complex 3D metal–organic framework 11 and the 2D coordination polymers 3 and 4 to the 1D coordination polymers 5–10, as well as the discrete 0D monomers 1 and 2. The structures of 1, 2, 5, and 7–10 are further extended [0D → 3D (1 and 2), 1D → 2D (5), and 1D → 3D (7–10)] into various supramolecular networks by strong hydrogen bonding interactions. Hence, the generated products 1–11 not only represent distinct dimensionalities but also disclose a variety of topological networks. In fact, one topologically unique multinodal net was identified in the very complex 3D MOF 11, thus also providing a contribution toward the search for new topologically unique networks in crystal engineering research. Further exploration of 4-(3-carboxyphenyl)picolinic acid as a poorly studied building block toward the construction of functional metal–organic networks is currently in progress.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Project 21201091). AMK acknowledges the Foundation for Science and Technology (FCT), Portugal (PEst-OE/QUI/UI0100/2013).
Notes and references
-
(a) W. X. Zhang, P. Q. Liao, R. B. Lin, Y. S. Wei, M. H. Zeng and X. M. Chen, Coord. Chem. Rev., 2015, 293-294, 263 Search PubMed;
(b) B. Y. Li, M. Chrzanowski, Y. M. Zhang and S. Q. Ma, Coord. Chem. Rev., 2016, 307, 106 Search PubMed;
(c) W. G. Lu, C. Y. Su, T. B. Lu, L. Jiang and J. M. Chen, J. Am. Chem. Soc., 2006, 128, 34 Search PubMed;
(d) Y. B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641 Search PubMed;
(e) X. D. Zheng and T. B. Lu, CrystEngComm, 2010, 12, 324 Search PubMed;
(f) G. Férey, Chem. Soc. Rev., 2008, 37, 191 Search PubMed;
(g) Z. Q. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315 Search PubMed;
(h) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 Search PubMed;
(i) G. Ferey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380 Search PubMed;
(j) A. Bhunia, S. Dey, M. Bous, C. Zhang, W. von Rybinski and C. Janiak, Chem. Commun., 2015, 51, 484 Search PubMed.
-
(a) S. D. Han, S. J. Liu, Q. L. Wang, X. H. Miao, T. L. Hu and X. H. Bu, Cryst. Growth Des., 2015, 15, 2253 Search PubMed;
(b) B. Gil-Hernández, S. Savvin, G. Makhloufi, P. Núňez, C. Janiak and J. Sanchiz, Inorg. Chem., 2015, 54, 1597 Search PubMed;
(c) M. L. Han, X. Q. Wu, G. W. Xu, G. X. Wen, D. S. Li and L. F. Ma, Inorg. Chem. Commun., 2016, 69, 31 Search PubMed;
(d) X. Jiang, C. M. Liu and H. Z. Kou, Inorg. Chem., 2016, 55, 5880 Search PubMed.
-
(a) S. S. Chen, L. Q. Sheng, Y. Zhao, Z. D. Liu, R. Qiao and S. Yang, Cryst. Growth Des., 2016, 16, 229 Search PubMed;
(b) J. Pang, F. Jiang, M. Wu, D. Yuan, K. Zhou, J. Qian, K. Su and M. C. Hong, Chem. Commun., 2014, 50, 2834 Search PubMed;
(c) R. Q. Zhong, Z. L. Xu, W. Z. Bi, S. B. Han, X. F. Yu and R. Q. Zou, Inorg. Chim. Acta, 2016, 443, 299 CrossRef CAS;
(d) P. T. K. Nguyen, H. T. D. Nguyen, H. Q. Pham, J. Kim, K. E. Cordova and H. Furukawa, Inorg. Chem., 2015, 54, 10065 Search PubMed.
-
(a) A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313 Search PubMed;
(b) L. W. Lee, T. T. Luo, C. M. Wang, G. H. Lee, S. M. Peng, Y. H. Liu, S. L. Lee and K. L. Lu, J. Solid State Chem., 2016, 239, 1 Search PubMed;
(c) J. Lee, Y. Kang, N. S. Cho and K. M. Park, Cryst. Growth Des., 2016, 16, 996 Search PubMed;
(d) M. K. Park, K. S. Lim, J. H. Park, J. H. Song, D. W. Kang, W. R. Lee and C. S. Hong, CrystEngComm, 2016, 18, 4349 Search PubMed.
