
Determination of silver in soft silicone wound dressings using dodecylbenzene sulfonic acid digestion and inductively coupled plasma optical emission spectroscopy
L. M.
Hallerstig
,
P.
Granath
,
L.
Lindgren
and
M.
Tranberg
*
Mölnlycke Health Care AB, Department of R&D, Gamlestadsvägen 3C, 41502 Gothenburg, Sweden. E-mail: mattias.tranberg@gmail.com
First published on 2nd December 2016
Abstract
Soft silicones are commonly used as a skin-friendly adhesive in wound care products. To prevent infection, silver (Ag) is frequently incorporated into wound dressings as an antimicrobial agent. Recently, wound dressings with Ag directly incorporated into soft silicone wound/skin contact layers have become commercially available. Thus there is a need for accurate Ag determination in these dressings. In this work the determination of Ag in soft silicones by inductively coupled plasma optical emission spectroscopy is described. This method utilises dodecylbenzene sulfonic acid for the digestion of silicones followed by extraction of Ag with hydrochloric acid (HCl). This method shows excellent accuracy (99–104%) and precision (0.3–4.4% CV) by combining matrix matched standards and internal standardization with indium. A short analysis time of 3 minutes per sample is required and the limit of quantification is 27 μg, corresponding to 540 mg kg−1 for a typical sample weight of 50 mg, which is sufficient to determine the Ag content in commercially available soft silicone wound dressings. The procedure is carried out as an open vessel digestion using a single polypropylene tube which minimizes the need for additional equipment and preparative work.
Introduction
Silicones are diverse materials which can be used in a variety of different applications. Due to their resistance to biodegradation and ageing, combined with their excellent biocompatibility, silicones are commonly used in biomedical devices.1 The use of silicones in wound dressings emerged in the 1990s. Silicones have primarily been used as a non-stick adhesive in wound/skin contact layers which minimises pain and trauma to the skin and wound tissue during dressing changes.2,3 The ability of a wound to heal is affected by a number of factors of which infection is a major concern. Ag has been known for centuries to prevent microbial growth and today a wide range of Ag-containing dressings have been developed. Ag is mostly incorporated into dressings in the form of small inorganic salts or complexes and also as metallic nanocrystalline Ag.4 More recently, Ag has been incorporated directly into the silicone components of dressings. Thus a need for accurate determination of Ag in these dressings is needed.In spite of silicones being a common material relatively few articles describe the determination of metals in silicones. The analysis of the metal content in silicones by inductively coupled plasma-optical emission spectroscopy (ICP-OES) has been described for breast implants5 and silicone oils.6 A number of articles have been published in Chinese journals that describe metal determination in different forms of silicones,7–10 but unfortunately only their abstracts are available in English. A common approach for digesting silicones would be microwave digestion with acids such as HCl, nitric acid (HNO3) and sulphuric acid (H2SO4). Apart from costs associated with investing in the appropriate equipment, typically these techniques require careful cleaning of digestion vessels and glassware between runs to avoid contamination and/or interference. In addition, incomplete dissolution of silicone gel using microwave digestion has been reported earlier.11 Another option for digestion is hydrofluoric acid (HF) which can be used for breaking silicon–oxygen bonds. However, HF is hazardous and requires appropriate safety considerations before use. The instrumentation also has to be compatible with HF-containing samples or procedures that remove the acid have to be performed before the analyte measurement. An extensive review of sample preparation for ICP-OES can be found elsewhere.12
Silicones can be swelled by organic solvents like xylene but will not fully dissolve due to cross linking of the polymer. Commercial liquid silicone digesters are available13 and removal of silicone resins with H2SO4 followed by washing with organic solvents has been described in patented applications.14 To our knowledge this approach has not been evaluated for analytical purposes.
