
Erythrocyte membrane based cationic polymer-mcDNA complexes as an efficient gene delivery system†
Ping
Huang‡
a,
Jing
Zhao‡
a,
Chiju
Wei
b,
Xiaohu
Hou
a,
Pingzhang
Chen
a,
Yan
Tan
c,
Cheng-Yi
He
a,
Zhiyong
Wang
*ac and
Zhi-Ying
Chen
*a
aCenter for Gene and Cell Engineering, Institute of Biomedicine and Biotechnology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P. R. China. E-mail: zy.chen1@siat.ac.cn
bMultidisciplinary Research Center, Shantou 515063, P. R. China. E-mail: chijuwei@stu.edu.cn
cPaul C. Lauterbur Research Center for Biomedical Imaging, Shenzhen Key Laboratory for MRI, Institute of Biomedical and Health Engineering, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P. R. China. E-mail: zy.wang@siat.ac.cn
First published on 28th November 2016
Abstract
Gene therapy has great promise for the treatment of obtained and inherited serious diseases. However, the lack of safe and efficient gene delivery systems remains a barrier for their clinical application. Here, we reported a potential gene delivery vehicle composed of the erythrocyte membrane and cationic polymers, for example the XtremeGENE from Roche and the ε-caprolactone modified polyethylenimine. In addition to high efficiency, this system showed negligible cytotoxicity compared to the two cationic polymers alone in various cell lines, including human embryonic kidney cells (293T), human liver cancer cells (Huh7 and HepG2), murine dendritic cells (DC2.4) and human umbilical cord mesenchymal stem cells (Hu-MSCs). Moreover, the results of confocal laser scanning microscopy and flow cytometry suggested that the cell uptake of this gene vector was improved and might be introduced by the fusion interaction between the erythrocyte membrane and targeted cells.Thus, all the results revealed that the erythrocyte membrane based gene delivery system might be able to serve as an excellent gene delivery system.
Introduction
Gene therapy has attracted much attention to program or enhance cell functions. Although viruses are one of the most effective gene carriers, uncertain problems such as immunogenicity, limited gene packaging capacity and manufacturing difficulties,1 are still obstacles for their clinical application. Minicircle DNA (mcDNA), a kind of novel non-viral vector, is prepared without the bacterium backbone from the traditional plasmid DNA. Therefore, mcDNA has many outstanding features, including low immunogenicity and excellent transgene expression.2 However, the bioavailability of mcDNA is limited by rapid clearance in vivo or by nuclease degradation in the lysosomes of cells.3 Thus, cationic lipids or polymers are prepared as the gene delivery system4–7 some of which with excellent gene transfection ability have been produced as commercial reagents, for example, LipofectamineR2000 (Invitrogen), XtremeGENE (Roche),11 polyethylenimine (PEI) and its derivatives.12 However, owing to their high electropositivity, these cationic lipids or polymers always exhibit severe cytotoxicity.8 Therefore, improving the biocompatibility of cationic transfection reagents becomes urgent. Due to its neutral charge, non-toxicity and non-immunogenicity, polyethylene glycol (PEG) is widely used to modify the surface of the delivery vehicle to avoid rapid clearance by the mononuclear phagocyte system (MPS).9 Moreover, some biodegradable chemical groups were introduced into polymer materials, for example amino ester groups, to enhance the materials’ biodegradability.10,11 Recently, some researchers also attempted to utilize proteins to increase the biocompatibility and targeting ability of delivery systems.12Natural erythrocyte has been explored as a drug carrier for a long time.13–15 However, the haemoglobin inside the erythrocyte would oxidise the cargos and make it unfit to deliver nucleic acids. Fortunately, by hypotonic-osmotic methods,16,17 erythrocyte membrane vehicles could be easily prepared from natural erythrocyte without haemoglobin while also maintaining the integrated membrane structure. In addition, various glycans and proteins on the cell surface endow its numerous bio-advantages and make it more stable under physiological conditions.18–22 Notably, integrated erythrocyte membrane vehicles can protect the encapsulated cargos from being cleared too early in systemic circulation.17,23,24 In recent years, these membrane based delivery vehicles have been widely used to deliver various functional bio-agents, such as nucleic acids, proteins, drugs and so on.25,26
In this paper, we have prepared a novel erythrocyte membrane based gene delivery system, encapsulating cationic polymer-mcDNA nanocomplexes. Herein, the erythrocyte membrane was adopted to improve the intrinsic biocompatibility and cationic polymers were used to condense mcDNA to enhance the interaction between the system and the treated cells. The loading efficiency, zeta potential, cytotoxicity, transfection efficiency and cell membrane fusion processes were systematically studied. All the results show that this system is a promising non-viral gene delivery vector and deserves further investigation.
