
Soluble lipoprotein receptor-related protein immunoreactive species in cell culture media and serum replacement supplements
Chantel
Fitzsimmons
*ab,
Daniel
Johnstone
 a,
Katherine
Conant
b,
Coryse
St Hillaire
b,
Carl H.
Parsons
c,
Monique
Stins
b,
Robert
Moir
d and
Elizabeth A.
Milward
ab
a,
Katherine
Conant
b,
Coryse
St Hillaire
b,
Carl H.
Parsons
c,
Monique
Stins
b,
Robert
Moir
d and
Elizabeth A.
Milward
ab
aSchool of Biomedical Sciences, University of Newcastle and Hunter Medical Research Institute, School of Biomedical Sciences (MSB 615), Department of Molecular Neurobiology University Dr, Callaghan, NSW 2308, Australia. E-mail: Chantel.fitzsimmons@newcastle.edu.au; Fax: +61 2 4921 7903; Tel: +61 2 4921 5549
bDepartment of Neurology, Johns Hopkins University, Baltimore, MD 21287, USA
cSchool of Medicine, Western Sydney University, Campbelltown, NSW 2560, Australia
dGenetics and Aging Research Unit, MassGeneral Institute for Neurodegenerative Disease, Department of Neurology, Massachusetts General Hospital, Charlestown 02129, USA
First published on 29th November 2016
Abstract
The low-density lipoprotein receptor-related protein (LRP) is a large multifunctional cell surface membrane receptor capable of binding over 50 ligands. These include molecules important in Alzheimer's disease such as the amyloid β-protein precursor (AβPP), the β-amyloid (Aβ) peptide and apolipoprotein E (ApoE). Full length LRP consists of a 515 kDa extracellular ligand binding α-chain and an 85 kDa membrane spanning β-chain. A soluble form of LRP (sLRP) present in human plasma retains the ability to bind ligands, including Aβ. This soluble form is an ectodomain fragment generated from the membrane bound form of the receptor by proteolytic cleavage. Here we report data demonstrating that some commercial ‘serum-free’ supplements and ‘serum-free’ media contain unlisted sLRP immunoreactive species that may reflect the presence of undefined serum protein extracts in these ‘serum-free’ preparations. This has the potential to interfere with experimental results and interpretation in a range of cell culture studies involving LRP or any of its ligands and possibly also other serum proteins.
Introduction
The low-density lipoprotein receptor-related protein (LRP) is a large transmembrane receptor expressed in a wide range of tissues including liver and brain, as reviewed elsewhere.1–4 It has both scavenging and signalling receptor functions. Over 50 ligands have been identified, including lipoproteins, proteases, growth factors, viral components and cytoplasmic adaptor and scaffolding proteins involved in regulating mitogen activated kinase signalling, cell adhesion, migration and proliferation, calcium signalling and neurotransmission in neural cells.1–4 The LRP receptor is proposed to participate in diverse physiological processes, including immune responses, signal transduction and memory. It is also thought to play a role in several diseases, notably Alzheimer's disease (AD), cardiovascular disease and HIV AIDS.1–4 The LRP ligands AβPP, Aβ and ApoE are implicated in AD pathogenesis. LRP is thought to play a key role in clearing Aβ from brain, both by direct binding of the peptide and through translocation of ApoE, its molecular chaperon.5–7Mature LRP contains two non-covalently associated subunits, the 515 kDa α-chain and the 85 kDa β-chain. The extracellular α-chain contains multiple ligand binding domains. The membrane spanning β-chain includes both extracellular and intracellular domains.8 Cleavage of mature membrane associated LRP within its extracellular domain releases soluble LRP (sLRP) fragments containing both the α and β chains into the extracellular environment.2,9,10 Soluble LRP immunoreactive species have been detected in human plasma,9 as well as in several other mammalian, avian and reptilian sera.11 The main sLRP immunoreactive species studied to date retain the ability to bind at least some of the ligands for membrane bound LRP, including the Aβ peptide. However, species bound to sLRP are not internalized and may provide a pool of competitive antagonists of ligand uptake by functional membrane bound LRP.12 The presence of sLRP in culture media supplements such as bovine fetal calf serum may be an issue for in vitro cell culture models. Exogenous sLRP in the culture media may interfere with the normal function of cell associated LRP since these species are likely to compete for ligand binding.
We have examined reagents commonly used for culturing neuronal cells and found that sLRP immunoreactive species are present not only in serum-containing cell culture media, but also in some commercial ‘defined’ or ‘serum-free’ media. The commercial ‘defined’, or ‘serum-free’ media do not list LRP as a component. We have identified a cell culture medium that does not contain detectable LRP immunoreactive species. In addition, we have demonstrated that this reagent can be used under serum-free experimental conditions for short to medium incubation periods with at least two neural cell lines.
Materials and methods
Antibodies
For Western blot, the goat polyclonal antibody (pAb) N20 and mouse monoclonal antibody (mAb) 8G1 (Santa Cruz, CA, USA) and mAb 3402 (American Diagnostica, Flanders, NJ, USA) were used against the LRP α-chain. The chain mouse mAbs 8B8 and 5A6 (Santa Cruz) and pAb mouse anti-LRP/A2MR (American Diagnostica) were used against the LRP β-chain. Western blots also used an isotype control for primary mouse mAbs (Invitrogen, Chicago, IL, USA) and the secondary donkey anti-goat IgG-HRP antibody (Santa Cruz and Promega, Annandale, NSW, Australia).Cell culture
The cells used were the SHSY-5Y line, derived from a human neuroblastoma,13 and the PC12 line, from a rat pheochromocytoma.14 Both cell lines were kindly provided by Dr Adam Carroll (The University of Newcastle, NSW, Australia). The cells lines were originally purchased from the American Type Culture Collection (ATCC). The SHSY-5Y cells were maintained in Dulbecco's modified Eagle medium (DMEM), containing 10% heat inactivated fetal bovine serum (FBS) (Invitrogen). The PC12 cells were maintained in DMEM containing 5% heat inactivated FBS and 10% heat inactivated fetal equine serum (FES; Hunter Antisera; Jesmond, NSW, Australia). Each growth media also contained 10 mM HEPES buffer, 0.15% sodium bicarbonate, 2 mM L-glutamine and 100 units per ml penicillin and 100 μg ml−1 streptomycin (Invitrogen). Cell lines were maintained under standard cell culture conditions (37 °C, 5% CO2 in a humidified incubator). Serum reduced or serum-free media supplements used in the study were B27 supplement, Neurobasal Medium and N2 supplement (Invitrogen), and ThermoTrace modified Eagle medium (TMEM) (ThermoTrace, Noble Park, VIC, Australia).Ion exchange chromatography
To improve visualization of sLRP species, in accordance with the published protocol for sLRP detection8 unconditioned media and ‘serum-free’ supplements were concentrated up to 10-fold by ion exchange chromatography on DEAE Sephacel gel (GE Healthcare; Pittsburgh, PA, USA), according to the manufactures instructions. The columns were eluted with 100 μl aliquots of high salt MES, consisting of 0.5 M NaCl in 20 mM MES (pH 6.0). The eluted fractions were analysed by Western immunoblotting, with or without prior immunoprecipitation, as described below.Immunoprecipitation (IP)
For IP, samples comprising 500 μl of DEAE sephacel concentrated fractions or cell culture medium or supplements were pre cleared with 50 μl of 50% protein G Sepharose 4B beads slurry (Invitrogen) for 1 hour at 4 °C. Samples were then incubated overnight at 4 °C in the presence of 10 μg of various anti-LRP antibodies (N20, 8G1, 5A6 or 8B8) in the presence of the isotype control (Invitrogen). After incubation with 100 μl of 50% protein G Sepharose 4B beads slurry (4 h, 4 °C), the beads were pelleted by micro centrifugation (61g, 1 min, 4 °C). The supernatant was removed and the beads washed twice with ice cold IP buffer (50 mM Tris HCL, 1% Triton-X 100, protease inhibitor cocktail, pH 8.0), twice with ice cold IP wash buffer (10 mM Tris HCL pH 8.0, 0.2% Triton-X 100, protease inhibitor cocktail, pH 8.0), once with ice cold phosphate buffered saline (PBS; 0.15 M NaCl, 0.01 M NaPO4, pH 7.2) containing 0.1% Triton-X and once with PBS without detergent (0.15 M NaCl, 0.01 M NaPO4). All washes were in 1 ml buffer for 5 min, at 4 °C. The final pellet was resuspended in non-reducing SDS-PAGE buffer, heated 5 min at 95 °C then microfuged (61g, 1 min, RT) and the supernatant collected for Western blot analysis.SDS PAGE and Western blot analysis
Supernatants obtained by IP, as above, were applied directly to gels for electrophoresis, as below. The protein content of all other samples was determined using a bicinchoninic acid assay (Pierce; Rockford, IL, USA), according to the manufacturer's instructions, and 40 μg total protein was applied per gel lane. Electrophoresis was performed in running buffer (25 mM Tris HCl, 192 mM glycine, 0.1% SDS, pH 8.3) at 100–150 V for 1–2 h on 7.5% or 10% SDS-PAGE mini gels under non-reducing conditions. Dual colour Precision plus protein standards (BioRad, Richmond, CA, USA) were used for molecular weight estimation in the range of 10 kDa to 250 kDa. HiMark pre-stained protein standards (Invitrogen) were used for estimations in the range of 250 kDa to 450 kDa.To provide an additional high molecular weight marker to assist in estimating the size of high molecular weight LRP immunoreactive species some gels also included a preparation of sheep cardiac ryanodine receptor (RyR; molecular weight ∼ 560 kDa), detected by immunolabeling using mouse anti-RyR antibody C333 recognizing sheep RyR, generously provided by A./Prof. Derek Laver (University of Newcastle, Australia) and Dr Paul Allen (Brigham and Women's Hospital, Boston, USA).
The gels were electroblotted onto Hybond P polyvinylidene fluoride (PVDF) membranes in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) at 0.35A, 4–6 °C for 1–2 h. The membranes were blocked in 5% non fat dried milk in PBS containing 0.1% Tween 20 (PBS-T) at RT for 1 h prior to incubation in anti-LRP antibodies diluted in 0.1% PBS-T (v/v). A different primary anti-LRP antibody was used for Western blot analyses from that used for immunoprecipitation (see Results). After incubation with primary antibody overnight at 4 °C, membranes were washed 3 × 5 min in 0.1% PBS-T and incubated at RT for 1 h with the appropriate secondary antibody diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000 (v/v) in 0.1% PBS-T. Membranes were then washed 3 × 5 min in 0.1% PBS-T and treated with enhanced chemiluminescence (ECL+) solution (GE Healthcare), according to the manufacturer's instructions, and scanned with the FUJI LAS 1000 imaging system (Fuji, Tokyo) or the Typhoon 9410 Variable Mode Imager (Molecular Dynamics, USA).
:5000 (v/v) in 0.1% PBS-T. Membranes were then washed 3 × 5 min in 0.1% PBS-T and treated with enhanced chemiluminescence (ECL+) solution (GE Healthcare), according to the manufacturer's instructions, and scanned with the FUJI LAS 1000 imaging system (Fuji, Tokyo) or the Typhoon 9410 Variable Mode Imager (Molecular Dynamics, USA).
Amino acid sequencing
For amino acid sequencing, after transfer to PVDF, proteins were stained with 0.5% (w/v) Coomassie blue R 250/15% acetic acid (v/v)/30% methanol (v/v). Bands of ∼500 kDa, ∼275 kDa, ∼65 kDa and ∼55 kDa were excised and sequenced by automated Edman degradation at the Synthesis & Sequencing Facility, Johns Hopkins University, Baltimore, MD, USA or the Biomolecular Research Facility, The University of Newcastle, NSW, Australia. For immunoblotting prior to N-terminal protein sequencing by the Biomolecular Research Facility, a Tris borate transfer buffer (50 mM Tris base, 50 mM boric acid, pH 8.5) was used.Cell viability assay
The PC12 or SHSY-5Y cells were grown overnight in the appropriate serum-containing growth media to ∼100% confluence in 96 well plates. Once confluence was reached, the serum-containing growth medium was removed and the cell monolayers were washed three times with sterile PBS. Various serum-free media (see Results) were added at a volume of 100 μl per well. The cells were incubated under standard cell culture conditions for selected time periods (1 h, 2 h, 4 h, 6 h, 8 h or 24 h). Cell viability in the serum-free media at each time period was determined according to the manufacturer's instructions using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega), based on reduction of a tetrazolium compound (3-(4,5-dimethylthiazol 2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; MTS). The absorbance at 490 nm was measured using a Bio Rad Model 550 Microplate Reader.RNA interference
The 25-base oligonucleotide Stealth siRNA™ CCGCUCUGAUGAGUCUGCUUCUGU targeting LRP-1, together with the complementary sequence and Negative Control Med GC duplex, were pre-designed, synthesised and validated by Invitrogen. Transfection of SHSY-5Y cells was performed using 100 μM siRNA, Opti-MEM® reduced serum medium (Invitrogen) and Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. The cells were co-transfected with 1 μg of pEGFP-N1 plasmid to estimate transfection efficiency and were incubated under standard cell culture conditions (37 °C, 5% CO2) for 6 h. After 6 h the complexes were replaced with serum-containing growth media and the cells were left to incubate for a further 18 h. The serum-containing growth medium was replaced with Neurobasal medium supplemented with N2 (Invitrogen) and after a further 4 h the conditioned medium was harvested for LRP analysis.RNA extraction
Total RNA extraction was performed using TRIzol reagent (Invitrogen), according to the manufacturer's instructions. Cells were harvested for RNA extraction at 28 h. The RNA was separated from remaining cellular macromolecules by differential extraction and precipitation with isopropanol. The pellet was washed with 75% ethanol and centrifuged (7500g, 5 min, 4 °C). The pellet was air-dried and dissolved in UltraPure™ DNase/RNase-free distilled water. Total RNA was quantitated by UV spectrophotometric analysis using the Beckman DU-640 spectrophotometer (Beckman Coulter Inc, Fullerton, CA, USA).Real-time PCR
The RNA were reverse transcribed using the high capacity cDNA reverse transcription kit (Applied Biosystems; Foster City, CA, USA), according to the manufacturer's instructions. Each reaction contained 2 μl diluted RNA (500 ng μl−1), 2 μl 10× RT buffer, 0.8 μl 25× dNTP mix (100 mM), 2 μl 10× random primers, 1 μl MultiScribe™ reverse transcriptase and 12.2 μl UltraPure™ DNase/RNase-free distilled water. For PCR, the oligonucleotide primers were designed to have an optimal annealing temperature of 60 °C. Primers were designed using Primer Express III software (Applied Biosystems) and purchased through Sigma-Aldrich. The primers are as follows: LRP-1: forward primer (5′-3′) CCCATCTTTGAGATCCGAATGT; LRP-1: reverse primer (5′-3′) ATTGTTCACCCGGCATTTGT.Each 20 μl reaction contained 1× Power SYBR Green PCR master mix (Applied Biosystems), 200 nM forward oligonucleotide primers, 200 nM reverse oligonucleotide primers and 6 μl cDNA. Each reaction was performed in triplicate in a 96-well plate format. UltraPure™ DNase/RNase-free distilled water was substituted for cDNA to serve as a negative control. A negative control (no cDNA template) was also included for each primer set to ensure reagents were free from nucleic acid contamination. The housekeeping gene, β-actin, was used as the reference gene and SYBR green was used as the detector. The cDNA was amplified using the ABI 7500 PCR machine (Applied Biosystems) under the following conditions; 1 cycle 50 °C/2 min, 1 cycle 95 °C/10 min and 40 cycles 95 °C/15 s; 60 °C s−1. As a form of quality control, the dissociation curve generated following amplification was examined to ensure the observed fluorescence was not due to primer dimers, the co-amplification of genomic DNA or other non-specific products. Gene expression was quantitated relative to β-actin.
Statistical analysis
Statistical significance was assessed by two-tailed t-test where p < 0.05 was required for significance.Results
sLRP immunoreactive species on immunoblots of ‘serum-free’ supplements and media
On Western blots, signal for several sLRP-α immunoreactive species was detected in unconditioned samples of two serum-containing media (PC12 medium containing 10% FES, 5% FBS and SHSY-5Y medium containing 10% FBS; Fig. 1A, Lanes 1, 2 respectively) but not in serum-free DMEM or PBS (Fig. 1A, Lanes 7, 8 respectively). The presence of soluble anti-LRP-α immunoreactive species in serum-containing media is consistent with previous reports of sLRP-α immunoreactive species in mammalian plasma.9 Signal included immunoreactive bands at ∼275 kDa and ∼500 kDa, the size of the sLRP-α form previously reported in plasma.9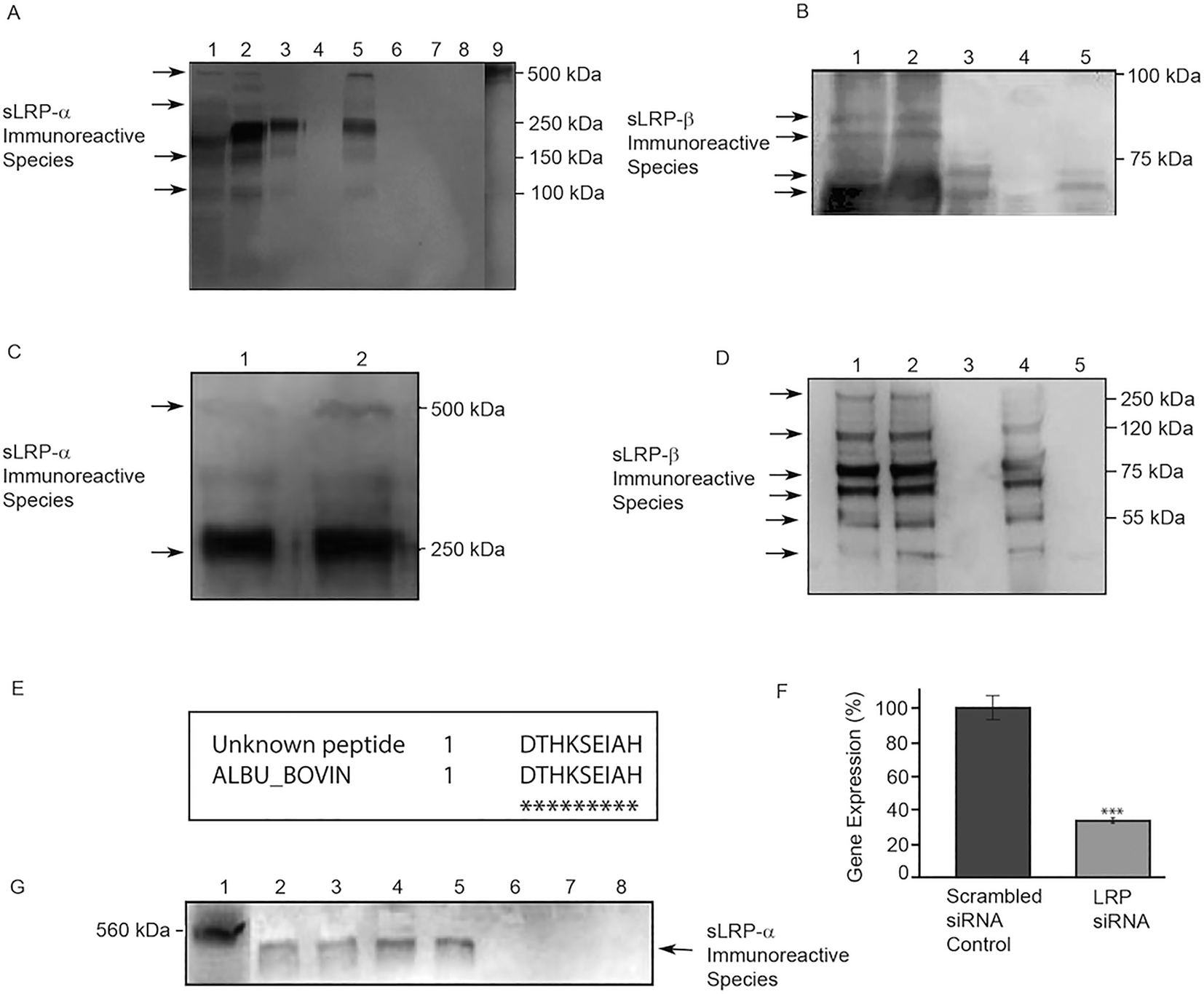 | ||
| Fig. 1 Presence of sLRP immunoreactive species in cell culture media and supplements. (A) Western blot of sLRP-α immunoreactive species (3402 antibody). (Lane 1) DMEM + 5% FBS, 10% FES (PC12 growth medium), (2) DMEM + 10% FBS (SHSY-5Y growth medium), (3) TMEM, (4) Neurobasal + N2, (5) Neurobasal + B27, (6) Neurobasal, (7) DMEM, (8) PBS, (9) RyR (labelled separately with C333); (B) Western blot of sLRP-β immunoreactive species (anti-LRP/A2MR antibody). (Lane 1) DMEM + 5% FBS, 10% FES (PC12 growth medium), (2) DMEM + 10% FBS (SHSY-5Y growth medium), (3) TMEM, (4) Neurobasal + N2, (5) Neurobasal + B27. (C) Western blot of sLRP-α immunoreactive species in B27. (Lane 1) B27 immunoprecipitated with N20 and probed with 8G1, 2. B27 immunoprecipitated with 8G1 and probed with N20. (D) Western blot of sLRP-β immunoreactive species in B27 and in Neurobasal medium detected by IP with 5A6 and Western immunoblotting with 8B8. (Lane 1) B27, (Lane 2) B27 (purified by DEAE Sephacel ion exchange chromatography), (Lane 3) mouse IgG mAb isotype control, (Lane 4) B27 (purified using an albumin removal column), (Lane 5) Neurobasal medium. (E) The amino acid sequence of the unknown ∼65 kDa peptide (single letter amino acid code) was found to correspond with peptides within the published sequence of serum albumin from Bos taurus (bovine) (accession no. P02769). The stars indicate amino acid matches and flanking numbers indicate the position of amino acids within the sequence. (F) Real time RT-PCR analysis of LRP gene expression after gene silencing by siRNA. The level of LRP mRNA transcripts relative to transcripts for the β-actin reference gene was quantitated by real time RT-PCR after LRP siRNA treatment. Cells transfected with scrambled siRNA served as the negative control. Data are presented as mean ± SEM (n = 4 biological replicates). Statistical analysis was performed using a two-tailed t-test where *** indicates a significant difference in comparison to the scrambled siRNA control at a level of p < 0.001. (G) The effect of LRP gene knockdown on sLRP-α expression. Representative Western immunoblot of LRP immunoreactive species detected after gene silencing using siRNA targeted against human LRP. Western immunoblots were probed with the anti-LRP-α antibody N20. | ||
Unexpectedly, defined ‘serum-free’ medium TMEM also contained sLRP-α immunoreactive species (Fig. 1A, Lane 3). The major sLRP-α immunoreactive species in TMEM co-migrated with the strongest immunoreactive band in fresh, unconditioned, serum-containing media (∼275 kDa). The ∼500 kDa anti-LRP-α immunoreactive form was not detected in defined medium TMEM but two other minor bands were detected (∼130 kDa and ∼200 kDa), co-migrating with minor bands present in the serum-containing media (Fig. 1A, Lanes 1, 2). Signal was also observed in different batches of fresh, unconditioned medium. Identity of sLRP-α immunoreactive bands was further verified using different commercial primary and secondary antibodies from independent sources.
Serum also contains soluble forms of the LRP β-chain.10 Two major sLRP-β immunoreactive bands were detected in TMEM at ∼60 kDa and ∼65 kDa (Fig. 1B, Lane 3), that co-migrated with bands in fresh, unconditioned serum-containing media (Fig. 1B, Lane 1, 2).
We next tested if Neurobasal medium or the two serum-free medium additives B27 and N2 contain sLRP-α or sLRP-β immunoreactive species. Neurobasal medium alone or supplemented with N2 (Neurobasal + N2) did not contain any detectable sLRP-α immunoreactive species (Fig. 1A, Lanes 6, 4 respectively). There were also no detectable sLRP-β immunoreactive species in Neurobasal + N2 medium (Fig. 1B, Lane 4). In contrast, when B27 was added to Neurobasal medium, strong signals were observed with both anti-sLRP-α and anti-sLRP-β probes. The major anti sLRP-α immunoreactive species in B27 (∼275 kDa) co-migrated with the major band detected in serum supplemented media and TMEM. However the ∼500 kDa form was also detected in B27, unlike in TMEM (Fig. 1A, Lanes 5, 3 respectively). In addition, B27 supplemented Neurobasal medium contained a ∼65 kDa anti-LRP-β immunoreactive species (Fig. 1B, Lane 5) that co-migrated with a band in fresh, unconditioned serum-containing media (Fig. 1B, Lanes 1, 2).
Detection of sLRP immunoreactive species by immunoprecipitation and immunoblotting
Fresh B27 supplement was then examined directly, without dilution in Neurobasal medium. Experiments used IP in combination with Western blot analysis to further investigate specificity and reproducibility across commercial batches. Samples were first immunoprecipitated with either N20 (α-chain) or 5A6 (β-chain) antibodies. Western blots of immunoprecipitated samples were then probed with a different α-chain (8G1) or β-chain (8B8) anti-LRP antibody. Experimental controls included tests in which the IP and probing antibodies were switched. High signals for both anti-LRP-α (Fig. 1C) and anti-LRP-β (Fig. 1C) confirmed immunoreactive species were present in all B27 supplement batches tested (Fig. 1C). Two major bands of sLRP-α immunoreactivity were observed at ∼500 kDa and ∼275 kDa (Fig. 1C, Lanes 1, 2), with six visible bands of sLRP-β immunoreactivity at ∼275 kDa, ∼120 kDa, ∼75 kDa, ∼65 kDa, ∼55 kDa and ∼35 kDa (Fig. 1D, Lanes, 1, 2). Precipitation with a mouse IgG isotype control gave no signal (Fig. 1D, Lane 3).Amino acid sequencing
LRP immunoreactive bands at ∼500 kDa, ∼65 kDa and ∼55 kDa from ‘serum-free’ B27 supplement were excised and subjected to N-terminal amino acid sequencing. The ∼500 kDa and ∼55 kDa species were found to be N-terminally blocked. Sequence data were obtained for the ∼65 kDa band however BLAST searching indicated that the predominant peptide contributing to this band corresponded to a region within bovine serum albumin (accession no. P02769; Fig. 1E). Albumin is the major protein component of B27. Treatment of B27 using the ProteoExtract™ Albumin Removal kit did not eliminate sLRP-β immunoreactivity (Fig. 1D).Short term viability of cells in serum-containing or serum-free media
Our findings suggested that Neurobasal + N2 medium is free of detectable levels of anti-LRP immunoreactive protein and may be a suitable culture medium for studies on LRP. However cells can be sensitive to culture conditions and could potentially be adversely affected by the absence of unidentified trace serum components in our proposed alternative medium. We compared different media for cell viability by MTS assay. Media tested for the SHSY-5Y cell line were (i) SHSY-5Y growth media, (ii) SHSY-5Y growth media without serum (comprising DMEM supplemented with HEPES, sodium bicarbonate, glutamine and antibiotics, as detailed in Methods), (iii) Neurobasal medium supplemented with N2 and (iv) DMEM alone (Fig. 2).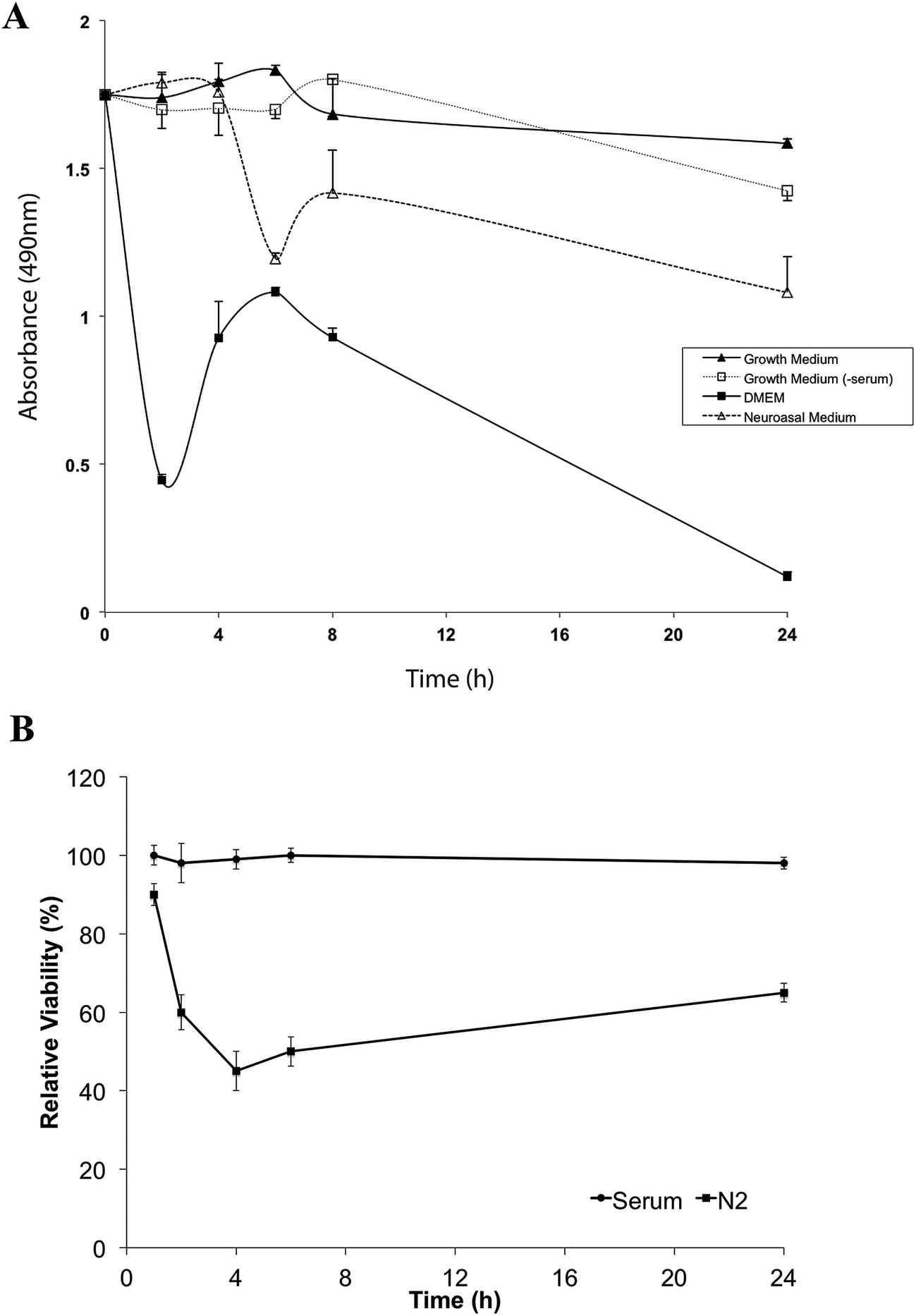 | ||
| Fig. 2 Short term viability of SHSY-5Y or PC12 cells in serum-containing or serum-free media. Confluent (A) SHSY-5Y or (B) PC12 cell cultures were maintained in serum-containing or serum-free media for periods up to 24 h. Cell viability was determined at 2 h, 4 h, 6 h, 8 h and 24 h using the MTS assay. Data are presented as mean ± SEM (n = 5 biological replicates). | ||
Cell viability fell markedly in DMEM alone and at 24 h fewer than 10% of cells remained viable. Viability of SHSY-5Y cells did not differ significantly at 4 h between serum-containing media, serum-free medium and Neurobasal + N2 medium. By 6 h, cell viability in Neurobasal + N2 had begun to decline and by 24 h only just over 50% of the cells remained viable. In SHSY-5Y growth media without added serum, cell viability was maintained for at least 8 h, declining to around 70% viability by 24 h (Fig. 2). Serum-free conditions resulted in rapid decline in the viability of the PC12 cell line with less than 60% of PC12 cells viable after 1 h in Neurobasal + N2 (Fig. 2B).
RNA interference
RNA interference was used to confirm the identity of the major ∼500 kDa secreted species detected by antibodies to the LRP-alpha chain. As described in Materials and methods, siRNA targeting human LRP-1 was used to silence cellular LRP-1 gene expression, with transcript reduction by ∼70% confirmed by real time RT-PCR (Fig. 1F). After LRP-1 RNA depletion, the soluble 500 kDa species was barely detectable in immunoblots of conditioned cell medium (Fig. 1G).Discussion
Serum contains many substances that affect cell survival, proliferation, growth and functions and often vary greatly between batches. Serum-free media can be used to regulate cell culture conditions more stringently but usually require supplementation with defined sets of proteins and other factors to preserve the growth and long-term viability of cells. ‘Serum-free’ media and additives are widely assumed to be devoid of undefined serum proteins. However, as our data show, the commercial ‘serum-free’ medium TMEM and ‘serum-free’ supplement B27 contain albumin and considerable quantities of sLRP-α and sLRP-β immunoreactive species that have the potential to compromise studies of LRP in cell culture models.The liver may be the main source of the sLRP in blood since it is a major site of LRP synthesis and plasma sLRP concentrations alter in some patients with impaired liver function.8 Soluble LRP-α species ranging between ∼200 kDa and ∼500 kDa that retain ligand binding capabilities are present in media conditioned by primary rat hepatocytes.8 Although most ligand binding domains in LRP are within the α-chain, at least one sLRP-β chain form might also have ligand binding capabilities. Specifically, a prominent species of ∼65 kDa has been detected with an antibody to a β-chain epitope in human plasma after concentration using an affinity matrix comprising Sepharose coupled to recombinant receptor-associated protein (RAP), an LRP ligand.8 While we observed a sLRP-β immunoreactive species of ∼65 kDa, we were unable to confirm this by N-terminal sequencing, due to the presence in B27 of large amounts of albumin, which has a similar molecular weight.
There were also two relatively large species of 275 kDa and 120 kDa detected by antibodies to the β-chain, which is generally considered to be present as smaller species of ∼55–85 kDa. Proteins were concentrated under non-denaturing conditions that would be predicted to maintain tertiary structure and Western immunoblots were done under non-reducing conditions likely to maintain disulfide bonds. The LDL receptor can form homodimers in mouse neuroblastoma and human neuroglioma cell models and primary cortical cultures from adult mouse brain15 and it is possible that sLRP species can also form dimeric aggregates under some conditions. The 275 kDa and 120 kDa bands could therefore represent dimeric species that include one or more epitopes recognized by β-chain antibodies. For example, the 275 kDa band could represent a heterodimeric species where part of the α-chain is still associated with part of the β-chain, resulting in a soluble form recognised by β-chain antibodies. It has been reported that truncated β-chain can co-purify with the LRP α-chain.8,10 Similarly it is possible that the 120 kDa band could correspond to a glycosylated homodimer derived from two 55 kDa β-chains.
Due to N-terminal blockage, we were unable to confirm by sequencing the identity of bands corresponding in size to the two main previously reported sLRP immunoreactive species of ∼500 kDa and ∼55 kDa, corresponding to sLRP-α and sLRP-β respectively, but the identity of the ∼500 kDa species has been further confirmed by LRP gene silencing. As above, both of these forms are likely to retain ligand-binding capabilities.9,10
As noted above, the various bands were immunoreactive to multiple anti-LRP antibodies that are widely used in studies of LRP. The presence of LRP immunoreactive species in the serum-free supplements compromises in vitro studies of LRP with these antibodies. It is therefore important to ensure that use of these supplements is not affecting results. One way of testing this is to perform comparison studies using alternative media preparations that do not contain cross-reactive species. We have been able to identify commercially available serum-free media and supplements that do not contain LRP immunoreactive species that maintain cell viability over time courses sufficient for many experimental purposes. Our data suggest that either SHSY-5Y growth medium alone in the absence of added serum or Neurobasal medium supplemented with N2 are best suited for cell culture based studies of LRP involving short to medium incubation periods.
In conclusion, our findings demonstrate that caution needs to be exercised with cell culture reagents marked as serum-free. These products often contain substantial quantities of undocumented serum proteins with the potential to introduce experimental artefacts. Overlooking this possibility may lead to inaccurate or misinterpreted results. We recommend that researchers perform simple checks on cell culture media to identify relevant contaminating biomolecules before conducting long and expensive experiments. Our findings provide an important guide for avoiding one potentially serious pitfall for LRP studies in neural cell culture models and should also be kept in mind in any research involving ‘serum-free’ reagents.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.Abbreviations
| Aβ | Amyloid β |
| AβPP | Amyloid β precursor protein |
| ApoE | Apolipoprotein E |
| DMEM | Dulbecco's modified eagle medium |
| FBS | Fetal bovine serum |
| FES | Fetal equine serum |
| HRP | Horseradish peroxidase |
| IP | Immunoprecipitation |
| LRP | Low density lipoprotein receptor related protein |
| mAb | Monoclonal antibody |
| MES | 2-(N-Morpholino)ethanesulfonic acid |
| MTS | 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| pAb | Polyclonal antibody |
| PBS-T | 0.1% Tween 20 in phosphate buffered saline |
| PVDF | Polyvinylidene fluoride |
| RT | Room temperature |
| RyR | Ryanodine receptor |
| sLRP | Soluble forms of LRP |
| TMEM | ThermoTrace modified eagle medium |
Acknowledgements
We thank Kelly Marquardt and Jason Woods for technical assistance.References
- J. Herz and D. K. Strickland, J. Clin. Invest., 2001, 108, 779–784 CrossRef CAS PubMed.
- A. P. Lillis, I. Mikhailenko and D. K. Strickland, J. Thromb. Haemostasis, 2005, 3, 1884–1893 CrossRef CAS PubMed.
- W. J. Schneider and J. Nimpf, Cell. Mol. Life Sci., 2003, 60, 892–903 CrossRef CAS PubMed.
- C. V. Zerbinatti and G. Bu, Rev. Neurosci., 2005, 16, 123–135 CAS.
- S. Ito, S. Ohtsuki, J. Kamiie, Y. Nezu and T. Terasaki, J. Neurochem., 2007, 103, 2482–2490 CrossRef CAS PubMed.
- M. Z. Kounnas, R. D. Moir, G. W. Rebeck, A. I. Bush, W. S. Argraves, R. E. Tanzi, B. T. Hyman and D. K. Strickland, Cell, 1995, 82, 331–340 CrossRef CAS PubMed.
- C. V. Zerbinatti, S. E. Wahrle, H. Kim, J. A. Cam, K. Bales, S. M. Paul, D. M. Holtzman and G. Bu, J. Biol. Chem., 2006, 281, 36180–36186 CrossRef CAS PubMed.
- T. E. Willnow, K. Orth and J. Herz, J. Biol. Chem., 1994, 269, 15827–15832 CAS.
- K. A. Quinn, P. G. Grimsley, Y. P. Dai, M. Tapner, C. N. Chesterman and D. A. Owensby, J. Biol. Chem., 1997, 272, 23946–23951 CrossRef CAS PubMed.
- K. A. Quinn, V. J. Pye, Y. P. Dai, C. N. Chesterman and D. A. Owensby, Exp. Cell Res., 1999, 251, 433–441 CrossRef CAS PubMed.
- P. G. Grimsley, K. A. Quinn and D. A. Owensby, Trends Cardiovasc. Med., 1998, 8, 363–368 CrossRef CAS PubMed.
- R. Deane, A. Sagare and B. V. Zlokovic, Curr. Pharm. Des., 2008, 14, 1601–1605 CrossRef CAS PubMed.
- J. L. Biedler, L. Helson and B. A. Spengler, Cancer Res., 1973, 33, 2643–2652 CAS.
- L. A. Greene and A. S. Tischler, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2424–2428 CrossRef CAS.
- A. Makarova, K. K. Bercury, K. W. Adams, D. Joyner, M. Deng, R. Spoelgen, M. Koker, D. K. Strickland and B. T. Hyman, Neurosci. Lett., 2008, 442, 91–95 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |