
Rapid determination of the Mn average oxidation state of Mn oxides with a novel two-step colorimetric method
Yanhua
Zhu
,
Xinran
Liang
,
Huaiyan
Zhao
,
Hui
Yin
,
Mingming
Liu
,
Fan
Liu
and
Xionghan
Feng
*
Key Laboratory of Arable Land Conservation (Middle and Lower Reaches of Yangtse River), Ministry of Agriculture, College of Resources and Environment, Huazhong Agricultural University, Wuhan 430070, China. E-mail: fxh73@mail.hzau.edu.cn; Fax: +86 27 87288618; Tel: +86 27 87280271
First published on 18th November 2016
Abstract
The Mn average oxidation state (Mn AOS) of Mn oxides has a significant impact on their reactivity towards trace metals and organic contaminants via sorption, catalysis and oxidation processes. Accurate determination of the Mn AOS is a key step to understanding the structures, composition, physicochemical properties, environmental behaviors and potential applications of Mn oxides. Here, a rapid two-step colorimetric method was developed to determine the Mn AOS of various Mn oxides, and it was tested on several Mn oxides (vernadite, acid birnessite and hausmannite). We also determined the Mn AOS of these Mn oxides with the conventional oxalic acid–permanganate back titration method and X-ray absorption near-edge spectroscopy (XANES) for comparison. In this rapid two-step colorimetric method, leucoberbelin blue I (LBB) and formaldoxime colorimetry were employed to obtain the oxidation numbers of high-valent Mn and total Mn, respectively, which were then used to calculate the Mn AOS. The colorimetric measurements are of considerable color stability and high sensitivity, thus enabling rapid, convenient and highly accurate determination of the Mn AOS compared with conventional methods. In addition, the required sample amount is greatly reduced (from ∼0.05 g to ∼0.005 g), making the proposed method an appropriate strategy for micro-volume samples.
1. Introduction
Manganese (Mn) (hydro)oxide (generally termed as Mn oxide) minerals are widely distributed in soils and sediments, and play a crucial role in the transport and fate of both contaminants and nutrients in the environment through sorption, catalysis and oxidation processes.1–7 Synthetic analogs of various natural Mn oxides have many applications including but not limited to adsorbents,6 oxidants,8 catalysts and electrode materials.9 However, natural and synthetic Mn oxides are usually characterized by mixed Mn valence states, i.e. Mn2+, Mn3+ and Mn4+. The Mn AOS represents the average oxidation state of manganese, and can affect the sorption capacities of metal ions by vacancy sites, and particularly influence the mechanism of redox reactions and the topological transformation of manganese oxide minerals.10 It is essential to know the Mn AOS of these Mn oxides with mixed valences to understand their structures, stability and reactivity. Therefore, accurate determination of the Mn AOS of Mn oxides is of great importance.Currently, there are several methods to determine the Mn AOS, which can be generally divided into chemical titration methods11–17 and spectroscopic analysis.18–22 The titration methods involve the reduction of high-valent Mn (Mn4+ and Mn3+) by appropriate reductants. The most commonly used reductants are oxalate,14,16,23 iodide15,23 and (NH4)2FeSO4 (Mohr salt). In the oxalic acid–permanganate back titration method, oxalic acid or sodium oxalate is reacted with Mn oxides, and the excess oxalate is then determined using titration of KMnO4 solution, the concentration of which is predetermined with standard Na2C2O4. Based on the total Mn in the sample, the volume of oxalate consumed and the concentration of KMnO4 solution, the Mn AOS can be calculated. However, the titration operation is usually time consuming, and the reaction conditions must be strictly controlled to ensure that the reaction occurs at the desired stoichiometry with negligible impacts of side reactions. Firstly, the titration must be conducted under strict heating conditions at temperatures between 75 and 85 °C to accelerate the reaction between oxalate and KMnO4, and prevent thermal decomposition of C2O42−. Besides, the acidity of the reactions should be delicately controlled because too low pH would promote the decomposition of KMnO4 to MnO2 and too high pH would lead to the decomposition of H2C2O4. In the iodide method, excess NaI is used to reduce MnOx to Mn2+, and quantitatively generate I3−via the reaction of I− with produced I2. Then, I3− in the solution is titrated with standardized Na2S2O3 solution to obtain the oxidation numbers of high-valent Mn. This method can be conducted at room temperature. However, it requires a high concentration of iodide/iodine in the resulting solution for total Mn determination because a large amount of iodide is required to completely transform I2 into I3− species; besides, Fe3+ interference must be excluded when using this method. The potentiometric titration method was clearly described by Grangeon et al.17 The Mn oxides are reduced by (NH4)2Fe(SO4)2·6H2O to Mn2+ and the excess reductant is titrated by KMnO4, and then the total Mn is determined by the oxidation of Mn2+ to Mn3+ which is stabilized by P2O74−. In this method, there is no need to accurately determine the solution concentration, but a titrator is required to monitor the titration end point of the redox reaction.
For the spectroscopy methods, X-ray photoelectron spectroscopy (XPS),18 electron energy loss spectroscopy22,24 and X-ray absorption spectra19–21,25 are used to determine the Mn AOS directly. For instance, the narrow scans of Mn 2p3/2, Mn 3p or Mn 3s are used in XPS analysis.8,18,26,27 The relative proportions of Mn4+, Mn3+ and Mn2+ can be determined by fitting the Mn 2p3/2 spectrum either using three peaks representing each valence Mn or using the multiple peak parameters including 5–6 multiplet peaks for Mn2+, 5 multiplet peaks for Mn3+ and 5–6 multiplet peaks for Mn4+. The multiple peak parameters were first calculated by Gupta and Sen28 and then developed and modified by Nesbitt et al.,18 Banerjee and Nesbitt29 and Biesinger et al. (2011).26 The Mn 3s splitting value can also be used for determining the Mn AOS.27,30 Electron energy-loss spectroscopy (EELS) has been widely used to measure the oxidation state of metals, such as Mn, and meaningful results can be obtained by proper conduction of the experiments, such as safe dose fluence of the beam which is different for Mn oxides with different structures.22 As the position of the edge of Mn K-edge X-ray absorption near edge spectroscopy (XANES) is sensitive to Mn oxidation states, it is commonly used to determine the Mn AOS by using a standard curve using linear combination fitting of the standards.20 However, different coordination environments of Mn will also affect the position of the absorption edge. Based on the compilation of the XANES spectra of a series of naturally occurring manganates, synthetic analogs with known structure and chemical composition and pure-valence phase species, Manceau et al.21 proposed a Combo method to determine the Mn AOS of various Mn oxide materials.21 However, the accuracy decreases when the proportion of Mn2+ is higher than 15%, and when there is a large amount of layer Mn3+, the amount and distribution of layer Mn3+ will non-additively affect the XANES features.21
In the present study, a novel method for rapid determination of the Mn AOS was proposed. Leucoberbelin blue I (LBB) is a synthetic triphenyl compound (C23H26N2O3S). LBB of reduced state is colorless and can be easily oxidized to a blue compound by the high-valent Mn in Mn oxides, i.e. Mn3+ and Mn4+. The reaction is written as the following equation.31
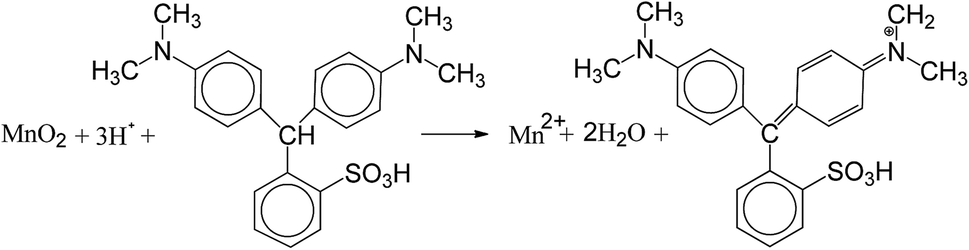
The colored compound can be quantitatively detected by using a visible spectrophotometer with a significant absorption peak at 620 nm. The LBB method is commonly used for the qualitative and quantitative detection of high-valent Mn in the microbial manganese oxidation process.28 In this study, this method was modified to accurately determine the content of high-valent Mn. In addition, a linear standard curve of the spectrophotometric absorbance at 620 nm (A620) versus the concentration of transferred electrons (CTE) was established through the reaction of KMnO4 solution with LBB. Then, LBB and the formaldoxime colorimetric method were employed to obtain the oxidation numbers of high-valent Mn and total Mn, respectively, based on which the Mn AOS was calculated. The novel two-step colorimetric method is characterized by rapid detection, high sensitivity and color stability. In addition, it can be more efficient to determine the Mn AOS in a wide range of pH 3.5–10.
2. Materials and methods
2.1. Apparatus and reagents
X-ray powder diffraction (XRD) analysis was performed using a Bruker D8 Advance diffractometer operated at a tube voltage of 40 kV and a tube current of 40 mA. Intensities were collected at a step-scan of 5–85° with a scan rate of 1° min−1 per 0.02° 2θ step. The K and Na elemental analysis of samples was measured with a flame spectrophotometer (UK, Sherwood M 410). The absorbance was measured with an Agilent (Santa Clara, CA, USA) 8454 UV-vis spectrophotometer. The XANES spectra of samples were measured in transmission mode at room temperature on the 1W1B beamline at the Beijing Synchrotron Radiation Facility (BSRF).5LBB was purchased from Sigma Chemical Co. (USA). The other chemical reagents were all of analytical reagent grade and obtained from Sinopharm Chemical Reagent Co. (China). All aqueous solutions were prepared with distilled deionized water (18 MΩ, from Aquapro AJY-1001-U).
2.2. Preparation of Mn oxides
For the synthesis of vernadite, 1.28 L 0.2 mol L−1 KMnO4 solution was slowly added to 1.44 L 0.5 mol L−1 NaOH solution with vigorous stirring. 0.3 mol L−1 MnCl2·4H2O solution was then added dropwise to the above mixture. The obtained slurry was continuously stirred for 12 h and left to settle for 4 h. The resulting dark brown precipitate was mixed into 1 mol L−1 NaCl solution and then shaken for 1 h.For the synthesis of hausmannite, 2 L MnCl2·4H2O solution (0.015 mol L−1) was prepared, and the solution pH was adjusted to 9 using 1 mol L−1 NaOH with a titrator (Metrohm 907). The solution was centrifuged after reaction for 6 h.
Acid birnessite was synthesized by heating a 500 mL KMnO4 solution (0.4 mol L−1) to 100–110 °C at an oil bath temperature. With vigorous stirring, 20 mL of 6 mol L−1 HCl solution was added at a rate of 1 mL min−1, followed by 30 min refluxing.33 Then, the suspension was cooled down to room temperature and aged for 12 h at 60 °C.
The obtained precipitate was washed repeatedly with distilled deionized water until the supernatant conductivity was lower than 20 μS cm−1. Finally, the sample was freeze-dried, ground, and stored in a desiccator after 100-mesh sieving.
2.3. Elemental analysis and oxalic acid–permanganate back titration method
The elemental analysis of the samples was performed as follows: 0.02 g of sample was dissolved in 50 mL 0.02 mol L−1 hydroxylamine hydrochloride. The contents of K and Na were measured using a flame spectrophotometer.The Mn AOS of vernadite, hausmannite and acid birnessite was determined with the oxalic acid–permanganate back titration method as a comparison.16 0.2 g of the manganese minerals was dissolved in 5 mL of 0.5 mol L−1 H2C2O4 and 10 mL of 1 mol L−1 H2SO4 to reduce the Mn oxide sample to Mn2+ solution. The excess C2O42− was back-titrated with standardized KMnO4 solution at 75–85 °C to obtain the oxidation number of Mn with a valence higher than two, i.e. Mn3+ and Mn4+.
The total Mn content was determined with the formaldoxime colorimetric method.34 0.01 g of the sample was dissolved in 50 mL of 0.01 mol L−1 hydroxylamine hydrochloride. After the solution was diluted 10 times, 2 mL sample solution mixed with 2 mL buffer and 2 mL formaldoxime solution was added to a 25 mL colorimetric tube followed by the addition of 2 mL EDTA-2Na. Then, the absorbance of the solution was determined at 450 nm after 20 min. The Mn AOS was calculated according to both the total Mn content and the titration result. The procedure was repeated three times for each sample.
2.4. X-ray absorption near-edge spectroscopy (XANES)
The XANES spectra of vernadite, hausmannite and acid birnessite were measured. Mn K-edge XANES data were acquired over the energy range of 6.35–7.23 keV. Before every sample was detected, a Mn metal foil reference was collected (6539 eV) to calibrate the monochromator. SIXPack was used for reduction and analysis of the XANES spectra.35 In the data-reduction step, replicate spectra were aligned to a common energy scale and averaged. For Mn K-edge spectra, averaged spectra were background-subtracted using the following parameters: E0 = 6556 eV, Rbkg = 0.9 Å and k-weight = 3.2.5. Novel two-step colorimetric method
The procedures of the rapid two-step colorimetric method are illustrated in Fig. 1. Briefly, two-step colorimetry, i.e. LBB and formaldoxime colorimetry, were employed to obtain the oxidation numbers of high-valent Mn and total Mn, respectively, from which the Mn AOS was calculated.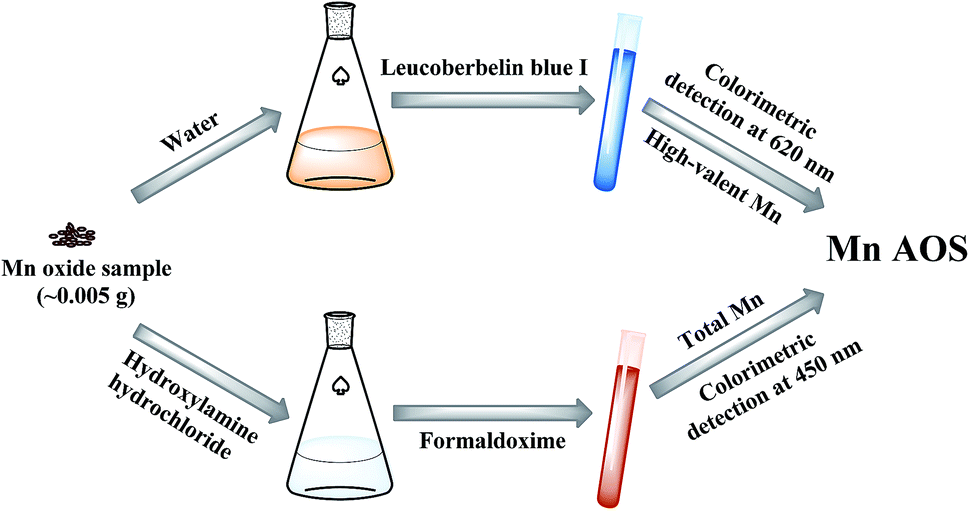 | ||
| Fig. 1 The diagram of the procedures of the rapid two-step colorimetric method. | ||
(2) 5, 10, 15, 20, 25, 30, and 35 μL of the KMnO4 stock solutions were respectively added to 1.5 mL tubes and then was filled up to 1 mL with distilled deionized water;
(3) 100 μL of the diluted solution and 500 μL of LBB solution were respectively added into 1.5 mL tubes and were allowed to react for 15–20 min in the dark;
(4) The absorbance of the reacted solution was determined on a diode array spectrophotometer at 620 nm.
The CTE (concentration of transferred electrons) was calculated by multiplying the KMnO4 concentration with a factor of 5. According to the absorbance measured at 620 nm (A620) and CTE, a standard curve of A620-CTE was obtained.
3. Results and discussion
3.1. Powder XRD patterns of samples
The phases of these obtained samples were confirmed by powder XRD analysis. The XRD patterns of vernadite, hausmannite and acid birnessite are shown in Fig. 2. The patterns of samples are in good agreement with Drits et al. (1997)36 for vernadite and birnessite, JCPDS-24-0734 for hausmannite, respectively.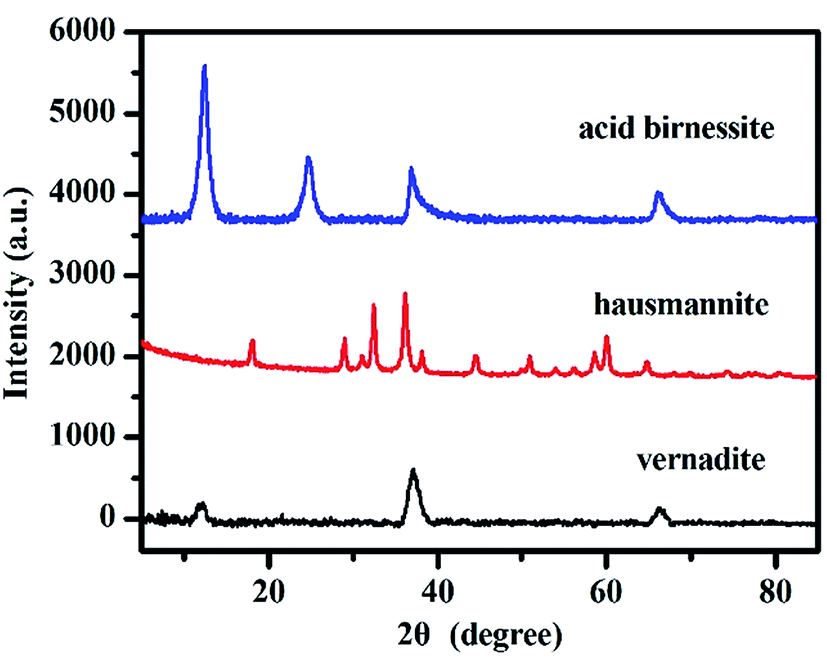 | ||
| Fig. 2 Powder XRD patterns of vernadite, hausmannite and birnessite samples (based on Drits et al. (1997)36 for vernadite and birnessite, JCPDS-24-0734 for hausmannite | ||
This result indicates that the three synthetic manganese oxides are single-phase pure minerals.
3.2. Mn AOS determined with the oxalic acid–permanganate back titration method
The redox reaction equation of the oxalic acid–permanganate back titration method can be denoted as follows:| (x − 1)C2O42− + 2xH+ + MnOx = Mn2+ + (2x − 2)CO2 + xH2O |
The obtained total Mn content and the Mn AOS of vernadite, hausmannite and acid birnessite are shown in Table 1. The percentages of total Mn in vernadite, hausmannite and acid birnessite are 47.16 ± 1%, 67.86 ± 1%, and 48.58 ± 2%, respectively. The Mn AOS was also calculated by the reaction equation. As a result, vernadite has a Mn AOS of 3.98 ± 0.04, and that of hausmannite and acid birnessite is 2.69 ± 0.03 and 4.05 ± 0.03, respectively.
| Sample | Element content (%) | AOSa | AOSb | ||
|---|---|---|---|---|---|
| Mn | K | Na | |||
| a Mn AOSs of the samples determined with oxalic acid–permanganate back titration method. b Mn AOSs determined through fitting of the XANES spectra with the Combo method. | |||||
| Vernadite | 47.16 ± 1 | 2.44 ± 0.01 | 2.11 ± 0.02 | 3.98 ± 0.04 | 3.80 ± 0.04 |
| Hausmannite | 67.86 ± 1 | 0.031 ± 0.01 | 0.055 ± 0.01 | 2.69 ± 0.03 | 3.20 ± 0.05 |
| Acid birnessite | 48.58 ± 2 | 7.66 ± 0.02 | 0.102 ± 0.01 | 4.05 ± 0.03 | 3.85 ± 0.04 |
The oxalic acid–permanganate back titration method has an advantage of high precision (±0.05) due to good repeatability of titration, and thus is widely used in the chemical determination of the Mn AOS of natural and synthetic Mn oxide samples.16,19,37 However, the titration must be conducted under strict heating conditions at a temperature between 75 and 85 °C to accelerate the reaction between oxalate and KMnO4, and prevent oxalate in the solution from thermal decomposition, making the operation of this method time-consuming and complicated.
3.3. Mn AOS calculated by fitting of Mn K-edge XANES
Mn AOS can be reflected by the line shapes and the white line position of K-edge XANES spectra.19 Mn K-edge XANES spectra of vernadite, hausmannite and acid birnessite are depicted in Fig. 3. The relative percentages of Mn2+, Mn3+ and Mn4+ of the samples were determined by LCF fitting with the Combo method which was performed in Manceau et al.21 The fitting results are presented in Fig. 3. The spectrum of vernadite is best-fitted with 3.22% Mn2+ + 13.3% Mn3+ + 83.48% Mn4+, that of hausmannite is best-fitted with 16.3% Mn2+ + 46.9% Mn3+ + 36.8% Mn4+, and that of acid birnessite is best fitted with 4.34% Mn2+ + 6.48% Mn3+ + 89.18% Mn4+.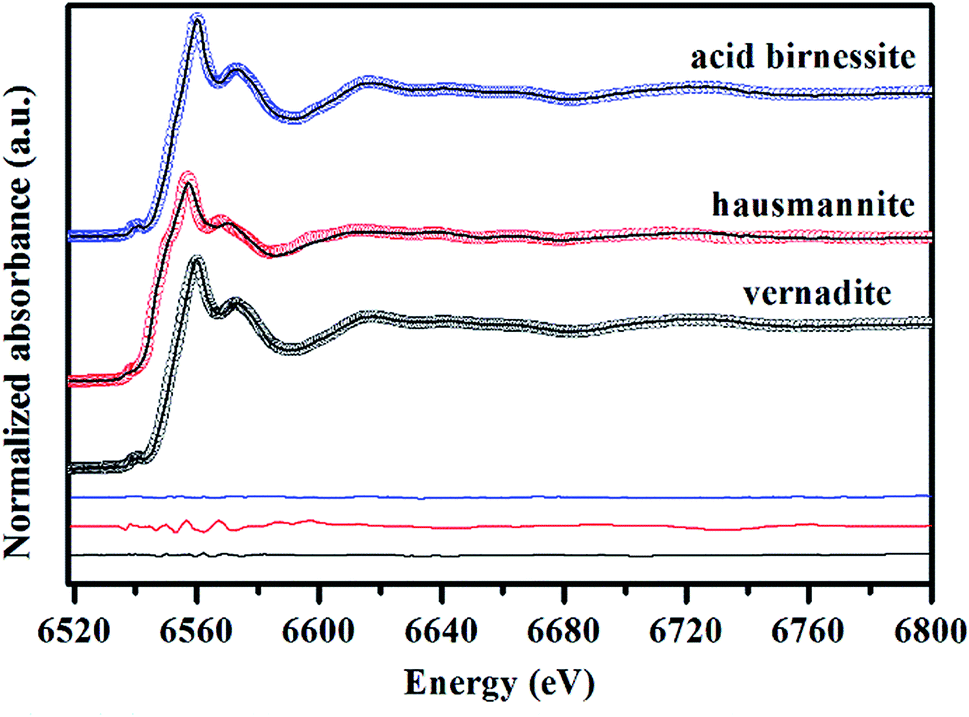 | ||
| Fig. 3 Fitting of Mn K-edge XANES spectra of vernadite, hausmannite and acid birnessite using the Combo method.21 Circles are experimental data, and lines are the best-fit linear combination of 17 references. Difference plots are shown at the bottom. | ||
According to the above results, the bulk Mn AOS of vernadite, hausmannite and acid birnessite is 3.80, 3.20 and 3.85, respectively. Generally speaking, the Combo method has a precision of ±0.05 in Mn AOS analysis. However, in the above results obtained using the Combo method, there is a quite larger amount of Mn3+ than expected for hausmannite, whose ideal Mn AOS is 2.67. It was also demonstrated by Manceau et al. that the accuracy of the Combo method is greatly decreased as the content of layer Mn3+ in manganates increases, which is due to the strong Jahn–Teller distortion effect of Mn3+ that affects the shape of the XANES spectra.21 Furthermore, the obtained Mn AOSs of vernadite and acid birnessite were about 0.2 lower than those derived from the oxalic acid–permanganate back titration method (Table 1), which might be ascribed to the apparent X-ray induced partial reduction due to their low crystallinity. In addition, for the XANES spectra method, sufficient standard samples and access to the synchrotron facilities are required for Mn AOS determination.
3.4. Mn AOS determination using the novel two-step colorimetric method
LBB solution can react with high-valent manganese to form a blue compound, oxidized LBB with a maximal adsorption at 620 nm in the spectrum (Fig. 4). Therefore, the absorption peak intensity of the blue complex at 620 nm is proportional to the concentration of high-valent manganese.28,31 In terms of KMnO4 solution, Mn7+ is reduced to Mn2+ by LBB with the simultaneous transference of five electrons, suggesting that the concentration of transferred electrons is five times the consumed KMnO4 concentration. Fig. 4 shows the standard curve of A620-CTE obtained by the reaction of KMnO4 with 0.04% LBB solution.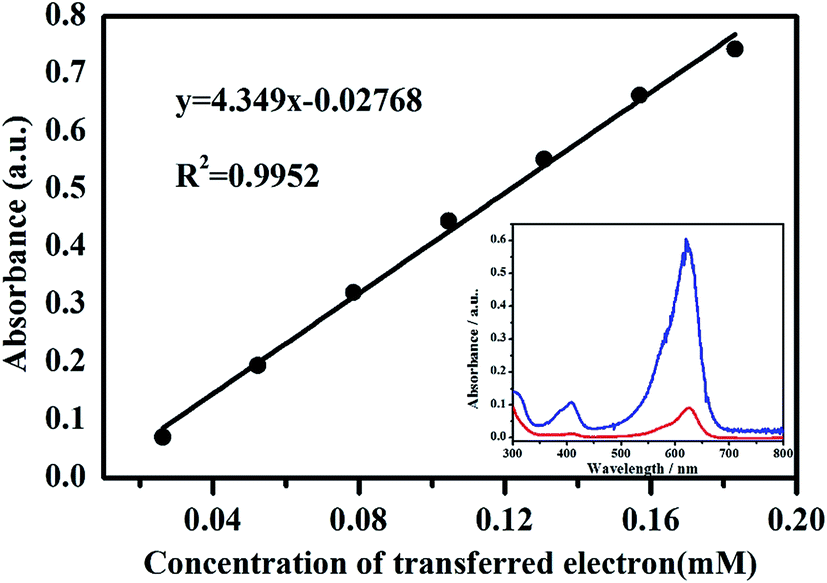 | ||
| Fig. 4 Standard curve of A620versus the transferred electrons concentration. The inset is the spectra of original LBB solution (red), and oxidized LBB solution (blue) after reaction with acid birnessite. | ||
It can be observed that A620 has a significantly positive linear relationship with the concentration of transferred electrons (R2 = 0.9952, n = 7). According to the standard curve of A620-CTE, the concentration of transferred electrons was calculated by:
| c(CTE) = (A620 + 0.02768)/4.349 | (1) |
In terms of manganese oxide minerals, Mn3+ and Mn4+ are reduced to Mn2+ quantitatively by LBB. The number of moles of CTE (n(CTE)) in the reaction system can be calculated as:
| n(CTE) = c(CTE) × V/1000 | (2) |
The percentages of total Mn of the samples are shown in Table 1, and the number of moles of total Mn was calculated by:
| n(Mntotal) = m × (Mntotal)%/MMn | (3) |
In the equations, m and V are the weight of Mn oxide mineral and the total volume of the mineral suspension, respectively, and MMn is the molecular weight of Mn.
The Mn AOS was calculated using the following equation:
| AOS(Mn) = n(CTE)/n(Mntotal) + 2, | (4) |
By combining eqn (1)–(4), the final equation for calculating the Mn AOS was established as follows:
| AOS(Mn) = 0.01265V(A620 + 0.02768)/m × (Mntotal)% + 2 | (5) |
Subsequently, the Mn AOS of the samples was calculated using eqn (5), and the results are shown in Table 2.
| Sample | m/g | A 620 | n (CTE)/mol | n (Mntotal)/mol | AOS |
|---|---|---|---|---|---|
| Vernadite | 0.0051 | 0.7197 | 8.593 | 4.373 | 4.00 ± 0.02 |
| 0.0050 | 0.7214 | 8.612 | 4.287 | ||
| 0.0050 | 0.7301 | 8.712 | 4.287 | ||
| Hausmannite | 0.0051 | 0.7052 | 4.213 | 6.292 | 2.67 ± 0.01 |
| 0.0051 | 0.7318 | 4.366 | 6.292 | ||
| 0.0052 | 0.6987 | 4.176 | 6.416 | ||
| Acid birnessite | 0.0052 | 0.7747 | 9.225 | 4.593 | 4.00 ± 0.01 |
| 0.0051 | 0.7528 | 8.973 | 4.505 | ||
| 0.0051 | 0.7652 | 9.116 | 4.505 |
Table 2 shows that the Mn AOS of vernadite, hausmannite and acid birnessite is 4.00 ± 0.02, 2.67 ± 0.01 and 4.00 ± 0.01, respectively. The precision of the two-step colorimetric method was estimated to be 0.01–0.02, which is close to that of the titration methods and higher than that of the Combo method; besides, this method shows good reproducibility. The better performance of this method than the Combo method may be ascribed to the partial reduction of high-valent Mn on the mineral surfaces by X-ray radiation during the spectrum collection.21
The above results showed that this two-step colorimetric method, the combination of leucoberbelin blue I (LBB) and formaldoxime colorimetry, is applicable to measure the Mn AOS in a rapid, convenient and highly accurate way relative to current Mn AOS determination methods. In contrast, the original colorimetric LBB method cannot directly provide the Mn AOS value. It is commonly used to detect whether and how much high-valent Mn (over +2 valence) is produced through an oxidation process in an experimental system, especially in a biotic system.32,33 However, besides the determination of the concentrations of high-valent Mn, it is more important to measure the Mn average oxidation state (AOS) of Mn oxides formed in a system with mixed valences in many scenarios, and chemical titration and spectroscopic analysis methods are usually used for the determination of the Mn AOS instead.18,19,21,22
In addition, it should be noted that both the Mn AOS and accurate mole fraction of each Mn oxidation state are important parameters for the properties of Mn oxides. When the latter is known, the former can be readily calculated. In some scenarios where only the value of the Mn AOS is required, such as the evaluation of oxidation degree/efficiency of a Mn2+ containing system during biotic or abiotic oxidation, or the determination of the chemical formula of a Mn oxide sample, either the conventional titration approach or the proposed two-step colorimetric method can be applied. But when it is important to know the proportion of each Mn oxidation state, such as the determination of the composition of various Mn oxides in a mixed Mn oxide sample or determination of the crystallochemical formula of a Mn oxide sample with mixed Mn valences, spectroscopic analysis (such as XPS or XANES) should be performed.
4. Conclusion
A novel two-step colorimetric method was developed for Mn AOS determination with LBB and formaldoxime colorimetric measurement. Using this method, the Mn AOS of synthetic vernadite, hausmannite and acid birnessite was determined to be 4.00 ± 0.02, 2.67 ± 0.01, 4.00 ± 0.01, respectively. These values are consistent with those obtained with the conventional oxalate reduction-KMnO4 back titration method. The results show that this two-step colorimetric method has the advantages of rapid detection, high sensitivity, color stability and being time saving. The precision of this method (about 0.01–0.02) are close to or higher than that of the oxalic acid–permanganate back titration method and XANES spectroscopy method. Besides, this method could eliminate the interference of Fe3+ on the Mn AOS, and the determination could be conducted at room temperature without the application of expensive apparatus. In addition, this novel method only requires a small amount of sample (0.01 g) for Mn AOS determination compared with other methods. Therefore, it is more suitable for trace micro-biological and chemical synthetic manganese oxide samples.Acknowledgements
The authors gratefully acknowledge the anonymous reviewers for their insightful comments. We thank the National Natural Science Foundation of China (Grant No. 41471194 & 41171197) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB15020402) for financial support of this research. We sincerely acknowledge Prof. Zuo Xiong Liu of the College of Foreign Languages of Huazhong Agricultural University for his kind help to review the final draft and improve the English.References
- A. T. Stone, K. L. Godtfredsen and B. Deng, Chemistry of Aquatic Systems: Local and Global Perspectives, in Sources and Reactivity of Reductants Encountered in Aquatic Environments, ed. Giovanni Bidoglio(s), Springer, Netherlands, 1994, pp. 337–374 Search PubMed.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS.
- J. T. Kay, M. H. Conklin, C. C. Fuller and P. A. O'Day, Environ. Sci. Technol., 2001, 35, 4719–4725 CrossRef CAS PubMed.
- B. M. Tebo, J. R. Bargar, B. G. Clement, G. J. Dick, K. J. Murray, D. Parker, R. Verity and S. M. Webb, Annu. Rev. Earth Planet. Sci., 2004, 32, 287–328 CrossRef CAS.
- H. Yin, W. F. Tan, L. R. Zheng, H. J. Cui, G. H. Qiu, F. Liu and X. H. Feng, Geochim. Cosmochim. Acta, 2012, 93, 47–62 CrossRef CAS.
- M. Villalobos, J. Barger and G. Sposito, Environ. Sci. Technol., 2005, 39, 569–576 CrossRef CAS PubMed.
- M. Villalobos, M. Carrillo-Cárdenas, R. Gibson, N. R. López-Santiago and J. A. Morales, Environ. Chem., 2014, 11, 279–288 CrossRef CAS.
- H. Yin, X. H. Feng, G. H. Qiu, W. F. Tan and F. Liu, J. Hazard. Mater., 2011, 188, 341–349 CrossRef CAS PubMed.
- S. H. Lee, T. W. Kim, D. H. Park, J. H. Choy, S. J. Hwang, N. Z. Jiang, S. E. Park and Y. H. Lee, Chem. Mater., 2007, 19, 5010–5017 CrossRef CAS.
- W. Zhao, H. Cui, F. Liu, W. F. Tan and X. H. Feng, Clays Clay Miner., 2009, 57, 513–520 CrossRef CAS.
- J. J. Lingane and R. Karplus, Ind. Eng. Chem., Anal. Ed., 1946, 18, 191–194 CrossRef CAS.
- K. J. Vetter and N. Jaeger, Electrochim. Acta, 1966, 11, 401–419 CrossRef CAS.
- J. W. Murray, L. S. Balistrieri and B. Paul, Geochim. Cosmochim. Acta, 1984, 48, 1237–1247 CrossRef CAS.
- D. Z. Piper, J. R. Basler and J. L. Bischoff, Geochim. Cosmochim. Acta, 1984, 48, 2347–2355 CrossRef CAS.
- J. H. Carpenter, Limnol. Oceanogr., 1965, 10, 135–143 CrossRef CAS.
- N. Kijima, H. Yasuda, T. Sato and Y. Yoshimura, J. Solid State Chem., 2001, 159, 94–102 CrossRef CAS.
- S. Grangeon, A. Manceau, J. Guilhermet, A. C. Gaillot, M. Lanson and B. Lanson, Geochim. Cosmochim. Acta, 2012, 85, 302–313 CrossRef CAS.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- M. Villalobos, B. Toner, J. Bargar and G. Sposito, Geochim. Cosmochim. Acta, 2003, 67, 2649–2662 CrossRef CAS.
- S. Grangeon, B. Lanson, N. Miyata, Y. Tani and A. Manceau, Am. Mineral., 2010, 95, 1608–1616 CrossRef CAS.
- A. Manceau, M. A. Marcus and S. Grangeon, Am. Mineral., 2012, 97, 816–827 CrossRef CAS.
- K. J. T. Livi, B. Lafferty, M. Zhu, S. Zhang, A. C. Gaillot and D. L. Sparks, Environ. Sci. Technol., 2012, 46, 970–976 CrossRef CAS PubMed.
- J. W. Murray, L. S. Balistrieri and B. Paul, Geochim. Cosmochim. Acta, 1984, 48, 1237–1247 CrossRef CAS.
- J. L. Mansot, P. Leone, P. Euzen and P. Palvadeau, Microsc., Microanal., Microstruct., 1994, 5, 79–90 CrossRef CAS.
- I. Saratovsky, P. G. Wightman, P. A. Pastén, J. F. Gaillard and K. R. Poeppelmeier, J. Am. Chem. Soc., 2006, 128, 11188–11198 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- R. P. Gupta and S. K. Sen, Phys. Rev. B: Solid State, 1975, 12, 15–19 CrossRef CAS.
- D. Banerjee and H. W. Nesbitt, Geochim. Cosmochim. Acta, 2001, 65, 1703–1714 CrossRef CAS.
- V. R. Galakhov, M. Demeter, S. Bartkowski, M. Neumann, N. A. Ovechkina, E. Z. Kurmaev, N. I. Logachevskaya, Y. M. Mukovskii, J. Mitchell and D. L. Ederer, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 113102 CrossRef.
- H. J. Altmann, Fresenius' Z. Anal. Chem., 1972, 262, 97–99 CrossRef CAS.
- W. E. Krumbein and H. J. Altmann, Helgoland Mar. Res., 1973, 25, 347–356 CAS.
- R. M. McKenzie, Mineral. Mag., 1971, 38, 493–502 CAS.
- E. Burle and W. W. Kirby-Smith, Estuaries, 1979, 2, 198–201 CrossRef.
- S. M. Webb, Phys. Scr., 2006, 2005, 1011–1014 Search PubMed.
- V. A. Drits, E. Silvester, A. I. Gorshkov and A. Manceau, Am. Mineral., 1997, 82, 946–961 CAS.
- D. S. Freeman and W. G. Chapman, Analyst, 1971, 96, 865–869 RSC.
| This journal is © The Royal Society of Chemistry 2017 |