-
(a) Y. Wu, J. Wang, L. Zou, F. C. Zeng, J. S. Feng, X. R. Wu, J. W. Xu and S. R. Batten, Inorg. Chem. Commun., 2016, 69, 13 Search PubMed;
(b) Y. L. Gai, K. C. Xiong, L. Chen, Y. Bu, X. J. Li, F. L. Jiang and M. C. Hong, Inorg. Chem., 2012, 51, 13128 Search PubMed;
(c) R. B. Lin, S. Y. Liu, J. W. Ye, X. Y. Li and J. P. Zhang, Adv. Sci., 2016, 3, 1500434 Search PubMed;
(d) K. M. Wang, L. Du, Y. L. Ma and Q. H. Zhao, Transition Met. Chem., 2016, 41, 573 Search PubMed;
(e) L. Jaremko, A. M. Kirillov, P. Smoleński and A. J. L. Pombeiro, Cryst. Growth Des., 2009, 9, 3006 Search PubMed;
(f) M. V. Kirillova, A. M. Kirillov, A. N. C. Martins, C. Graiff, A. Tiripicchio and A. J. L. Pombeiro, Inorg. Chem., 2012, 51, 5224 Search PubMed.
-
(a) C. K. Wang, F. F. Xing, Y. L. Bai, Y. M. Zhao, M. X. Li and S. R. Zhu, Cryst. Growth Des., 2016, 16, 2277 Search PubMed;
(b) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 Search PubMed;
(c) I. H. Choi, Y. Kim, D. N. Lee and S. Huh, Polyhedron, 2016, 105, 96 Search PubMed;
(d) R. A. Agarwal and S. Mukherjee, Polyhedron, 2016, 105, 228 Search PubMed;
(e) L. Zhang, Z. Su, F. Jiang, L. Yang, J. Qian, Y. Zhou, W. Li and M. Hong, Nanoscale, 2014, 6, 6590 Search PubMed;
(f) M. Yadav, A. Bhunia, S. K. Jana and P. W. Roesky, Inorg. Chem., 2016, 55, 2701 Search PubMed;
(g) M. S. Deenadayalan, N. Sharma, P. K. Verma and C. M. Nagaraja, Inorg. Chem., 2016, 55, 5320 Search PubMed;
(h) D. De, T. K. Pal, S. Neogi, S. Senthilkumar, D. Das, S. S. Gupta and P. K. Bharadwaj, Chem. – Eur. J., 2016, 22, 3387 CrossRef CAS PubMed.
-
(a) G. Ye, K. Y. Zou, Y. Yang, J. J. Wang, X. F. Gou and Z. X. Li, J. Solid State Chem., 2015, 225, 31 Search PubMed;
(b) S. Parshamoni and S. Konar, CrystEngComm, 2016, 18, 4395 Search PubMed;
(c) Y. Q. Chen, S. J. Liu, Y. W. Li, G. R. Li, K. H. He, Z. Chang and X. H. Bu, CrystEngComm, 2013, 15, 1613 Search PubMed;
(d) S. S. Chen, Q. Liu, Y. Zhao, R. Qiao, L. Q. Sheng, Z. D. Liu, S. Yang and C. F. Song, Cryst. Growth Des., 2014, 14, 3727 Search PubMed.
-
(a) J. Z. Gu, Z. Q. Gao and Y. Tang, Cryst. Growth Des., 2012, 12, 3312 Search PubMed;
(b) X. X. Xu, Y. Lu, E. B. Wang, Y. Ma and X. L. Bai, Cryst. Growth
Des., 2006, 6, 2029 Search PubMed;
(c) C. P. Li and M. Du, Chem. Commun., 2011, 47, 5958 Search PubMed;
(d) L. Hou, Y. Y. Lin and X. M. Chen, Inorg. Chem., 2008, 47, 1346 Search PubMed.
-
(a) A. M. Stepenson and D. Ward, Chem. Commun., 2012, 48, 3605 Search PubMed;
(b) R. Singh and P. K. Bharatdwaj, Cryst. Growth Des., 2013, 13, 3722 Search PubMed;
(c) Q. Gao, F. L. Jiang, M. Y. Wu, Y. G. Huang, W. Wei and M. C. Hong, Cryst. Growth Des., 2010, 10, 184 Search PubMed;
(d) M. Du, C. P. Li, C. S. Liu and S. M. Fang, Coord. Chem. Rev., 2013, 257, 1282 Search PubMed;
(e) P. Smoleński, S. W. Jaros, C. Pettinari, G. Lupidi, L. Quassinti, M. Bramucci, D. Petrelli, L. A. Vitali, A. Kochel and A. M. Kirillov, Dalton Trans., 2013, 42, 6572 Search PubMed.
-
(a) S. B. Miao, B. M. Ji, Y. F. Wang, Z. H. Li, D. S. Deng, C. Y. Xu and L. Zhou, Inorg. Chem. Commun., 2015, 62, 47 Search PubMed;
(b) L. Li, X. Y. Cao and R. D. Huang, Chin. J. Chem., 2016, 34, 143 Search PubMed;
(c) J. Zhao, Y. N. Wang, W. W. Dong, Y. P. Wu, D. S. Li and Q. C. Zhang, Inorg. Chem., 2016, 55, 3265 Search PubMed;
(d) L. Z. Chen, Q. J. Pan, X. X. Cao and F. M. Wang, CrystEngComm, 2016, 18, 1944 Search PubMed;
(e) Y. L. Shao, Y. H. Cui, J. Z. Gu, J. Wu, Y. W. Wang and A. M. Kirillov, CrystEngComm, 2016, 18, 765 Search PubMed;
(f) J. Z. Gu, A. M. Kirillov, J. Wu, D. Y. Lv, Y. Tang and J. C. Wu, CrystEngComm, 2013, 15, 10287 Search PubMed.
-
(a) H. Erer, O. Z. Yeşilel and M. Arıcı, Cryst. Growth Des., 2015, 15, 3201 CrossRef CAS;
(b) B. B. Ding, Y. Q. Weng, Z. W. Mao, C. K. Lan, X. M. Chen and B. H. Ye, Inorg. Chem., 2005, 44, 8836 Search PubMed;
(c) L. Chen, S. H. Gou and J. Q. Wang, J. Mol. Struct., 2011, 991, 149 Search PubMed;
(d) W. L. Liu, L. H. Ye, X. F. Liu, L. M. Yuan, J. X. Jiang and C. G. Yan, CrystEngComm, 2008, 10, 1395 Search PubMed;
(e) J. Z. Gu, Y. H. Cui, X. X. Liang, J. Wu, D. Y. Lv and A. M. Kirillov, Cryst. Growth Des., 2016, 16, 4658 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 Search PubMed.
-
(a) V. A. Blatov, IUCr CompComm Newsletter, 2006, 7, 4 Search PubMed;
(b) V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 Search PubMed.
-
(a) M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 Search PubMed;
(b) M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343 Search PubMed.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 Search PubMed.
- Y. S. Wang and Z. M. Zhou, J. Solid State Chem., 2015, 228, 117 Search PubMed.
- S. Pal, T. K. Pal and P. K. Bharadwaj, CrystEngComm, 2016, 18, 1825 Search PubMed.
- V. W. W. Yam, Acc. Chem. Res., 2002, 35, 555 Search PubMed.
- S. L. Zheng, J. H. Yang, X. L. Yu, X. M. Chen and W. T. Wong, Inorg. Chem., 2004, 43, 830 Search PubMed.
-
O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
-
C. Y. Weng, Thesis, Carnegie Mellon University, Pittsburgh, PA, 1969 Search PubMed.
- W. Hiller, J. Strähle, A. Datz, M. Hanack, W. E. Hatfield, L. W. Ter Haar and P. Gütlich, J. Chem. Soc., 1984, 329 Search PubMed.
- J. Bonner and M. Fisher, Phys. Rev., 1964, 134, A640 Search PubMed.
- P. Muthuselvam, R. Sankar, V. N. Singh, G. N. Rao, W. L. Lee, G. Y. Guo and F. C. Chou, Inorg. Chem., 2015, 54, 4303 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and analytical data, description of TGA results, Tables S1 and S2 containing selected bonding parameters, additional structural representation (Fig. S1), TGA curves (Fig. S2) and PXRD patterns of 1–11 (Fig. S3); crystallographic data in CIF format. CCDC 1496266–1496268, 1498139, 1498140 and 1505548–1505553. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce02115h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b
b