Experimental section
Materials and methods
XtremeGENE HP DNA Transfection Reagent was purchased from Roche, Sweden. SYBR® Safe DNA gel stain was purchased from Invitrogen (USA). Trypan blue and Cell Counting Kit-8 assay were purchased from Dojindo, Kumamoto (Japan). DiIC1(5) iodide was obtained from Bridgen (China). Hoechst 33342 and 5(6)-carboxy fluoresceindiacetate N-hydroxysuccinimidylester (CFSE) Cell Labeling Kit V2 was obtained from Abcam® Discover More (UK). Cell culture media Dulbecco's Modified Eagle Medium (DMEM) high glucose with L-glutamine and sodium pyruvate, DMEM-F12, penicillin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1), and streptomycin (10 mg mL−1), 0.25% trypsin, Phosphate Buffered Saline (PBS) and Fetal Bovine Serum (FBS) were obtained from Invitrogen (USA). A kind of polyethylenimine derivative, ε-caprolactone modified polyethylenimine (PEICL) was prepared via our previously reported method.27 Minicircle DNA (mcDNA) for which encoding enhanced the green fluorescence protein (EGFP) reporter gene, was amplified and purified in our own laboratory.28 The erythrocyte was obtained from Balb/C female mice (4–6 weeks). All the Balb/C mice were purchased from Guangdong Province Laboratory Animal Center (Guangzhou, China). All animal experiments were approved by the Institutional Animal Care and Usage Committee (Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, file number: SIAT-IRB-140304-YYS-CZY-A0039) and carried out in accordance with Regulations for the Administration of Affairs Concerning Experimental Animals (Guangdong Province, China).
000 U mL−1), and streptomycin (10 mg mL−1), 0.25% trypsin, Phosphate Buffered Saline (PBS) and Fetal Bovine Serum (FBS) were obtained from Invitrogen (USA). A kind of polyethylenimine derivative, ε-caprolactone modified polyethylenimine (PEICL) was prepared via our previously reported method.27 Minicircle DNA (mcDNA) for which encoding enhanced the green fluorescence protein (EGFP) reporter gene, was amplified and purified in our own laboratory.28 The erythrocyte was obtained from Balb/C female mice (4–6 weeks). All the Balb/C mice were purchased from Guangdong Province Laboratory Animal Center (Guangzhou, China). All animal experiments were approved by the Institutional Animal Care and Usage Committee (Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, file number: SIAT-IRB-140304-YYS-CZY-A0039) and carried out in accordance with Regulations for the Administration of Affairs Concerning Experimental Animals (Guangdong Province, China).
Cell culture
Three types of human carcinoma cells (293T, Huh7, HepG2) and one kind of immune cell line (DC2.4) were obtained from the cell bank of the Chinese Academy of Science, Shanghai. The primary cells, human umbilical cord mesenchymal stem cells, Hu-MSCs were separated from fresh umbilical cord, which was gifted from Hornetcorn Biotechnology (Shenzhen, China). All the cell lines were maintained in the cell culture media which was supplemented with 10% FBS and 1% penicillin streptomycin solution in a humidified atmosphere containing 5% CO2 at 37 °C. 293T, Huh7, HepG2 and DC2.4 cells were cultured in DMEM and Hu-MSCs cells were cultured in DMEM-F12.Preparation of gene delivery system
The erythrocyte membrane based gene delivery system, containing cationic polymer-DNA complexes (EM-cationic polymer-mcDNA) was prepared according to the osmotic shock method.16 The entire procedure was carried out at 4 °C in a sterile environment and utilized sterile solutions. About 1 mL of heparinised blood was centrifuged at 13000 rpm for 3 min to remove serum and was washed three times with cold PBS to remove white cells. The erythrocyte pellets were resuspended in 1.5 mL of cold hypotonic solution29 (15 min, 4 °C) to swell cells and remove haemoglobin. Then the supernatant was removed after centrifugation (13000 rpm, 5 min, 4 °C) and the residual pellet was the erythrocyte membrane. Hypotonic solutions containing cationic polymer-mcDNA complexes (0, 0.5, 1, 2, 4, 8 and 10 μg μL−1) were utilized to resuspend the erythrocyte membrane softly. Here XtremeGENE and PEICL were selected to form cationic complexes with the following appropriate ratio: 1 μL (1 μg μL−1) mcDNA for 1 μL XtremeGENE; 1 μL (1 μg μL−1) mcDNA for 4 μL PEICL (1 μg μL−1).27 After 30 min incubation at 4 °C in darkness, the whole hypotonic solution system was then adjusted to isotonic with resealing solution (50% glucose solution). Through previous formulation optimization experiments, we obtained the final appropriate hypotonic solution formula: 10 mM Tris, 0.1 mM EDTA, 1 mM CaCl2.
Characterization
The loading efficiency of mcDNA in the erythrocyte membrane based gene delivery system was quantified by using flow cytometry. Here, mcDNA was stained by SYBR Green. It was performed in 10 μL reaction volumes containing with 0.1 μL of SYBR Green, 1 μg of mcDNA and 9.9 μL distilled water. The redundant SYBR Green was removed by centrifugation (13000 rpm, 5 min, 4 °C) three times. The residual pellets were then suspended in 0.3 mL of PBS. The erythrocyte membrane without the polymer-mcDNA complexes was used as a negative control. Moreover, the morphologies of the system were observed under an inverted fluorescence microscope (Nikon Eclipse Ti-E, Japan). In order to evaluate the integrity of the erythrocyte membrane structure, all the samples were stained by trypan blue30 and measured by flow cytometry (BD FACS CantoTM II, USA).
The zeta potentials of the cationic polymer-mcDNA complexes and the EM-cationic polymer-mcDNA were measured by a dynamic light scattering system (DLS, Malvern Zetasizer Nano ZS, USA). Furthermore, the cationic polymer-mcDNA complexes and the EM-cationic polymer-mcDNA system were electrophoresed in 1% (W/V) agarose gel with EB and tris-acetate (TAE) as running buffer at 90 V for 30 min. The result was visualized using a Bio-Rad Universal Hood II. Herein, 10 μL of sample solution containing 0.1 μg mcDNA was utilized for gel electrophoresis.
Transfection in vitro
In vitro gene transfection was investigated in 293T, Huh7, HepG2, DC2.4 and Hu-MSCs cells. The cells were seeded at a density of 1.0 × 104 cells in 96-well plates with complete FBS containing media and were incubated at 37 °C for 24 h prior to sample addition. Then, the media were removed and 100 μL FBS free DMEM was added followed by the addition of the EM-cationic polymer-mcDNA system (with the same 0.5 μg mcDNA per well). The cells were incubated for 4 h and the samples containing media were replaced with fresh FBS containing media. After 24, 48, 72, 96 and 168 h, the EGFP expression was observed by using an inverted fluorescence microscope. To measure the gene transfection efficiency, flow cytometry (BD FACS CantoTM II, USA) was performed. In all the experiments, the XtremeGENE-mcDNA complexes and PEICL-mcDNA complexes were used as controls. Free erythrocyte membrane was used as the negative control. The amount of mcDNA condensed with XtremeGENE and PEICL in the studies was the same (0.5 μg per well).Cytotoxicity assays
In vitro cytotoxicity of EM-cationic polymer-mcDNA was investigated using Cell Counting Kit-8 (CCK-8). 293T cells were seeded at a density of 1.0 × 104 cells in 96-well plates in complete FBS containing media and were incubated at 37 °C for 24 h prior to sample addition. The media were then removed and 100 μL of FBS free DMEM containing different delivery systems was added for 4 h. The media was then replaced with 100 μL of fresh FBS containing media and then the cells were allowed to grow for 24, 48, 72, 96 and 168 h. At different incubation times, 10 μL of CCK8 solution was added to each well and the plates were incubated at 37 °C for another 2 h. Then, the absorbance of each sample was measured using a Bioradmicroplate reader (Synergy 4, BioTek) at a wavelength of 450 nm.31,32 The cell viability was obtained according to the manufacturer's instructions. XtremeGENE-mcDNA and PEICL-mcDNA were used as the positive controls. An erythrocyte membrane was used as the negative control. The amounts of mcDNA condensed with XtremeGENE and PEICL in the studies were the same (0.5 μg per well).Cellular membrane fusion assay
The cellular membrane fusion of EM-XtremeGENE-mcDNA in 293T cells was investigated by using a confocal laser scanning microscope (CLSM) and quantified by flow cytometry analysis. 293T cells were seeded on the Lab-Tek®Chambered# 1.0 Borosilicate Coverglass System (Thermo Scientific Nunc) and incubated overnight at 37 °C. When the cells reached 85% confluency, the media was removed. The cytoplasm of 293T cells were stained with 5,6-carboxyfluorescein diacetate, a succinimidyl ester (CFSE, 10 μM in PBS) for 10 min in darkness at 37 °C and washed three times with PBS. The erythrocyte membrane of EM-XtremeGENE-mcDNA was stained with DiIC1(5) iodide (5 μM in PBS for 20 min in darkness at 37 °C). The media was then removed and FBS free DMEM media with DiIC1(5) iodide stained EM-XtremeGENE-mcDNA were added into the wells. After two hours of incubation, 293T cells were washed with PBS three times to remove the adherent samples. The FBS containing media was added to each well for the next incubation. At the presupposed time points (2, 4, 6 and 24 h), the cells were washed with PBS three times and then fixed with paraformaldehyde (4%) for 15 min in darkness at room temperature. After staining with Hoechst 33342 (10 μg mL−1 in PBS for 5 min in darkness at room temperature), the cells were examined under a CLSM (LEICA TCS SP5-II, Germany). The excitation/emission wavelengths were 495/517 nm for CFSE (green), 638/658 nm for DiIC1(5) iodide (red) and 346/460 nm for Hoechst 33342 (blue). Moreover, the presence of DiIC1(5) iodide stained cells was quantified by flow cytometry. mcDNA used in this study was an empty mcDNA with a similar molecular weight to the mcDNA EGFP reporter gene.Statistical analysis
The data are expressed as the means ± standard deviation (X ± SD). Statistically significant differences among the multiple groups are analyzed by the Tukey test.Results and discussion
Characterization
The Erythrocyte membrane based gene delivery system was prepared by loading the cationic polymer-mcDNA complexes into the erythrocyte membrane. The packaging capacity of mcDNA is an important index for evaluation of this system. Therefore, different concentrations of mcDNA were used to prepare EM-cationic polymer-mcDNA. As shown in Fig. 1, increasing the mcDNA concentration from 0 to 10 μg μL−1 led to an increment of the loading efficiency. Furthermore, loading curve saturation was approached at the mcDNA concentration of 8 μg μL−1. So the gene delivery system used in the following experiments was performed at a mcDNA concentration of 8 μg μL−1. | ||
| Fig. 1 The loading ratio of mcDNA by the membrane system with different concentrations of mcDNA (0, 0.5, 1, 2, 4, 8 and 10 μg μL−1) in 100 μL volumes. | ||
The morphologies of erythrocyte, the erythrocyte membrane and the EM-cationic polymer-mcDNA system, were observed by optical microscopy. As shown in Fig. 2A, the erythrocyte and membrane system presented typical double concave disc-like structures with smooth surfaces and clear edges.33 Compared to erythrocyte, the colour of the membrane system was much lighter due to the lack of hemoglobin.16 However, after combining with the cationic polymer-mcDNA complexes, there was no significant change in the size and structure.
 | ||
| Fig. 2 The morphology and integrity assays of EM-cationic polymer-mcDNA. (A) The microscopic morphology and integrity ratio (%) of erythrocyte, the erythrocyte membrane (EM), EM-XtremeGENE-mcDNA and EM-PEICL-mcDNA system. (B) FSC-SSC signals which were measured by flow cytometry. | ||
As is known, the intact membrane system structure is crucial for cargo delivery.18,19,21 In order to investigate the integrity of our erythrocyte membrane systems, trypan stain analysis was carried out. Trypan blue, as a diazo dye, is usually applied to colour dead cells, but the intact membrane structure would prevent the staining process. Hence, all the samples were tested by trypan blue. After the observation under an optical microscope, the integrity ratio was calculated as [(uncoloured cells/total cells) × 100%]. As shown in Fig. 2A, more than 90% of the samples maintained membrane integrity even with the loading of cationic polymer mcDNA complexes.
Flow cytometry was also used to characterize the integrity of the systems. The forward scattering signal (FSC) indicates the size of samples and the side scattering signal (SSC) shows the internal structural complexity. In Fig. 2B, the membrane system exhibited the same FSC and a slightly weaker SSC signal intensity compared to erythrocyte. This result indicated that the loading of the cationic polymers–mcDNA complexes did not affect the membrane structure and maintained the original morphology of erythrocyte. These results were consistent with the optical microscopy results and proved that the loading process of the cationic polymer-mcDNA complexes did not damage the integrity of the membrane.
The surface charge of gene delivery systems is an important factor for their cytotoxicity. Hence, the DLS method was used to analyse the zeta potential of the samples. As shown in Fig. 3A, the free mcDNA molecules and the erythrocyte membrane presented negative charges, while the XtremeGENE-mcDNA and PEICL-mcDNA nanocomplexes exhibited highly positive charges, which is in accordance with previous reports.34 After binding with the erythrocyte membrane, the zeta potentials of the XtremeGENE-mcDNA and PEICL-mcDNA nanocomplexes were neutralized significantly. As is known, cationic polymers with strong positive charge usually result in cytotoxicity owing to the strong interaction with the targeted cell.8,35 Now the erythrocyte membrane based delivery system which carried slightly positive charges might have better biocompatibility.
