
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Clearing the undergrowth: detection and quantification of low level impurities using 19F NMR†
Pinelopi
Moutzouri
a,
Peter
Kiraly
a,
Andrew R.
Phillips
b,
Steven R.
Coombes
c,
Mathias
Nilsson
a and
Gareth A.
Morris
 *a
*a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK. E-mail: g.a.morris@manchester.ac.uk
bPharmaceutical Sciences, AstraZeneca, Silk Road Business Park, Macclesfield, SK10 2NA, UK
cPharmaceutical Technology and Development, AstraZeneca, Silk Road Business Park, Macclesfield, SK10 2NA, UK
First published on 23rd November 2016
Abstract
A new method for the analysis of low level impurities in sparsely fluorinated species allows measurement of clean high dynamic range 19F spectra, fully decoupled and free of interfering signals from 13C isotopomers.
The high sensitivity and wide chemical shift range of 19F NMR1–4 make it potentially very attractive for characterising fluorine-containing impurities. In pharmaceutical chemistry, for example, a quarter of current drugs contain one or more fluorines,5 and regulatory authorities require all impurities above 0.1% of a main active pharmaceutical ingredient to be identified and quantified.6 Both 1D 19F NMR and 19F DOSY have been used for the detection of minor fluorinated impurities.7 One major technical problem is the difficulty of exciting quantitatively the very wide chemical shift range of 19F, but solutions now exist for both 1D8 and DOSY9 experiments. However, there remains the problem of 13C isotopomer signals. At around 0.54% of the intensity of 12C isotopomer signals, these are in the same range as impurity signals of interest and often have similar chemical shifts, and therefore complicate their identification and quantitation. The obvious solution is to use broadband 13C decoupling to collapse the heteronuclear J-couplings. This can work well for 1H spectra, albeit at the expense of some sample heating.10–15 However, 19F is exquisitely sensitive to chemical environment and its large secondary isotope shift means that the decoupled (19F–13C) signals have slightly different chemical shifts from the parent (19F–12C) signals, so decoupling just halves the number of 19F–13C signals, rather than hiding them all under the parent. Here we show how to acquire clean 19F spectra without interference from 13C isotopomers and with no heteronuclear (1H or 13C) splittings. The new method does not use 13C decoupling, minimising sample heating, and should greatly facilitate the detection and quantification of low-level impurities by 19F NMR.
Fig. 1 shows 19F spectra of a slightly degraded sample of rosuvastatin (1, Scheme 1), used for treating dyslipidaemia, spiked with small amounts of precursors 2 and 3. The proton-decoupled spectrum of Fig. 1a (multiplet structure renders the proton coupled spectrum, shown in Fig. S4 of the ESI,† uninformative) is complicated by the presence of both one-bond and long-range 13C satellites; one of the two satellite signals due to the presence of 13C at the ortho position with respect to fluorine is almost degenerate with (8 ppb from) the signal of 2.
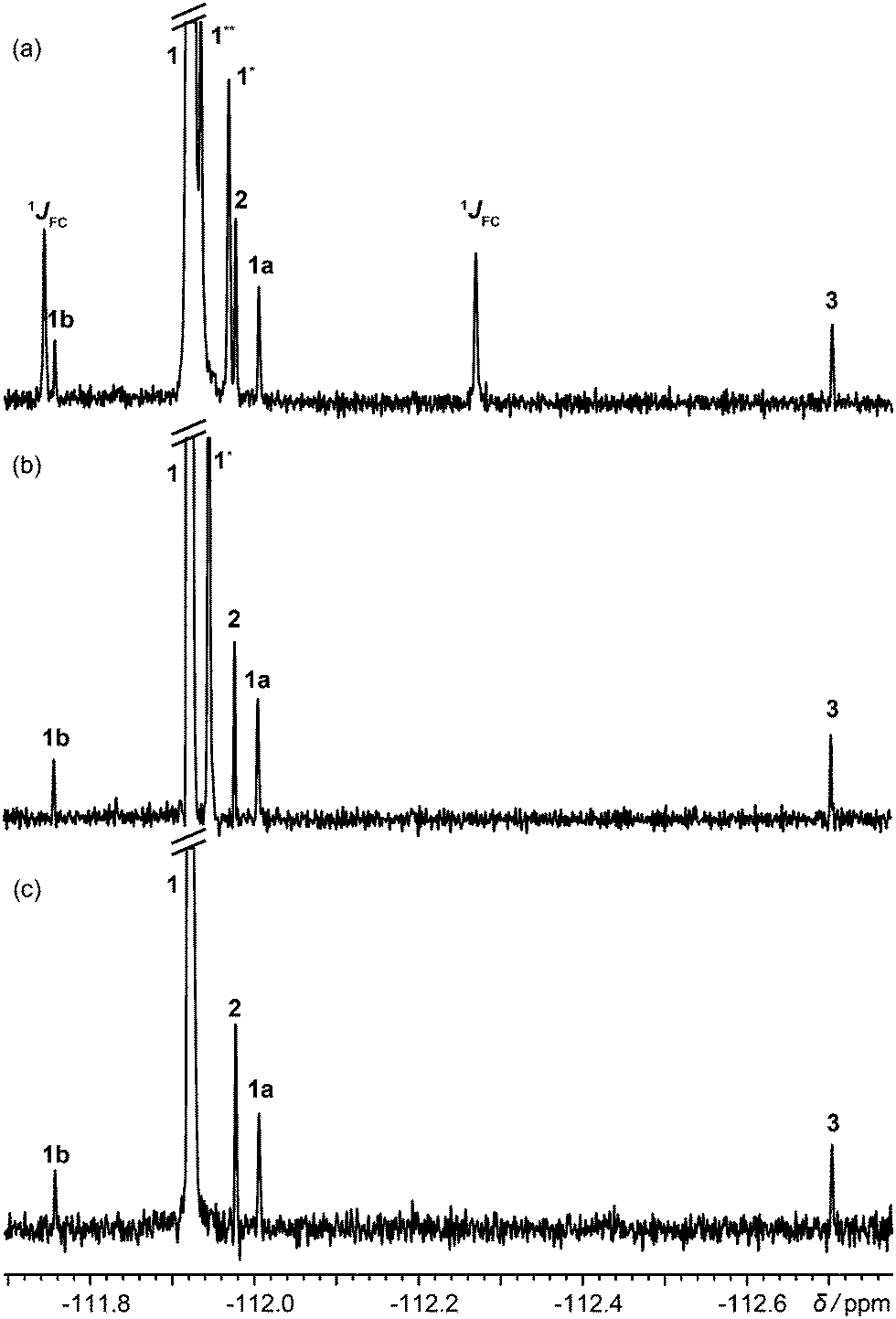 | ||
| Fig. 1 (a) 1H decoupled 19F spectrum; (b) 1H decoupled 19F spectrum acquired with the pulse sequence of Fig. S1a of the ESI,† with one-bond satellites filtered out and long-range couplings decoupled; (c) 1H decoupled, 13C isotopomer-suppressed 19F spectrum acquired with the pulse sequence of Fig. 2. Assignments are shown for rosuvastatin (1), its ipso, ortho and meta 13C isotopomers (1JCF, 1* and 1**), BEM (2), DPPO (3), a diastereomeric impurity of 1 (1a), and a degradation product (1b). All spectra used the same acquisition time of 13.5 h. | ||
 | ||
| Scheme 1 Rosuvastatin (1), two of its precursors, BEM (2) and DPPO (3), and fluconazole (4). | ||
Acquiring a spectrum with this resolution with full broadband decoupling is uncomfortably close to the limits of many instruments, because of the long high-power irradiation required, but if the one-bond 13C satellite signals are suppressed (see Section S1 of the ESI†), low power irradiation can be used to decouple the remaining longer-range (≥two-bond) couplings. This gives the spectrum of Fig. 1b, in which a singlet signal is seen for the 2.2% of ortho-13C 1. Had full 13C decoupling been used, the ipso-13C signal of 1, midway between the one-bond satellites in Fig. 1a, would have been degenerate with that of impurity 1a (a diastereomer). In the spectrum of Fig. 1c, in contrast, which was obtained with the new method, no resolvable signals at all are seen from 13C isotopomers, and there is no interference with the signals of the minor components of the sample.
The new method, using the pulse sequence of Fig. 2, is compatible with several different hardware configurations; the results shown here used a single high band radiofrequency (RF) amplifier and a (1H/19F),13C triple-resonance probe with a double-tuned high band coil. The experiment consists of three parts: a low-pass filter to suppress one-bond 13C satellite signals; a JCF – modulated spin echo; and time-shared acquisition during which the 19F signal is recorded under 1H decoupling.
 | ||
| Fig. 2 ODYSSEUS (optimal decoupling yielding satellite suppression-edited ultraclean spectra) pulse sequence for the acquisition of 1H decoupled, 13C isotopomer-suppressed 19F spectra. Closed narrow rectangles represent 90° hard RF pulses, and open wide rectangles 180° hard RF pulses. The delay Δ is set to 1/(2 1JFC). Adiabatic bilevel 1H decoupling during time-shared acquisition uses two types of WURST pulse with different durations and amplitudes. In systems with 19F–19F coupling, both 180° 19F pulses should be selective. Further experimental details are given in the Experimental section of the ESI.† | ||
The low-pass J filter,16–20 which converts 19F antiphase signals into unobservable heteronuclear multiple quantum coherences when Δ = 1/(2 1JCF), suppresses the one-bond 13C satellite signals. Since a 19F spin echo is needed to refocus the fluorine chemical shift, there is time to use two 13C 90° pulses in a two-stage filter; if a wide range of 1JCF values is present, further stages can be added.
The modulated spin echo, which is analogous to a heteronuclear 2D J resolved experiment,21–23 makes the phases of the remaining 13C satellite signals depend on the evolution time t1, while the desired signals from the 12C isotopomers are unaffected. Weighted averaging of experiments with different t1 cancels the modulated signals, leaving a clean spectrum. In practice the most effective way to perform this averaging is by double Fourier transformation and integral projection onto F2 of the F1 range spanned by the lineshape of the parent signal. This suppresses all satellite signals that would be resolvable in the 1D spectrum, while preserving the quantitative character of the spectrum. The final 13C 90° pulse deals with the problem of the phasetwist lineshape24–26 of a 2D J spectrum by suppressing the sine-modulated dispersive part of the signal. The remaining cosine-modulated signal can then be selected by zeroing the imaginary component after the first Fourier transformation, leading to signals that are doubled in F1 but have 2D absorption mode lineshapes. The choice of increment 1/sw1 in t1 is determined by the range of couplings to be suppressed (sw1 > nJCH), and the number of increments ni by the T2 of the parent signal (ni > sw1 T2). Relaxation losses during t1 lead to a small sensitivity penalty for the new method, about a factor of 2 here (apparent on comparing Fig. 1a and c).
The data acquisition section of the pulse sequence uses time-shared decoupling because the 1H and 19F channels share the same coil in the probe used. In normal circumstances, a simple WALTZ27,28 or similar decoupling waveform would suffice to decouple 1H from 19F, but the very high dynamic range of the sample means that the weak systematic signal modulations such methods induce would here give rise to significant decoupling sidebands (see Fig. S3, ESI†). These are suppressed very effectively here by the use of bilevel adiabatic decoupling.29
As well as decoupling 1H from 19F during acquisition, it can be helpful to decouple in the earlier parts of the sequence, to suppress any echo modulation caused by strong 1H–1H coupling. This is common in aromatic spin systems (as for example in Fig. S2 of the ESI†).21,30,31 Here the quality of decoupling is less critical, so bilevel decoupling is not needed.
Fig. S5 (ESI†) shows the intermediate stage in the production of Fig. 1c at which the F2 projection of the 2D is calculated. Each 13C isotopomer gives four symmetrically-disposed signals, with frequency coordinates (±JCF/2, δ ± JCF/2); in Fig. S5 (ESI†) both of the less shielded satellites overlap in F2 with t1-noise from the parent peak. Integration between the dotted lines produces the spectrum of Fig. 1c.
To test the quantification performance of the new method, the relative percentages of the impurities compared to the main drug substance were measured using the spectrum (Fig. S6 of the ESI†) of a fresh, undegraded, sample. Since the dynamic range of the spectrum is very high, lineshape fitting32–36 was used instead of conventional integration. As shown in Table 1, the relative percentages measured agree well with those expected.
| Expected (%) | Measured (%) | |
|---|---|---|
| 2 | 0.33 | 0.37 ± 0.03 |
| 3 | 0.17 | 0.18 ± 0.03 |
| 1a | 0.28 | 0.26 ± 0.03 |
In systems with mutually coupled fluorines, homonuclear J modulation interferes with 13C satellite suppression if hard 180° 19F pulses are used in Fig. 2. Selective 180° pulses avoid this problem, as shown in Fig. 3 for the antifungal drug fluconazole, which has JFF = 8.1 Hz. Fig. 3b and c were acquired separately using the selective analogue of Fig. 2 to excite the regions around −107 and −111 ppm respectively, revealing the degradation products 4a, 4b and 4c.
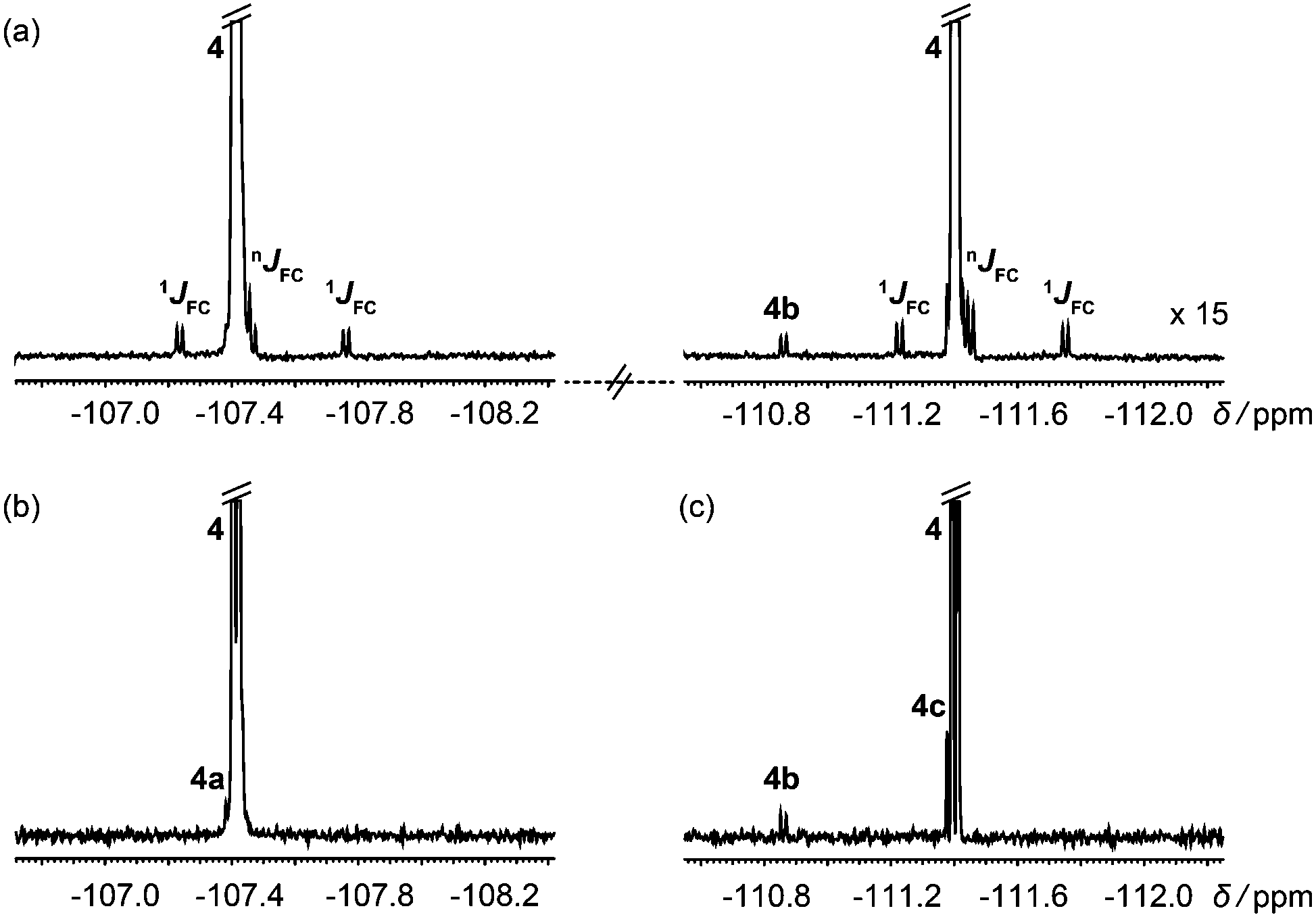 | ||
| Fig. 3 (a) 1H decoupled 19F spectrum of a degraded sample of the antifungal drug fluconazole (4); (b and c) 1H decoupled, 13C isotopomer-suppressed 19F spectra acquired separately for each parent signal using the pulse sequence of Fig. 2 with selective 19F 180° pulses. | ||
13C isotopomer signals can pose significant challenges in identifying and quantifying impurities down to the 0.1% level. The novel approach introduced here of filtering out, rather than decoupling, these signals offers the possibility of acquiring clean, high dynamic range 19F spectra without interference from species containing 13C. A slightly simpler approach can be used in proton spectra.
This work was supported by AstraZeneca and by the Engineering and Physical Sciences Research Council (grant number EP/N033949/1). All raw experimental data, and the pulse sequence code, can be downloaded from DOI: http://10.15127/1.304823.
Notes and references
- S. Trefi, V. Gilard, S. Balayssac, M. Malet-Martino and R. Martino, J. Pharm. Biomed. Anal., 2008, 46, 707–722 CrossRef CAS PubMed.
- J. C. Lindon and I. D. Wilson, eMagRes, John Wiley & Sons, Ltd, 2007 DOI:10.1002/9780470034590.emrstm1479.
- S. Trefi, V. Gilard, M. Malet-Martino and R. Martino, J. Pharm. Biomed. Anal., 2007, 44, 743–754 CrossRef CAS PubMed.
- W. He, F. Du, Y. Wu, Y. Wang, X. Liu, H. Liu and X. Zhao, J. Fluorine Chem., 2006, 127, 809–815 CrossRef CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- ICH, Impurities in new drug substances Q3A (R2) International Conference on Harmonisation, IFPMA, Geneva, Switzerland, 2006 Search PubMed.
- N. Mistry, I. M. Ismail, R. Duncan Farrant, M. Liu, J. K. Nicholson and J. C. Lindon, J. Pharm. Biomed. Anal., 1999, 19, 511–517 CrossRef CAS PubMed.
- J. E. Power, M. Foroozandeh, R. W. Adams, M. Nilsson, S. R. Coombes, A. R. Phillips and G. A. Morris, Chem. Commun., 2016, 52, 2916–2919 RSC.
- J. E. Power, M. Foroozandeh, P. Moutzouri, R. W. Adams, M. Nilsson, S. R. Coombes, A. R. Phillips and G. A. Morris, Chem. Commun., 2016, 52, 6892–6894 RSC.
- A. J. Shaka, P. B. Barker and R. Freeman, J. Magn. Reson., 1985, 64, 547–552 CAS.
- T. Fujiwara, T. Anai, N. Kurihara and K. Nagayama, J. Magn. Reson., Ser. A, 1993, 104, 103–105 CrossRef CAS.
- R. Fu and G. Bodenhausen, Chem. Phys. Lett., 1995, 245, 415–420 CrossRef CAS.
- L. S. Simeral, Appl. Spectrosc., 1995, 49, 400–402 CrossRef CAS.
- Ē. Kupče and R. Freeman, Chem. Phys. Lett., 1996, 250, 523–527 CrossRef.
- R. Freeman and Ē. Kupče, NMR Biomed., 1997, 10, 372–380 CrossRef CAS PubMed.
- A. Bax, R. H. Griffey and B. L. Hawkins, J. Magn. Reson., 1983, 55, 301–315 CAS.
- A. Bax and M. F. Summers, J. Am. Chem. Soc., 1986, 108, 2093–2094 CrossRef CAS.
- H. Kogler, O. W. Sørensen, G. Bodenhausen and R. R. Ernst, J. Magn. Reson., 1983, 55, 157–163 CAS.
- L. Müller, J. Am. Chem. Soc., 1979, 101, 4481–4484 CrossRef.
- N. T. Nyberg and O. W. Sørensen, Magn. Reson. Chem., 2006, 44, 451–454 CrossRef CAS PubMed.
- G. Bodenhausen, R. Freeman, G. A. Morris and D. L. Turner, J. Magn. Reson., 1977, 28, 17–28 CAS.
- G. Bodenhausen, R. Freeman, R. Niedermeyer and D. L. Turner, J. Magn. Reson., 1976, 24, 291–294 CAS.
- L. Müller, A. Kumar and R. R. Ernst, J. Magn. Reson., 1977, 25, 383–390 Search PubMed.
- P. Bachmann, W. P. Aue, L. Müller and R. R. Ernst, J. Magn. Reson., 1977, 28, 29–39 CAS.
- A. Bax, A. F. Mehlkopf and J. Smidt, J. Magn. Reson., 1979, 35, 373–377 CAS.
- R. Freeman, S. P. Kempsell and M. H. Levitt, J. Magn. Reson., 1979, 34, 663–667 CAS.
- A. J. Shaka, J. Keeler, T. Frenkiel and R. Freeman, J. Magn. Reson., 1983, 52, 335–338 CAS.
- A. J. Shaka, J. Keeler and R. Freeman, J. Magn. Reson., 1983, 53, 313–340 CAS.
- Ē. Kupče, R. Freeman, G. Wider and K. Wüthrich, J. Magn. Reson., Ser. A, 1996, 122, 81–84 CrossRef.
- R. Freeman, G. A. Morris and D. L. Turner, J. Magn. Reson., 1977, 26, 373–378 CAS.
- A. Kumar and R. R. Ernst, Chem. Phys. Lett., 1976, 37, 162–164 CrossRef CAS.
- M. Ala-Korpela, Y. Hiltunen, J. Jokisaari, S. Eskelinen, K. Kiviniitty, M. J. Savolainen and Y. A. Kesäniemi, NMR Biomed., 1993, 6, 225–233 CrossRef CAS PubMed.
- Y. Hiltunen, M. Ala-Korpela, J. Jokisaari, S. Eskelinen, K. Kiviniitty, M. Savolainen and Y. A. Kesäniemi, Magn. Reson. Med., 1991, 21, 222–232 CrossRef CAS PubMed.
- V. V. Mihaleva, S. P. Korhonen, J. van Duynhoven, M. Niemitz, J. Vervoort and D. M. Jacobs, Anal. Bioanal. Chem., 2014, 406, 3091–3102 CrossRef CAS PubMed.
- H. M. Parsons, C. Ludwig and M. R. Viant, Magn. Reson. Chem., 2009, 47, S86–S95 CrossRef CAS PubMed.
- P. Soininen, J. Haarala, J. Vepsäläinen, M. Niemitz and R. Laatikainen, Anal. Chim. Acta, 2005, 542, 178–185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc08836h |
| This journal is © The Royal Society of Chemistry 2017 |