
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A one-pot route to thioamides discovered by gas-phase studies: palladium-mediated CO2 extrusion followed by insertion of isothiocyanates†
Asif
Noor
a,
Jiawei
Li
a,
George N.
Khairallah
a,
Zhen
Li
a,
Hossein
Ghari
b,
Allan J.
Canty
 c,
Alireza
Ariafard
*b,
Paul S.
Donnelly
*a and
Richard A. J.
O'Hair
*a
c,
Alireza
Ariafard
*b,
Paul S.
Donnelly
*a and
Richard A. J.
O'Hair
*a
aSchool of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Melbourne, Victoria 3010, Australia. E-mail: pauld@unimelb.edu.au; rohair@unimelb.edu.au
bDepartment of Chemistry, Faculty of Science, Central Tehran Branch, Islamic Azad University, Shahrak Gharb, Tehran, Iran. E-mail: ariafard@yahoo.com
cSchool of Physical Sciences, University of Tasmania, Hobart, Tasmania 7001, Australia
First published on 10th March 2017
Abstract
A new palladium mediated “one pot” synthesis of thioamides from aromatic carboxylic acids (ArCO2H + RNCS → ArC(S)NHR + CO2) was discovered by gas-phase experiments and theoretical studies. Palladium-mediated decarboxylation of aromatic carboxylic acids followed by addition of substituted isothiocyanates leads to the corresponding thioamide derivatives.
A desire to prepare new and known materials by efficient chemical synthesis using methods that are as environmentally benign as possible stimulates continued efforts to discover new reactions.1–3 Computational chemistry is often used to probe the mechanisms of new reactions4 and mass spectrometry (MS) is often used to characterise