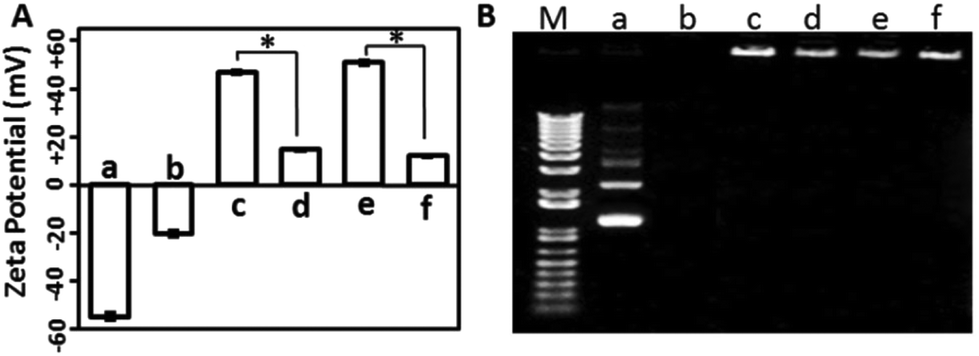 | ||
| Fig. 3 (A) Zeta potential measurement results by DLS (n = 3). (B) Agarose gel electrophoresis retardation assay. (M: marker; a: Naked mcDNA; b: erythrocyte membrane; c: XtremeGENE-mcDNA; d: EM-XtremeGENE-mcDNA; e: PEICL-mcDNA; f: EM-PEICL-mcDNA). Here, 0.1 μL (1 μg μL−1) mcDNA for 0.1 μL XtremeGENE; 0.1 μL (1 μg μL−1) mcDNA for 0.4 μL PEICL (1 μg μL−1). | ||
For effective gene delivery, the delivery system should effectively compact and condense DNA.36 Agarose gel electrophoresis was performed to investigate the DNA complexing ability. As shown in Fig. 3B, the cationic polymers, XtremeGENE and PEICL, compacted mcDNA and then hindered the mobility of mcDNA in an electric field. Interestingly, the mcDNA was also retarded completely in the membrane-based system, EM-XtremeGENE-mcDNA and EM-PEICL-mcDNA, which demonstrated that the negative surface charge of the erythrocyte membrane did not influence the combination between the mcDNA and cationic polymers (XtremeGENE and PEICL).
Transfection and cytotoxicity in 293T cells
The highly transfectable 293T cells were used to investigate the gene transfection efficiencies.37 As shown in Fig. 4A, the cells treated with the EM-XtremeGENE-mcDNA and EM-PEICL-mcDNA samples, presented a much stronger green fluorescence signal than the cationic polymer based groups. Transfection efficiencies were furthermore confirmed via flow cytometry (Fig. 4B). Compared with the XtremeGENE-mcDNA group (30.15%), about 50.88% of the cells expressed a green fluorescence protein in the EM-XtremeGENE-mcDNA group, even in a short culture time (24 h). With the extension of time, the transfection efficiency improved. After 48 h of incubation, gene expression in the EM-XtremeGENE-mcDNA treated group was 1.8 fold higher than that of XtremeGENE-mcDNA (43.61% vs. 79.42%). Even 7 days (168 h) later, the superiority of transfection efficiency was still presented. In addition to XtremeGENE, PEICL is another kind of gene delivery agent.27 As shown in Fig. 4A and B, the EM-PEICL-mcDNA system also showed obviously high gene transfection efficiency comparing with the PEICL–mcDNA nanocomplexes (e.g., 24 h, P < 0.001). These results demonstrated that the erythrocyte membrane based gene delivery system can increase the transfection efficiency of cationic polymers. Biocompatibility is a prerequisite for a gene vehicle. In order to assess the cytotoxicity of the erythrocyte membrane based system, CCK-8 assay was performed in 293T cells in vitro. As shown in Fig. 4C, all of the cationic polymer-mcDNA complexes presented obvious cytotoxicity.38 However, when the nanocomplexes were loaded onto the erythrocyte membrane, the cell viability was obviously improved. After transfection for 72 h and 96 h, the cell viabilities of EM-XtremeGENE-mcDNA remained above 95%. A similar result was also noted in the EM-PEICL-mcDNA group. It is also worth noting that the cell viability of EM-PEICL-mcDNA even exceeded 100% (115.0%) after transfection for 168 h. And the negative control groups treated with the erythrocyte membrane also showed good biocompatibility (e.g., 24 h, 118.3%; 72 h, 105.5%; 96 h, 108.8%). These results demonstrated that the erythrocyte membrane gene delivery system could obviously decrease the cytotoxicity of cationic polymers, and even promote the cell proliferation. | ||
| Fig. 4 The in vitro gene transfection and cytotoxicity of 293T cells after treatment with the erythrocyte membrane (EM), XtremeGENE-mcDNA, EM-XtremeGENE-mcDNA, PEICL-mcDNA and EM-PEICL-mcDNA. (A) Fluorescence microscope images of EGFP expression for 48 h, scale bar 100 μm. (B) Transfection efficiency of 293T cells for 24, 48, 72, 96 and 168 h. (C) Cell viability of 293T cells for 24, 48, 72, 96 and 168 h. *P < 0.001. | ||
This showed that the erythrocyte membrane could improve the biocompatibility and gene transfection efficiency of mcDNA/polymer nanocomplexes. We hypothesize two main reasons to explain these results. One is due to the fact that the erythrocyte membrane could protect the nanocomplexes from nuclease degradation in lysosomes.3 Another reason is that there are various natural glycans and proteins (for example CD47) on the complete erythrocyte membrane biostructure25,26 which make the nanocomplexes more biocompatible,18,19 and beneficial for cellular uptake.
Cellular membrane fusion in 293T cells
The gene delivery system introducing the cellular membrane into the targeted cells is thought to be the first and one of the rate-limiting steps for the transfection process, which determines the transfection efficiency.39Qualitative analysis of the cell membrane fusion of EM-XtremeGENE-mcDNA was evaluated by CLSM. CFSE (green) and DiI1C(5) were applied in the experiment. CFSE (green) is a cell permeant dye40 and can covalently bind with the intracellular proteins of living cells and then emit green fluorescence.40 DiI1C(5) iodide is a kind of lipophilic fluorescent dye and can combine with the cell membrane. DiI1C(5) iodide stained the erythrocyte membrane based delivery system emitting red fluorescence.41 These stained systems were treated with targeted cell for two hours, and then the redundant materials were removed from the cell culture. As shown in Fig. 5, there were a lot of red fluorescence points around 293T cells after two hours’ incubation. Four hours later, the red fluorescence intensity decreased a little. However, the green fluorescence began to co-localize with the red fluorescence in the cytoplasm, and the merged fluorescence imaging presented a yellow colour that showed that the erythrocyte membrane based gene delivery system could be fused into the cell membrane of 293T. With the extension of incubation time, the red fluorescence intensity and green fluorescence of CFSE became weaker due to the partitioned dyes between the daughter cells.
 | ||
| Fig. 5 CLSM images of 293T cells treated with EM-XtremeGENE-mcDNA for different time (2, 4, 6 and 24 h) at 37 °C. The cytoplasm of 293T cell appears in green fluorescence (CFSE, Ex/Em = 495/517 nm); the nucleus of 293T cell appears in blue fluorescence (Hoechst, Ex/Em = 346/460 nm); EM-XtremeGENE-mcDNA appears in red fluorescence (DiIC1(5) iodide, Ex/Em = 638/658 nm). Scale bar 25 μm. | ||
In order to further investigate the time-dependent cell membrane fusion process, quantitative analysis was undertaken. The cell proliferation can be analyzed by tracking the mean green fluorescence intensity per cell by using flow cytometry.42 Similarly, by measuring the red fluorescence intensity of DiI1C(5) iodide41 per cell, the distribution and variation of the membrane based system in the 293T cells could be evaluated. As shown in Fig. 6A, the live cells in gating (P1) were used in the analyses. It showed that the CFSE fluorescence intensity was gradually reduced with the prolonging incubation time (Fig. 6B). Moreover, DiIC1(5) iodide fluorescence (erythrocyte membrane labeled) presented similar decreased trends to CFSE fluorescence (293T cell labeled). These results indicated that the uptake of this gene vector might be introduced by the fusion between the membrane systems and the targeted cells. Moreover, after the fusion by the 293T cells, the erythrocyte membrane became a part of the 293T cells membrane and could be partitioned between the daughter cells from the original ones. Meanwhile, membrane fusion could induce the cell proliferation.43 This hypothesis could also explain the high cell viability of EM-PEICL-mcDNA (Fig. 4C).
 | ||
| Fig. 6 FCM analyses of CFSE and DiIC1(5) iodide. (A) The FSC-SSC signal of EM-XtremeGENE-MC·DNA treated 293T cells (P1 is the live cell gating used in this analysis). (B) Plot of CFSE fluorescence intensity (black) and DiIC1(5) iodide fluorescence intensity (blue) at different time points; 2, 4, 6 and 24 h. | ||
Transfection in hard-transfect cell lines
Using the traditional transfection agents, the transfection results were usually unsatisfactory in some special cell types, such as immune cells (DC2.4),37 metabolic types of tumor cells (Huh7, HepG2), and primary cells (human umbilical cord mesenchymal stem cells, Hu-MSCs).44 Nevertheless, the cells treated with the erythrocyte membrane based system showed higher gene expression (Fig. 7). | ||
| Fig. 7 The microscopic images of EGFP fluorescent and bright field of DC2.4, Huh7, HepG2 and Hu-MSCs cells after the treatment with XtremeGENE-mcDNA (a) and EM-XtremeGENE-mcDNA (b) for 24, 48, 72 and 96 h. Scale bar 100 μm. | ||
Conclusions
In this study, a novel non-viral erythrocyte membrane based gene delivery system was prepared. Compared with the commercial Roche gene transfection reagent and the polyethylenimine derivative, this system exhibited much higher transfection efficiency and minimal cytotoxicity, not only in the highly transfectable cell line (293T), but also in many hard-transfect cell lines (DC2.4, Huh7, HepG2 and Hu-MSCs). All the results demonstrated that this system might be a very promising gene delivery system. Furthermore, the transfection in vivo of the EM-CA-mcDNA gene delivery system is under research now.Acknowledgements
The authors gratefully acknowledge the support for this research by Natural Science Foundation of GuangDong Province of China (2014A030310335), the National Natural Science Foundation of China (NSFC) (Grant no. 51203177, 81602499, 81471778, 51602338 and 81602499), the National High Technology Research and Development Program (863 Program) of China (Grant no. 2014AA020708) and Science and Technology Foundation of Shenzhen, China (Grant No. SFG2012.566, JCYJ20140417113430662, JCYJ20150521094519466 and SKC2012.237).Notes and references
- X. Guo and L. Huang, Acc. Chem. Res., 2012, 45, 971–979 CrossRef CAS PubMed.
- M. A. Kay, C. Y. He and Z. Y. Chen, Nat. Biotechnol., 2010, 28, 1287–1289 CrossRef CAS PubMed.
- F. Liu, L. M. Shollenberger, C. C. Conwell, X. Yuan and L. Huang, J. Gene Med., 2007, 9, 613–619 CrossRef CAS PubMed.
- M. C. Moran, N. Rosell, G. Ruano, M. A. Busquets and M. P. Vinardell, Colloids Surf., B, 2015, 134, 156–168 CrossRef CAS PubMed.
- D. Costa, W. H. Briscoe and J. Queiroz, Colloids Surf., B, 2015, 133, 156–163 CrossRef CAS PubMed.
- M. E. Hwang, R. K. Keswani and D. W. Pack, Pharm. Res., 2015, 32, 2051–2059 CrossRef CAS PubMed.
- A. K. Jain, H. Yusuf, A. Pattani, H. O. McCarthy, D. M. McDonald and V. L. Kett, J. Pharm. Biomed. Anal., 2014, 100, 236–242 CrossRef CAS PubMed.
- Y. Q. Wang, J. Su, F. Wu, P. Lu, L. F. Yuan, W. E. Yuan, J. Sheng and T. Jin, Int. J. Nanomed., 2012, 7, 693–704 CAS.
- Y. Zhang, N. Kohler and M. Zhang, Biomaterials, 2002, 23, 1553–1561 CrossRef CAS PubMed.
- J. Zhao, P. Huang, Z. Wang, Y. Tan, X. Hou, L. Zhang, C. Y. He and Z. Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 19284–19290 Search PubMed.
- J. Zhao, L. Yang, P. Huang, Z. Wang, Y. Tan, H. Liu, J. Pan, C. Y. He and Z. Y. Chen, J. Colloid Interface Sci., 2016, 463, 93–98 CrossRef CAS PubMed.
- L. Tang, Q. Yin, Y. Xu, Q. Zhou, K. Cai, J. Yen, L. W. Dobrucki and J. Cheng, Chem. Sci., 2015, 6, 2182–2186 RSC.
- C. Lizano, S. Sanz, J. Luque and M. Pinilla, Biochim. Biophys. Acta, 1998, 1425, 328–336 CrossRef CAS.
- K. K. Phua, D. Boczkowski, J. Dannull, S. Pruitt, K. W. Leong and S. K. Nair, Adv. Healthcare Mater., 2014, 3, 837–842 CrossRef CAS PubMed.
- Q. Dong and W. Jin, Electrophoresis, 2001, 22, 2786–2792 CrossRef CAS PubMed.
- R. Flower, E. Peiretti, M. Magnani, L. Rossi, S. Serafini, Z. Gryczynand and I. Gryczynski, Invest. Ophthalmol. Visual Sci., 2008, 49, 5510–5516 Search PubMed.
- C. G. Millan, M. L. Marinero, A. Z. Castaneda and J. M. Lanao, J. Controlled Release, 2004, 95, 27–49 CrossRef CAS PubMed.
- B. T. Luk, C.-M. J. Hu, R. H. Fang, D. Dehaini, C. Carpenter, W. Gao and L. Zhang, Nanoscale, 2014, 6, 2730–2737 RSC.
- S. Aryal, C.-M. J. Hu, R. H. Fang, D. Dehaini, C. Carpenter, D.-Er Zhang and L. Zhang, Nanomedicine, 2013, 8, 1271–1280 CrossRef CAS PubMed.
- W. Gao, C. M. Hu, R. H. Fang, B. T. Luk, J. Su and L. Zhang, Adv. Mater., 2013, 25, 3549–3553 CrossRef CAS PubMed.
- H. Cheng, Z. Fan and Z. Hl, J. Mater. Chem. B, 2014, 2, 8231–8238 RSC.
- C.-M. J. Hu, R. H. Fang, B. T. Luk, K. N. H. Chen, C. Carpenter, W. Gao, K. Zhang and L. Zhang, Nanoscale, 2013, 5, 2664–2668 RSC.
- M. Hamidi, A. Zarrin, M. Foroozesh and S. Mohammadi-Samani, J. Controlled Release, 2007, 118, 145–160 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang and L. Zhang, Adv. Healthcare Mater., 2012, 1, 537–547 CrossRef CAS PubMed.
- A. Stier, S. Reichert, F. Criscuolo and P. Bize, Exp. Gerontol., 2015, 71, 118–134 CrossRef PubMed.
- M. Shaillender, R. Luo, S. S. Venkatraman and B. Neu, Int. J. Pharm., 2011, 415, 211–217 CrossRef CAS PubMed.
- Y. Tan, L. Xie, Z. Wang, N. Zhang, C. Zou, Z. Y. Chen, X. Liu, J. Lu, G. Liu and H. Zheng, Biomaterials, 2015, 56, 219–228 CrossRef CAS PubMed.
- X. H. Hou, X. Y. Guo, Y. Chen, C. Y. He and Z. Y. Chen, Mol. Ther.–Methods Clin. Dev., 2015, 1, 14062–14068 CrossRef PubMed.
- S. H. Kim, E. J. Kim, J. H. Hou, J. M. Kim, H. G. Choi, C. K. Shim and Y. K. Oh, Biomaterials, 2009, 30, 959–967 CrossRef CAS PubMed.
- T. He, G. Chi, B. Tian, T. Tang and K. Dai, Mol. Med. Rep., 2015, 12, 4063–4070 CAS.
- H. Jia, L. Xiang, Z. Wang and Q. Zhou, Cell Biochem. Biophys., 2015, 73, 393–397 CrossRef CAS PubMed.
- J. Feng, X. Chen, Y. Wang, Y. Du, Q. Sun, W. Zang and G. Zhao, Mol. Cell. Biochem., 2015, 408, 163–170 CrossRef CAS PubMed.
- G. Benga, B. E. Chapman, T. Romeo, G. C. Cox and P. W. Kuchel, Protoplasma, 2015, 252, 1181–1185 CrossRef CAS PubMed.
- R. Lindemann, Scand. J. Haematol., 1975, 14, 216–225 CrossRef CAS PubMed.
- M. Gujrati, A. Malamas, T. Shin, E. Jin, Y. Sun and Z. R. Lu, Mol. Pharm., 2014, 11, 2734–2744 CrossRef CAS PubMed.
- Y. Shan, T. Luo, C. Peng, R. Sheng, A. Cao, X. Cao, M. Shen, R. Guo, H. Tomas and X. Shi, Biomaterials, 2012, 33, 3025–3035 CrossRef CAS PubMed.
- J. Ju, M. L. Huan, N. Wan, Y. L. Hou, X. X. Ma, Y. Y. Jia, C. Li, S. Y. Zhou and B. L. Zhang, Bioorg. Med. Chem. Lett., 2016, 26, 2401–2407 CrossRef CAS PubMed.
- E. Junquera and E. Aicart, Adv. Colloid Interface Sci., 2016, 233, 161–175 CrossRef CAS PubMed.
- T. K. Prasad, N. Rangaraj and N. M. Rao, FEBS Lett., 2005, 579, 2635–2642 CrossRef CAS PubMed.
- A. Rodriguez and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2010, 92, 214–220 CrossRef PubMed.
- C. Weir, M. C. Morel-Kopp, A. Gill, K. Tinworth, L. Ladd, S. N. Hunyor and C. Ward, Heart, Lung Circ., 2008, 17, 395–403 CrossRef PubMed.
- G. Bocharov, T. Luzyanina, J. Cupovic and B. Ludewig, Front. Immunol., 2013, 4, 264–271 Search PubMed.
- F. Lang, M. Föller, K. Lang, P. Lang, M. Ritter, A. Vereninov, I. Szabo, S. M. Huber and E. Gulbins, Methods Enzymol., 2007, 428, 209–225 Search PubMed.
- Q. Yu, J. Cao, B. Chen, W. Deng, X. Cao, J. Chen, Y. Wang, S. Wang, J. Yu, X. Xu and X. Gao, Int. J. Nanomed., 2015, 10, 7097–7107 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: 10.1039/c6bm00638h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |