
Electrospun poly(lactic acid) fibers containing novel chlorhexidine particles with sustained antibacterial activity†
Dong
Luo
*a,
Xi
Zhang
a,
Saroash
Shahid
b,
Michael J.
Cattell
b,
David J.
Gould
c and
Gleb B.
Sukhorukov
a
aSchool of Engineering and Materials Science, Queen Mary University of London, E1 4NS, UK. E-mail: d.luo@qmul.ac.uk
bBarts and The London School of Medicine and Dentistry, Queen Mary University of London, E1 2AD, UK
cWilliam Harvey Research Institute, Queen Mary University of London, EC1M 6BQ, UK
First published on 25th November 2016
Abstract
The treatment of persistent infections often requires a high local drug concentration and sustained release of antimicrobial agents. This paper proposes the use of novel electrospinning of poly(lactic acid) (PLA) fibers containing uncoated and encapsulated chlorhexidine particles. Chlorhexidine particles with a mean (SD) diameter of 17.15 ± 1.99 μm were fabricated by the precipitation of chlorhexidine diacetate with calcium chloride. Layer-by-layer (LbL) encapsulation of the chlorhexidine particles was carried out to produce encapsulated particles. The chlorhexidine particles had a high chlorhexidine content (90%), and when they were electrospun into PLA fibers a bead-in-string structure was obtained. The chlorhexidine content in the fibers could be tuned and a sustained release over 650 h was produced, via chlorhexidine particle encapsulation. Chlorhexidine release was governed by the polyelectrolyte multilayer encapsulation as demonstrated by SEM and confocal imaging. The incorporation of uncoated and encapsulated chlorhexidine particles (0.5% and 1% wt/wt chlorhexidine) into the fibers did not cause toxicity to healthy fibroblasts or affect cell adhesion to the fibers over a period of 5 days. The chlorhexidine-containing fibers also demonstrated sustained antibacterial activity against E. coli via an agar diffusion assay and broth transfer assay. Therefore, the chlorhexidine-containing PLA fibers may be useful in the treatment of persistent infections in medicine and dentistry.
Introduction
Local and sustained delivery of antibacterial agents is a novel concept for the treatment of recurrent and chronic infections, as an increased drug concentration at the site of infection could reduce the need for systemic drug administration, and avoid side effects.1,2 Over-use of antibiotics may also potentially lead to bacterial resistance,1 so other strategies are required. A high local drug concentration is essential to eliminate bacteria initially and a sustained drug release in the long term prevents secondary infection.3 A lack of this can lead to the formation of biofilms which are often more drug-resistant.4,5 Therefore, many delivery systems, such as microparticles, nanogels, foams, films, and nanofibers, have been developed to deliver antibacterial agents,6–9 among which the nanofibers are advantageous for drug delivery because of their small diameter, high specific surface area, and surface functionalities.9,10 PLA is particularly attractive to produce fibers due to its good mechanical strength, excellent biocompatibility, and biodegradability and it is currently used in biomedical applications.11The fibers are commonly produced by electrospinning and there are various approaches to load the fibers with antibacterial agents. The most straightforward method is directly mixing the drug in the electrospinning solution. The antimicrobial effect of the fibers made using this approach is not durable, as the release cannot be controlled,12,13 although core-sheath fibers fabricated by coaxial electrospinning are reported to be able to prolong the course of drug release.14 Chemical binding of antibacterial agents to fibers can however improve the controllability of release. For instance, cyclodextrins were utilized in fibers to complex with chlorhexidine digluconate,15 and chitosan fibers were functionalized with thiol groups to covalently bind with gentamicin-loaded liposomes.9 Drug carriers, such as mesoporous silica nanoparticles,16 Ca-alginate microspheres,17 chitosan nanoparticles,18 and clay nanotubes,19 were combined with electrospinning fibers to facilitate controllable drug release.20 The drug loading ratio is however always a concern with these methods, as it is vital to achieve a high local drug concentration to avoid re-application of the treatment. For example, in the treatment of periodontal pockets, fibers, nanocapsules, gels or chips placed in the pockets must release the drug effectively for 2 weeks.15 Therefore, if drug particles/crystals could be directly incorporated into electrospun fibers, the local drug concentration could be increased and the release profile tuned by fiber thickness or drug particle encapsulation.
Chlorhexidine is a broad spectrum antimicrobial agent widely used in dentistry and for other antibacterial purposes.21 It is not likely to crystallize,22 but due to the coordination capability of the biguanide structure the molecules tend to form complexes with metal ions.23 Thus this property was utilized to synthesize other formulations of chlorhexidine compounds.24,25 It was reported that chlorhexidine nanoparticles could be synthesized by instantaneous reaction between chlorhexidine digluconate and sodium hexametaphosphate. The nanoparticles produced were however not homogenous, and the chlorhexidine nanoparticles could rapidly adhere onto an elastomeric wound dressing which enabled long-term release of chlorhexidine.26 More interestingly, in the authors’ recent work, a novel formulation of homogeneous chlorhexidine particles was produced by precipitation of chlorhexidine diacetate and calcium chloride, the diameter of which could be tuned by adjusting the reaction temperature.27 Furthermore, these particles could be encapsulated using layer-by-layer (LbL) self-assembly to form microcapsules.28,29 The LbL self-assembly technique is extensively used for drug delivery and there are many reports which directly used drug crystals for LbL encapsulation.30,31 The release profile of drugs from these capsules could be tuned by the shell architectures.32 In previous studies, chlorhexidine was incorporated into electrospun fibers by diffusion into the fiber mesh, or by directly electrospinning chlorhexidine and polymer mixtures.33,34 These fibers however resulted in the rapid exhaustion of chlorhexidine within two days,33 a lack of sustained long-term effect, and the increased risk of toxicity. Therefore, combining the LbL encapsulated chlorhexidine particles with electrospun fibers may be a promising approach to overcome these problems.
In this work, we developed a novel electrospun PLA fiber with antibacterial properties by combining uncoated or encapsulated chlorhexidine particles into the fibers. Chlorhexidine particles were synthesized by precipitation of chlorhexidine diacetate with CaCl2, and the particles were further encapsulated using polyelectrolytes. The uncoated and encapsulated chlorhexidine particles were freeze-dried and incorporated into PLA fibers by electrospinning. Chlorhexidine release was carried out in different media over 650 hours and the fibers were examined before and after release by SEM and confocal microscopy. Their biocompatibility was evaluated using a MTT assay and by a fibroblast adhesion study. The antibacterial properties were tested using an agar diffusion assay and broth transfer experiment.
Materials and methods
Materials
Chlorhexidine diacetate (C6143, Lot 19H0417), poly(allylamine hydrochloride) (PAH, 56 kDa, 283223, Lot MKBJ4274V), poly(sodium 4-styrenesulfonate) (PSS, 70 kDa, 243051, Lot BCBF6120V), Rhodamine B (RhB, 283924, Lot 063K3407), fluorescein isothiocyanate isomer I (FITC, Lot 020M5305), calcium chloride (C8106, Lot SLBF7416 V), phosphate buffered saline (PBS, Lot RNBD7772), DMEM (41966, Lot 1513243), and thiazolyl blue tetrazolium bromide (MTT, M5655, Lot 052K5328) were all purchased from Sigma-Aldrich and were used directly without further purification. Poly(lactic acid) (PLA, 2002D) was from Nature works. Agar (BP1423, Lot 127054), tryptone (BPE9726, Lot 21012), yeast extract (BCE800, Lot 4381720), and sodium chloride (S3160, Lot 1333838) were bought from Fisher Bio reagents.Chlorhexidine particle synthesis
Chlorhexidine particles were made by precipitation of 15 mg mL−1 chlorhexidine diacetate with 0.33 M CaCl2 in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume at room temperature, which was reported in our previous work.27 The mixtures were shaken for 1 min, then centrifuged at 2000 rpm for 1 min (Eppendorf centrifuge 5417C, Germany). The precipitates were then washed three times with corresponding concentrated CaCl2 solutions. For visualization of the chlorhexidine spheres using confocal microscopy, the particles were labelled by RhB. The yield of chlorhexidine compounds was characterized using scanning electron microscopy (SEM, FEI inspect-F, USA) with an accelerating voltage of 10 kV, working distance of 10 mm, and spot size of 3.5. Confocal microscopy (Leica TS confocal scanning system, Germany) was also used for characterization.