products5 but rarely have these techniques been used together as tools for the discovery of new reactions.6 Here, we report their use to direct the hypothesis driven discovery of a new method to synthesise thioamides, an important class of compounds with applications in medicinal chemistry.7
The method involves ‘swapping’ the carboxylate functional group of readily available aromatic carboxylic acids (with the loss of CO2) for a heterocumulene (eqn (1)). This new method could prove to be an attractive alternative to conventional methods that require multiple steps and/or harsh reaction conditions.8
| ArCO2H + RNCS → ArC(S)NHR + CO2 | (1) |
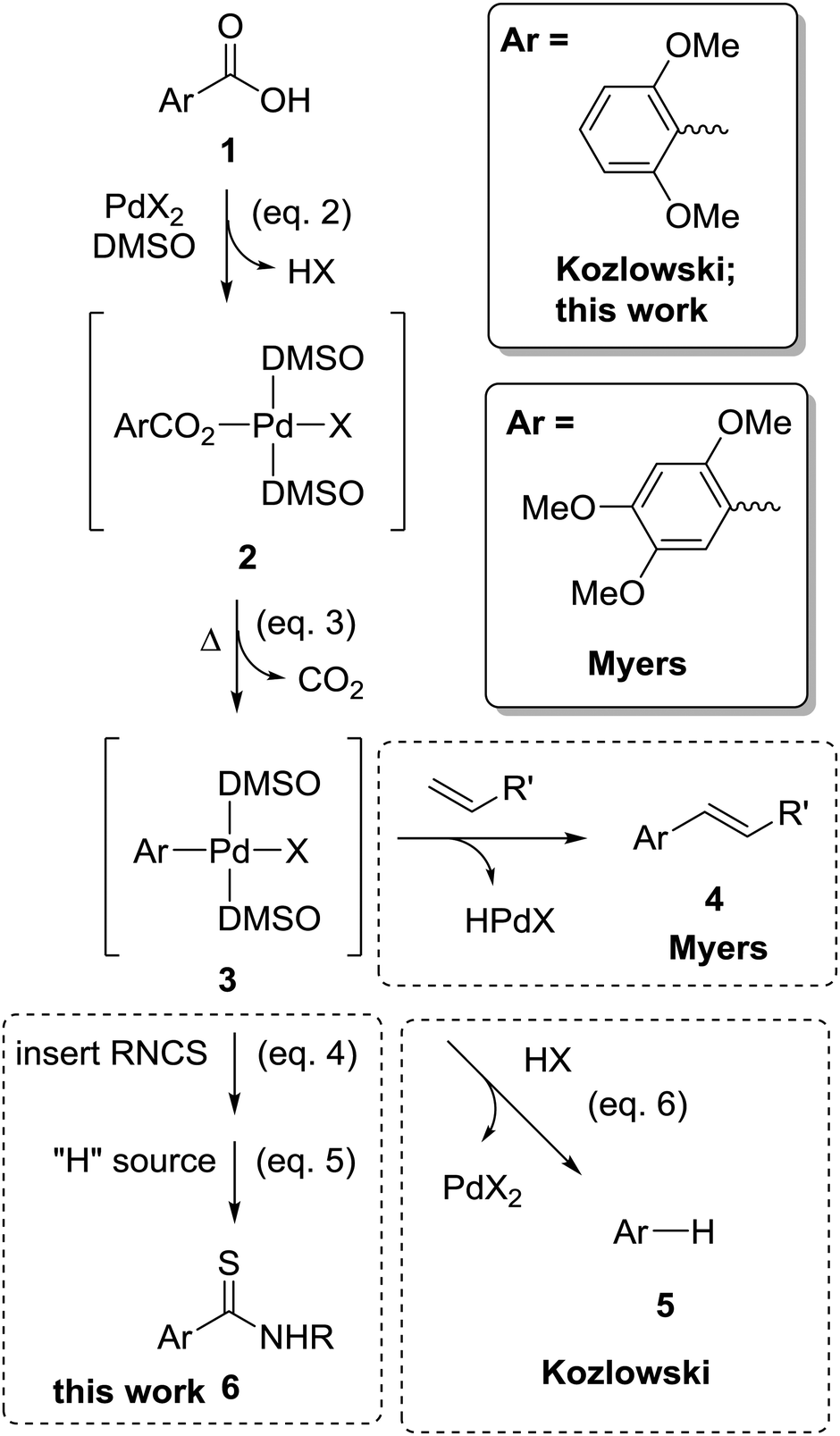 | ||
| Scheme 1 Key reactions of relevance to palladium catalysed decarboxylative transformation of aromatic carboxylic acids into thioamides (eqn (1)): the decarboxylative Heck reaction 1 → 2 → 3 → 4 (Myers);12 protodecarboxylation 1 → 2 → 3 → 5 (Kozlowski);13 and thioamide synthesis 1 → 2 → 3 → 6 (this work) share the same steps for the formation of the organopalladium intermediate (3), but differ in its subsequent reactions. | ||
To model the key decarboxylation and insertion steps associated with transformation of coordinated benzoate to coordinated thiobenzamides shown in Scheme 1, the two DMSO ligands were replaced by the phen ligand and loss of the anionic group X provided the requisite charge for the MS studies. Thus eqn (3) and (4) below were investigated using multistage mass spectrometry (MS) experiments and DFT calculations (Fig. 1).