In this work we used a long alkyl-chain sulfonic acid, dodecylbenzene sulfonic acid (DBSA, Fig. 1), in order to combine the swelling effect of an organic solvent with the hydrolytic activity of an acid for the digestion of silver-containing soft silicones. The silver is extracted using 32% (w/w) HCl and is then determined by ICP-OES. The whole procedure is carried out in a single-use plastic tube in order to minimize time-consuming steps such as rinsing and washing of glassware. Validation was performed with matrix matched spiked samples at different levels and with different operators. The method was applied to two commercially available silver-containing silicone wound dressings.
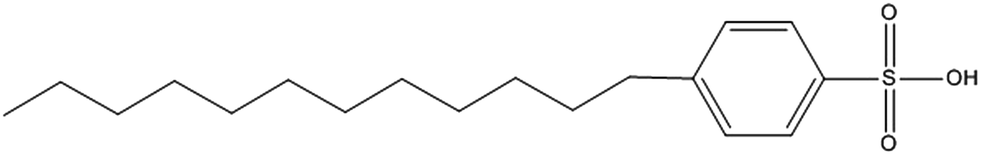 | ||
| Fig. 1 Structure of dodecylbenzene sulfonic acid (DBSA). | ||
Experimental
Instrumentation
For analysis, a ThermoFisher iCAP 6200 DUO equipped with a glass concentric pneumatic nebulizer (Thermo Scientific, MA, USA) coupled to a glass cyclonic spray chamber (Thermo Scientific, MA, USA) was used. An ASX-520 autosampler (Cetac Technologies Inc., NE, USA) was used for sample introduction with 10% (w/w) HCl as wash solution. The software controlling the system was Qtegra (Thermo Scientific, MA, USA). Argon (≥99.998%, Air Liquide, Malmö, Sweden) was used as the plasma, the auxiliary and the nebuliser gas. The nebuliser gas flow was regulated by varying the nebuliser gas pressure. The settings for the ICP-OES are cited in Table 1. Background correction was performed by subtracting the average area of two pixels on the left and right of the analytical peak with the area of the analytical peak.| RF power | 1150 W |
| Auxiliary gas flow rate | 1.0 L min−1 |
| Ar plasma flow rate | 12 L min−1 |
| Nebuliser gas pressure | 0.5 Pa |
| Ag(I) line/measure mode | 338.289 nm/axial |
| In(I) line/measure mode | 325.609 nm/axial |
| Integration time | 10 s |
| Number of repeats per sample | 3 |
| Sample uptake time | 50 s |
| Sample uptake rate | 1.5 mL min−1 |
| Wash time | 60 s |
Preparation of calibration solutions, standards and control samples
To be able to accurately evaluate a method it is of importance to have representative control samples. For this purpose silver-containing cured silicone samples were prepared. Calibration standards were prepared in the same way. Calibration solutions were prepared by the dilution of liquid stock silver standards (1000 mg L−1, Merck, Darmstadt, Germany and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1, Teknolab AB, Kolbotn, Norway) in 0.5 M HNO3 (Merck, Darmstadt, Germany). Type II water (reverse osmosis + electrodeionisation) was used for all dilutions (Millipore, Molsheim, France). Samples were then prepared directly in the digestion tube as outlined in Table 2. First, 4.5–6.0 mg of carboxymethylcellulose (CMC) was added to a 50 mL Digitube. 400 μL of the corresponding calibration solution was then added and the mixture was swirled gently. The fluid was evaporated in a heating block at 90 °C and silicone components A and B (Mölnlycke Health Care, Gothenburg, Sweden) were added. The samples were mixed thoroughly with a plastic pipette to distribute the silver-containing cellulose evenly in the silicone. The tip of the pipette was carefully cut off and left at the bottom of the tube. The silicone was then cured at 125 °C for 45–60 minutes.
000 mg L−1, Teknolab AB, Kolbotn, Norway) in 0.5 M HNO3 (Merck, Darmstadt, Germany). Type II water (reverse osmosis + electrodeionisation) was used for all dilutions (Millipore, Molsheim, France). Samples were then prepared directly in the digestion tube as outlined in Table 2. First, 4.5–6.0 mg of carboxymethylcellulose (CMC) was added to a 50 mL Digitube. 400 μL of the corresponding calibration solution was then added and the mixture was swirled gently. The fluid was evaporated in a heating block at 90 °C and silicone components A and B (Mölnlycke Health Care, Gothenburg, Sweden) were added. The samples were mixed thoroughly with a plastic pipette to distribute the silver-containing cellulose evenly in the silicone. The tip of the pipette was carefully cut off and left at the bottom of the tube. The silicone was then cured at 125 °C for 45–60 minutes.
| Calibration standard/control sample (mg Ag) | Calibration solution conc. (400 μL added) | Silicone component A (mg) | Silicone component B (mg) | CMC (mg) |
|---|---|---|---|---|
| 0.027 | 67.5 mg L−1 | 45–60 | 45–60 | 4.5–6 |
| 0.094 | 235 mg L−1 | 45–60 | 45–60 | 4.5–6 |
| 0.33 | 825 mg L−1 | 45–60 | 45–60 | 4.5–6 |
| 1.15 | 2875 mg L−1 | 45–60 | 45–60 | 4.5–6 |
| 4.0 | 10000 mg L−1 |
45–60 | 45–60 | 4.5–6 |
| Blank | — | 45–60 | 45–60 | 4.5–6 |
Digestion
Digestion was carried out in polypropylene tubes (Digitubes, Perkin Elmer, Waltham, USA) by adding 2 mL of DBSA (70% in propanol, Sigma Aldrich, St. Louis, USA) and 250 μL of indium (10000 mg L−1, Inorganic Ventures, Christiansburg, USA) to the samples. The tubes were placed in a heating block (SPB 50-48, Perkin Elmer, Waltham, USA) at 150 °C for one hour. The tubes were removed from the heating block, allowed to cool and 18 mL of 32% (w/w) HCl (Merck, Darmstadt, Germany) was added. The tubes were vortexed for 30 seconds to extract the silver. After allowing the phases to separate the black upper organic phase was removed by using a plastic pipette and the volume of the sample was further reduced, again using a plastic pipette, until about 5 mL remained. The 5 mL of sample were then diluted to 50 mL with 10% (w/w) HCl. As a precaution the samples were filtered before analysis. Filtration was performed using 0.45 μm polypropylene filters (Digifilters, Perkin Elmer, Waltham, USA). After attachment to a filtration manifold (PerkinElmer, Waltham, USA) the fluid was passed through the filters using a vacuum pump (KNF, Neuberg, Germany). The filtered solution was then analysed by ICP-OES.
Determination of silver in silicone wound dressings
Silver was determined in two commercially available silicone wound dressings, Mepitel Ag (Mölnlycke Health Care) and SurgiClear (Covalon). Both these dressings have a soft silicone adhesive attached either to a mesh (Mepitel Ag) or to a plastic film (SurgiClear). For analysis, 45–55 mg of the silicone adhesive was removed using a spatula and put in a 50 mL Digitube. The samples were then treated and analysed as described above.Results and discussion
Line selection and merit parameters
During the exploratory phase of the method development four Ag lines were initially considered, 224.874 nm, 243.779 nm, 328.068 nm and 338.289 nm, with yttrium, lanthanum, indium and rhodium as potential internal standards. The spectral line of 338.289 nm and indium were found to be the best with respect to interference, the calibration range and robustness.Calibration samples were prepared in a similar matrix as the authentic samples to minimise possible matrix effects.15,16 Calibration curves were constructed by plotting the ratio of the analyte signal to internal standard signal and then using least squares linear regression. To lower the overall error of the method 1/SD2-weighting was used for quantification, where SD is the standard deviation of the three repeats analysed per sample. The 3s- and 10s-criteria were used to determine the limit of detection (LOD) and the limit of quantification (LOQ), respectively. The LOD and LOQ were calculated from four blank samples and the values of 0.0035 mg and 0.012 mg for the LOD and LOQ were obtained, respectively. The calibration range was set to 0.027–4.0 mg with calibration samples at 5 levels (Table 2) since this resulted in calibration curves with better linearity. A typical calibration curve had a R2 value of 1.000 and the equation was y = 2.79x + 0.006 where y = the Ag/In signal ratio and x = the amount of Ag (mg).
Digestion
Initially two different digestion approaches were tested. The first approach used organic solvents (hexamethyldisiloxane, toluene and heptane) in order to swell the silicone and make the Si–O bonds more susceptible to acid attack. H2SO4 was then added for digestion. The other approach utilised microwave digestion of the silicone with HNO3. Both of these approaches failed to completely degrade the silicone. In an effort to improve the digestion we speculated that by combining a long alkyl chain with a strong acid it would be possible to penetrate the cross-linked silicone polymer and degrade the Si–O bonds completely. In terms of acidity, sulfonic acids (pKa of about −3) are comparable to HNO3 (pKa of −1.4) and H2SO4 (pKa of −3) and DBSA was chosen since it had previously been suggested to be able to remove and dissolve cured silicone.17 When silver-containing silicone samples were digested with DBSA a homogenous black phase formed with a distinct silver-coloured phase on top (Fig. 2). | ||
| Fig. 2 Glass tube with the digested silicone with a silver-coloured phase on top. | ||
Identifying the exact mechanism of the degradation was beyond the scope of this work. However, it can be said that silicone chains have been shown to undergo degradation at elevated temperatures in the presence of acids.18 DBSA can act as a Brønsted acid and a catalyst during the hydrolysis of silcones19 and acids under anhydrous conditions have been shown to catalyse the cleavage of siloxane bonds in cyclosiloxanes.20 In addition, sulfonic acids, including DBSA, have been shown to catalyse the hydrolysis of alkoxysilanes in an alkoxysilane-grafted ethylene–propylene copolymer. The rate of the reaction was dependent on the sulfonic acid's ability to diffuse into polymer matrices.21 In a similar manner it is believed that hydrophobic DBSA can reach and catalyse the hydrolysis of the siloxane bonds in silicones more effectively than a water-based acid (Scheme 1).
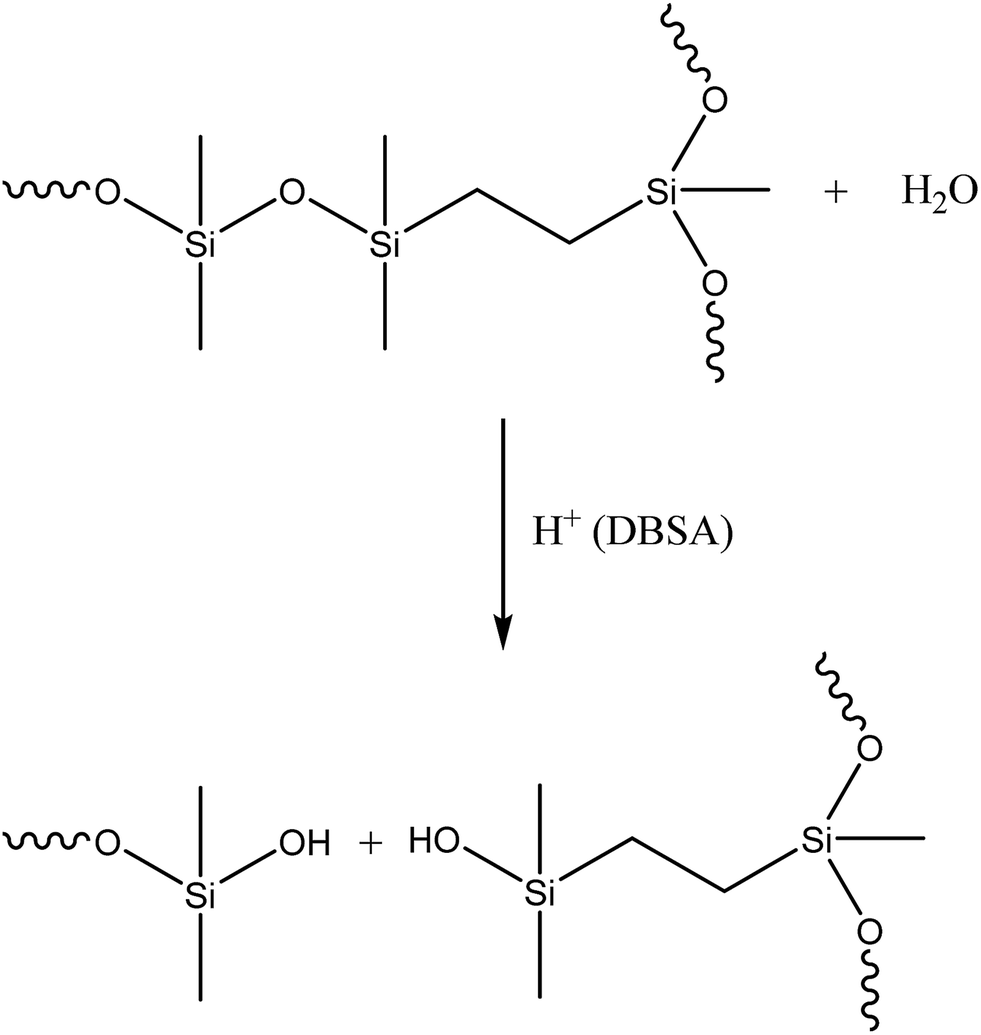 | ||
| Scheme 1 Hydrolysis of the siloxane bond catalysed by DBSA. | ||
In order to extract the silver from the digested silicone, HCl (32%, w/w) was used. This was to avoid the formation of insoluble AgCl by possible trace Cl− present in the sample. Ag+ will form AgCl in contact with Cl− but excess Cl− will give rise to AgClx1−x-species that are readily soluble.22 However, HCl will make the Ag+ more susceptible to photo-reduction to Ag(s) and the samples were therefore stored in the dark.
By including an internal standard during the digestion step the practical work was facilitated since the internal standard compensated for small variations in volume that were inevitable during the digestion procedure (e.g. the dilution step). The concentration of the internal standard was chosen so that a good and robust signal intensity was obtained which also allowed for the dilution of the samples above the LOQ.
In order to avoid time-consuming procedures often associated with ICP-OES analysis such as acid rinsing and washing of glassware, single-use plastic tubes were used for the digestion procedure. Polypropylene is commonly used as an acid-resistant material. The melting point of polypropylene is between 130 and 170 °C which can be regarded as a relatively low digestion temperature. However, by using DBSA and polypropylene tubes it was possible to fully digest the silicone at 150 °C within one hour. The boiling point of DBSA is 204.5 °C,23 higher than that of common acids used for digestions such as HNO3 and HCl, thus making it more compatible with open vessel digestion.
Method validation
The accuracy, precision and linearity of the method were determined using control samples at three levels: low (0.027 mg Ag, n = 3), mid (0.33 mg Ag, n = 3) and high (4.0 mg Ag, n = 3). In addition, the complete analysis was performed with three different operators to account for operator-dependent variability. The amount of Ag in the control samples was determined against calibration curves obtained on the day of the analyte quantification. The stability of the digested control samples was also evaluated by storing the samples in the dark and analysing them after one week and one month.Validation was carried out according to the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) and International Organization for Standardization (ISO) guidelines.24–26 The instrument precision was determined to be 0.1% (CV) by performing 10 consecutive measurements of Ag on a mid control sample. The accuracy and repeatability (intra-operator precision) were determined for each operator (Table 3). The accuracy was within ±5% of the target values and the coefficient of variance (Cvr) was below 5%. The average accuracy and reproducibility (inter-operator precision) were determined for all three operators. The average accuracy was within ±5% of the target values and the coefficient of variance (CvR) was below 5% (Table 4). The stability of the treated control samples was also evaluated by storing the samples in the dark and analysing them after one week and one month (Table 5). The accuracy of the stored samples was within ±10% of the target values with CVs below 5%.
| Parameter | Operator A | Operator B | Operator C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Low | Mid | High | Low | Mid | High | Low | Mid | High | |
| Ag (mg) | 0.0276 | 0.337 | 4.09 | 0.0282 | 0.330 | 3.93 | 0.0277 | 0.342 | 4.09 |
| Accuracy (%) | 104.6 | 102.5 | 102.2 | 104.3 | 102.0 | 99.4 | 102.8 | 103.3 | 102.1 |
| Repeatability, Cvr (%) | 4.3 | 0.6 | 0.6 | 0.3 | 1.9 | 0.9 | 1.3 | 1.5 | 0.6 |
| Parameter | Low | Mid | High |
|---|---|---|---|
| Ag (mg) | 0.028 | 0.34 | 4.1 |
| Average accuracy (%) | 103.9 | 102.6 | 101.2 |
| Reproducibility, CvR (%) | 2.6 | 1.4 | 1.7 |
| Parameter | 1 week | 1 month | ||||
|---|---|---|---|---|---|---|
| Low | Mid | High | Low | Mid | High | |
| Ag (mg) | 0.026 | 0.33 | 3.9 | 0.025 | 0.32 | 3.8 |
| Accuracy (%) | 94.5 | 98.7 | 97.8 | 91.3 | 96.2 | 95.3 |
| CV (%) | 4.3 | 0.4 | 0.3 | 4.0 | 0.4 | 0.4 |
The dilution of samples above the upper LOQ was evaluated by preparing control samples with a silver content of 6 mg. The samples were diluted twice as much as the regular samples (2.5 mL was left before dilution instead of 5 mL). The calculated average accuracy for these samples was 100.8% (CV 0.2%, n = 3).
No apparent memory effects were identified during the method development or validation. During the analysis the middle calibration sample (0.5 mg L−1) was run every 10th sample to detect possible drifts in the system.
Determination of silver in wound dressings
The developed method was used to determine the silver content in two commercially available silver-containing silicone dressings. For SurgiClear the silver content was determined to be 0.44% (w/w) silver salt (n = 3, CV = 0.6%), which is in agreement with the specification of 0.5% (w/w) silver salt (as informed by the manufacturer). The form of the silver salt is an intellectual property of Covalon and cannot be disclosed. For Mepitel Ag, the Ag content was first converted from weight percent to weight/area using a known amount of silicone per cm2 (Mölnlycke Health Care, internal communication). The result was 2.36 mg Ag per cm2 (n = 10, CV = 0.7%), which is in agreement with the specification of 1.8–3.0 mg Ag per cm2 (as informed by the manufacturer).Conclusion
The results presented in this work show that soft silicones can be digested using DBSA. Ag can be extracted from the digest with HCl and quantified by ICP-OES. The developed method was applied to soft silicone wound dressings but should in principle be applicable to any type of soft silicone. A novel approach was to use DBSA for digestion which was able to completely degrade the silicone in contrast to common acids such as HNO3 and H2SO4. It has to be noted that soft silicones have a relatively low degree of crosslinking, which makes it easier for DBSA to degrade them. Silicones with more rigid cross-linking may require other approaches for digestion. The digestion procedure was carried out in a single-use polypropylene tube requiring only a heating block as the additional equipment in this step of the procedure. This approach minimised the need for cleaning of digestion vessels and the risk of memory effects and cross contamination but in principle any digestion system compatible with DBSA and HCl could be used for the digestion procedure. The validated method showed excellent accuracy and precision and a fast analysis time (3 min per sample). Whether the excellent accuracy and precision were due to the use of matrix matched calibration standards was not investigated further. Thus, if non-matrix matched standards need to be used these parameters need to be re-evaluated. In conclusion, the current method is a simple and reliable choice for the determination of Ag and potentially other metals in soft silicones.Acknowledgements
The authors acknowledge the experimental support of Magnus Persson and technical input of Gabriel Kaszonyi.References
- R. Yoda, J. Biomater. Sci., Polym. Ed., 1998, 9, 561–626 CrossRef CAS PubMed.
- M. Waring, S. Bielfeldt, K. Matzold, K. P. Wilhelm and M. Butcher, J. Wound Care, 2011, 20, 412 CrossRef CAS PubMed.
- H. Matsumura, R. Imai, M. Gondo and K. Watanabe, Int. Wound J., 2012, 9, 451–455 CrossRef PubMed.
- S. Thomas, in Surgical Materials and Wound Management, Medetech Publications, Cardiff, 2010, ch. 16, pp. 473–523 Search PubMed.
- S. V. Maharaj, Anal. Bioanal. Chem., 2004, 380, 84–89 CrossRef CAS PubMed.
- K. A. Das, R. Chakarborty, M. L. Cervera and M. de la Guardia, Spectrosc. Lett., 1998, 31, 1245–1253 CrossRef.
- S. Lu, Y. Ni and S. Fu, Youjigui Cailiao, 2010, 24, 45–49 CAS.
- H. Liu, Y. Tang, L. Fu and X. Nie, Xiangjiao Gongye, 2009, 56, 58–59 CAS.
- C.-X. Wan and X.-Y. Xiao, Guangpu Shiyanshi, 2007, 24, 844–847 CAS.
- H. Xie and L. Li, Xiangjiao Gongye, 2003, 50, 678–680 CAS.
- A. El-Jammal and D. M. Templeton, Anal. Proc. incl. Anal. Commun., 1995, 32, 293–295 RSC.
- Z. Mester and R. Sturgeon, Sample Preparation for Trace Element Analysis, Elsevier, 1st edn, 2003 Search PubMed.
- M. Velderrain, Adv. Packag., 2007, 16, 27–28 CAS.
- P.-F. Fu, G. Kim and S. Wang, WIPO Pat., 2014205285, 2014.
- J. L. Todolí, L. Gras, V. Hernandis and J. Mora, J. Anal. At. Spectrom., 2002, 17, 142–169 RSC.
- G. L. Scheffler and D. Pozebon, Anal. Methods, 2015, 7, 5180–5185 RSC.
- H. Haas and M. Snyder, WIPO Pat., 2002044261, 2002.
- A. M. Brook, in Silicon in Organic, Organometallic, and Polymer Chemistry, John Wiley & Sons, Inc., 2000, ch. 9, p. 295 Search PubMed.
- M. A. Bigdeli, G. Gholami and E. Sheikhhosseini, Chin. Chem. Lett., 2011, 22, 903–906 CrossRef CAS.
- L. Wilczek and J. Chojnowski, Macromolecules, 1981, 14, 9–17 CrossRef CAS.
- K. Adachi and T. Hirano, J. Sol-Gel Sci. Technol., 2009, 49, 186–195 CrossRef CAS.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef CAS PubMed.
- US Coast Guard, CHRIS – Hazardous Chemical Data 1984-5, 1984, vol. 2 Search PubMed.
- ICH Harmonized tripartite guideline, Text on validation of analytical procedures, Q2A.
- ICH Harmonized tripartite guideline, Validation of analytical procedures: Methodology, Q2B.
- ISO 5725-1 Accuracy (trueness and precision) of measurements methods and results. General principles and definitions.
| This journal is © The Royal Society of Chemistry 2017 |