:1 by volume at room temperature, which was reported in our previous work.27 The mixtures were shaken for 1 min, then centrifuged at 2000 rpm for 1 min (Eppendorf centrifuge 5417C, Germany). The precipitates were then washed three times with corresponding concentrated CaCl2 solutions. For visualization of the chlorhexidine spheres using confocal microscopy, the particles were labelled by RhB. The yield of chlorhexidine compounds was characterized using scanning electron microscopy (SEM, FEI inspect-F, USA) with an accelerating voltage of 10 kV, working distance of 10 mm, and spot size of 3.5. Confocal microscopy (Leica TS confocal scanning system, Germany) was also used for characterization.
Chlorhexidine particle encapsulation
LbL self-assembly of PAH and PSS was carried out to encapsulate the chlorhexidine particles. Both the PAH and PSS solutions were 2 mg mL−1 with 0.33 M CaCl2 added to reduce the dissolution of chlorhexidine particles during LbL encapsulation. The PAH was added to the spherical chlorhexidine particles as the first layer and the mixture was shaken (Vortex-Genie 2, Germany) for 10 min. The mixture was then centrifuged (Eppendorf centrifuge 5417C, Germany) at 2000 rpm and washed three times with 0.33 M CaCl2 solution. The second PSS layer was assembled using the same procedure. After assembling six layers, the encapsulated chlorhexidine particles with the structure chlorhexidine/(PAH/PSS)3 was produced. The supernatant during each layer assembly and after washing was collected for UV measurement to determine the chlorhexidine encapsulation rate (Lambda 35, Perkin Elmer, USA). To allow the use of fluorescence imaging, one of the PAH layers was labelled with FITC and the chlorhexidine core was labelled with RhB. SEM and confocal microscopy were used to characterize the capsules. All the uncoated chlorhexidine particles and encapsulated chlorhexidine particles were freeze-dried (ScanVac Cool Safe Freeze Drying, Denmark) at −107 °C and 0.009 mbar for 1 day and the proportion of chlorhexidine in the powder was calculated.Electrospinning of PLA fibers
PLA fibers were fabricated by electrospinning at room temperature, with a working distance of 15 cm, pumping rate of 1 mL h−1, and a voltage of 18 kV. PLA was dissolved in a mixed solvent of chloroform and acetone (3:1 by volume) at 7%. All the chlorhexidine particles were added at 0.5, 1, and 5% (wt/wt) to the PLA and mixed using a Rotomix (ESPE RotoMix, USA). PLA fibers were collected on foil and characterized using SEM and their mean (SD) diameter was analysed using Nano Measure software (version 1.2). Confocal microscopy and FTIR (Bruker, Billerica, MA) were also used to confirm the presence of chlorhexidine.
Release kinetics of chlorhexidine from the fibers
The release of chlorhexidine from the fibers was carried out in H2O and buffer (PBS). Fibers containing 5% (wt/wt) chlorhexidine particles (both types) were collected from the foil and weighed (Salter ANDER-180A weighing scale, UK). They were divided into cuvettes, and each sample was 25 mg (n = 3). 2 mL of deionized H2O or PBS was added to each cuvette and the fibers were kept immersed at room temperature. At each time point (from 1 h to 650 h), fibers were transferred into fresh medium and the solutions were measured using UV-vis absorption (Lambda 35, Perkin Elmer, USA). The chlorhexidine released into the solutions was determined at 254 nm according to the established calibration.27 After 650 hours all the fibers were collected and characterized again with SEM and confocal microscopy.Cytotoxicity of the chlorhexidine-containing fibers
The cytotoxicity of the chlorhexidine-containing fibers to fibroblasts was determined using the MTT assay. Both fibers with 0.5, 1, and 5% (wt/wt) chlorhexidine particles, and PLA fibers without chlorhexidine, were rinsed with 70% ethanol and sterilized by exposing to UV for 2 h. The fibers were then immersed in culture medium at 1 mg mL−1 and were kept in an incubator for 24 h or 48 h. The fibers were next removed and the medium was used for cell culture. The 3T3 cells (from William Harvey Research Institute) were seeded in 96-well plates at 1 × 104 per well, and cultured with fresh medium for 24 h. The medium was then removed and replaced with the chlorhexidine fiber release medium, and cells were cultured for 5 days, after which the medium was removed and 20 μL of MTT solution was added to each well and cells were cultured for 4 h. Then 150 μL MTT solvent (4 mM HCl, 0.1% Nondet P-40 in isopropanol) was added to each well. The plate was read in a Multiskan Ascent Plate Reader (Thermo Fisher Scientific, UK) with the filter at 570 nm.Fibroblast adhesion to the fibers
Both fibers with 0.5, 1, and 5% (wt/wt) chlorhexidine particles were cut into a 0.8 cm × 1 cm mesh, and sterilized using the method described before. PLA fibers without chlorhexidine particles were used as a control. Prior to cell seeding, the fiber meshes were fixed at the bottom of a removable cell culture chamber (Lab-Tek® II, Chamber Slide™). An Engineered 3T3 cell (expressing EGFP)35 suspension was added to each well at 2 × 104 cells per well, and then cultured for 24 h. The fiber meshes were collected and rinsed with PBS to remove any of the dead or detached cells. Confocal microscopy images were acquired with a confocal microscope using a 63×/Oil DIC (WD = 0.19 mm) objective. All the fiber meshes were spread on glass slides and scanned in x–y–z mode. The interval between sections was set as 0.3 μm. All the images along the z position were stacked and three-dimensionally displayed using Imaris software (Bitplane, version7.7).Antibacterial assay
The antibacterial properties of the chlorhexidine particle-loaded electrospun PLA fibers were tested by growth inhibition of Escherichia coli (E. coli, DH5a), using both an agar diffusion assay and broth transfer assay. The LB broth base solution was prepared (with 0.5 g NaCl, 10 g tryptone, and 5 g yeast extract per liter). To make the LB agar plates, agar was added to the LB broth base solution at 15 g L−1. Bacterial suspension in LB broth base solution was cultured at 37 °C/200 rpm shaking, and the density of the suspensions was adjusted to that of a McFarland 0.5 turbidity, which corresponded to 1.5 × 108 cells per mL using a spectrophotometer (Cecil CE2021, USA) at 625 nm. 0.4 mL of the bacterial suspension was spread on the surface of the LB agar plates. The sensitivity of E. coli to inhibition with chlorhexidine was firstly demonstrated using filters (diameter 7 mm) treated with uncoated or encapsulated chlorhexidine particles with chlorhexidine concentrations from 5 mg mL−1 to 50 μg mL−1 and placed on agar plates pre-spread with E. coli. The fibers were cut into discs (diameter 7 mm, thickness 0.1 mm) and rinsed with 70% ethanol and H2O, and then placed on LB agar plates which were incubated at 37 °C for 24 h and then the diameter (SD) (n = 3) of the zones of inhibition was measured in mm.The sustained antibacterial effect of the chlorhexidine-containing fibers was also examined in a transfer experiment. Bacterial suspensions in LB broth base with McFarland 0.5 turbidity were diluted 200 times. Each of the fiber discs (fibers with uncoated or encapsulated chlorhexidine particles containing 0.5, 1, and 5% (wt/wt) chlorhexidine, n = 3) were immersed into the bacterial suspensions (1 mL) and cultured at 37 °C/200 rpm shaking. After each hour, the fiber discs were transferred into fresh bacterial suspensions. After 9 hours, all the fiber discs were discarded and all the bacterial suspensions were incubated for another 24 hours. The optical absorptions were measured at 625 nm (Cecil CE2021, USA).
Statistical methods
The Student's t test (Microsoft Excel 2016 software) was used to analyze statistically significant differences between groups. Significance refers to p values less than 0.05 and 0.01.Results
Chlorhexidine particles and LbL encapsulation
Chlorhexidine particles were synthesized by precipitation. To get homogeneous spherical particles the concentration of both chlorhexidine diacetate and CaCl2 was tuned, as described in our previous study.27 As shown in Fig. 1a the synthesized chlorhexidine particles had an interconnected structure with a porous surface morphology. The spheres had an average diameter (SD) of 17.15 (1.99) μm (Fig. S1, ESI†). Their inner structure could be seen by RhB labelling as shown in Fig. 1c, and a radiating dendritic structure was identified. When encapsulated with polyelectrolytes the porous interconnected morphology could not be seen, and flake shells were present on the surface (Fig. 1b). The LbL encapsulation was further confirmed by confocal images and the FITC-labelled shell appeared as green/yellow circles surrounding the chlorhexidine spheres (Fig. 1d) (uncoated and encapsulated chlorhexidine particles at low magnification are shown in Fig. S1, ESI†). In addition, the chlorhexidine spheres were still intact after encapsulation, indicating that minimum dissolution of chlorhexidine occurred as the encapsulation efficiency was 97%. The chlorhexidine content in the uncoated and encapsulated chlorhexidine particles was calculated to be 90% and 46% respectively, according to the remaining chlorhexidine in the supernatant and weight of the freeze-dried powder.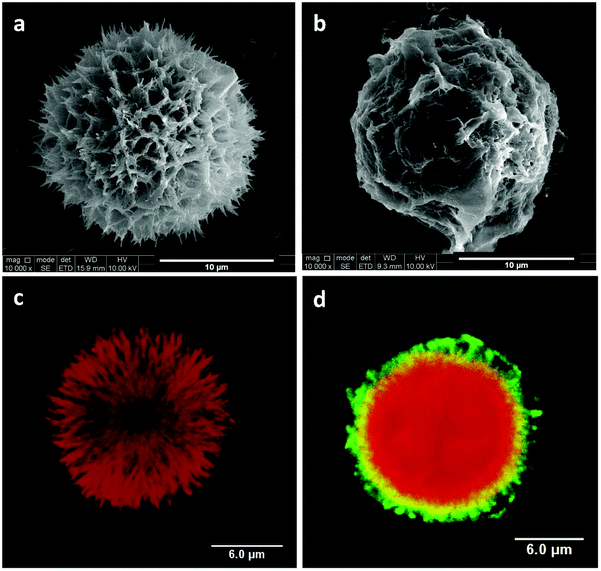 | ||
| Fig. 1 Chlorhexidine particles. SEM images of (a) chlorhexidine particle without coating and (b) encapsulated chlorhexidine particle with PAH/PSS layers. Confocal images of (c) chlorhexidine particle labelled with RhB and (d) encapsulated chlorhexidine particle with one of the PAH layers labelled with FITC. | ||
Electrospun PLA fibers with chlorhexidine particles
Chlorhexidine was incorporated into the PLA fibers by electrospinning. The SEM images of the fabricated fibers (Fig. 2) indicate a regular fibrous structure. When the chlorhexidine particles were incorporated (5%), a bead-in-string structure was demonstrated, in which the spheres were connected by two ends, with one fiber end much thicker than the other. For the fibers with chlorhexidine particles, a very thin layer of PLA covered the particle surface and the interconnected structure could still be seen (Fig. 2b). In contrast, when the chlorhexidine particles were encapsulated and electrospun into fibers, a rougher appearance was observed (Fig. 2c). The mean diameter (SD) of the PLA fibers (control) was 1.35 ± 0.06 μm. When the content of the chlorhexidine was increased (from 0.5% to 25%) the diameter of the fibers decreased, as demonstrated in Fig. 3. There was no significant difference when the chlorhexidine content was below 10%. On further increasing the ratio to 25% (wt/wt), the fiber diameter significantly (p < 0.01) reduced to 0.96 ± 0.06 μm (SEM images of fibers containing different amounts of chlorhexidine particles and their diameter distributions are shown in Fig. S2 and S3, ESI†). After incorporating CHX, the mechanical properties of the PLA electrospun fiber mat were also decreased (Table S1, ESI†). Meanwhile, the contact angle test showed that the wettability of the fibers was not affected by particle incorporation (123.6 ± 3.7° for control and 121.6 ± 7.2° for 5.0 wt/wt CHX fibers). The uncoated and encapsulated chlorhexidine particles within the fibers could be clearly seen in the confocal images (Fig. 4). The black shadow around the chlorhexidine particles in the merged images indicates the PLA layer (Fig. 4a2). The chlorhexidine particles were still intact after electrospinning and the FTIR spectra confirmed the presence of chlorhexidine in the fibers (Fig. S4, ESI†). | ||
| Fig. 2 SEM images of electrospun fibers containing 5% chlorhexidine: (a) PLA fibers as a control; (b) fibers with uncoated chlorhexidine particles; and (c) fibers with encapsulated chlorhexidine particles, at a magnification of (1) 1000× and (2) 4000×. | ||
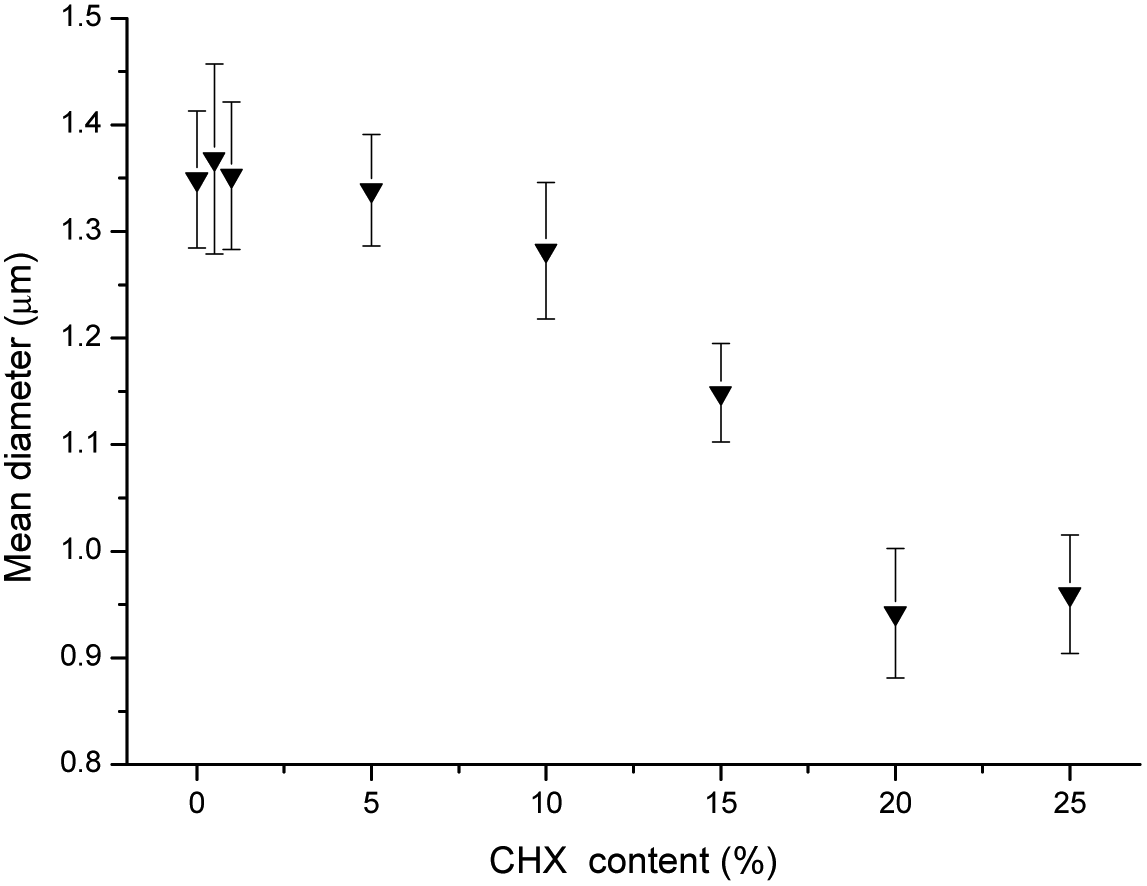 | ||
| Fig. 3 Mean (SD) diameter of the PLA fibers as a function of chlorhexidine particle content. | ||
 | ||
| Fig. 4 Confocal images of (a) PLA fibers with uncoated chlorhexidine particles and (b) fibers with encapsulated chlorhexidine particles, (1) in the red channel and (2) merged images with the transmitted channel. Chlorhexidine particles were labelled with RhB and one of the PAH layers was labelled with FITC. The yellow colour indicates the overlap of the core and shell. | ||
Release kinetics of chlorhexidine
The release profile of chlorhexidine from the PLA fibers containing chlorhexidine particles was monitored over 650 hours (Fig. 5). In H2O the fibers with uncoated chlorhexidine particles showed a burst release during the first day, with over 60% of the chlorhexidine released, after which a sustained release was observed. In contrast, the fibers with encapsulated particles displayed a lower (20–25%) sustained release of chlorhexidine. In PBS, both of the fibers had much lower release rates compared to those in H2O, and chlorhexidine encapsulation again produced a lower but sustained release rate (Fig. 5b).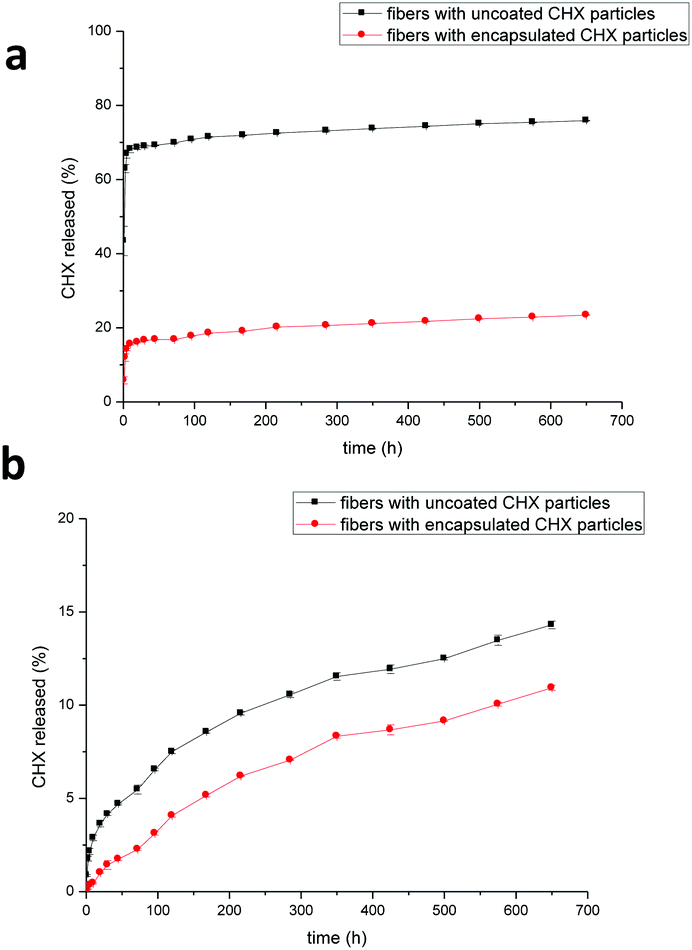 | ||
| Fig. 5 Release kinetics of chlorhexidine particle-containing fibers in (a) H2O and (b) PBS. The PLA fibers with uncoated and encapsulated chlorhexidine particles had a chlorhexidine content of 5% (wt/wt). | ||
The morphology of the fibers after the release test in H2O is shown in Fig. 6a–f. For the fibers containing chlorhexidine particles, most of the chlorhexidine was released and collapsed spheres were observed (Fig. 6a). The porous surface of the collapsed sphere revealed the original morphology, indicating that the PLA layer had been penetrated. Fibers containing encapsulated chlorhexidine particles retained more of their surface coating and chlorhexidine content in the collapsed spheres (Fig. 6d). These observations were confirmed by confocal imaging, as in the fibers with uncoated chlorhexidine particles virtually no chlorhexidine was identified (little signal was detected in the red channel), and voids could be seen in the transmitted channel (Fig. 6b and c). However, for the fibers with encapsulated particles some chlorhexidine remained inside the fiber, although small voids could be seen (Fig. 6e and f). As for fibers release in PBS, most of the chlorhexidine remained in both of the fibers and the corresponding SEM images and confocal images are shown (Fig. S5, ESI†).
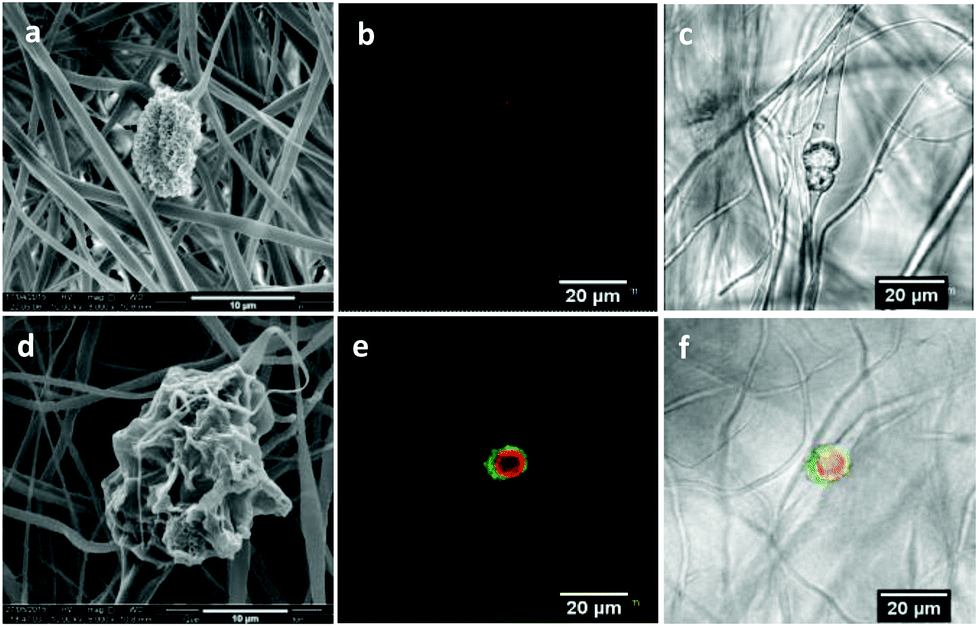 | ||
| Fig. 6 SEM and confocal images of electrospun fibers after the 650 h release test: SEM images of fibers with uncoated (a) and encapsulated (d) chlorhexidine particles; and confocal images of fibers containing uncoated (b) and encapsulated (e) chlorhexidine particles in the red channel, and uncoated (c) and encapsulated (f) chlorhexidine particles in the merged transmitted channel. | ||
Cytotoxicity of fibers and cell adhesion
The cytotoxicity of the fibers (containing particles) to the fibroblasts is shown in Fig. 7. The fibers containing uncoated and encapsulated chlorhexidine particles with 0.5% and 1% (wt/wt) chlorhexidine were not toxic to cells over a period of 5 days, as there was no statistical difference (p > 0.05) in cell viability between the PLA control and the two test groups. When the chlorhexidine content in the fibers was increased to 5% (wt/wt), the fibers containing the uncoated chlorhexidine particles reduced the cell viability significantly over 1 day (p < 0.05) and more than 2 days (p < 0.01). Meanwhile, the fibers with encapsulated particles (5% chlorhexidine loading ratio) showed better compatibility with cells, with a slight cell viability reduction at day 2 and day 5 (p < 0.05).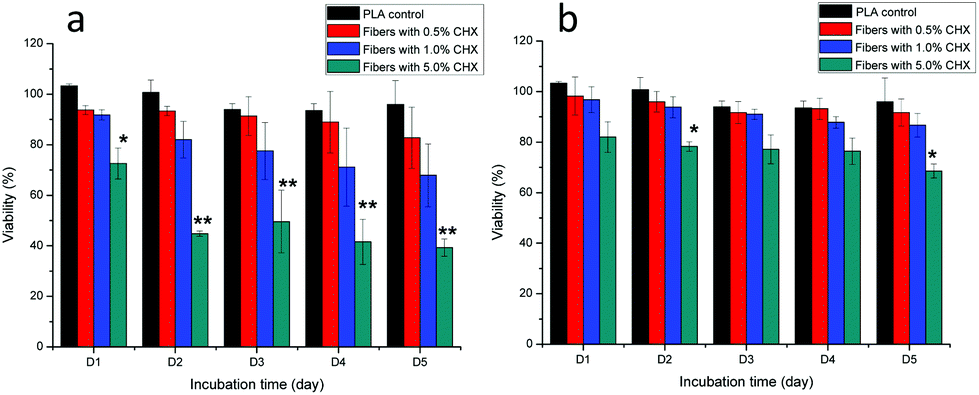 | ||
| Fig. 7 Cytotoxicity of fibers containing chlorhexidine with fibroblast cells (3T3). PLA fibers containing uncoated chlorhexidine particles (a) and encapsulated chlorhexidine particles (b). Values are the mean of 5 wells and vertical lines are SD, and *p < 0.05 and **p < 0.01 indicate statistical differences compared to the control PLA fiber treated cells. | ||
Cell adhesion to the fibers is another indicator of potential cytotoxicity, as demonstrated in Fig. 8. The number of fluorescent cells on the PLA fibers directly reflected the influence of the chlorhexidine content, with fewer cells on the 5% (wt/wt) chlorhexidine-containing fibers than on the control PLA fibers. The cells appeared less spread out on the fibers and more round shaped cells were observed, which may suggest toxicity at 5% (wt/wt) chlorhexidine content. Fibers with less chlorhexidine (0.5 and 1% wt/wt) in the fibers encouraged cell adherence to the fibers, with typically elongated cell filopodia extensions.19 A similar observation was made for fibers with encapsulated chlorhexidine particles (Fig. S6, ESI†).
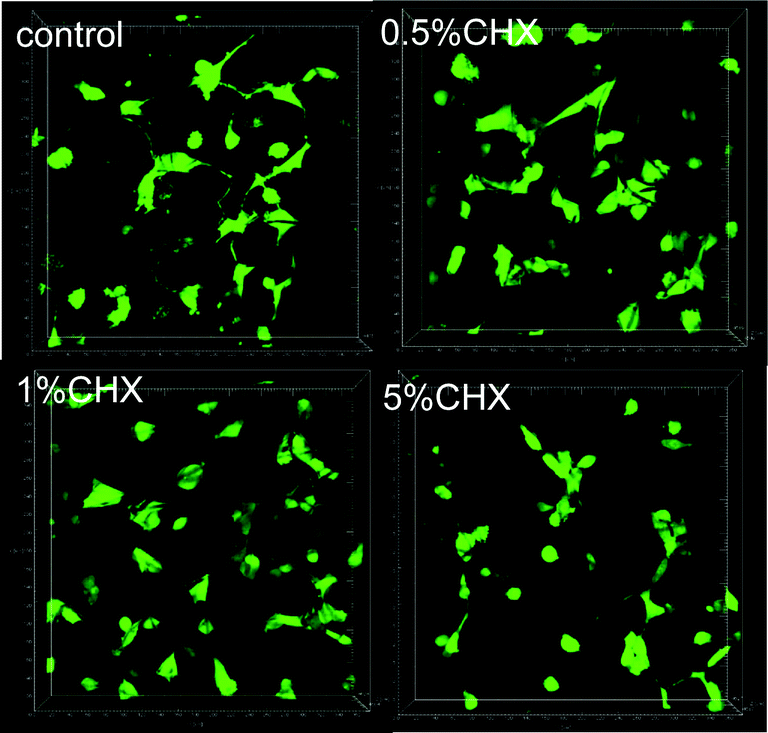 | ||
| Fig. 8 3D images of fibroblast adhesion on PLA fibers containing uncoated chlorhexidine particles with a chlorhexidine loading ratio of 0.5, 1, and 5% (wt/wt). | ||
Antibacterial activity
The antibacterial activity of the fibers against E. coli is presented in Fig. 9. The diameter of the clear zone is an indication of the inhibitory effect of the chlorhexidine-containing fibers, with a higher chlorhexidine ratio in the fibers leading to large inhibition zones. No inhibition against E. coli was observed for the control PLA discs. For fibers with chlorhexidine particles, large inhibition zones were observed for all the fiber discs with the chlorhexidine ratio at 0.5, 1, and 5% (wt/wt), and higher chlorhexidine loading rates resulting in larger inhibition zones. In contrast, the inhibitory zones were only observed at 1 and 5% (wt/wt) for the fibers with encapsulated chlorhexidine particles, which resulted in much smaller inhibitory diameters (p < 0.01).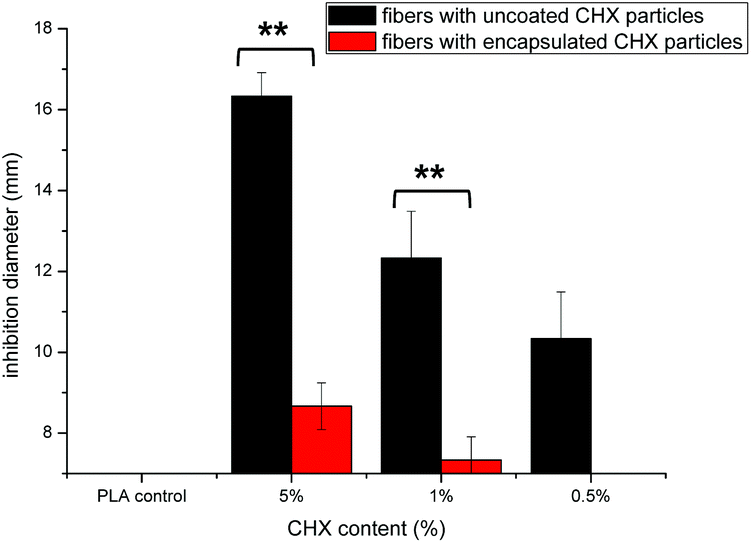 | ||
| Fig. 9 In vitro inhibition of chlorhexidine-containing fibers against E. coli using an agar diffusion assay: PLA fibers containing uncoated chlorhexidine particles (black), encapsulated chlorhexidine particles (red), and fibers without chlorhexidine used as a control. **p < 0.01 indicates statistical differences between the two types of fibers (n = 3). | ||
The sustained inhibition effect against E. coli was demonstrated by a transfer experiment (Fig. 10). The chlorhexidine burst release (uncoated particles) within one hour inhibited proliferation of E. coli in LB broth suspensions over the next 24 hours. For the control (untreated bacterial suspensions), the bacteria proliferated and a turbidity around 0.8 was found after 24 h (green line). However, 5% (wt/wt) chlorhexidine in both the fiber-containing uncoated and encapsulated chlorhexidine particles completely inhibited bacterial growth even after nine transfers (black line, Fig. 10a and b). When the chlorhexidine content in the fibers was reduced to 1% (wt/wt) (red line), no inhibition was observed for the fibers with encapsulated particles and the inhibition effect against E. coli depleted quickly after 4 transfers for fibers containing the uncoated chlorhexidine particles. Similar observations were displayed for the fibers with 0.5% (wt/wt) encapsulated chlorhexidine particles (blue line).
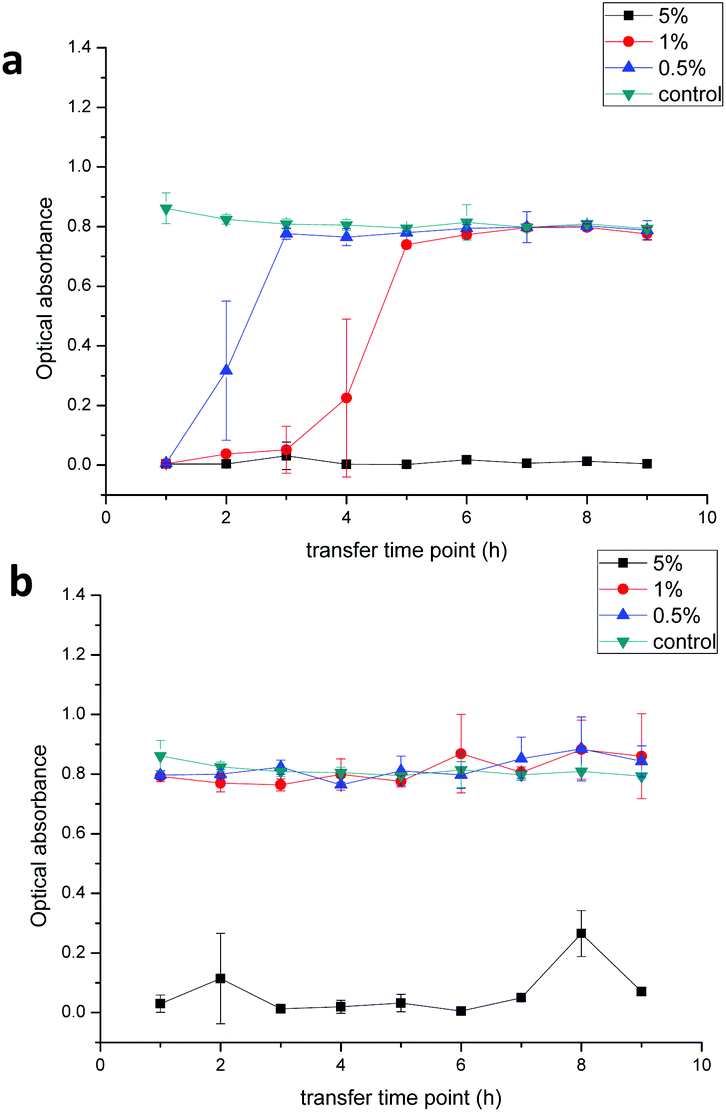 | ||
| Fig. 10 In vitro inhibition of chlorhexidine-containing PLA fibers against E. coli using a broth transfer assay. The chlorhexidine loading rate in fibers containing (a) uncoated chlorhexidine particles and (b) encapsulated chlorhexidine particles was 0.5% (blue), 1% (red), and 5% (black) (wt/wt). PLA fibers without chlorhexidine were used as a control (green) (n = 3). | ||
Discussion
Herein, we propose a strategy to produce electrospun PLA fibers with a high content of an antibacterial agent, by incorporating uncoated or encapsulated chlorhexidine particles into the fibers. Chlorhexidine has a low solubility in water and is commonly used in salts such as chlorhexidine digluconate and chlorhexidine diacetate,3 however these crystals are usually large and irregular. They can either be ground into fine powders,36 or like most of the other insoluble drugs can be sonicated to produce more homogeneous particles.37,38 The novel method used in the current study utilized the co-precipitation of chlorhexidine diacetate and CaCl2 to produce homogeneous spherical chlorhexidine particles. These spheres had a very high drug content (90%), which is advantageous when incorporating them into any drug delivery system, since traditional incorporation strategies of physically soaking or chemically bonding13,39 have a very low drug loading efficiency. The formation mechanism of the novel chlorhexidine particles was due to the remarkable coordination capability of the biguanide group of the chlorhexidine molecule with Ca2+ and the solubility reduction effect of Cl−, as described in previous work.27 The concentration of Ca2+ and Cl− in solution was vital to form the interconnected spherical structure, as less optimized ion concentrations in solution resulted in disordered precipitation. The diameter of the chlorhexidine particles could also be tuned via temperature during the synthesis.27 The properties of chlorhexidine molecules have also been utilized by other authors to produce chlorhexidine complexes or nanoparticles.24–26The uncoated chlorhexidine particles produced a rapid and high chlorhexidine release rate in H2O and PBS, so LbL encapsulation was utilized to tailor the sustained release rate (Fig. 5b). This type of polymer encapsulation has also been used for many other drugs, such as ibuprofen, curcumin, furosemide, and paclitaxel.40 The driving force for polyelectrolyte assembly on the drug crystals was electrostatic because usually the drug crystals are charged. The porous surface morphology of the chlorhexidine spheres allowed the polymers to be absorbed and to penetrate their surface, which is illustrated in the confocal images of the encapsulated particles (Fig. 1). The incorporation of particles in the fibers resulted in a typical bead-in-string structure.41 According to previous research, the embedding of microparticles results in defects and notches in the fibers, but the diameter of the fibers was increased and the mechanical properties were enhanced.41,42 The addition of chlorhexidine particles in PLA fibers in the current study did not lead to obvious defects in the fibers, but it did have a significant effect (when the CHX loading was over 15% wt/wt) on the fiber diameter as a narrowing effect was observed at the distal end, adjacent to the particle. The addition of microparticles (calcium carbonate and HA particles until 5% addition) into electrospun polymer solutions previously increased their viscosity, and thus led to an increase of fiber diameter.41,42 However, the average fiber diameter was dramatically reduced by adding the chlorhexidine particles, which may be associated with the size and volume fraction of the introduced particles, as the diameter of chlorhexidine particles was much larger (17.15 ± 1.99 μm) than the electrospun fibers.20 Significant amounts of PLA were required to coat and penetrate these larger particles which may have drawn the fibers thinner. The shear force generated during electrospinning may be another explanation, which is strong enough to lead to the deformation of spherical particles into ellipsoidal particles.43 Thus the proximal side of the fiber was always thicker than the distal side. These characteristics of the fibers also led to a mechanical property decrease (Table S1, ESI†). As for the wettability of fibers, it is assumed that all the chlorhexidine particles were covered by a thin layer of PLA, so the hydrophilicity of the fiber mesh was not affected. The chlorhexidine was also concentrated in the particles instead of distributed freely all over the fibers, which may minimize the effect on the wettability of the fibers.
The encapsulation of chlorhexidine spheres by PLA or polyelectrolytes had a fundamental influence on the chlorhexidine release kinetics in H2O, although both kinds of fibers displayed a burst release at the beginning and sustained release afterwards. In order to avoid dissolution of chlorhexidine crystals, the electrospun solutions were freshly prepared, and the fibers were immediately spun. The dissolved chlorhexidine in the fibers may have caused the burst release at the beginning of the release experiment in water. Comparing the difference in the fibers containing uncoated and encapsulated chlorhexidine particles, it is clear that with polyelectrolyte shells it was harder for chlorhexidine to diffuse out. Mixing metronidazole with the polymer solutions and electrospinning gave over 90% metronidazole release from the fibers within the first two days. However, by pre-loading metronidazole in halloysite clay nanotubes, a sustained release was achieved.19 Various carriers such as mesoporous silica or ZnO nanoparticles have been employed to load drugs before electrospinning into fibers to achieve sustained release.16,44,45 In this paper electrospinning the LbL encapsulated drug crystals demonstrated another approach, as the LbL shells were already proven to be effective to reduce drug dissolution and diffusion,38,46 and their drug loading rate is much higher than that of other carriers. Moreover, the release can be further tuned by adjusting the number of deposited polyelectrolyte layers and the type of polyelectrolytes.32
The cytotoxicity of chlorhexidine to cells is dose- and time-dependent, and it was reported that a chlorhexidine concentration over 1 μg mL−1 was toxic to 3T3 cells.39,47 The chlorhexidine content in the fibers with 5% (wt/wt) encapsulated particles was 50 μg mL−1, and they showed no cytotoxicity after 24 hours. Although the cell viability slightly decreased over the 5 days, it still showed much better compatibility over fibers with uncoated particles. We can assume that by chlorhexidine particle encapsulation, the release kinetics is better governed, which ensures a safer performance. Previous work reported that drug crystals encapsulated by LbL shells could reduce toxicity and side effects,31,48 and this is in agreement with the current study. Adherence of cells to the fibers was also evidence of good biocompatibility, as cells could adhere, bridge, and communicate with the neighbouring cells on the fiber membranes, as revealed with SEM and confocal 3D imaging.19,49 Therefore, the PLA fibers produced here are advantageous to ensure a safe performance.
Substantial inhibition of the proliferation of bacteria was demonstrated by the agar diffusion inhibition assay. Different bacterial strains have different sensitivities to chlorhexidine concentration, and the minimum inhibitory concentration (MIC) for E. coli is 2–8 μg mL−1.13 At lower concentrations, chlorhexidine only interferes with the metabolism of the bacteria.50 As demonstrated, fibers with 0.5% (wt/wt) chlorhexidine particles still indicated inhibition against E. coli, and the inhibition diameters were substantially affected by the diffusion manner of chlorhexidine in agar. For the fibers containing encapsulated chlorhexidine particles, the dissolution and diffusion of chlorhexidine was effectively hindered, and thus the inhibitory zone of bacterial growth was significantly smaller than that with the fibers with chlorhexidine particles. Similarly, the sensitivity against E. coli was reduced, as proven by comparing the uncoated and encapsulated chlorhexidine particles at equivalent doses (Fig. S7, ESI†). This was consistent with the LB broth transfer results that indicated a sustained inhibitory effect against E. coli for the fibers containing encapsulated chlorhexidine particles at a high chlorhexidine loading ratio (5% wt/wt). However, for 0.5% or 1% wt/wt chlorhexidine concentrations, no inhibition against E. coli was observed within one hour transfers (Fig. 10b). Bacterial inhibition may be therefore based on contact between chlorhexidine and the bacterial cell membrane, with complete isolation of chlorhexidine particles through encapsulation reducing the chance of direct contact between chlorhexidine particles and E. coli, regardless of whether it was dissolved or not. For localized treatment of infections, a sustained release of antibacterial agents is preferred, and thus controlled release by chlorhexidine encapsulation could be beneficial. More interestingly, a combination of uncoated chlorhexidine particles and encapsulated chlorhexidine particles in the same fibers may lead to a burst release at the beginning and more sustained release in the long term, which is attractive for the treatment of bacterial infections.4 However, to validate the performance of chlorhexidine PLA fibers in our study, further in vivo testing is needed.
Conclusions
In summary, novel uncoated and encapsulated chlorhexidine particles electrospun in PLA fibers with sustained antibacterial properties have been demonstrated in this work. The incorporation of chlorhexidine particles into fibers greatly improved the drug loading ratio, and more sustained release was achieved by pre-encapsulating chlorhexidine particles with polyelectrolyte multilayers. Both fibers showed no cytotoxicity to fibroblasts at a low chlorhexidine loading rate (from 0.5% to 1% wt/wt), and by encapsulating the particles, the biocompatibility was significantly improved at 5% (wt/wt). Similar results were illustrated by 3D imaging of cell adhesion on the fibers. Both fibers showed good inhibition against E. coli growth in the agar diffusion assay and sustained antibacterial effect via broth transfer experiments (5% wt/wt). Therefore, the developed PLA fibers containing chlorhexidine particles may have promising applications for the localized treatment of infections. The encapsulated particles in the fibers should also allow for tailored drug release, which might be matched to the disease stages, to allow more effective and responsive treatment.Acknowledgements
The authors thank Mr Russell Bailey of Nanovision (QMUL) for help with SEM, and Dr Dongsheng Wu for assistance in CLSM. Dong also thanks the financial support from China Scholarship Council for his Ph.D. study.References
- T. S. Kashi, S. Eskandarion, M. Esfandyari-Manesh, S. M. Marashi, N. Samadi, S. M. Fatemi, F. Atyabi, S. Eshraghi and R. Dinarvand, Int. J. Nanomed., 2012, 7, 221–234 Search PubMed.
- J. Xue, M. He, H. Liu, Y. Niu, A. Crawford, P. D. Coates, D. Chen, R. Shi and L. Zhang, Biomaterials, 2014, 35, 9395–9405 CrossRef CAS PubMed.
- P. Zeng, G. Zhang, A. Rao, W. Bowles and T. S. Wiedmann, Int. J. Pharm., 2009, 367, 73–78 CrossRef CAS PubMed.
- B. Duncan, X. Li, R. F. Landis, S. T. Kim, A. Gupta, L. S. Wang, R. Ramanathan, R. Tang, J. A. Boerth and V. M. Rotello, ACS Nano, 2015, 9, 7775–7782 CrossRef CAS PubMed.
- Z. Wang, Y. Shen and M. Haapasalo, Dent. Mater., 2014, 30, e1–16 CrossRef CAS PubMed.
- I. C. Yue, J. Poff, M. E. Cortes, R. D. Sinisterra, C. B. Faris, P. Hildgen, R. Langer and V. P. Shastri, Biomaterials, 2004, 25, 3743–3750 CrossRef CAS PubMed.
- S. Maya, S. Indulekha, V. Sukhithasri, K. T. Smitha, S. V. Nair, R. Jayakumar and R. Biswas, Int. J. Biol. Macromol., 2012, 51, 392–399 CrossRef CAS PubMed.
- T. Phaechamud and J. Charoenteeraboon, AAPS PharmSciTech, 2008, 9, 829–835 CrossRef CAS PubMed.
- N. Monteiro, M. Martins, A. Martins, N. A. Fonseca, J. N. Moreira, R. L. Reis and N. M. Neves, Acta Biomater., 2015, 18, 196–205 CrossRef CAS PubMed.
- R. Sridhar, R. Lakshminarayanan, K. Madhaiyan, V. A. Barathi, K. Lim and S. Ramakrishna, Chem. Soc. Rev., 2015, 44, 790–814 RSC.
- E. Llorens, S. Calderon, L. J. Del Valle and J. Puiggali, Mater. Sci. Eng., C, 2015, 50, 74–84 CrossRef CAS PubMed.
- E. R. Kenawy, G. L. Bowlin, K. Mansfield, J. Layman, D. G. Simpson, E. H. Sanders and G. E. Wnek, J. Controlled Release, 2002, 81, 57–64 CrossRef CAS.
- L. Chen, L. Bromberg, T. A. Hatton and G. C. Rutledge, Polymer, 2008, 49, 1266–1275 CrossRef CAS.
- E. Jo, S. Lee, K. T. Kim, Y. S. Won, H.-S. Kim, E. C. Cho and U. Jeong, Adv. Mater., 2009, 21, 968–972 CrossRef CAS.
- N. Tabary, F. Chai, N. Blanchemain, C. Neut, L. Pauchet, S. Bertini, E. Delcourt-Debruyne, H. F. Hildebrand and B. Martel, Acta Biomater., 2014, 10, 318–329 CrossRef CAS PubMed.
- B. Song, C. Wu and J. Chang, Acta Biomater., 2012, 8, 1901–1907 CrossRef CAS PubMed.
- H. Qi, P. Hu, J. Xu and A. Wang, Biomacromolecules, 2006, 7, 2327–2330 CrossRef CAS PubMed.
- Y. Wang, B. Wang, W. Qiao and T. Yin, J. Pharm. Sci., 2010, 99, 4805–4811 CrossRef CAS PubMed.
- J. Xue, Y. Niu, M. Gong, R. Shi, D. Chen, L. Zhang and Y. Lvov, ACS Nano, 2015, 9, 1600–1612 CrossRef CAS PubMed.
- C. L. Zhang and S. H. Yu, Chem. Soc. Rev., 2014, 43, 4423–4448 RSC.
- C. Farrugia and J. Camilleri, Dent. Mater., 2015, 31, e89–e99 CrossRef CAS PubMed.
- N. Dupont, A. N. Lazar, F. Perret, O. Danylyuk, K. Suwinska, A. Navaza and A. W. Coleman, CrystEngComm, 2008, 10, 975 RSC.
- P. J. Bailey and S. Pace, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS.
- M. Călinescu, T. Negreanu-Pîrjol, R. Georgescu and O. Călinescu, Cent. Eur. J. Chem., 2010, 8, 543–549 Search PubMed.
- M. Badea, R. Olar, M. Iliş, R. Georgescu and M. Călinescu, J. Therm. Anal. Calorim., 2012, 111, 1763–1770 CrossRef.
- M. E. Barbour, S. E. Maddocks, N. J. Wood and A. M. Collins, Int. J. Nanomed., 2013, 8, 3507–3519 CrossRef PubMed.
- D. Luo, S. Shahid, R. M. Wilson, M. J. Cattell and G. B. Sukhorukov, ACS Appl. Mater. Interfaces, 2016, 8, 12652–12660 CAS.
- K. Ariga, Y. M. Lvov, K. Kawakami, Q. Ji and J. P. Hill, Adv. Drug Delivery Rev., 2011, 63, 762–771 CrossRef CAS PubMed.
- M. F. Bedard, B. G. De Geest, A. G. Skirtach, H. Mohwald and G. B. Sukhorukov, Adv. Colloid Interface Sci., 2010, 158, 2–14 CrossRef CAS PubMed.
- A. A. Antipov, G. B. Sukhorukov and E. Donath, J. Phys. Chem. B, 2001, 105, 2281–2284 CrossRef CAS.
- A. Agarwal, Y. Lvov, R. Sawant and V. Torchilin, J. Controlled Release, 2008, 128, 255–260 CrossRef CAS PubMed.
- H. Ai, S. A. Jones, M. M. de Villiers and Y. M. Lvov, J. Controlled Release, 2003, 86, 59–68 CrossRef CAS PubMed.
- J. Song, S. J. Remmers, J. Shao, E. Kolwijck, X. F. Walboomers, J. A. Jansen, S. C. Leeuwenburgh and F. Yang, Nanomedicine, 2016, 12, 1357–1364 CAS.
- J. G. Fernandes, D. M. Correia, G. Botelho, J. Padrão, F. Dourado, C. Ribeiro, S. Lanceros-Méndez and V. Sencadas, Polym. Test., 2014, 34, 64–71 CrossRef CAS.
- D. Gould, N. Yousaf, R. Fatah, M. C. Subang and Y. Chernajovsky, Arthritis Res. Ther., 2007, 9, R7 Search PubMed.
- L. Cheng, M. D. Weir, H. H. Xu, A. M. Kraigsley, N. J. Lin, S. Lin-Gibson and X. Zhou, Dent. Mater., 2012, 28, 573–583 CrossRef CAS PubMed.
- Z. Zheng, X. Zhang, D. Carbo, C. Clark, C. Nathan and Y. Lvov, Langmuir, 2010, 26, 7679–7681 CrossRef CAS PubMed.
- A. C. Santos, P. Pattekari, S. Jesus, F. Veiga, Y. Lvov and A. J. Ribeiro, ACS Appl. Mater. Interfaces, 2015, 7, 11972–11983 CAS.
- L. J. del Valle, M. Roa, A. Díaz, M. T. Casas, J. Puiggalí and A. Rodríguez-Galán, J. Polym. Res., 2012, 19, 1–13 CrossRef.
- L. J. De Cock, S. De Koker, B. G. De Geest, J. Grooten, C. Vervaet, J. P. Remon, G. B. Sukhorukov and M. N. Antipina, Angew. Chem., Int. Ed., 2010, 49, 6954–6973 CrossRef CAS PubMed.
- H. A. Metwally, R. V. Ardazishvili, A. N. Severyukhina, A. M. Zaharevich, A. A. Skaptsov, S. B. Venig, G. B. Sukhorukov and D. A. Gorin, J. Bionanosci., 2014, 5, 22–30 CrossRef.
- J. C. Sy, A. S. Klemm and V. P. Shastri, Adv. Mater., 2009, 21, 1814–1819 CrossRef CAS.
- C. Herrmann, A. Turshatov and D. Crespy, ACS Macro Lett., 2012, 1, 907–909 CrossRef CAS.
- K. Qiu, C. He, W. Feng, W. Wang, X. Zhou, Z. Yin, L. Chen, H. Wang and X. Mo, J. Mater. Chem. B, 2013, 1, 4601–4611 RSC.
- J. Wei, J. Hu, M. Li, Y. Chen and Y. Chen, RSC Adv., 2014, 4, 28011–28019 RSC.
- P. Pattekari, Z. Zheng, X. Zhang, T. Levchenko, V. Torchilin and Y. Lvov, Phys. Chem. Chem. Phys., 2011, 13, 9014–9019 RSC.
- N. Naghavi, J. Ghoddusi, H. R. Sadeghnia, E. Asadpour and S. Asgary, Dent. Mater., 2014, 33, 64–69 CrossRef CAS.
- J. Zhao, Y. Cui, A. Wang, J. Fei, Y. Yang and J. Li, Langmuir, 2011, 27, 1499–1504 CrossRef CAS PubMed.
- U. Stachewicz, T. Qiao, S. C. Rawlinson, F. V. Almeida, W. Q. Li, M. Cattell and A. H. Barber, Acta Biomater., 2015, 27, 88–100 CrossRef CAS PubMed.
- Y.-T. Chen, S.-L. Hung, L.-W. Lin, L.-Y. Chi and L.-J. Ling, J. Periodontol., 2003, 74, 1652–1659 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental results. See DOI: 10.1039/c6bm00646a |
| This journal is © The Royal Society of Chemistry 2017 |