| [(L)Pd(X)2] + ArCO2H → [(L)Pd(X)(O2CAr)] + HX | (2) |
| [(phen)Pd(O2CPh)]+ → [(phen)Pd(Ph)]+ + CO2 | (3) |
| [(phen)Pd(Ph)]+ + RNCS → [(phen)Pd(SNRCPh)]+ | (4) |
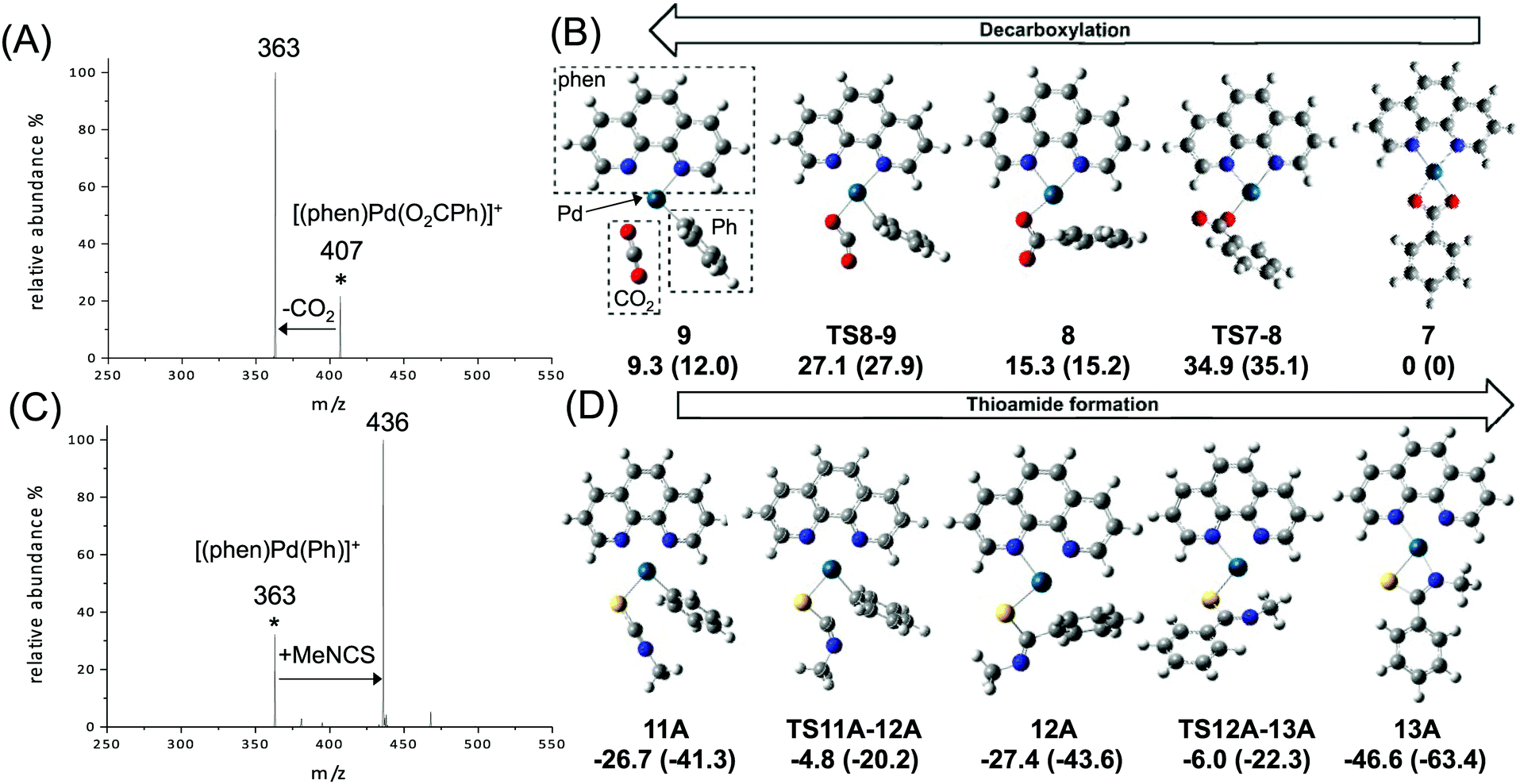 | ||
| Fig. 1 Gas-phase experiments (LTQ ion trap) and DFT calculations (B3LYP-gd3bj/SDD6-31+G(d)//M06/SDD6-31+G(d)) on: (A and B) decarboxylation (eqn (3), ΔG = 10.8 kcal mol−1, ΔH = 23.7 kcal mol−1, MS2 CID, normalised collision energy of 14% and reaction time of 10 ms); (C and D) insertion of MeNCS into the Pd–C bond (eqn (4), MS3 IMR, concentration of MeNCS is 2.8 × 109 molecule cm−3 and reaction time is 250 ms). DFT calculated species are orientated to highlight the direct relationship between carbon dioxide extrusion and isothiocyanate insertion for isoelectronic CO2 and SCNR. Relative Gibbs and enthalpy energies (in parentheses, in kcal mol−1). For (B) and (D) carbon = grey, nitrogen = blue, oxygen = red, palladium = turquoise, sulphur = yellow. | ||
Stimulated by the proof of concept gas-phase studies, we explored the conversion of aromatic carboxylic acids into thioamides using a stoichiometric amount of palladium acetate. Individual reaction steps shown in Scheme 1 (eqn (2)–(5)) were examined via1H NMR spectroscopy. Successful conversion relies on the insertion reaction (eqn (4)) being faster than protonation of the organometallic (eqn (6)) that leads to protodecarboxylation (eqn (7)).
| [(L)Pd(X)(SNRCAr)] + “H source” → ArC(S)NHR | (5) |
| [(L)Pd(X)(Ar)] + HX → [(L)Pd(X)2] + ArH | (6) |
| ArCO2H → ArH + CO2 | (7) |
The effect of replacing the phen ligand used in the MS experiments with coordinated DMSO was investigated by DFT calculations.17 It is possible for the [Pd(DMSO)2] complexes to exists as either cis or trans isomers (Fig. S41 and 44, ESI†). The presence of a ‘reservoir’ of acetic acid in solution (absent in the gas-phase) can lead to the formation of 1,3-dimethoxybenzene so competition between insertion and protonation of the organopalladium was considered. The solvation effect of DMSO on the optimised structures required the use of a conductor polarizable continuum model (CPCM). The reactivity of 14 for insertion of MeNCS and protonation was considered by DFT (Fig. 2). Insertion starts with a simple associative substitution reaction in which one of the coordinating O atoms of the bidentate acetate ligand is replaced by a sulfur atom of the isothiocyanate, TS14–15, to give κ1-acetate complex 15 followed by the insertion of MeNCS into the Pd–Ar bond to afford 16. An alternative orientation of MeNCS in TS15–16′ is higher in energy. For protonation, 14 is substituted by acetic acid to form κ1-acetate intermediate 17. Proton transfer forms 18. The transition structures for insertion and protonation steps (TS15–16 and TS17–18, respectively) both lie lower in energy than those for substitution. TS14–15 is more stable than TS14–17 by 1.1 kcal mol−1, suggesting that the insertion reaction would be faster than protonation, a result substantiated by the NMR studies. DFT calculations revealed how the nature of the R groups of RNCS affects the ease of the insertion process (Scheme S3, ESI†). The TS15–16 is always more stable than TS15–16′ and the energy barrier dependence of the RNCS substitution (TS14–15) on the R group is insignificant. The insertion transition structure (TS15–16) is lower in energy than TS14–15, with the exception of R = tBu, so the chemoselectivity is dictated by the substitution. For R = tBu, the steric repulsion between the tBu substituent and the aryl ligand in TS15–16 results in this transition structure being destabilised considerably and lies above TS14–15. Thus, for R = tBu, the insertion step, and not substitution, is likely to play the crucial role in determining the chemoselectivity. Thus the insertion and protonation pathways become more competitive, which explains the slow reaction of 14 with tBuNCS observed experimentally.
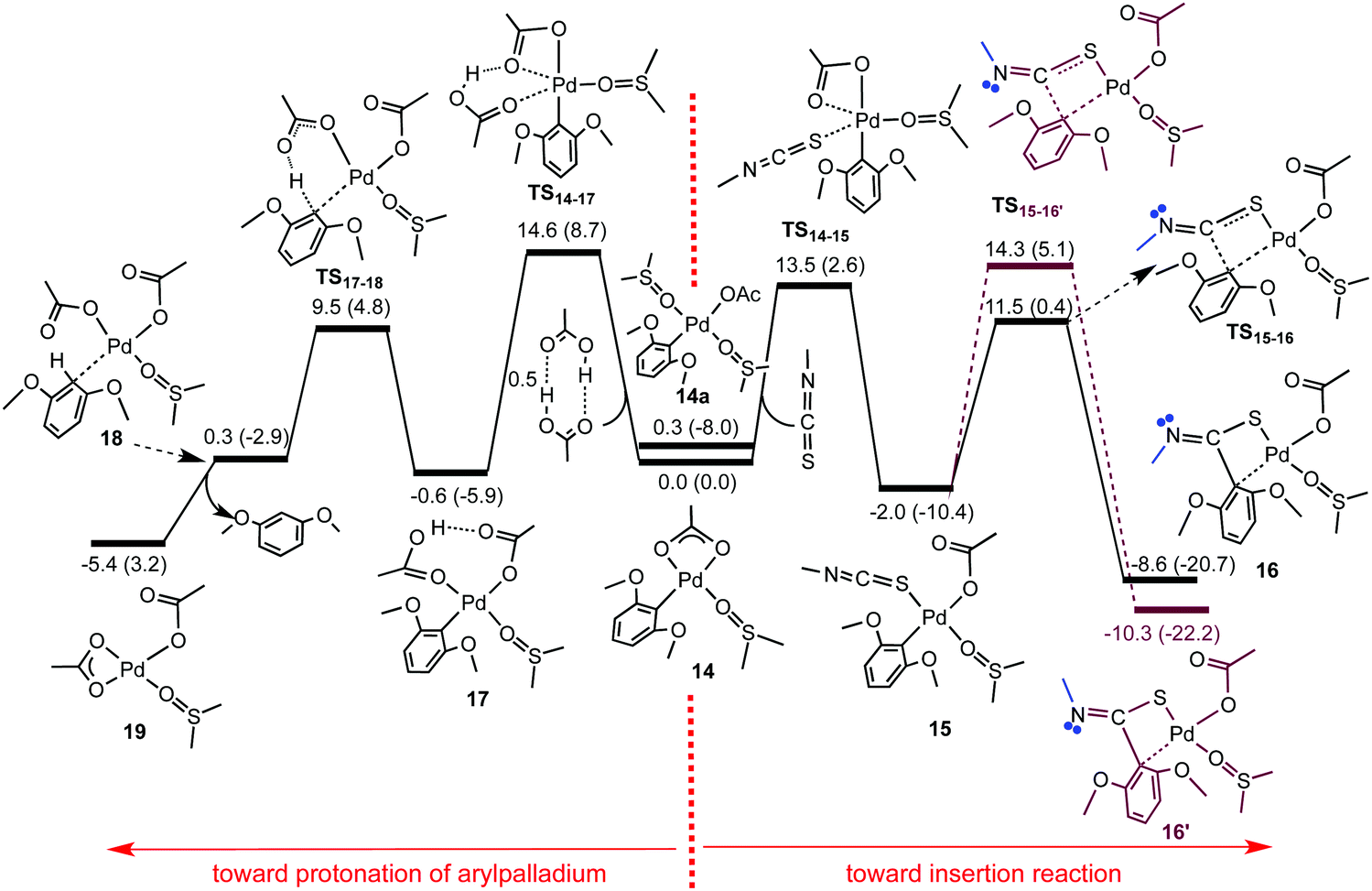 | ||
| Fig. 2 Energy profile showing competition between protonation and insertion of MeNCS for [(DMSO)Pd(O2CCH3)(Ar)], 14. Relative Gibbs and enthalpy energies (in parentheses) are given in kcal mol−1, calculated at the B3LYP-D3BJ/BS3//M06/BS1 level of theory in DMSO using the CPCM approach. | ||
The reaction scope was investigated with different RNCS substrates (Table 1). The resulting N-aryl(alkyl)-2,6-dimethoxythiobenzamide products were characterised by HRMS, 1H and 13C NMR spectroscopy (Fig. S8 and S25, ESI†) and, where possible X-ray crystallography (Fig. S26 and S40, ESI†). The acetic acid formed in eqn (2) can promote protonation (eqn (6)) or other side-reactions so for the reactions with aryl isothiocyanates, K2CO3 was added after decarboxylation. The faster insertion of alkyl isothiocyanates meant that addition of base was not required. Following addition of NaBH4 products 6a–c could be isolated in modest yields (47–57%, Table 1, entries 1–3). The reaction with sterically demanding tBuNCS required a longer reaction time, resulting in more protodecarboxylated and amide products as compared to thioamide 6d. Addition of K2CO3 after decarboxylation is essential for reactions with arylisothiocyanates to prevent partial conversion of the thioamides to amides (identified by ESI-MS and isolated and characterised by 1H NMR for 2,6-dimethoxy-N-phenylbenzamide, Fig. S13 and S22, ESI†). While the exact mechanism of this transformation is unknown, soft metal ions such as Pd2+ promote hydrolysis of thioamides to amides18 and amides have been noted as unwanted side products in palladium catalysed transformations of thioamides.19 In the case of PhNCS, the yield of 6e improved with the use of both K2CO3 and NaBH4 (Table 1). The yield of isolated thioamide was found to depend on the “H” source added. Addition of 5 equiv. of NaBH4 gave the optimum yield of 6f and 6g. The potential reduction of the nitro functional group in 6h was avoided by not including a “H” source.
In summary, a mechanism-based approach using gas-phase ion chemistry was used to uncover a new transformation of aromatic carboxylic acids to substituted thioamides. This same transformation can be readily achieved as a “one-pot” method in solution using stoichiometric amounts of organic precursors and palladium acetate. Preliminary studies suggest that a range of other aryl carboxylates can be used as substrates.20 The two crucial steps of decarboxylation of the coordinated carboxylate in [(phen)Pd(O2CC6H5)]+ (eqn (3)) and insertion of the isothiocyanate into the Pd–C bond of [(phen)Pd(C6H5)]+ (eqn (4)) are directly related to each other by the isoelectronic nature of CO2 and RNCS. MacMillan has recently classified decarboxylation reactions in which two fragments recombine as CO2ExR (ExR = Extrusion–Recombination).21 By analogy, the chemistry described here is part of group of CO2ExIn (ExIn = Extrusion–Insertion) reactions.22 It is possible that this approach is compatible with other small molecules that are isoelectronic with CO2 and this is currently under investigation.
We acknowledge: Financial support from Australian Research Council (DP1096134 (GNK), DP120101540 (AJC), DP150101388 (AJC, RAJO), DP160100288 and FT130100204 (PSD), computing time from the University of Tasmania and National Computing Infrastructure, Profs. Marisa Kozlowski, Craig Hutton and Jon Tunge for useful discussions and Mr Yang Yang for experimental assistance.
References
- R. M. Roberts, Serendipity: Accidental Discoveries, Science, Wiley, 1989 Search PubMed.
- (a) A. McNally, C. K. Prier and D. W. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed; (b) K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859 CrossRef CAS PubMed.
- (a) Inventing Reactions, ed. L. J. Gooßen, Springer Berlin Heidelberg, 2013 Search PubMed; (b) K. N. Houk and P. H.-Y. Cheong, Nature, 2008, 455, 309–313 CrossRef CAS PubMed; (c) Q. N. N. Nguyen and D. J. Tantillo, Chem. – Asian J., 2013, 9, 674–680 CrossRef PubMed; (d) Y. Wang and Z. X. Yu, Acc. Chem. Res., 2015, 18, 2288–2296 CrossRef PubMed; (e) S. Mitsumori, H. Zhang, P. H.-Y. Cheong, K. N. Houk, F. Tanaka and C. F. Barbas, J. Am. Chem. Soc., 2006, 128, 1040–1041 CrossRef CAS PubMed.
- G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706 CrossRef CAS PubMed.
- C. Iacobucci, S. Reale and F. De Angelis, Angew. Chem., Int. Ed., 2016, 55, 2980 CrossRef CAS PubMed.
- S. A. Kunzi, J. M. S. Toro, T. den Hartog and P. Chen, Isr. J. Chem., 2016, 56, 53 CrossRef.
- (a) F. Wang, R. Langley, G. Gulten, L. G. Dover, G. S. Besra, W. R. Jacobs, Jr and J. C. Sacchettini, J. Exp. Med., 2007, 204, 73 CrossRef CAS PubMed; (b) M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442 CrossRef CAS PubMed.
- (a) E. Schaumann, Synthesis of Thioamides and Thiolactams, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, Netherlands, 2014, vol. 6, p. 411 Search PubMed; (b) B. Kaboudin, V. Yarahmadi, J.-Y. Kato and T. Yokomatsu, RSC Adv., 2013, 3, 6435 RSC.
- The formation of CO2(g) is likely to be a driving force. DFT calculations estimate the energetics as: ΔG = −15.5 kcal mol−1 to form Z-PhC(S)NHCH3; ΔG = −13.3 kcal mol−1 Z-PhC(S)NHPh. See ESI† Scheme S1.
- R. A. J. O'Hair and N. J. Rijs, Acc. Chem. Res., 2015, 48, 329 CrossRef PubMed.
- (a) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS; (b) L. J. Gooßen and K. Gooßen, Top. Organomet. Chem., 2013, 44, 121–142 CrossRef.
- D. Tanaka, S. P. Romeril and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 10323 CrossRef CAS PubMed.
- J. S. Dickstein, J. M. Curto, O. Gutierrez, C. A. Mulrooney and M. C. Kozlowski, J. Org. Chem., 2013, 78, 4744 CrossRef CAS PubMed.
- J. Louie, Curr. Org. Chem., 2005, 9, 605 CrossRef CAS.
- J. Vicente, J.-A. Abad, R. Bergs, M. C. Ramirez de Arellano, E. Martínez-Viviente and P. G. Jones, Organometallics, 2000, 19, 5597 CrossRef CAS.
- (a) M. A. Ortuño, S. Conejero and A. Lledós, Beilstein J. Org. Chem., 2013, 9, 1352 CrossRef PubMed; (b) M. Woolley, A. Ariafard, G. N. Khairallah, K. H.-J. Kwan, P. S. Donnelly, J. M. White, A. J. Canty, B. F. Yates and R. A. J. O'Hair, J. Org. Chem., 2014, 79, 12056 CrossRef CAS PubMed.
- G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834 RSC.
- D. P. N. Satchell and R. S. Satchell, in The Chemistry of Sulphur-Containing Functional Groups, ed. S. Patai, Z. Rappoport, Wiley, Chichester, 1993, ch 12, p. 599 Search PubMed.
- (a) K. Inamoto, C. Hasegawa, K. Hiroya and T. Doi, Org. Lett., 2008, 10, 5147 CrossRef CAS PubMed; (b) T. Yamauchi, F. Shibahara and T. Murai, Org. Lett., 2015, 17, 5392 CrossRef CAS PubMed.
- Substituted coordinated benzoates [(phen)Pd(O2CC6H4X)]+ (e.g. X = o-OMe, p-OMe and o-NO2) also undergo decarboxylation followed by insertion in both the gas and condensed phase consistent with N. Rameau, S. Cadot, A. Paquet, C. Pinel and L. Djakovitch, Top. Catal., 2014, 57, 1430 CrossRef CAS ).
- C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 11938 CrossRef CAS PubMed.
- For “intercepted decarboxylative allylation” see: J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1468845–1468850. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc00865a |
| This journal is © The Royal Society of Chemistry 2017 |