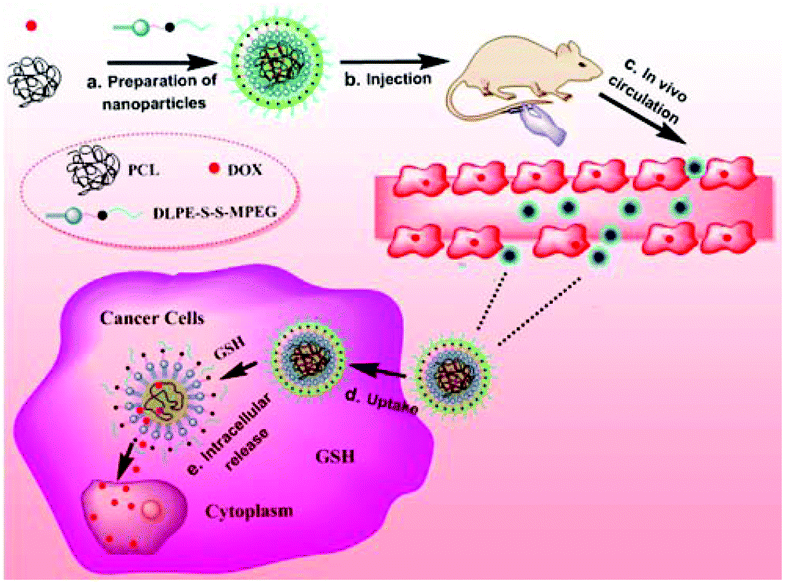
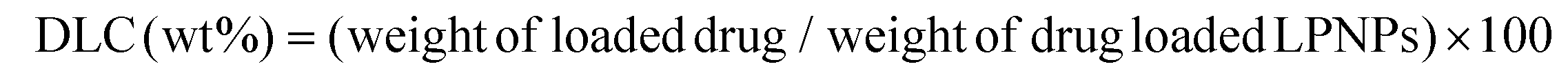Two-component reduction-sensitive lipid–polymer hybrid nanoparticles for triggered drug release and enhanced in vitro and in vivo anti-tumor efficacy
Received
20th September 2016
, Accepted 2nd November 2016
First published on 14th November 2016
Abstract
An amphiphilic polymer DLPE-S-S-MPEG was synthesized and employed with PCL to prepare two-component reduction-sensitive lipid–polymer hybrid nanoparticles (SLPNPs) for in vitro and in vivo delivery of a hydrophobic anticancer drug (Doxorubicin, DOX). Insensitive lipid–polymer hybrid nanoparticles (ILPNPs) were prepared as a control. The mean sizes of the LPNPs ranged from 100 nm to 120 nm. The TEM observations showed that the LPNPs have spherical morphologies with homogeneous distribution. The disulfide bond of DLPE-S-S-MPEG was cleaved by dithiothreitol (DTT), which resulted in the disassembly of SLPNPs and triggered the release of encapsulated DOX. The in vitro cytotoxicities of DOX/LPNPs against HeLa cells, HepG2 cells and COS-7 cells were studied. It was demonstrated that DOX/SLPNPs showed higher cytotoxicity against HeLa cells and HepG2 cells than DOX/ILPNPs, but showed a slight difference in the case of COS-7 cells. CLSM observation and FCM measurement further confirmed that the introduction of S–S bonds caused fast intracellular release of DOX from SLPNPs. Moreover, compared with DOX/ILPNPs and free DOX, DOX/SLPNPs exhibited higher antitumor activity. Both DOX/SLPNPs and DOX/ILPNPs showed lower cardiac toxicity and kidney toxicity than free DOX, which were confirmed by histological and immunohistochemical analyses. The tissue distribution of DOX in mice exhibited that two kinds of DOX/LPNPs accumulated extensively in the liver and spleen, while free DOX accumulated mainly in the heart and kidney 12 h after injection. Two-component SLPNPs may be a promising drug delivery carrier for reduction-triggered delivery of DOX.
Introduction
In the past decades, nanosized drug delivery carriers, mainly including polymeric nanoparticles, liposomes, micelles, and inorganic nanoparticles, have shown great potential in cancer treatment.1–5 Although some nanoparticle-based formulations, such as Doxil and Abraxane, have been approved for clinical application, most nanoparticulate drug carriers are at the research stage. Different kinds of nanoscale drug carriers have their own advantages and disadvantages. For example, liposomes demonstrated excellent biocompatibility, however, drug leakage from liposomes and the stability problem with liposomal formulations limited their clinical applications. In contrast, the advantages of polymeric nanoparticles include high drug loading and stability, but the complex chemical structures and synthetic procedures of many polymers may result in difficulties translating them into the clinic. To combine the advantages of two kinds of important nanocarriers, liposomes and polymeric nanoparticles, in drug delivery, Zhang et al. developed a creative single-step nanoprecipitation strategy to construct lipid–polymer hybrid nanoparticles (LPNPs) by self-assembly of poly D,L-lactic-co-glycolic acid (PLGA), lecithin and PEG-DSPE.6–8 LPNPs usually consists of three parts: (i) the biodegradable hydrophobic core (PLGA, PCL, etc.), which was used to load hydrophobic drugs and sustainedly release the encapsulated drug; (ii) the lipid monolayer around the hydrophobic core; and (iii) the hydrophilic shell usually containing a linear polyethylene glycol (PEG), which can improve the carrier stability and extend the body circulation lifetime. Due to the advantages of LPNPs, including easy preparation, favorable stability in biological environments and sustained drug release ability, LPNPs have been widely used to deliver bioactive molecules for cancer chemotherapy,9–16 photothermal therapy17,18 and theranostics.19
On the other hand, tumor microenvironment responsive nanocarriers have attracted much attention in cancer therapy. The intrinsic stimuli of tumor tissue or tumor cells, such as tumor extracellular or intracellular pH, matrix metalloproteases (MMP), and intracellular reductive molecules, were utilized to trigger the release of drug encapsulated in responsive nanocarriers.20–22 Glutathione (GSH) is the most abundant low-molecular-weight biological thiol, which is abundant in the cell cytoplasm (1 mM to 10 mM) but is rarely present in blood plasma (∼2 μM). The concentration level of cytosolic GSH in some tumor cells is reportedly several times higher than that in normal cells. Due to the unique cleavage property of disulfide bond with biological thiols inside the tumor cells, a variety of disulfide-based reduction-sensitive nanocarriers were developed for delivery of chemotherapeutics,23–31 nucleic acids32,33 and photosensitizers.34
Although the traditional LPNPs, typically composed of PLGA, lecithin and PEG-DSPE, have shown great potential in drug delivery, these LPNPs are insensitive in drug release. Sensitive LPNPs have seldom been reported in the literature.35 We previously reported a new kind of reduction-sensitive LPNPs prepared from PLGA, lecithin and disulfide-containing amphiphilic PEG derivative mPEG-S-S-C16. Doxorubicin (DOX), a hydrophobic anticancer drug, was encapsulated in the PLGA core of the LPNP.36–38 The DOX-loaded LPNPs were stable in the blood and tumor tissue, however, the biological thiols, such as GSH, triggered the release of DOX from LPNPs once the DOX-loaded nanocarriers entered into tumor cells. In this work, we prepared two-component reduction-sensitive LPNPs (SLPNPs) from PCL and amphiphilic DLPE-S-S-MPEG (Scheme 1). In this hybrid nanosystem, lecithin is unnecessary, which is significantly different from traditional LPNPs. They are simpler than traditional three-component LPNPs, which is important in practical application. We also prepared two-component insensitive LPNPs (ILPNPs) from PCL and DLPE-C-C-MPEG as a control. DOX was efficiently encapsulated in both SLPNPs and ILPNPs. Both DOX/SLPNPs and DOX/ILPNPs were stable in water and PBS buffer (pH 7.4), however, only DOX/SLPNPs were destabilized under reductive conditions and rapidly released DOX. The comprehensive in vitro and in vivo evaluation further demonstrated the significant advantages of DOX/SLPNPs in cancer treatment in comparison to DOX/ILPNPs.
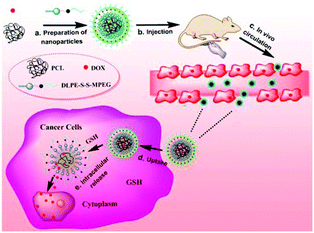 |
| | Scheme 1 Illustration of the composition of DOX/SLPNPs and reduction-triggered release of DOX in cancer cells. | |
Experimental section
Materials and methods
1,2-Dilauroyl-3-sn-phosphatidylethanolamine (DLPE, 98%) and 4-dimethylaminopyridine (DMAP, 99%) were purchased from J&K Chemical. 3,3′-Dithiodipropionic acid (DDA, >99.0%) and DL-dithiothreitol (DTT, >98%) were purchased from TCI and Merck, respectively. Polycaprolactone (PCL, MW 6500) was purchased from Tianjin Heowns Biochem LLC. Dicyclohexylcarbodiimide (DCC, ≥95%) was purchased from Shanghai Chemical Co. and doxorubicin hydrochloride (DOX·HCl) was purchased from Shanghai Yingxuan Co. Methoxypolyethylene glycol (MPEG, Mn 2000) was purchased from Sigma-Aldrich and dried in toluene by azeotropic distillation before use. Chloroform (CHCl3) and N,N-dimethyl formamide (DMF) were purchased from Shanghai Chemical Co. and dried before use. All other chemicals were of analytical grade and were used as received. HeLa cells, HepG2 cells and COS-7 cells were purchased from the China Center for Type Culture Collection (Wuhan University) and incubated at 37 °C in a humidified 5% CO2 atmosphere. Female athymic BALB/c-nu mice (4–5 weeks old, 16 ± 2 g) were purchased from Beijing HFK Bioscience Co. Ltd (Beijing, China). All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and the procedures were approved by the Wuhan University of China Animal Care and Use Committee.
Fourier transform infrared (FTIR) spectra were recorded on a Bio-Rad FTS 6000 spectrometer (Bio-Rad Company, Hercules, California, USA) using KBr pellets at room temperature. The 1H NMR spectra were obtained on VX-300 spectrometer at 300 MHz in deuterated chloroform (CDCl3) and tetramethylsilane (TMS) as an internal reference.
Synthesis of MPEG-S-S-COOH and MPEG-C-C-COOH
3,3′-Dithiodipropionic acid (636 mg, 3 mmol), DCC (741 mg, 3.6 mmol) and DMAP (36.6 mg, 0.3 mmol) were dissolved in 20 mL of anhydrous DMF under an argon atmosphere. Then, a solution of pre-dried MPEG (4.0 g) in 20 mL of anhydrous DMF was added dropwise into the above solution at 0 °C and stirred for 24 h at room temperature. After filtration, the filtrate was concentrated and dried under reduced pressure. The obtained product was dissolved in water and extracted with dichloromethane thrice. The organic layer was collected and concentrated under reduced pressure. The concentrated solution was then precipitated in diethyl ether and purified by dissolution–precipitation in dichloromethane/diethyl ether twice. The product was dried overnight in vacuum and MPEG-S-S-COOH was obtained with 90.9% yield (4.18 g). FTIR: 2907, 2741, 1735, 1105 cm−1. 1H NMR (300 MHz, CDCl3): δ (ppm) 2.71–2.75 (t, 2H, CH2COO), 2.75–2.79 (t, 2H, CH2COO), 2.90–2.92 (t, 4H, CH2SS), 3.40 (s, 3H, OCH3), 3.55–3.82 (m, CH2CH2O), 4.24–4.26 (t, 2H, COOCH2). The synthesis procedure of MPEG-C-C-COOH was similar to that of MPEG-S-S-COOH, but 3,3′-dithiodipropionic acid was replaced with octanedioic acid.
Synthesis of DLPE-S-S-MPEG and DLPE-C-C-MPEG
MPEG-S-S-COOH (1.085 g), DCC (126 mg, 0.6 mmol) and DMAP (6.3 mg, 0.05 mmol) were dissolved in 30 mL of anhydrous CHCl3 at room temperature under a nitrogen atmosphere. Then a solution of DLPE (290 mg, 0.5 mmol) and Et3N (100 μL) in 20 mL of anhydrous CHCl3 was added dropwise, and stirred for 24 h. The mixture was filtered and the filtrate was concentrated under reduced pressure. Subsequently, the concentrated solution was precipitated in diethyl ether and purified by dissolution–precipitation in dichloromethane/diethyl ether twice. The product was collected by filtration and dried under vacuum with a yield of 89.4% (1.22 g). FTIR: 3427, 2741, 1736, 1627, 1535, 1115 cm−1. 1H NMR (300 MHz, CDCl3): δ (ppm) 0.86 (s, 3H, CH3), 1.26 (s, 36H, CH2CH2), 2.75–2.77 (t, 2H, CH2COO), 2.79–2.80 (t, 2H, CH2COO), 2.90–2.92 (t, 4H, CH2SS), 3.41 (s, 3H, OCH3), 3.55–3.71 (m, CH2CH2O), 4.24–4.26 (t, 2H, COOCH2). The synthesis procedure of DLPE-C-C-MPEG polymer was similar to that of DLPE-S-S-MPEG polymer, but MPEG-S-S-COOH was replaced with MPEG-C-C-COOH.
Preparation of blank LPNPs
DLPE-S-S-MPEG polymer (20 mg) or DLPE-C-C-MPEG polymer (20 mg) were dissolved in 10 mL of 4% (v/v) ethanol water and stirred for 6 min at 50 °C. Then, a solution of PCL (40 mg) in 10 mL of DMF was added dropwise and whirled violently for 3 minutes. Subsequently, the mixture solution was transferred to a dialysis tube (MWCO: 8000 Da) and dialyzed against twice reverse osmosis water at 25 °C for 24 h. During dialysis, water was exchanged every four hours.
Preparation of DOX loaded LPNPs
4 mg of DOX·HCl and 10 μL of Et3N were dissolved in 5 mL of DMF and stirred overnight in the dark to obtain the DOX base, and then 5 mL of a DMF solution of PCL (40 mg) was added and stirred in the dark for another 4 h. Subsequently, DLPE-S-S-MPEG polymer (20 mg) or DLPE-C-C-MPEG polymer (20 mg) were dissolved in 10 mL of 4% (v/v) ethanol water, and then the DMF solution of DOX and PCL was added dropwise. The mixture was whirled violently for 3 minutes and transferred to a dialysis tube (MWCO: 8000 Da) to dialyze against twice reverse osmosis water. The final suspension was filtered through a syringe filter (pore size = 0.45 μm) and was stored at 4 °C.
Characterization of LPNPs and DOX/LPNPs
The mean size and polydispersity index (PDI) of LPNPs and DOX/LPNPs were measured by dynamic light scattering (DLS) with a ZETA-SIZER Nano Series ZEN3600 (Malvern Instruments Ltd, UK) at 25 °C. Each measurement was repeated three times. The physical stabilities of LPNPs were also evaluated by DLS. Briefly, samples were stored at 4 °C, and assessed by DLS at predetermined intervals for changes in mean size and PDI. The morphologies of the particles were observed with a Jeol JEM-100CXII transmission electron microscope (TEM) (Tokyo, Japan) at an acceleration voltage of 100 kV. To prepare the TEM samples, a drop of each LPNPs suspension was placed on a copper grid with formvar film, stained with a drop of 1% phosphotungstic acid and dried. The drug loading content (DLC) was measured on a RF-5301 PC spectrofluorophotometer (λex = 485 nm and λem = 590 nm) using a calibration curve constructed from DOX/DMSO solutions. Typically, 1 mL of each DOX/LPNPs suspension was freeze-dried and dissolved in DMSO to measure the DOX concentration according to the calibration curve. The DLC was calculated according to the following formula:
Reduction-triggered disassembly behavior and in vitro DOX release
DLS was used to monitor the size and PDI change of SLPNPs in response to 10 mM DTT in phosphate-buffered saline (PBS, pH 7.4). Briefly, DTT in PBS was added to a SLPNPs solution and the final concentration of DTT was 10 mM. At predetermined intervals, the size and PDI of SLPNPs were measured by DLS. The equal concentration of SLPNPs solution without DTT was measured as a control.
In vitro DOX release behaviors were studied in PBS with or without DTT. After diluting to the same concentration, 2 mL DOX/SLPNPs or DOX/ILPNPs was transferred to a dialysis tube (MWCO: 8000 Da). Then the dialysis tubes were immersed in 40 mL of PBS or PBS with 10 mM DTT and kept at 37 °C in a shaking water bath at 100 rpm. At predetermined time intervals, 3 mL of the external buffer was taken out and replaced with an equal volume of fresh corresponding buffer. The samples were analyzed by spectrofluorophotometer to determine the DOX content.
In vitro cytotoxicity assay
The cytotoxicities of DOX/SLPNPs and DOX/ILPNPs against HeLa cells, HepG2 cells and COS-7 cells were evaluated by MTT assay, and free DOX was used as a control. Cells were seeded in a 96-well plate at a density of 5.0 × 103 cells per well in 100 μL of DMEM containing 10% FBS. After 24 h incubation at 37 °C in a 5% CO2 atmosphere, 100 μL of DMEM medium containing of DOX/SLPNPs, DOX/ILPNPs or free DOX was added, and the cells were cultured for 24 h. The medium was removed and 200 μL of fresh medium was added to each well, and then the cells were incubated for another 24 h. For cytotoxicities of SLPNPs and ILPNPs, the incubation time was continuous 48 h after adding samples. Subsequently, 20 μL of MTT solution (5 mg mL−1 in PBS) was added to each well except the background group (20 μL of PBS buffer for each well). The mixture was further incubated for 4 h. Then, the medium was carefully removed and 150 μL of DMSO was added. The absorbance of each well was measured at 570 nm using a microplate reader (Bio-Rad, Model 550, USA). The relative cell viability was calculated according to the following equation:
| Cell viability (%) = [(Asample − A0)/(Acontrol − A0)] × 100, |
where Acontrol was obtained in the presence of MTT and no sample, and Asample was obtained in the presence of both sample and MTT.
CLSM
HeLa cells or HepG2 cells were seeded on a confocal dish at a density of 1.5 × 105 cells per dish in 2 mL of complete DMEM containing 10% FBS, and incubated for 24 h. DOX/SLPNPs or DOX/ILPNPs were added to each dish at a final DOX concentration of 2 μg mL−1. The cells were further incubated for 24 h and the culture medium was removed. The cells were rinsed three times with PBS, and then fixed with 4% (w/v) paraformaldehyde aqueous solution for 15 min at room temperature. The nuclei of the cells were stained with Hoechst 33258 (5 μg mL−1 in PBS) for 12 min at 37 °C, and then rinsed with PBS three times. Finally, 1 mL of PBS was added to each dish and the cellular uptake of DOX/SLPNPs and DOX/ILPNPs was observed by CLSM (Nikon, TE2000, EZ-C1, Japan).
Flow cytometry studies
HeLa cells were seeded in 6-well plates at a density of 4.0 × 105 cells per well, and incubated for 24 h. Then DOX/SLPNPs or DOX/ILPNPs were added to each well with a final DOX concentration of 2 μg mL−1. Cells without any treatment were used as a control. After 24 h incubation, the medium was removed, and the cells were rinsed with PBS. Then 0.5 mL of trypsin was added and the cells were blown down with DMEM containing 10% FBS after 1 min incubation in a 5% CO2 incubator at 37 °C. Subsequently, the cell suspensions were centrifuged at 1000 rpm for 5 min. The cells were washed three times with PBS and analyzed with a flow cytometer (CyAN-ADP, Beckman) after resuspension in 500 μL of PBS.
CLSM observation of endocytosis inhibition
HeLa cells were seeded on a confocal dish at a density of 1.5 × 105 cells per dish in 2 mL of complete DMEM containing 10% FBS, and incubated for 24 h. The cells were pre-treated for 1 h with three different endocytosis inhibitors (5 mM MBCD, 0.45 M sucrose or 5 μM cytochalasin D) separately. Then DOX/SLPNPs or DOX/ILPNPs were added at a DOX dose of 2 μg mL−1 and the cells were further incubated for 4 h. The medium was removed and the cells were rinsed three times with PBS. Subsequently, the cells were fixed with 4% (w/v) paraformaldehyde aqueous solution and the nuclei of the cells were stained with Hoechst 33258 (5 μg mL−1 in PBS). After rinsing three times with PBS, the cells were observed by CLSM.
FCM analysis of endocytosis inhibition
HeLa cells were seeded in 6-well plates at a density of 4.0 × 105 cells per well. After 24 h incubation, the cells were pre-incubated with three different endocytosis inhibitors (5 mM methyl-β-cyclodextrin (MBCD), 0.45 M sucrose or 5 μM cytochalasin D) for 1 h separately. Cells without treatment by any endocytosis inhibitor were used as a control. Then the cells were incubated for another 4 h with media containing DOX/SLPNPs or DOX/ILPNPs at a final DOX concentration of 2 μg mL−1. The medium was removed and the cells were rinsed with PBS. Then 0.5 mL of trypsin was added and the cells were blown down with DMEM containing 10% FBS after 1 min incubation in a 5% CO2 incubator at 37 °C. Subsequently, the cell suspensions were centrifuged at 1000 rpm for 5 min. The cells were washed three times with PBS and re-suspended in 500 μL of PBS for analysis using a CyAN-ADP flow cytometer.
In vivo anti-tumor effect
HepG2 cells (5 × 106 cells) were injected subcutaneously into the right back fat pad of the BALB/c nude mice to establish a tumor model. When the tumors reached an approximate volume of 100 mm3, the mice were randomly divided into 4 groups and numbered; each group had 5 mice. Then DOX/SLPNPs, DOX/ILPNPs, free DOX at a dose of 7 mg DOX per kg and equivalent PBS were injected through the tail vein. The day was identified as day 0. The tumor volume (V) was measured using a caliper every 2 days for up to 16 days and calculated as V = a × b2/2, where a and b are the longest and shortest diameters, respectively. The weights of mice were also monitored. After 16 days, mice were all sacrificed by cervical vertebra dislocation and their tumors were immediately harvested. Tissues such as the heart, kidney, liver, lungs and spleen were immediately removed, rinsed with saline and fixed with 4% (w/v) paraformaldehyde solution.
Histological and immunohistochemical analyses
Tissues were fixed in 4% (w/v) paraformaldehyde solution overnight, and then embedded in paraffin. The paraffin-embedded tissues were cut at ∼4 μm thickness for H&E (hematoxylin and eosin) staining and immunohistochemical staining of the proliferating cell nuclear antigen (PCNA) and the terminal transferase dUTP nick-end labeling (TUNEL) assay. The histological alterations and pathological examination were detected using a microscope (Olympus IX51/Q-Imaging Micro Publisher).
In vivo tissue distribution studies
DOX/SLPNPs, DOX/ILPNPs and free DOX were intravenously injected into mice bearing HepG2 xenograft tumors via the tail vein. The DOX dose was 7 mg kg−1. After 12 h, mice were sacrificed by cervical vertebra dislocation, and the tumors and tissues such as the heart, kidney, liver, lungs and spleen were immediately collected, rinsed with saline and fixed with 4% (w/v) paraformaldehyde solution. Subsequently, the tissues were dried and weighed, homogenized with an Ultra-Turrax Homogenizer (IKA T25, 12500 rpm) in 1.5 mL of KH2PO4 solution (20 × 10−3 M). Then 200 μL of the tissue homogenate was exposed to 50 μL of HCl (5 M) at 60 °C for 3 h. Next, 50 μL of 1 M NaOH was added and DOX in each mixture was extracted using 300 μL chloroform and isopropanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The organic layer was collected by centrifugation (12000 rpm, 8 min) and evaporated to dryness at 60 °C overnight. The residue was then dissolved in 300 μL of acetonitrile and filtered through a membrane with pore size of 200 nm. The samples were detected by HPLC system (SHIMADZU, LC-15C, Kyoto, Japan) equipped with a reversed phase C18 column (4.6 mm × 250 mm, 5 μm) and a fluorescence detector at 35 °C. The detection wavelength was 485 nm, the mobile phase was acetonitrile and water (45:55, v/v) and the flow rate was 1 mL min−1. To generate the control group, free DOX was added to the tissues from the drug-free mice before homogenization.
:1, v/v). The organic layer was collected by centrifugation (12000 rpm, 8 min) and evaporated to dryness at 60 °C overnight. The residue was then dissolved in 300 μL of acetonitrile and filtered through a membrane with pore size of 200 nm. The samples were detected by HPLC system (SHIMADZU, LC-15C, Kyoto, Japan) equipped with a reversed phase C18 column (4.6 mm × 250 mm, 5 μm) and a fluorescence detector at 35 °C. The detection wavelength was 485 nm, the mobile phase was acetonitrile and water (45:55, v/v) and the flow rate was 1 mL min−1. To generate the control group, free DOX was added to the tissues from the drug-free mice before homogenization.
Results and discussion
Synthesis of amphiphilic PEG derivatives
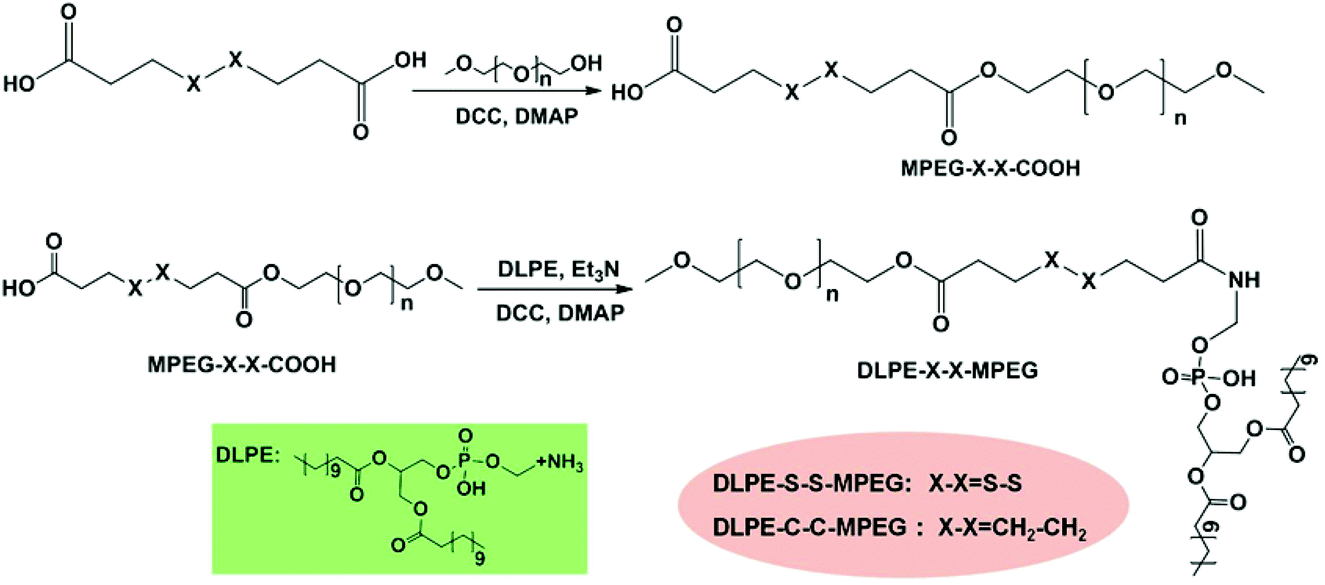
Disulfide-containing MPEG mono carboxylic acid (MPEG-S-S-COOH) was prepared via the esterification reaction of 3,3′-dithiodipropionic acid and MPEG in anhydrous DMF in the presence of DCC and DMAP. DLPE-S-S-MPEG was then synthesized by the coupling of DLPE with MPEG-S-S-COOH. The synthesis procedure is illustrated in Scheme 2. As a control, reduction-insensitive DLPE-C-C-MPEG was similarly prepared from MPEG, octanedioic acid and DLPE.
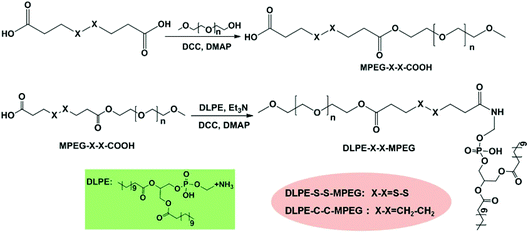 |
| | Scheme 2 Synthesis of DLPE-S-S-MPEG and DLPE-C-C-MPEG. | |
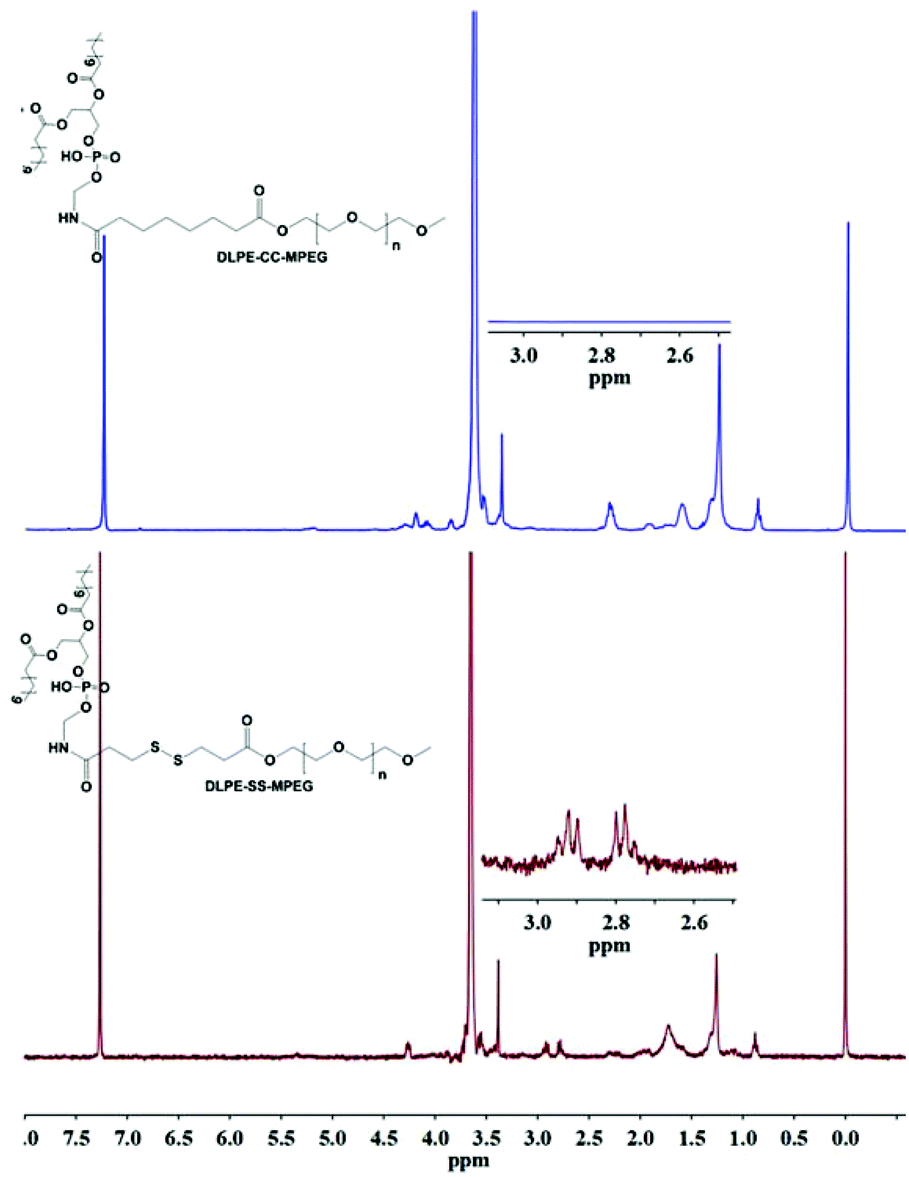
The successful coupling of MPEG-S-S-COOH and DLPE was verified by 1H NMR. As shown in Fig. 1, the characteristic peaks at 3.38–3.71 ppm were assigned to the protons in CH2CH2O of MPEG, and the peaks at 2.78–2.92 ppm belong to –CO–CH2–C–S–S–C–CH2–CO–. The signals at 4.26 ppm and 1.26 ppm suggested the successful conjugation of DLPE with MPEG-S-S-COOH.
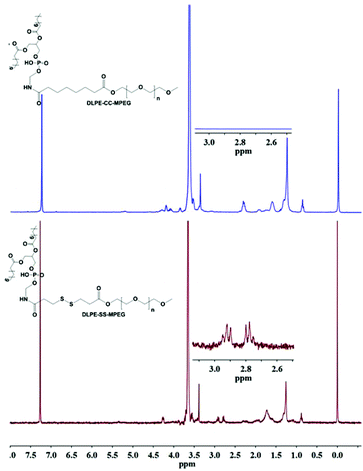 |
| | Fig. 1
1H NMR spectra of DLPE-S-S-MPEG and DLPE-C-C-MPEG. | |
Stability of LPNPs and DOX/LPNPs
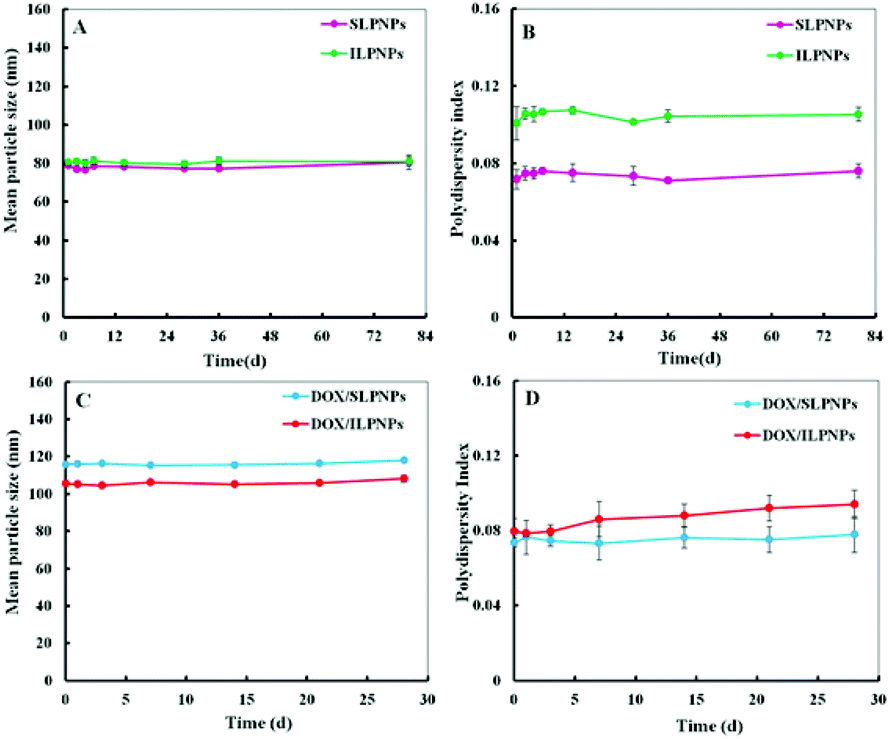
Two-component lipid–polymer hybrid nanoparticles (LPNPs) were prepared from PCL and DLPE-MPEG via a modified single-step assembly method. Blank LPNPs and DOX-loaded LPNPs (DOX/LPNPs) were separately prepared in the absence or in the presence of DOX. The DLC of DOX/SLPNPs and DOX/ILPNPs was 3.46% and 3.92%, respectively. The storage stabilities of LPNPs and DOX/LPNPs were evaluated by dynamic light scattering (DLS) measurement. The results are shown in Fig. 2A and B. SLPNPs, prepared from PCL and DLPE-S-S-MPEG, showed an average size of around 80 nm and a polydispersity index (PDI) of 0.075. Similarly, ILPNPs, prepared from PCL and DLPE-C-C-MPEG, showed an average size of around 80 nm and a PDI of 0.105. Both SLPNPs and ILPNPs were stable with homogeneous size distribution. Even after storage for 80 days at 4 °C, there was no significant increase in both the mean particle size and PDI of SLPNPs and ILPNPs. In the case of DOX-loaded LPNPs, the mean particle size of both DOX/SLPNPs (115 nm) and DOX/ILPNPs (105 nm) became slightly larger in comparison to those of blank LPNPs. Both DOX/SLPNPs and DOX/ILPNPs were also stable with homogeneous size distribution even after storage for 30 days at 4 °C.
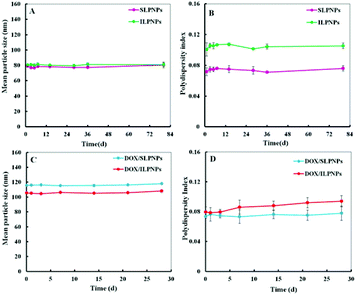 |
| | Fig. 2 Storage stability of SLPNPs and ILPNPs (A. mean particle size and B. polydispersity index) after 80 days storage at 4 °C measured by DLS. Storage stability of DOX/SLPNPs and DOX/ILPNPs (C and D) after 28 days storage at 4 °C. Data were presented as a mean ± SD (n = 3). | |
Reduction-triggered disassembly of SLPNPs
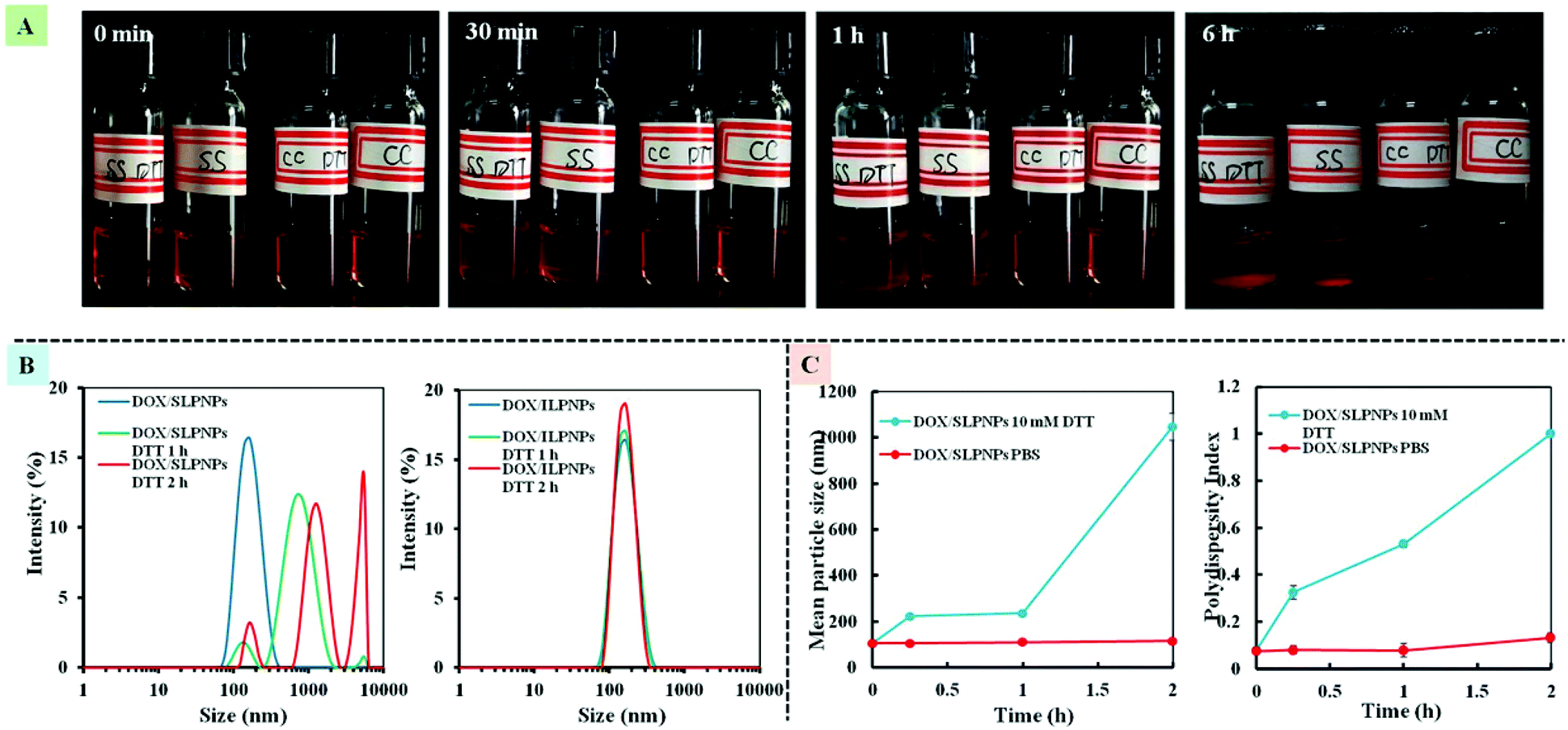
We have previously demonstrated that the disassembly of three-component disulfide-containing lipid–polymer hybrid nanoparticles (LPNPs) occurred under reductive conditions,37,38 which triggered the release of encapsulated hydrophobic drug in the core of LPNPs. To demonstrate the responsiveness of the disulfide bond in DLPE-S-S-MPEG, DTT was separately added into DOX/SLPNPs and DOX/ILPNPs solution with a final DTT concentration of 10 mM. The visual photos showing the change of DOX/SLPNPs (SS) and DOX/ILPNPs (CC) incubated in PBS 7.4 with 10 mM DTT or without DTT are shown in Fig. 3A. The images indicated that after adding DTT, DOX/SLPNPs gradually aggregated and precipitated after 6 h incubation, however, there was no significant change for DOX/ILPNPs even after 6 h incubation in PBS containing 10 mM DTT. In contrast, no changes were observed for both DOX/SLPNPs and DOX/ILPNPs incubated in PBS 7.4 without DTT. The average sizes and PDI changes of DOX/SLPNPs and DOX/ILPNPs in response to 10 mM DTT were also measured by DLS. As shown in Fig. 3B and C, the sizes and PDI of DOX/SLPNPs rapidly increased with the increase of incubation time in PBS containing 10 mM DTT, while there were no changes for DOX/ILPNPs under the same conditions. When DOX/SLPNPs and DOX/ILPNPs were separately incubated in PBS without DTT, there were no changes in average sizes and PDI. The results indicated that DOX/SLPNPs are stable in PBS without DTT, however possess excellent sensitivity under reductive conditions.
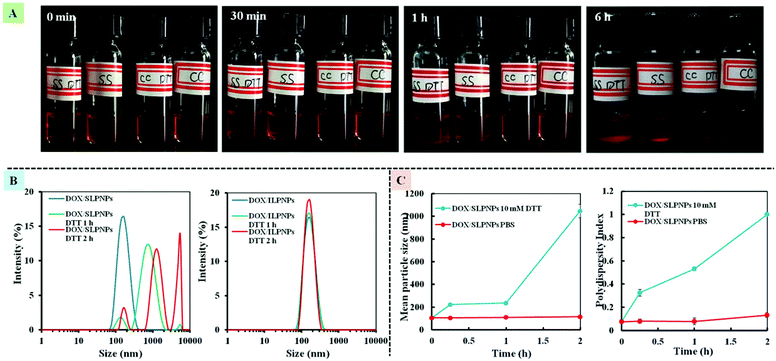 |
| | Fig. 3 The photo of DOX/SLPNPs (SS) and DOX/ILPNPs (CC) in response to PBS with 10 mM DTT or without DTT (A). The size distribution of DOX/SLPNPs and DOX/ILPNPs in response to 10 mM DTT in PBS measured by DLS (B). The average size and PDI changes of DOX/SLPNPs in response to DTT (C). | |
The size and morphology changes of DOX/SLPNPs and DOX/ILPNPs before and after treatment with DTT were observed by TEM. As shown in Fig. 4, without DTT treatment, both DOX/SLPNPs and DOX/ILPNPs were well dispersed as individual nanoparticles with spherical shape. After treatment with 10 mM DTT for 10 min, the DOX/SLPNPs aggregated and showed irregular morphologies, and after 1 h treatment, DOX/SLPNPs disintegrated. However, there were no significant changes for DOX/ILPNPs even after 1 h treatment with 10 mM DTT. These phenomena were consistent with the results of DLS measurement.
 |
| | Fig. 4 TEM images of DOX/ILPNPs (A), and DOX/ILPNPs with 10 mM DTT treatment for 10 min (A1) and 1 h (A2); TEM images of DOX/SLPNPs (B), and DOX/SLPNPs with 10 mM DTT treatment for 10 min (B1) and 1 h (B2). | |
In vitro reduction-triggered DOX release from SLPNPs
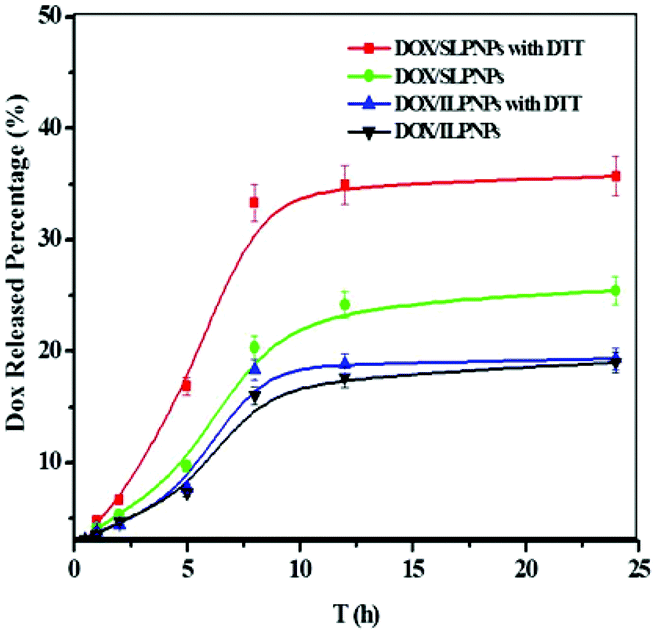
DOX is one of the most widely used chemotherapeutic drugs and is a popular research tool because of its inherent fluorescence. In this work, DOX, as an anticancer drug, was separately loaded into SLPNPs and ILPNPs. The release of DOX from DOX/SLPNPs and DOX/ILPNPs was evaluated using dialysis. As shown in Fig. 5, DOX release from the SLPNPs showed a rapid pattern in the first few hours in the presence of 10 mM DTT. 12 h later, 35.7% of DOX was released. However, at the same time point, only 20% of DOX was released in the absence of DTT. These results may be due to the cleavage of the disulfide bond in the DLPE-S-S-MPEG, which caused the dissociation of the LPNPs structure. By contrast, the addition of DTT had little effect on the release of DOX from the reduction-insensitive ILPNPs. These results suggested that reduction-sensitive SLPNPs may achieve site-specific drug delivery of drug in the presence of reducing agents.
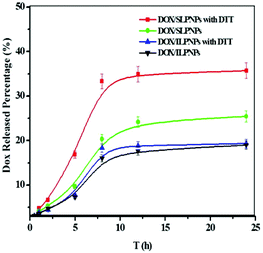 |
| | Fig. 5
In vitro release percentage of DOX from LPNPs to the outer dialysis medium of PBS (pH 7.4) with or without 10 mM DTT at 37 °C on a shaker table at 100 rpm. Data are presented as mean ± SD (n = 3). | |
In vitro cytotoxicity assay
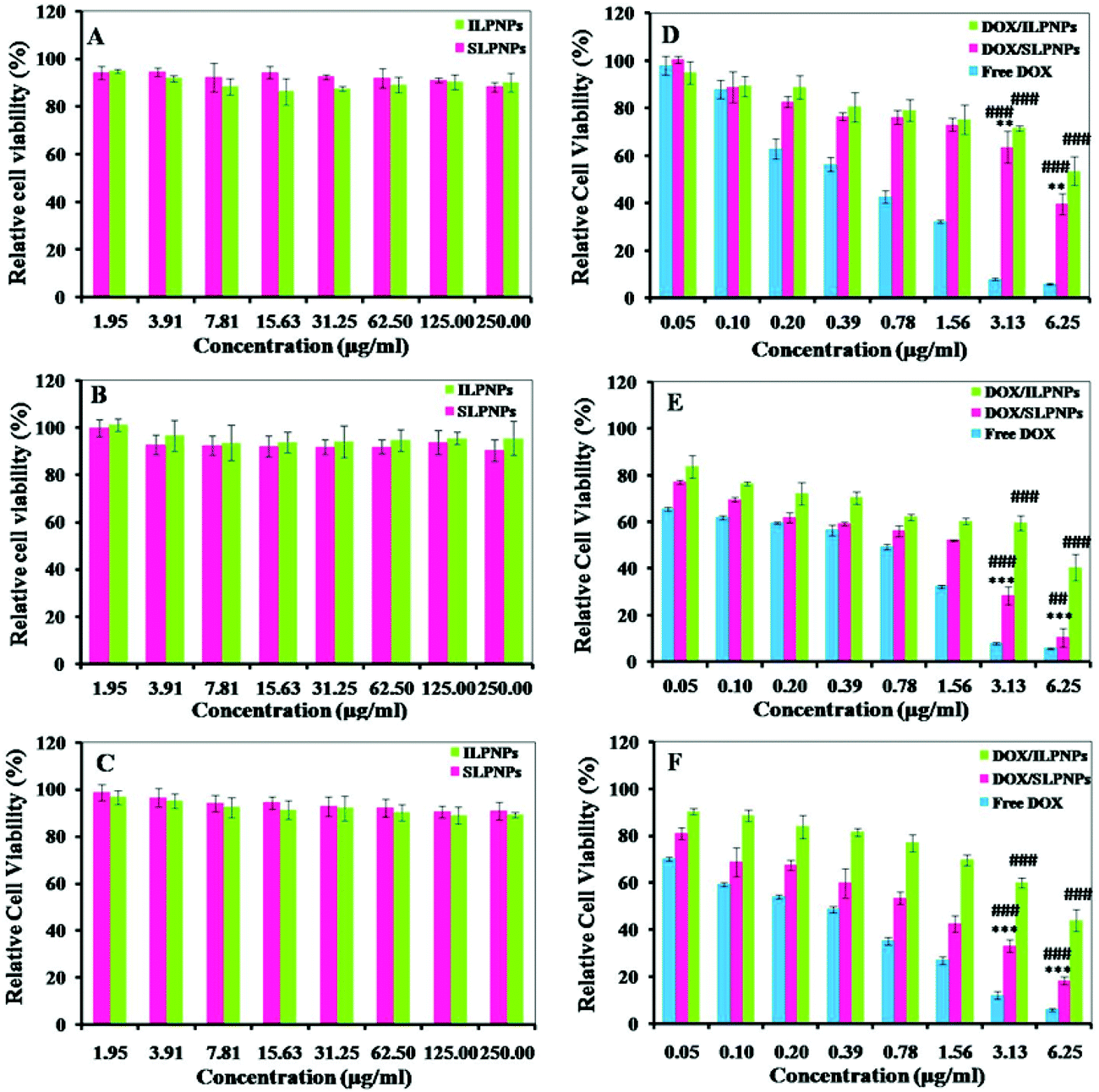
The in vitro cytotoxicities of LPNPs and DOX/LPNPs against HeLa cells, HepG2 cells or COS-7 cells were determined using MTT assay. To evaluate the toxicity of LPNPs without loading DOX, cells were separately cultured with SLPNPs and ILPNPs at concentrations from 1.95 to 250 μg mL−1. As shown in Fig. 6(A–C), even up to 250 μg mL−1, the activities of all kinds of cells were not significantly affected. This indicated that LPNPs are safe for application in drug delivery. The cytotoxicities of DOX/SLPNPs, DOX/ILPNPs and free DOX were further evaluated. As shown in Fig. 6(D–F), the inhibition of cell growth with three formulations was dose-dependent. DOX showed high toxicity to all tested cells, including normal cells (COS-7 cells) and cancer cells (HeLa cells and HepG2 cells). DOX/ILPNPs exhibited low toxicity to all three kinds of cells. Different from DOX and DOX/ILPNPs, DOX/SLPNPs showed lower toxicity to normal cells (COS-7 cells) but higher toxicity to cancer cells (HeLa cells and HepG2 cells). The unique characteristic of DOX/SLPNPs in selective inhibition of cancer cells growth is beneficial to cancer therapy. DOX, as a small molecular anticancer drug, is easily transported into cells and enters nuclei by passive diffusion. The antitumor efficacy of DOX encapsulated in nanocarriers is often determined by the intracellular release of DOX once drug-loaded nanoparticles enter into cells. As discussed above, the release of DOX from DOX/SLPNPs is much faster than that from DOX/ILPNPs in the presence of DTT. The fast drug release from DOX/SLPNPs was attributed to the cleavage of the disulfide bond by GSH inside cancer cells. As a result, DOX/SLPNPs exhibited higher toxicity to cancer cells than that of DOX/ILPNPs. In contrast, in the case of COS-7 cells, there is not enough reducing agent, such as GSH, which results in a slight difference between the slow DOX release from DOX/SLPNPs and DOX/ILPNPs. It is clear that both DOX/SLPNPs and DOX/ILPNPs are less toxic to COS-7 cells.
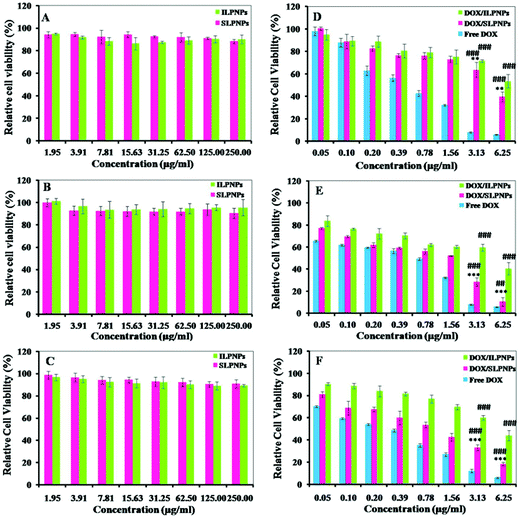 |
| | Fig. 6 Cytotoxicities of SLPNPs and ILPNPs against COS-7 cells (A), HeLa cells (B) and HepG2 cells (C). Cytotoxicities of DOX/ILPNPs, DOX/SLPNPs and free DOX against COS-7 cells (D), HeLa cells (E) and HepG2 cells (F). Data are presented as mean ± SD (n = 4). **P < 0.01 and ***P < 0.001 versus DOX/ILPNPs group, ##P < 0.01 and ###P < 0.001 versus free DOX group. | |
In vitro cellular uptake
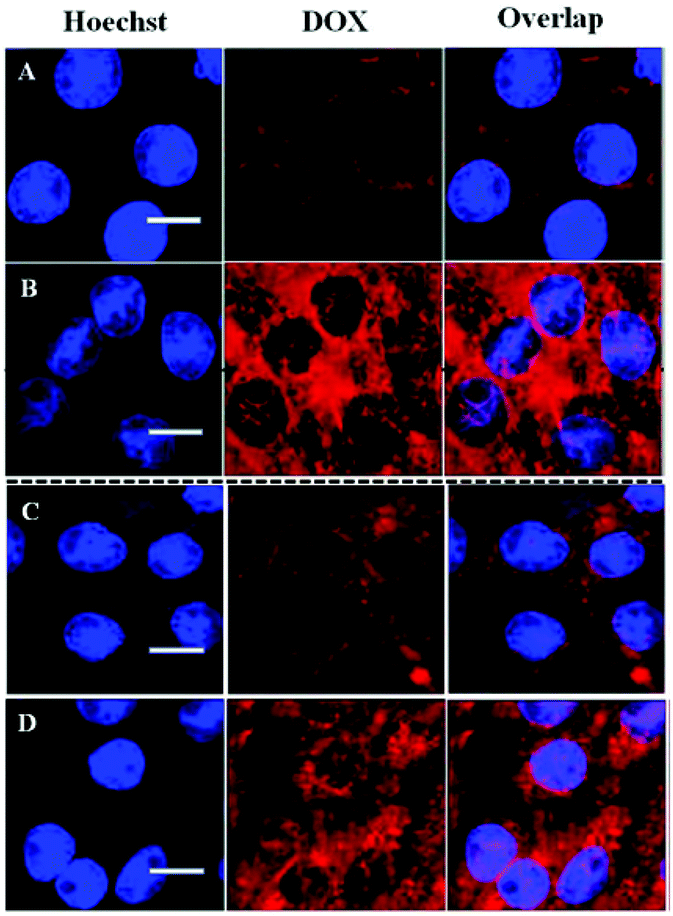
Cellular uptake and intracellular drug release of DOX/SLPNPs and DOX/ILPNPs inside HeLa cells and HepG2 cells were monitored by CLSM (Fig. 7). It should be pointed out that DOX encapsulated in nanoparticles is weakly fluorescent due to quenching effect. The red fluorescence intensity of DOX is related to the amount of free DOX inside the cells. The cells were imaged using CLSM after 24 h treatment with DOX/SLPNPs or DOX/ILPNPs at a DOX dosage of 2 μg mL−1. In the case of DOX/ILPNPs, weak red fluorescence was observed in both HeLa cells and HepG2 cells. Almost no fluorescence was found in the nuclei. Different from the case of DOX/ILPNPs, after treatment with DOX/SLPNPs at the same dosage of DOX, significantly strong red fluorescence was observed in both HeLa cells and HepG2 cells in comparison to that of DOX/ILPNPs. Red fluorescence of DOX was also found in the nuclei. The significant difference of intracellular fluorescence intensity between DOX/ILPNPs and DOX/SLPNPs was related to the faster release of DOX from DOX/SLPNPs than DOX/ILPNPs. These observations were consistent with the in vitro drug release and cytotoxicity results.
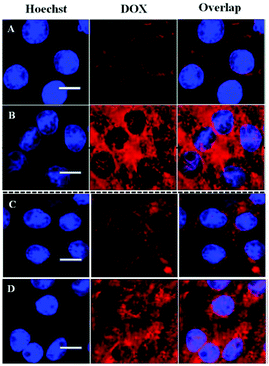 |
| | Fig. 7 CLSM images of HeLa cells after treatment with DOX/ILPNPs (A) and DOX/SLPNPs (B) for 24 h; CLSM images of HepG2 cells after treatment with DOX/ILPNPs (C) and DOX/SLPNPs (D) for 24 h. DOX dosage was 2 μg mL−1. Scale bar is 20 μm. | |
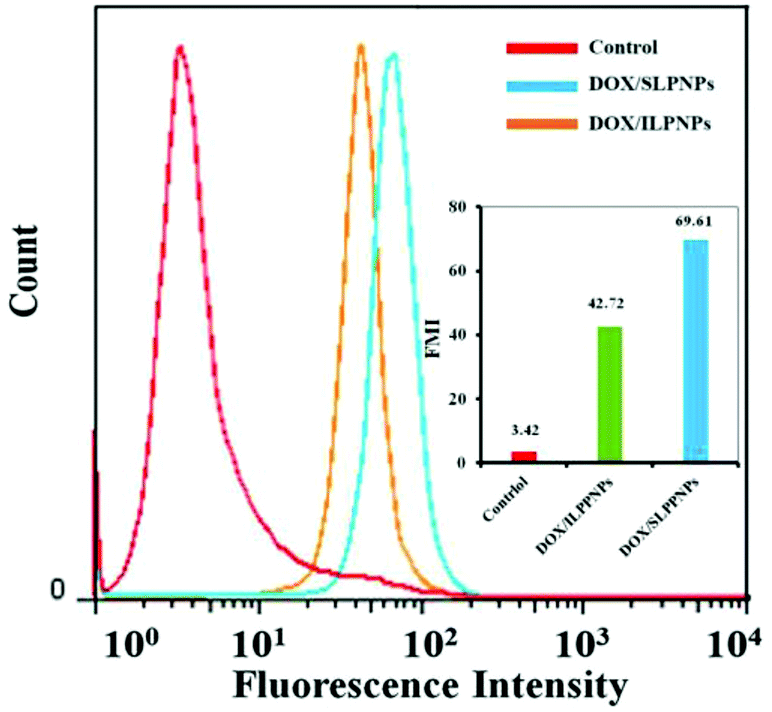
Flow cytometry (FCM) was used to further investigate the intracellular drug release from DOX/ILPNPs and DOX/SLPNPs. Fig. 8 shows the mean DOX fluorescence intensity in HeLa cells after separate incubation with DOX/SLPNPs or DOX/ILPNPs at a dosage of 2 μg mL−1 for 24 h. The MFI of cells treated with DOX/SLPNPs was much higher than the cells incubated with DOX/ILPNPs. The fluorescence intensity was associated with the amount of DOX released from the nanoparticles. The results were in accordance with the CLSM observations.
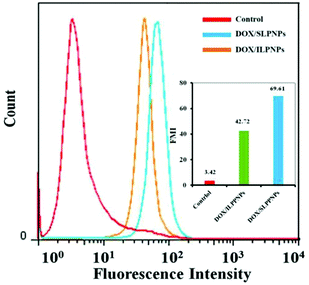 |
| | Fig. 8 Flow cytometry analyses of HeLa cells treated with DOX/SLPNPs and DOX/ILPNPs for 24 h. DOX dosage was 2 μg mL−1. The cells incubated with medium without any DOX/LPNPs were used as control. | |
Combining the results of in vitro drug release, in vitro cytotoxicity, CLSM observations and FCM analysis, it can be concluded that the better antitumor efficacy of DOX/SLPNPs than DOX/ILPNPs is attributed to the reduction-sensitive release of DOX inside tumor cells.
Endocytosis inhibition
Three different types of endocytosis inhibitors were used to investigate the endocytosis pathway of both DOX/SLPNPs and DOX/ILPNPs. 5 mM MBCD was used to inhibit caveolae-mediated endocytosis, 0.45 mM hypertonic sucrose to inhibit clathrin-mediated endocytosis, and 5 μM cytochalasin D to inhibit micropinocytosis.
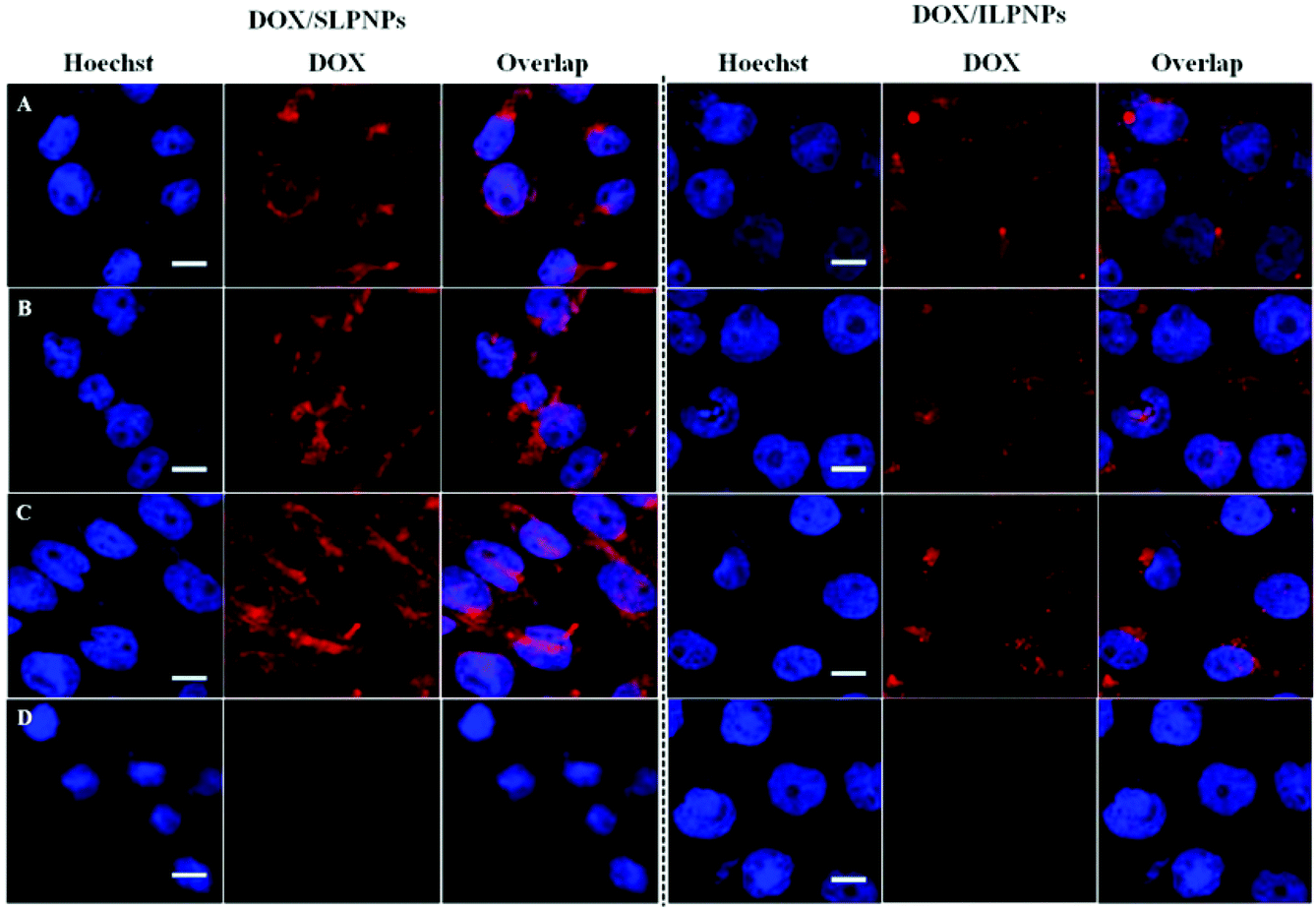
HeLa cells were separately incubated with DOX/SLPNPs and DOX/ILPNPs in the presence or absence of endocytosis inhibitors for 4 h. Fig. 9 shows the red fluorescence of DOX observed in HeLa cells using CLSM. In the cases of both DOX/SLPNPs and DOX/ILPNPs, the red fluorescence of DOX in HeLa cells treated with hypertonic sucrose was significantly weaker than that of cells incubated without inhibitor. Meanwhile, MBCD and cytochalasin D did not affect the fluorescence of DOX in the cells cultured with DOX/SLPNPs and DOX/ILPNPs, indicating that the entry of DOX/SLPNPs and DOX/ILPNPs into HeLa cells depended mainly on clathrin-mediated endocytosis.
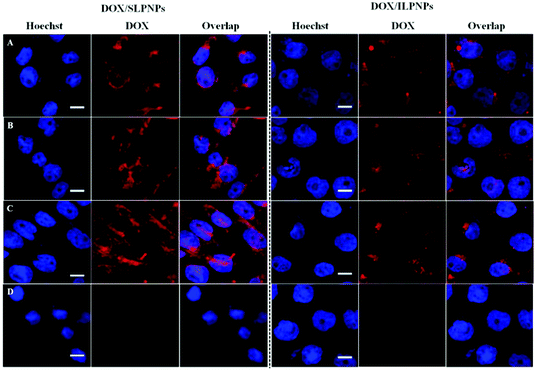 |
| | Fig. 9 CLSM images of HeLa cells incubated with DOX/SLPNPs or DOX/ILPNPs for 4 h without any endocytosis inhibitors (A), with 5 mM MBCD (B), with 5 μM cytochalasin D (C) and with 0.45 mM hypertonic sucrose (D). DOX dosage was 2 μg mL−1. Scale bar is 15 μm. | |
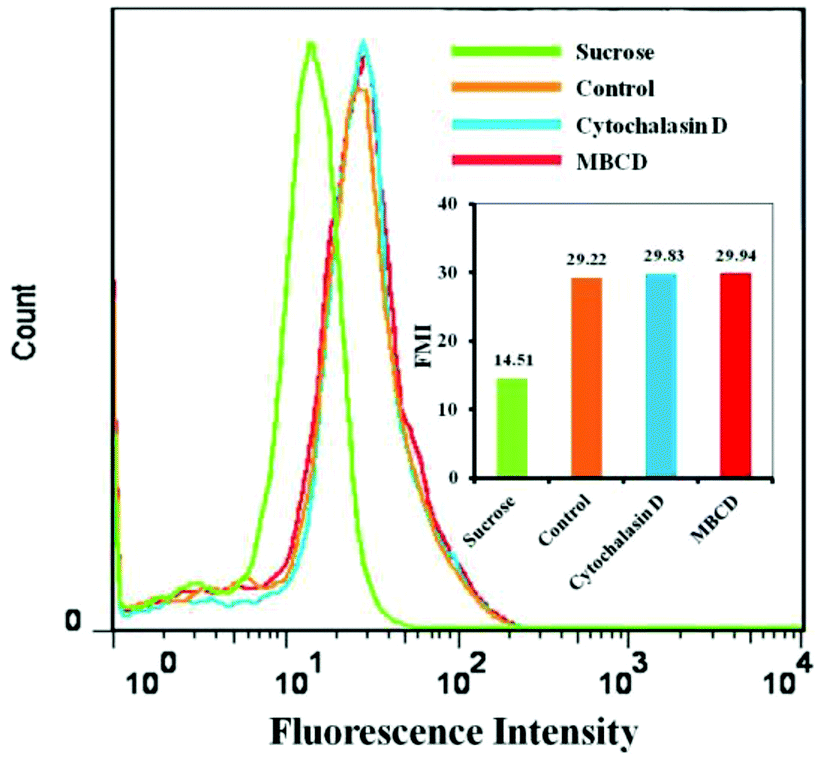
FCM analysis results were consistent with the CLSM observations. Fig. 10 shows the mean DOX fluorescence intensity in HeLa cells after incubation with DOX/SLPNPs with or without endocytosis inhibitors for 4 h. The entry of DOX/SLPNPs into HeLa cells was inhibited by hypertonic sucrose and the cellular uptake significantly decreased to 49.6% in comparison to that without inhibitor. However, the uptake of DOX/SLPNPs by HeLa cells was unaffected by MBCD and cytochalasin D. Both CLSM observations and FCM analysis demonstrated that the most prominent pathway for both DOX/SLPNPs and DOX/ILPNPs uptake by cancer cells was clathrin-mediated endocytosis, whereas caveolae-mediated endocytosis and micropinocytosis had minimal involvement. The different antitumor efficiencies of DOX/SLPNPs and DOX/ILPNPs were not determined by the uptake of DOX/SLPNPs and DOX/ILPNPs by cancer cells, but by the different drug release characteristics of DOX/SLPNPs and DOX/ILPNPs after internalization into the cancer cells.
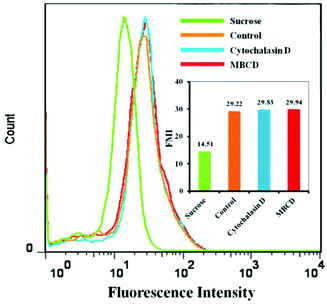 |
| | Fig. 10 Flow cytometry analyses of HeLa cells incubated with DOX/SLPNPs for 4 h with MBCD, cytochalasin D, hypertonic sucrose (sucrose) and without any endocytosis inhibitors (control). DOX dosage was 2 μg mL−1. | |
In vivo antitumor efficacy
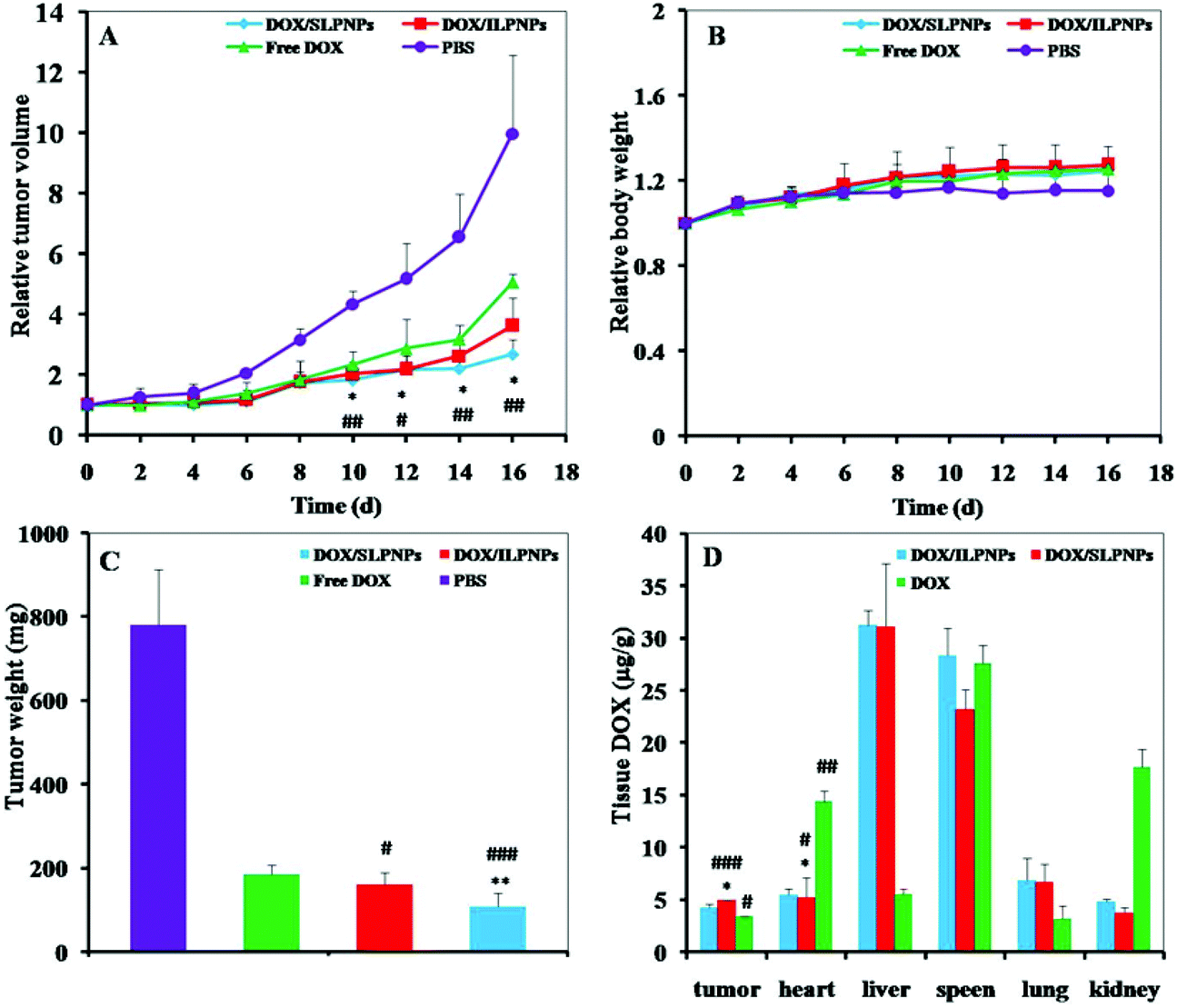
The in vivo antitumor efficacies were evaluated with female BALB/c nude mice bearing HepG2 tumors. Once the tumors reached approximately 100 mm3, the tumor-bearing mice were treated by intravenously injecting DOX/SLPNPs, DOX/ILPNPs, free DOX and PBS. As shown in Fig. 11A and B, the tumor volumes and body weights were monitored in real-time every two days. Compared to the control group, the tumor growths were significantly slower in the experimental group separately treated with DOX/SLPNPs, DOX/ILPNPs and free DOX. In the first four days, there were no significant differences among all the experimental groups; even the tumor growth of the group treated with free DOX was relatively slow. However, 6 d after injection, the tumor growth inhibition ability of free DOX became weaker than that of DOX/SLPNPs and DOX/ILPNPs. 16 d after injection, the relative tumor volume (RTV) of the negative control group treated with PBS was 9.97, while the RTV of the experimental groups treated with DOX/SLPNPs, DOX/ILPNPs and free DOX were 2.68, 3.62 and 5.05, respectively. DOX/SLPNPs showed the most effective in vivo antitumor efficacy among DOX/SLPNPs, DOX/ILPNPs and free DOX. The in vivo antitumor results were consistent with our expectations.
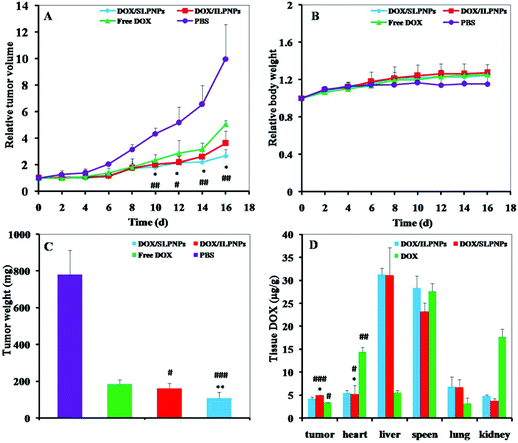 |
| | Fig. 11 Relative tumor volumes (A), relative body weights (B), and tumor weights (C) of HepG2 tumors after treatment with DOX/SLPNPs, DOX/ILPNPs, free DOX, and PBS. Data are presented as mean ± SD (n = 5). The DOX tissue distribution in female BALB/c nude mice bearing HepG2 tumors after tail vein injection of DOX/SLPNPs, DOX/ILPNPs, and free DOX for 12 h, respectively (D). Data were presented as mean ± SD (n = 3). *P < 0.05 and **P < 0.01 versus DOX/ILPNPs group, #P < 0.05, ##P < 0.01 and ###P < 0.001 versus free DOX group. | |
In vivo biodistribution
To further explain the in vivo antitumor difference among DOX/SLPNPs, DOX/ILPNPs and free DOX, the DOX content in various tissues of nude mice (tumor, heart, liver, spleen, lung and kidney) was measured by HPLC at 12 h after injection. As shown in Fig. 11D, the DOX concentrations in tumors injected with DOX/SLPNPs and DOX/ILPNPs were higher than those injected with free DOX. The higher DOX concentrations of DOX/SLPNPs and DOX/ILPNPs were ascribed to the EPR effect of nanoparticles. Free DOX was rapidly cleared from the bloodstream and eliminated through the kidneys after injection, which resulted in the low accumulation of DOX in the tumor. There were no significant difference in the DOX concentration in the tumors between the treatment of DOX/SLPNPs and DOX/ILPNPs. The more effective in vivo antitumor ability of DOX/SLPNPs than DOX/ILPNPs was ascribed to the reduction-responsive release of DOX from DOX/SLPNPs once DOX/SLPNPs entered into the tumor cells. In addition, significantly lower DOX concentration of DOX/SLPNPs and DOX/ILPNPs in the heart of mice than that with free DOX were detected, which implied that the side effect of DOX on the heart might be reduced by the encapsulation of DOX in the LPNPs.
Histological and immunohistochemical analyses
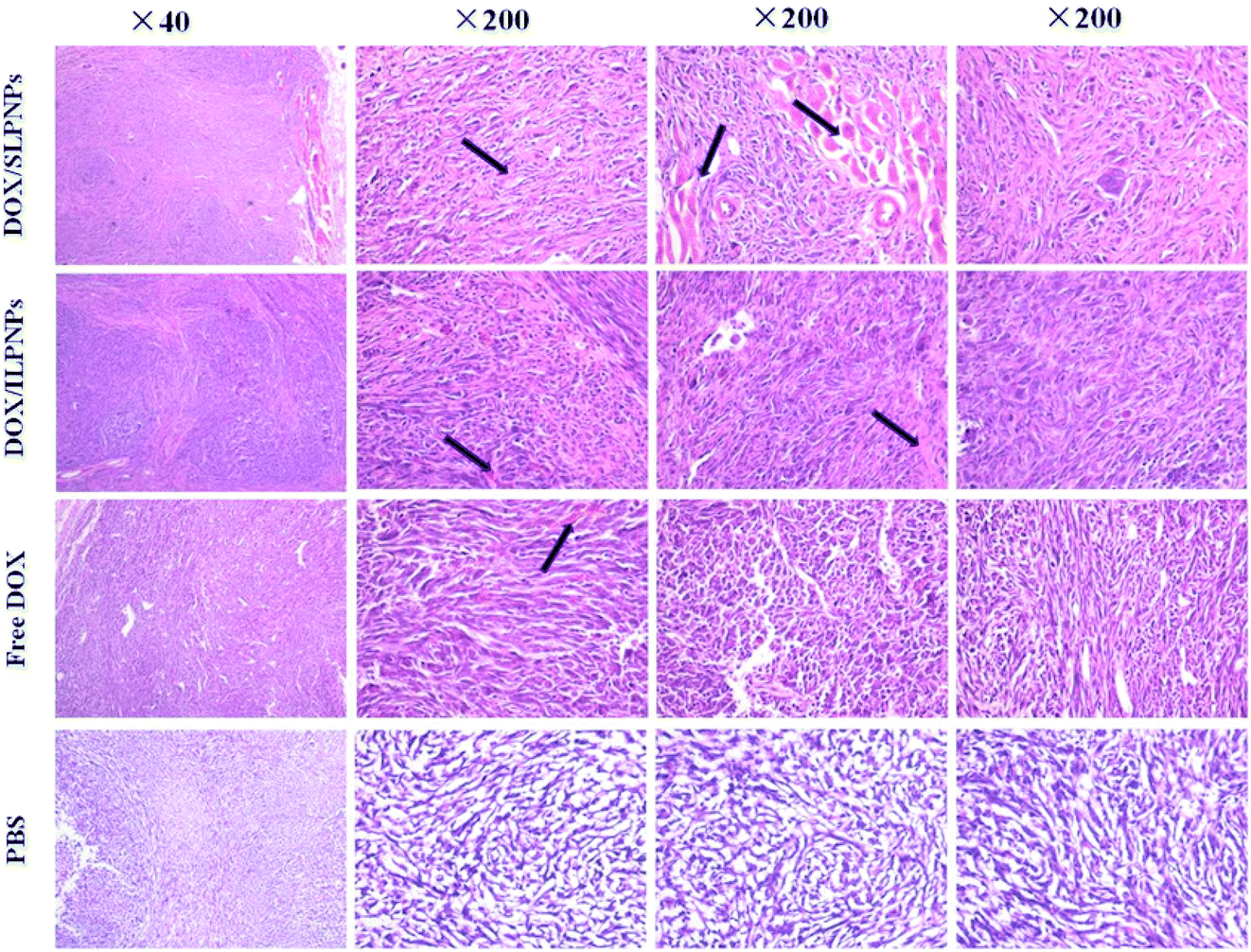
To observe and identify the apoptosis and necrosis of tumor cells, the tumors were dissected from mice and sectioned for pathology analysis (Fig. 12). Typically, in normal cells or tissues, nuclei are stained blue-purple with hematoxylin, whereas the cytoplasm and extracellular matrix have varying degrees of pink staining. In this work, tumor cells with a large nucleus and a spindle shape were observed in the tumor tissue treated with PBS, in which more chromatin and binucleolates were also observed, indicating rapid tumor growth. In contrast, in the case of the tumor cells treated with free DOX, DOX/SLPNPs and DOX/ILPNPs, extensive nuclear shrinkage, various degrees of tissue necrosis and fragmentation were observed. The chromatin was concentrated and nuclei became pyknotic, fragmented or absent, especially for the tumors treated with DOX/SLPNPs. The necrosis area in the DOX/SLPNPs group was the largest, the DOX/ILPNPs groups exhibited a relatively lower necrotic level, while the free DOX groups displayed the smallest necrosis area at the 16th day after injection.
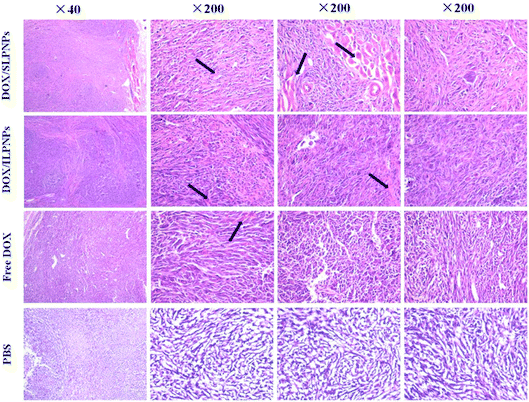 |
| | Fig. 12 H&E analyses of tumor tissues from mice treated with DOX/SLPNPs, DOX/ILPNP, free DOX and PBS for 16 days. The dose of DOX was 7.0 mg kg−1 (n = 5). | |
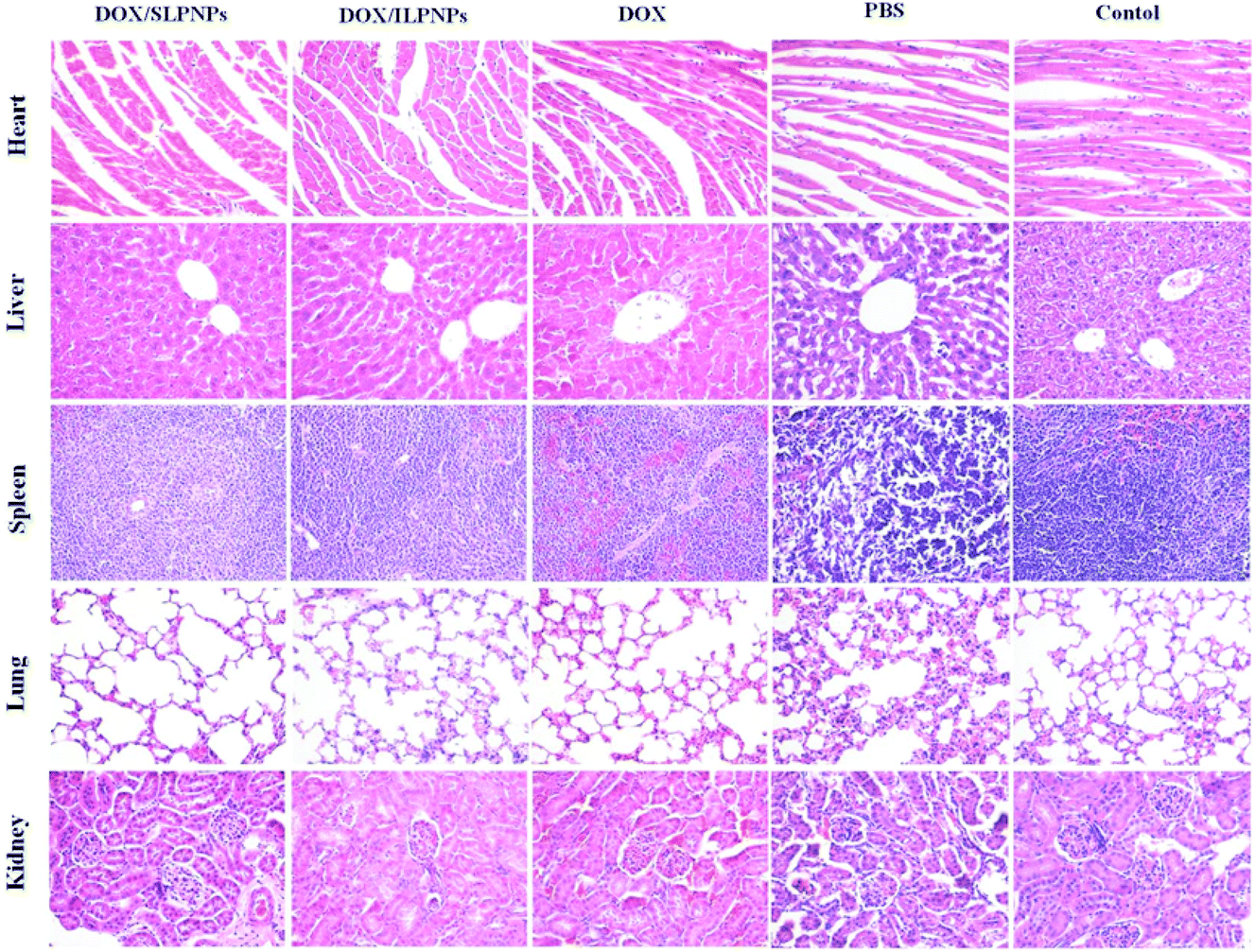
Long-term toxicity is a major concern for in vivo application of chemotherapy. The major organs (heart, liver, spleen, lung and kidney) sectioned from the mice bearing HepG2 tumors receiving PBS and various drug formulations at day 16 after the injections were harvested and stained by H&E. The organs sectioned from normal mice served as controls (Fig. 13). Compared with the normal control group, no significant morphological changes could be detected in all tested organs of the PBS groups. Similarly, no changes were observed in the livers and lungs of the mice treated with free DOX, DOX/SLPNPs and DOX/ILPNPs. However, the free DOX treated groups displayed noticeable damages in the heart, spleen and kidney. Different from the free DOX treated groups, only slight damages in the heart, spleen and kidney of DOX/SLPNPs or DOX/ILPNPs treated groups were observed. This was in accordance with in vivo DOX biodistribution results. The H&E analysis results indicated that both DOX/SLPNPs and DOX/ILPNPs can effectively reduce the cardiac toxicity and kidney toxicity of DOX in vivo.
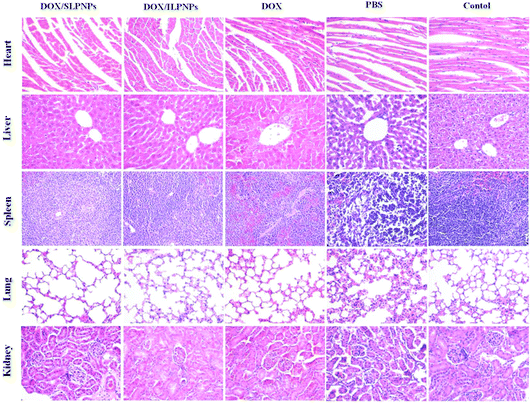 |
| | Fig. 13 H&E staining of major organs from the mice treated with DOX/SLPNPs, DOX/ILPNPs, free DOX and PBS. The organs from normal mice not bearing tumors were used as controls. Magnification: 200×. | |
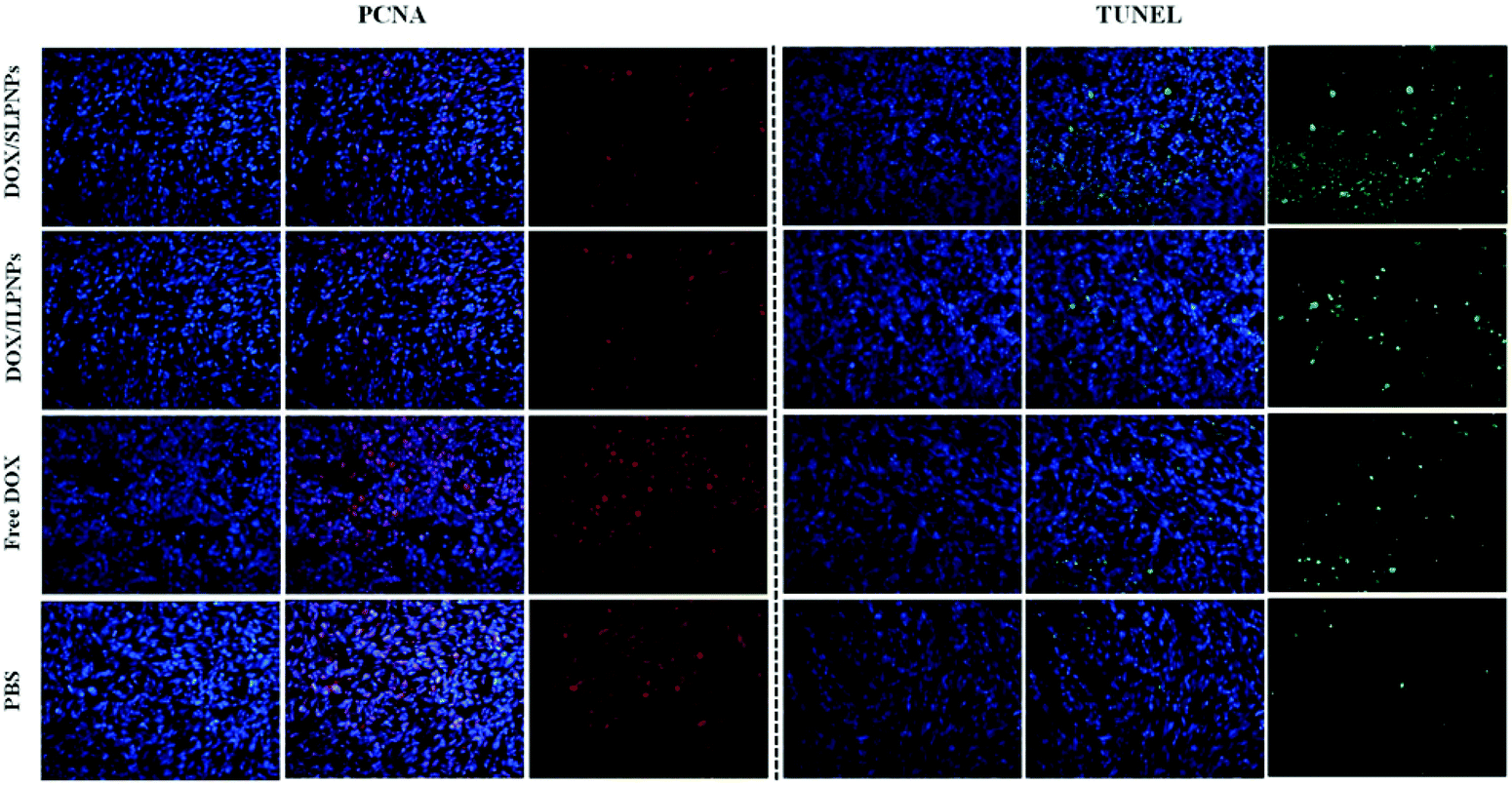
To further investigate the cell proliferation and cell apoptosis of tumors, the tumor sections treated with PBS, free DOX, DOX/ILPNPs and DOX/SLPNPs were evaluated by PCNA and TUNEL analysis. PCNA-positive proliferating cells are stained red, and TUNEL-positive apoptotic cells are stained green. As shown in Fig. 14, the cell proliferation signal of tumors treated with PBS was the strongest, second the free DOX group, next the DOX/ILPNPs group and the weakest was the DOX/SLPNPs group. In contrast, the cell apoptosis signal of tumors treated with PBS was the weakest, and that of the free DOX, DOX/ILPNPs and DOX/SLPNPs groups enhanced gradually, and the signal of DOX/SLPNPs group was the strongest. Compared with the other groups, the group treated with DOX/SLPNPs resulted in significantly reduced cell proliferation and increased cell apoptosis. The PCNA and TUNEL analysis results were consistent with the in vivo antitumor results and H&E stain results. In comparison to DOX/ILPNPs, the intracellular reduction-responsive release of DOX from SLPNPs enhanced the growth inhibition of tumor cells and increased tumor cell apoptosis. As a result, DOX/SLPNPs showed excellent in vivo antitumor efficacy.
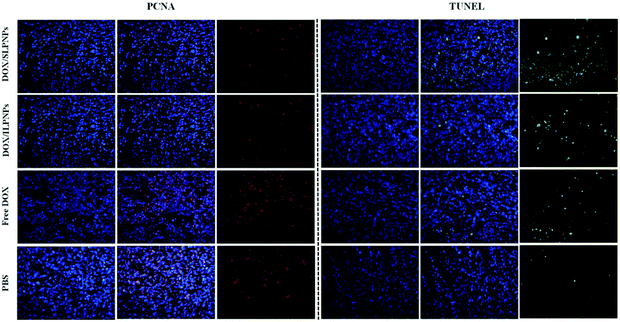 |
| | Fig. 14 PCNA and TUNEL analyses of tumor tissues from mice treated with DOX/SLPNPs, DOX/ILPNPs, free DOX and PBS. PCNA-positive proliferating cells are stained red, and TUNEL-positive apoptotic cells are stained green. Magnification: 200×. | |
Conclusions
Amphiphilic reduction-sensitive polymer DLPE-S-S-MPEG was synthesized and employed together with PCL to prepare two-component reduction-sensitive lipid–polymer hybrid nanoparticles (SLPNPs) via single-step self-assembly. Insensitive lipid–polymer hybrid nanoparticles (ILPNPs) were similarly prepared from PCL and DLPE-C-C-MPEG and used as a control. DOX-loaded SLPNPs rapidly released DOX under reduction conditions, efficiently delivered DOX to the cell nuclei, and displayed higher cytotoxicity against HeLa cells and HepG2 cells than DOX/ILPNPs. Endocytosis inhibition results indicated that both DOX/SLPNPs and DOX/ILPNPs entered cells mainly through the clathrin-mediated endocytosis pathway, and the different behaviors between DOX/SLPNPs and DOX/ILPNPs may have occurred after the LPNPs were internalized into the cells. Further in vivo biological evaluation confirmed that the introduction of reduction response ability in LPNPs can significantly enhance tumor cell growth inhibition ability, increase tumor cell apoptosis and improve in vivo antitumor efficacy. Combining the simple composition, easy preparation process, excellent antitumor efficacy and low side effects in vivo, two-component reduction-sensitive SLPNPs are a promising responsive nano drug delivery system for cancer therapy.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51473127, 51273150) and Opening Project of Key Laboratory of Biomedical Polymers of Ministry of Education at Wuhan University (20150103).
References
- A. Wicki, D. Witzigmann, V. Balasubramanian and J. Huwyler, J. Controlled Release, 2015, 200, 138–157 CrossRef CAS PubMed.
- M. Elsabahy, G. S. Heo, S. M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS PubMed.
- S. Eetezadi, S. Ekdawi and C. Allen, Adv. Drug Delivery Rev., 2015, 91, 7–22 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- L. F. Zhang, J. M. Chan, F. X. Gu, J. W. Rhee, A. Z. Wang, A. F. Radovic-Moreno, F. Alexis, R. Langer and O. C. Farokhzad, ACS Nano, 2008, 2, 1696–1702 CrossRef CAS PubMed.
- R. H. Fang, S. Aryal, C. M. J. Hu and L. F. Zhang, Langmuir, 2010, 26, 16958–16962 CrossRef CAS PubMed.
- R. H. Fang, K. N. H. Chen, S. Aryal, C. M. J. Hu, K. Zhang and L. F. Zhang, Langmuir, 2012, 28, 13824–13829 CrossRef CAS PubMed.
- S. Krishnamurthy, R. Vaiyapuri, L. F. Zhang and J. M. Chan, Biomater. Sci., 2015, 3, 923–936 RSC.
- J. M. Chan, L. F. Zhang, K. P. Yuet, G. Liao, J. W. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS PubMed.
- J. M. Chan, J. W. Rhee, C. L. Drum, R. T. Bronson, G. Golomb, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19347–19352 CrossRef CAS PubMed.
- L. H. Zhang, D. W. Zhu, X. Dong, H. F. Sun, C. X. Song, C. Wang and D. L. Kong, Int. J. Nanomed., 2015, 10, 2101–2114 CAS.
- M. B. Zheng, P. Gong, C. F. Zheng, P. F. Zhao, Z. Y. Luo, Y. F. Ma and L. T. Cai, J. Nanosci. Nanotechnol., 2015, 15, 4792–4798 CrossRef CAS PubMed.
- C. M. J. Hu, S. Kaushal, H. S. T. Cao, S. Aryal, M. Sartor, S. Esener, M. Bouvet and L. F. Zhang, Mol. Pharm., 2010, 7, 914–920 CrossRef CAS PubMed.
- J. M. Zhang, J. Hu, H. F. Cha, M. Skibba, G. Liang and M. W. Chen, Nanomedicine, 2016, 12, 1303–1311 CAS.
- Y. Zhang, P. Yu, L. Liu, B. Wu, C. Cui, M. Wu, L. J. Zhang, R. X. Zhuo and S. W. Huang, J. Controlled Release, 2015, 213, e128–e129 CrossRef PubMed.
- P. F. Zhao, M. B. Zheng, C. X. Yue, Z. Y. Luo, P. Gong, G. H. Gao, Z. H. Sheng, C. F. Zheng and L. T. Cai, Biomaterials, 2014, 35, 6037–6046 CrossRef CAS PubMed.
- M. B. Zheng, C. F. Yue, Y. F. Ma, P. Gong, P. F. Zhao, C. F. Zheng, Z. H. Sheng, P. F. Zhang, Z. H. Wang and L. T. Cai, ACS Nano, 2013, 7, 2056–2067 CrossRef CAS PubMed.
- M. B. Zheng, P. F. Zhao, Z. Y. Luo, P. Gong and L. T. Cai, Nanomedicine, 2016, 12, 515–515 Search PubMed.
- R. Mo and Z. Gu, Mater. Today, 2016, 19, 274–283 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- B. Deng, P. Ma and Y. Xie, Nanoscale, 2015, 7, 12773–12795 RSC.
- R. Cheng, F. H. Meng, C. Deng and Z. Y. Zhong, Nano Today, 2015, 10, 656–670 CrossRef CAS.
- X. B. Zhao and P. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 166–174 CAS.
- Y. C. Wang, F. Wang, T. M. Sun and J. Wang, Bioconjugate Chem., 2011, 22, 1939–1945 CrossRef CAS PubMed.
- F. Q. Chen, J. M. Zhang, Y. He, X. F. Fang, Y. T. Wang and M. W. Chen, Biomater. Sci., 2016, 4, 167–182 RSC.
- Y. C. Ma, J. X. Wang, W. Tao, C. Y. Sun, Y. C. Wang, D. D. Li, F. Fan, L. S. Qian and X. Z. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 26315–26325 CAS.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, L. Liu, R. X. Zhuo and S. W. Huang, Macromol. Biosci., 2013, 13, 1036–1047 CrossRef CAS PubMed.
- S. X. Li, L. Liu, L. J. Zhang, B. Wu, C. X. Wang, W. Zhou, R. X. Zhuo and S. W. Huang, Polym. Chem., 2016, 7, 5113–5122 RSC.
- C. Cui, P. Yu, M. Wu, Y. Zhang, L. Liu, B. Wu, C. X. Wang, R. X. Zhuo and S. W. Huang, Colloids Surf., B, 2015, 129, 137–145 CrossRef CAS PubMed.
- H. J. Han, H. B. Wang, Y. J. Chen, Z. H. Li, Y. Wang, Q. Jin and J. Ji, Nanoscale, 2016, 8, 283–291 RSC.
- Y. N. Xue, M. Liu, L. Peng, S. W. Huang and R. X. Zhuo, Macromol. Biosci., 2010, 10, 404–414 CrossRef CAS PubMed.
- S. Chen, L. Rong, H. Z. Jia, S. Y. Qin, X. Zeng, R. X. Zhuo and X. Z. Zhang, Biomater. Sci., 2015, 3, 753–763 RSC.
- T. T. Jing, L. Y. Fu, L. Liu and L. F. Yan, Polym. Chem., 2016, 7, 951–957 RSC.
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. F. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, L. Liu, R. X. Zhuo and S. W. Huang, Biomaterials, 2013, 34, 3858–3869 CrossRef CAS PubMed.
- C. Cui, Y. N. Xue, M. Wu, Y. Zhang, P. Yu, R. X. Zhuo and S. W. Huang, J. Controlled Release, 2013, 172, e17–e17 CrossRef CAS.
- B. Wu, P. Yu, C. Cui, M. Wu, Y. Zhang, L. Liu, C. X. Wang, R. X. Zhuo and S. W. Huang, Biomater. Sci., 2015, 3, 655–664 RSC.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.