
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the molecular determinants of fluorinase specificity†
W. L.
Yeo‡
a,
X.
Chew‡
b,
D. J.
Smith
cd,
K. P.
Chan
b,
H.
Sun
a,
H.
Zhao
ae,
Y. H.
Lim
*b and
E. L.
Ang
 *a
*a
aMetabolic Engineering Research Laboratory (MERL), Science and Engineering Institutes, Agency for Science, Technology and Research (A*STAR), 31 Biopolis Way, Nanos #01-01, Singapore 138669. E-mail: angel@merl.a-star.edu.sg
bInstitute of Chemical and Engineering Sciences (ICES), A*STAR, 8 Biomedical Grove, Neuros #07-01/02/03, Singapore 138665. E-mail: lim_yee_hwee@ices.a-star.edu.sg
cBioinformatics Institute, A*STAR, 30 Biopolis Street, Matrix #07-01, Singapore 138671
dBiotransformation Innovation Platform, A*STAR, 61 Biopolis Drive, Proteos #04-14, Singapore 138673
e215 Roger Adams Laboratory, Box C3, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue Urbana, IL 61801, USA
First published on 27th January 2017
Abstract
Molecular determinants of FlA1 fluorinase specificity were probed using 5′-chloro-5′-deoxyadenosine (5′-ClDA) analogs as substrates and FlA1 active site mutants. Modifications at F213 or A279 residues are beneficial towards these modified substrates, including 5′-chloro-5′-deoxy-2-ethynyladenosine, ClDEA (>10-fold activity improvement), and conferred novel activity towards substrates not readily accepted by wild-type FlA1.
Identification and characterization of a growing number of naturally occurring fluorinase enzymes (FlA,1,2 FlA1,3 FlA3,3 NobA3,4 and FlA45,6) offer an exciting prospect of producing fluorinated compounds under mild conditions.7 These fluorinases catalyze the conversion of S-adenosyl-L-methionine (SAM) and fluoride ion to generate 5′-fluoro-5′-deoxyadenosine (5′-FDA) via a nucleophilic substitution (SN2) mechanism.8–10 In order to expand the scope of application, substrate promiscuity was explored on non-native substrates. However, successful examples were only limited to 5′-chloro-5′-deoxyadenosine (5′-ClDA),11 2′-deoxyadenosine,12L-methionine (L-Met) analogs,13 methylaza-analog of SAM,14 and C-2 modified substrates (5′-chloro-5′-deoxy-2-ethynyladenosine, ClDEA,15,16 and di- and tetra-cyclic peptide constructs of ClDEA17). In addition, the catalytic efficiency of wild-type fluorinases on these non-native substrates is often reduced.13–16 There are only a handful of reports demonstrating limited success at enhancing substrate specificity through enyzme modification.4,6,10,13,18 The challenge here is the lack of understanding of the fluorinase–substrate interactions to enable a better engineered enzyme for fluorination.
Herein, we probed the molecular determinants of FlA1 fluorinase specificity towards non-native substrates which are modified at the adenine ring, as varying this moiety can give rise to a variety of useful fluorinated nucleosides.15,16,19–24 We have recently demonstrated that the catalytic efficiency of FlA1 fluorinase on a non-native substrate 5′-ClDA can be improved via directed evolution.18 With the improved variants as a starting point, we probed their promiscuity against substrates that are modified at the C-2 (R1 substitution) and the unexplored C-6 (R2 substitution) positions of the adenine ring (Table 1).
|
|
|||
|---|---|---|---|
| Substrates | Products | R1 | R2 |
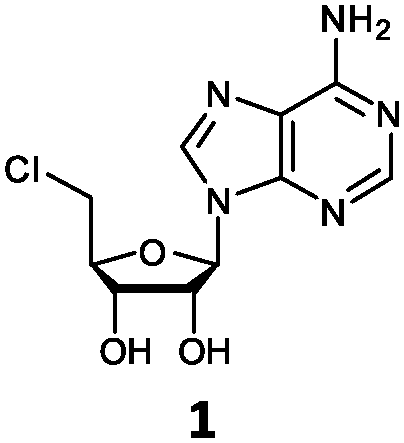
| 1 | 1a | H | NH2 |
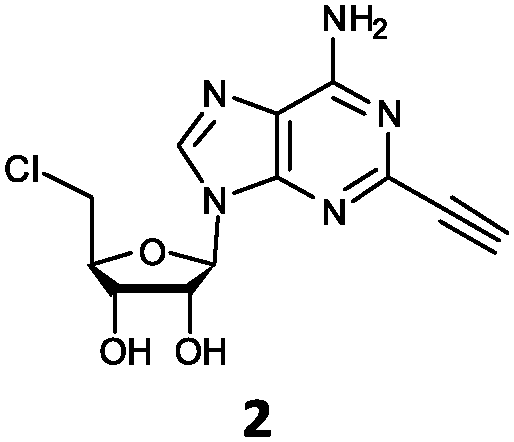
| 2 | 2a | C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH CH |
NH2 |
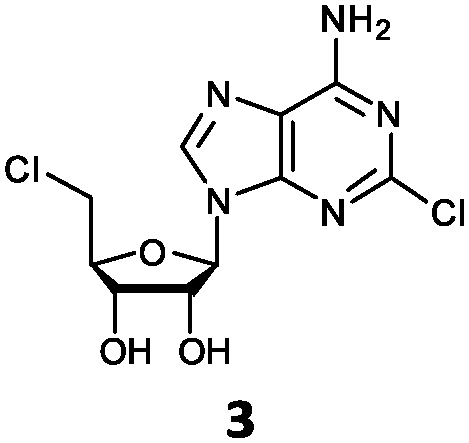
| 3 | 3a | Cl | NH2 |
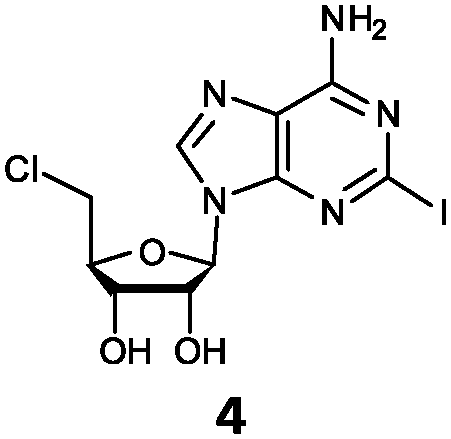
| 4 | 4a | I | NH2 |
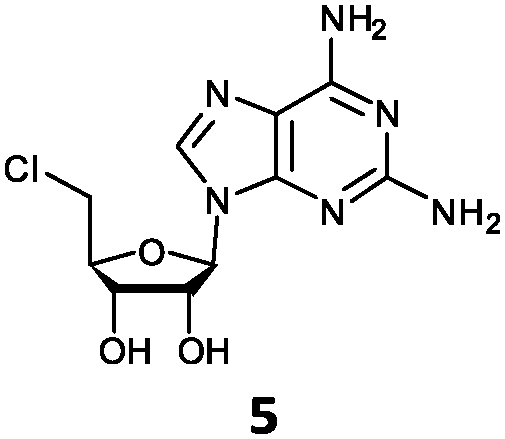
| 5 | 5a | NH2 | NH2 |
| 6 | 6a | NH2 | Cl |
| 7 | 7a | H | Cl |
| 8 | 8a | NH2 | H |
| 9 | 9a | H | H |
| 10 | 10a | NH2 | OMe |
| 11 | 11a | NH2 | O |

O'Hagan's group reported that the reaction of ClDEA 2 to 5′-fluoro-5′-deoxy-2-ethynyladenosine (FDEA 2a) using L-seleno-methionine progressed at about 60% of the rate of conversion of 5′-ClDA 1 to 5′-FDA 1a which demonstrated that the FlA fluorinase from Streptomyces cattleya has a specificity weakness at the C-2 position.15 Similarly, FlA1 fluorinase from Streptomyces sp. MA37 also showed a reduction in activity when 5′-ClDA 1 was substituted with ClDEA 2 in the L-Met reaction (Table 2A). However, using our previously identified variants of fah2047 (A279L) and fah2081 (A279Y), we found that their activities for CIDEA 2 were either maintained or improved, respectively as compared to 5′-ClDA 1. In particular, both variants also demonstrated >9-fold improvement in activity over FlA1 for ClDEA 2. These results suggest that position 279 could play a role in facilitating binding to the ethynyl group of the substrate at the C-2 position. When R1 substituents are bulkier groups (3 and 4), the % yield dropped further for all the enzyme variants, suggesting that the C-2 position is sensitive to steric bulk. Nonetheless, most of our enzyme variants showed improved activity over FlA1 for substrates 1–4, except for frh2066 (Y77W) which only showed improved activity for substrate 1. Thus, mutations F213Y and/or A279LY have led to improved activity against a wider range of R1 substituted substrates. When the R1 substituent is an amino group (5), the % yield for all the fluorinases increased as compared to that of 5′-ClDA 1, with all our enzyme variants still outperforming the fluorination efficiency of FlA1. This is an unprecedented improvement in the fluorination efficiency on 5′-ClDA analogs. Our results showed that having a hydrogen donor such as an amino group at the C-2 position of the adenine group enables the modified substrate to interact better with the fluorinase enzyme, thus achieving higher % yield of the corresponding fluorinated product.
| A | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
||||||
| Fluorinase | % yield | FI | % yield | FI | % yield | FI | % yield | FI | % yield | FI |
| a Standard deviation less than 0.1. | ||||||||||
| FlA1 | 6.6 ± 0.1 | — | 2.3 ± 0a | — | 3.5 ± 0.1 | — | 0 | — | 28.8 ± 0.3 | — |
| fah2019 | 12.5 ± 0.2 | 1.9 | 10.3 ± 0.1 | 4.5 | 7.8 ± 0a | 2.2 | 0.2 ± 0a | — | 35.4 ± 0.5 | 1.2 |
| fah2047 | 19.9 ± 0.3 | 3.0 | 20.5 ± 0.5 | 9.0 | 8.2 ± 0.1 | 2.3 | 0.2 ± 0a | — | 38.0 ± 0.2 | 1.3 |
| frh2066 | 13.9 ± 0.2 | 2.1 | 0.3 ± 0a | 0.1 | 0.1 ± 0a | 0 | 0 | — | 31.6 ± 0.4 | 1.1 |
| fah2081 | 15.3 ± 0.1 | 2.3 | 23.9 ± 0.6 | 10.5 | 8.5 ± 0.2 | 2.4 | 2.2 ± 0a | — | 37.7 ± 0.2 | 1.3 |
| fah2114 | 18.1 ± 0.3 | 2.7 | 13.5 ± 0.3 | 6.0 | 9.4 ± 0.1 | 2.7 | 0.1 ± 0a | — | 37.6 ± 0.5 | 1.3 |
| B | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|||||||
| Fluorinase | % yield | FI | % yield | FI | % yield | FI | % yield | FI | % yield | FI | % yield | FI |
| FlA1 | 0.2 ± 0a | — | 0 | — | 15.2 ± 0.3 | — | 0.1 ± 0a | — | 0 | — | 0 | — |
| fah2019 | 1.4 ± 0a | 6.6 | 0 | — | 33.2 ± 0.3 | 2.2 | 0.5 ± 0a | 6.5 | 0 | — | 0 | — |
| fah2047 | 2.0 ± 0.1 | 9.8 | 0 | — | 37.4 ± 0.7 | 2.5 | 0.1 ± 0a | 0.9 | 0 | — | 0 | — |
| frh2066 | 0 | 0 | 0 | — | 1.2 ± 0.1 | 0.1 | 0 | 0 | 0 | — | 0 | — |
| fah2081 | 0.4 ± 0a | 2.0 | 0 | — | 7.5 ± 0.1 | 0.5 | 0 | 0 | 0 | — | 0 | — |
| fah2114 | 5.8 ± 0.1 | 27.7 | 0.1 ± 0a | — | 30.2 ± 0.4 | 2.0 | 0.4 ± 0a | 4.7 | 0 | — | 0 | — |


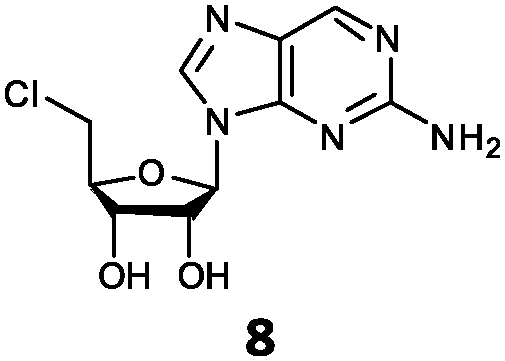


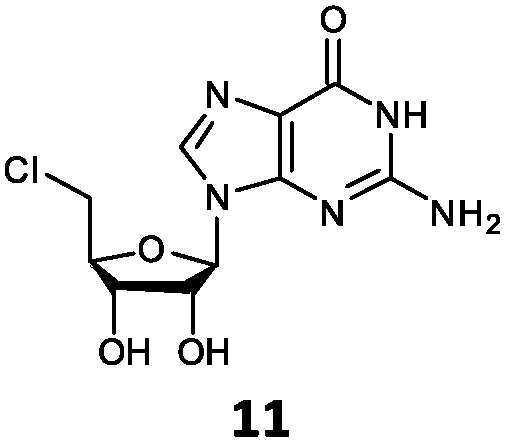
With the improvement in activity observed due to the amino group at the C-2 position (substrate 5), we went on to explore the changes at the C-6 position of the adenine group (R2 substitution). When R2 is either a chloro or hydrogen substituent (6 and 8 respectively), the % yield of all the fluorinases dropped significantly compared to substrate 5 after a 4-h reaction (Table S1, ESI†). To enable better comparison between the C-6 modified substrates, the reaction time was lengthened to 24 h (Table 2B). Three of our enzyme variants (fah2019, fah2047 and fah2114) showed improved activity (up to 27-fold) over FlA1 for substrates 6 and 8. However, no activity was seen for any of the fluorinases for substrates 10 or 11 even with an amino group at the C-2 position. These results suggested that F213Y and A279L mutations are beneficial for selected R2 substituents such as chlorine or hydrogen. When R1 was changed from an amino group to a hydrogen atom for substrates 6 and 8 (to give 7 and 9 respectively), there was a decrease in % yield similar to what was observed between 5 and 1. For substrate 7, there was no detectable activity for wild-type FlA1 whereas activity could be observed with fah2114 (F213Y, A279L). For substrate 9, marginal activity was observed for FlA1 and fah2047 (A279L) whereas improved activity (4–6-fold) was observed for fah2019 (F213Y) and fah2114 (F213Y, A279L). These observations suggest the benefits of a single F213Y mutation for improved activity on substrate 9.
To elucidate the rates of the best performing substrates, time-course reactions were conducted for FlA1 and fah2018 with substrates 1, 2 and 5 (Fig. 1). The initial rates for the substrates tested were significantly disparate, with FlA1 showing rates in the order 5 ≫ 1 ≫ 2 and fah2081 showing in the order 5 ≫ 1 ≈ 2. An interesting anomaly was substrate 2, which gave the highest % yield with fah2081 at 24 h, whereas the rates of the rest showed trends of considerable slowdown after ∼7 h. This suggests that tyrosine at the 279 position has imparted additional benefit towards substrate 2 even though it has a slower initial rate. As for FlA1, substrate 5 still gave the highest % yield after 24 h although the rate was plateaued after ∼7 h. The much lower activity seen for R2 substitution as compared to R1 substitution suggests that the C-6 position, which is largely unexplored, plays a more important role than the C-2 position in the binding of the substrate to the enzyme. Control experiments had ruled out the possibility that the inactivity observed was due to substrate or product degradation under the reaction conditions. It is also interesting to note that frh2066 (Y77W) showed improved activity over FlA1 for only two of the substrates (5′-ClDA 1 and 5). Dong et al. reported that the Y77 residue is involved in facilitating halide binding for FlA.9 This implies that the Y77 residue is critical for the fluorination reaction and should not be modified.6 The Y77 residue is conserved in FlA1, which showed 88% identity to FlA.3 For our previous work of directed evolution of FlA1 with 5′-ClDA 1 and L-Met substrates, saturation mutagenesis of the Y77 residue showed that Y77F (unpublished) and Y77W mutations displayed improvement over FlA1. This suggests that the Y77 residue may play an additional role in the FlA1 enzymatic reaction, although the model complexes for Y77W do not show any obvious reason for the improved activity towards substrates 1 and 5.
 | ||
| Fig. 1 Comparison of reactions rates for FlA1 and fah2081 with substrates 1, 2 and 5 at 37 °C over 24 h. Reaction conditions: Synthesized substrate (0.2 mM), L-Met (0.1 mM), NaF (80 mM), and fluorinase (50 μM) at different time points. % yield (mean of triplicates) = (concentration of product generated/concentration of substrate) × 100%, error bars = standard deviation of triplicates. | ||
To understand how the mutants impact the binding of substrates, structural models of the FlA1 mutants were generated using ROSETTA,25 with the recently solved crystal structure of FlA1 (PDB code 5B6I)18 as the template. Substrate coordinates were derived from the crystal structure of the Streptomyces cattleya fluorinase trimer complexed with substrate 2 (PDB code 4CQJ)15 and docked into the FlA1 mutant active site using PyMOL.26 All models were then subjected to energy minimisation using AMBER.27 The models identify key interactions that may have led to improved binding affinity translating to increased activity. The F213Y mutation is located in the binding pocket at a dimer interface, and is probably hydrogen bonded to D16 in the opposing monomer. This is likely to increase the stability of the enzyme, and may improve activity by increasing affinity towards the substrate. This is particularly noticeable with substrate 9 (Fig. 2A). A279Y also bridges the two monomers through a hydrogen bond between the tyrosine side-chain and the peptide carbonyl of W50 on an adjacent chain. These interactions translate into an overall improvement in activity for these mutations in their variants for most of the substrates.
 | ||
| Fig. 2 Novel substrate binding to wild-type FlA1 or FlA1 variants. (A) Substrate 9 (in light blue) bound to fah2019, showing the interaction between Y213 and D16 residues. (B) Substrate 2 (in orange and yellow) bound to fah2047 (in magenta) and fah2081 (in green), showing the interaction of the R1 substituent of the ethynyl group with L279 and Y279 residues respectively. (C) Substrate 5 (in blue) bound to wild-type FlA1, with hydrogen bonds shown between the substrate and enzyme. (D) Substrate 6 (in white) bound to FlA1 showing the loss of hydrogen bond interaction with the removal of the R2 substituent of the amino group. | ||
The improved activity observed for our enzyme variants for R1 substitutions on the adenine ring (2–5) is favourable for several reasons. Firstly, this C-2 position points into the bulk solvent, and therefore should have minimal effect on the binding to the FlA1 enzyme, similar to what was observed in the FlA enzyme.15 Secondly, the A279L mutation, where the leucine side-chain directly interacts with the substituent, is likely to form favourable van der Waals contacts with the substituent atoms (Fig. 2B). As for the A279Y mutation, the tyrosine side-chain brings the enzyme in closer contact to the ethynyl substituent of substrate 2, hence forming favourable van der Waals interactions. Thirdly, the amino group substituent is able to form favourable hydrogen bonds with the enzyme through the peptide carbonyl of P78 and the amide side-chain of N278 (Fig. 2C). The iodinated substrate 4 has much lowered activity for all mutants, and this might be because the P78 carbonyl and the N278 side-chain are unlikely to form any polar interactions or hydrogen bonds with the bulky iodine substituent.
The FlA1–5′-ClDA enzyme complex model when compared with the bound models for 6–11 identifies the main reason for diminished or loss of activity. The R2 substituent of the amino group is hydrogen-bonded to the enzyme through the amide side-chain of N215, and the peptide carbonyl of R277 (Fig. 2C). The loss of this group removes this interaction, which would lower the affinity of the enzyme for the substrate, and substituting with chlorine (6 and 7), hydrogen (8 and 9) or oxygen (11) will not form any stabilizing hydrogen bonds with N215 (Fig. 2D and Fig. S1A and B, ESI†). In the case of 10, the substituted methoxy group could be too bulky to fit into this pocket, and thus cannot form any stabilizing hydrogen bonds (Fig. S1C, ESI†). We tried to mutate N215 in order to explore potential alterations in specificity at the C-6 position, but so far the mutations we tested (N215LQ) have rendered the protein insoluble (data not shown). The addition of an amino group at the C-2 position helps in compensating for the loss of key interactions at the C-6 position and improving the binding of some substrates (e.g.6 and 8) due to the additional hydrogen bonding (vide supra), as well as the favourable van der Waals interactions from A279L and Y.
In conclusion, we found that substitutions at the C-2 position of the adenine group are sensitive to steric bulk whereas an amino group at the C-2 position led to enhanced fluorination yield. Additionally, our evolved FlA1 fluorinase variants are active against substrates that are modified at the C-2 and/or C-6 positions of the adenine ring despite being evolved against 5′-ClDA 1 and L-Met substrates. Modifications at F213 and/or A279 residues showed improved activity over FlA1 on the eight new substrates tested, including novel activity for two of the substrates, 4 and 7. Based on our previous kinetic studies,18 it is likely that the improvement in activity shown by our enzyme variants over FlA1 for these substrates is due to the enhancement of the first step, i.e. conversion of the substrate and L-Met to a SAM-analog in this two-step fluorination process. This work sheds light on the specificity of the FlA1 fluorinase with respect to modifications at the adenine moiety of the 5′-ClDA substrate and the mutations that can improve the promiscuity of the enzyme against non-native substrates. We believe that our work can serve as a starting point for engineering of enzymes to perform fluorination on more structurally diverse small molecules.
This work was funded by GlaxoSmithKline – Singapore Economic Development Board Partnership for Green and Sustainable Manufacturing and A*STAR ICES. We would like to thank the members of MERL for their suggestions and comments and Ms Doris Tan (ICES) for HRMS analyses.
Notes and references
- D. O'Hagan, C. Schaffrath, S. L. Cobb, J. T. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed.
- C. Schaffrath, H. Deng and D. O'Hagan, FEBS Lett., 2003, 547, 111 CrossRef CAS PubMed.
- H. Deng, L. Ma, N. Bandaranayaka, Z. Qin, G. Mann, K. Kyeremeh, Y. Yu, T. Shepherd, J. H. Naismith and D. O'Hagan, ChemBioChem, 2014, 15, 364 CrossRef CAS PubMed.
- Y. Wang, Z. Deng and X. Qu, F1000Research, 2014, 3, 61 Search PubMed.
- S. Huang, L. Ma, M. H. Tong, Y. Yu, D. O'Hagan and H. Deng, Org. Biomol. Chem., 2014, 12, 4828 CAS.
- L. Ma, Y. Li, L. Meng, H. Deng, Y. Li, Q. Zhang and A. Diao, RSC Adv., 2016, 6, 27047 RSC.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634 CrossRef PubMed.
- C. D. Cadicamo, J. Courtieu, H. Deng, A. Meddour and D. O'Hagan, ChemBioChem, 2004, 5, 685 CrossRef CAS PubMed.
- C. Dong, F. Huang, H. Deng, C. Schaffrath, J. B. Spencer, D. O'Hagan and J. H. Naismith, Nature, 2004, 427, 561 CrossRef CAS PubMed.
- X. Zhu, D. A. Robinson, A. R. McEwan, D. O'Hagan and J. H. Naismith, J. Am. Chem. Soc., 2007, 129, 14597 CrossRef CAS PubMed.
- H. Deng, S. L. Cobb, A. R. McEwan, R. P. McGlinchey, J. H. Naismith, D. O'Hagan, D. A. Robinson and J. B. Spencer, Angew. Chem., Int. Ed., 2006, 45, 759 ( Angew. Chem. , 2006 , 118 , 773 ) CrossRef CAS PubMed.
- S. L. Cobb, H. Deng, A. R. McEwan, J. H. Naismith, D. O'Hagan and D. A. Robinson, Org. Biomol. Chem., 2006, 4, 1458 CAS.
- M. Thomsen, S. B. Vogensen, J. Buchardt, M. D. Burkart and R. P. Clausen, Org. Biomol. Chem., 2013, 11, 7606 CAS.
- M. E. Sergeev, F. Morgia, M. R. Javed, M. Doi and P. Y. Keng, J. Mol. Catal. B: Enzym., 2013, 97, 74 CrossRef CAS.
- S. Thompson, Q. Zhang, M. Onega, S. McMahon, I. Fleming, S. Ashworth, J. H. Naismith, J. Passchier and D. O'Hagan, Angew. Chem., Int. Ed., 2014, 53, 8913 ( Angew. Chem. , 2014 , 126 , 9059 ) CrossRef CAS PubMed.
- S. Thompson, M. Onega, S. Ashworth, I. N. Fleming, J. Passchier and D. O'Hagan, Chem. Commun., 2015, 51, 13542 RSC.
- S. Thompson, I. N. Fleming and D. O'Hagan, Org. Biomol. Chem., 2016, 14, 3120 CAS.
- H. Sun, W. L. Yeo, Y. H. Lim, X. Chew, D. J. Smith, B. Xue, K. P. Chan, R. C. Robinson, E. G. Robins, H. Zhao and E. L. Ang, Angew. Chem., Int. Ed., 2016, 55, 14277 ( Angew. Chem. , 2016 , 128 , 14489 ) CrossRef CAS PubMed.
- H. Deng, S. L. Cobb, A. D. Gee, A. Lockhart, L. Martarello, R. P. McGlinchey, D. O'Hagan and M. Onega, Chem. Commun., 2006, 652 RSC.
- M. Onega, J. Domarkas, H. Deng, L. F. Schweiger, T. A. Smith, A. E. Welch, C. Plisson, A. D. Gee and D. O'Hagan, Chem. Commun., 2010, 46, 139 RSC.
- M. Winkler, J. Domarkas, L. F. Schweiger and D. O'Hagan, Angew. Chem., Int. Ed., 2008, 47, 10141 ( Angew. Chem. , 2008 , 120 , 10295 ) CrossRef CAS PubMed.
- S. D. Mills, A. E. Eakin, E. T. Buurman, J. V. Newman, N. Gao, H. Huynh, K. D. Johnson, S. Lahiri, A. B. Shapiro, G. K. Walkup, W. Yang and S. S. Stokes, Antimicrob. Agents Chemother., 2011, 55, 1088 CrossRef CAS PubMed.
- S. S. Stokes, H. Huynh, M. Gowravaram, R. Albert, M. Cavero-Tomas, B. Chen, J. Harang, J. T. Loch 3rd, M. Lu, G. B. Mullen, S. Zhao, C. F. Liu and S. D. Mills, Bioorg. Med. Chem. Lett., 2011, 21, 4556 CrossRef CAS PubMed.
- S. S. Stokes, M. Gowravaram, H. Huynh, M. Lu, G. B. Mullen, B. Chen, R. Albert, T. J. O'Shea, M. T. Rooney, H. Hu, J. V. Newman and S. D. Mills, Bioorg. Med. Chem. Lett., 2012, 22, 85 CrossRef CAS PubMed.
- Y. Song, F. DiMaio, R. Y. Wang, D. Kim, C. Miles, T. Brunette, J. Thompson and D. Baker, Structure, 2013, 21, 1735 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System (Version 1.8.2.2), Schrödinger, LLC Search PubMed.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary results, experimental methods, synthesis, characterization and NMR spectra of compounds. See DOI: 10.1039/c6cc09213f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |