DOI:
10.1039/C7RA03364H
(Paper)
RSC Adv., 2017,
7, 22802-22809
A sulfate radical based ferrous–peroxydisulfate oxidative system for indomethacin degradation in aqueous solutions†
Received
22nd March 2017
, Accepted 19th April 2017
First published on 25th April 2017
Abstract
The degradation of indomethacin (IM) by ferrous ion-activated potassium peroxydisulfate (Fe2+/PDS) was investigated. We aimed to determine the optimal conditions for the removal of IM under different concentrations of Fe2+and PDS, evaluate the effects of operational parameters (solution pH, humic acid (HA), N2 bubbling and persulfate species), and propose the degradation mechanism of IM by the Fe2+/PDS system. The sequential addition of Fe2+ led to an improvement in the IM degradation and TOC removal efficiency. When the molar ratio of IM/PDS/Fe2+ was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:2, the IM was almost completely degraded. Restrictions to the degradation efficiency of IM were caused by increasing the solution pH, bubbling with nitrogen, or through the addition of HA. A low concentration of Cl− had no effect on the reaction, while a high concentration led to a dramatic inhibitory effect. In addition, quenching experiments revealed that SO4˙− was the major active radical for the degradation of IM by ferrous ion-activated peroxydisulfate. Based on the identification of transformation products by liquid chromatography-mass spectrometry (LC-MS/MS), the pathways of the ferrous–peroxydisulfate oxidative system for the degradation of IM were tentatively proposed.
:1.5:2, the IM was almost completely degraded. Restrictions to the degradation efficiency of IM were caused by increasing the solution pH, bubbling with nitrogen, or through the addition of HA. A low concentration of Cl− had no effect on the reaction, while a high concentration led to a dramatic inhibitory effect. In addition, quenching experiments revealed that SO4˙− was the major active radical for the degradation of IM by ferrous ion-activated peroxydisulfate. Based on the identification of transformation products by liquid chromatography-mass spectrometry (LC-MS/MS), the pathways of the ferrous–peroxydisulfate oxidative system for the degradation of IM were tentatively proposed.
1. Introduction
Indomethacin (IM) is a typical non-steroidal anti-inflammatory drug, which inhibits the synthesis of prostaglandin in attaining an antipyretic, analgesic, and anti-inflammatory effect.1 It has been widely used due to its high efficacy, and the annual IM output in China was 246 tons in 2011. Since only a small portion of indomethacin is absorbed by the human body, most of this substance is excreted and makes its way to sewage treatment plants. In addition, there are no dedicated facilities that are specifically designed to remove IM in sewage treatment plants, thus the removal efficiency of IM is poor, at a rate of less than 10%.2,3 Ultimately, the remaining IM is discharged into ambient water bodies, such as rivers and lakes; hence, the IM content in the environment increases year by year. The concentrations of IM in surface waters can be up to several hundred ng L−1, IM has even been detected at a level of 0.53 ng L−1 in drinking water in Southern China.4 Therefore, the study of effective treatment technologies for the elimination of IM in wastewater and drinking water is of great scientific interest.
New technologies have been applied for the treatment of wastewater and drinking water in order to decrease the concentration of NASIDs. These include the use of membrane technologies and other AOPs, such as ozonation,5 permanganate,6 and chlorination.7 Currently, the study of transformation products in many WWTPs using conventional processes has received less attention. Therefore, although NASIDs emissions are reduced following treatment, the downstream transformation products generated have still not garnered the required level of awareness and urgency. Furthermore, in some cases these products may be even more toxic than their parent substances.8 Sulfate-radical based AOPs comprise a promising technology for practical applications in wastewater treatment plants.
The sulfate radical SO4˙− is very promising for the degradation of organic pollutants due to its high oxidation potential (E0 = 2.6 V), particularly when applied to aromatic pollutants. This radical may be generated via the activation of KHSO5 and K2S2O8 in conjunction with radiation, photolysis, microwave and heat.9–14 Moreover, transition metal ions are also able to activate K2S2O8 to form SO4˙−. Sulfate radicals have a longer half-life than hydroxyl radicals, primarily due to their preference for electron transfer reactions. Hydroxyl radicals may also react via hydrogen-atom abstraction and addition, along with electron transfer, which plays a less important role.15–17
In this paper, we attempted to evaluate the feasibility of employing ferrous-activated PDS to degrade IM in aqueous solution. Iron species were selected as a catalyst due to their low cost, abundant supply, and environmental compatibility. The oxidation process was optimized for soluble iron, oxidant concentration, and Fe2+ batch adding. The effects of several environmental factors were evaluated. Major active radicals that contributed to the degradation of IM were identified by quenching studies using ethanol and tert-butanol, with different reactivity toward hydroxyl and sulfate radicals. The mechanisms of IM degradation by ˙OH and SO4˙− were also revealed through liquid chromatography mass spectrometry (LC-MS/MS) analysis. To the best of our knowledge, this is the first study on detail mechanisms and transformation pathways of IM oxidation by ferrous-activated persulfate.
2.1. Chemicals
Indomethacin (IM), and humic acid were purchased from Aladdin Industrial Corporation (Shanghai), whereas potassium peroxydisulfate was obtained from Sinopharm Chemical Reagent (Shanghai, China). Ferrous sulfate, ferric sulfate, and ethanol, tert-butanol were purchased from Chengdu Kelong Chemical Reagent Co. Ltd. All of the reagents above were of analytical grade and required no further purification. The acetonitrile purchased from Anaqua Chemicals Supply Co. Ltd. (USA) was of HPLC grade. High purity (99.99%) compressed N2 or O2 was purchased from Jingong (Hangzhou, China), and all solutions were prepared with Milli-Q water of 18.25 MΩ cm−1.
2.2. Determination of degradation kinetics
A model organic compound (IM) was selected for all of the experiments, which were homogeneous batch based, and run for 32 min at ambient temperature. Prior to each experiment, the appropriate quantities of oxidant (0.01–0.04 mM) and iron (0.01–0.04 mM) were added to achieve the predefined molar ratios of contaminant, oxidant, and iron species. Initial pH was adjusted by 0.01 M H2SO4 or NaOH to desired value. The effects of humic acid (HA, 0–10 mg L−1, the TOC of 10 mg L−1 HA was 2.873 mg L−1) and chloride (Cl−, 0–20 mM) were evaluated. Following the addition of all the chemicals from their respective stock solutions, the initial volume of the reaction solution was 500 mL. All samples were quenched immediately with excess sodium nitrite to halt the chemical oxidation reaction. Most of the experiments were performed in triplicate in order to assure accurate data acquisition, and error bars in the figures represent standard deviations.
Liquid samples were obtained periodically and analyzed, and indomethacin was quantified by an Agilent 1100 series HPLC (Agilent, USA) system, which was equipped with a diode array detector (SPD-M20A). The column used was a Chromolith Performance XDB-C18 column (150 × 2.1 mm, 5 μm), and the injection volume was 4 μL. The mobile phase was a mixture of acetonitrile and 5 mM ammonium acetate in water (65:35, v/v) at a flow rate of 0.2 mL min−1 under isocratic conditions, and the UV wavelength for detection was 228 nm.
TOC was measured by a TOC-VCPH analyzer (Shimadzu) to identify the mineralization of the organic contaminants.
2.3. Identification of transformation products
In order to demonstrate the IM reaction mechanism, the identification of the intermediate products were carried out with an Agilent HPLC/MS system that was equipped with a Hypersil ODS column (250 × 4.6 mm, 5 μm). A mixture of 65% HPLC-grade acetonitrile and 35% Milli-Q water (containing 5 mM ammonium acetate) was used as the eluent at a flow rate of 0.6 mL min−1. A 10 μL of sample was injected by using an auto sampling device, with a 228 nm detection wavelength. The eluent from the chromatographic column entered the UV-vis detector, followed by electrospray ionization (ESI) and mass analyzer. Data acquisition was performed in the negative ion mode, and the optimized parameters were as follows: dry temperature, 350 °C; spray voltage, 3.5 kV; cone voltage, 40 V; dry gas flow, 10 L min−1. Nitrogen was used as an auxiliary gas at 40 psi, and argon was used as the collision gas. Full scan spectra were recorded in the range of from 100 to 600.
3. Results and discussion
3.1. Effect of Fe2+ and PDS concentrations in Fe2+/PDS system
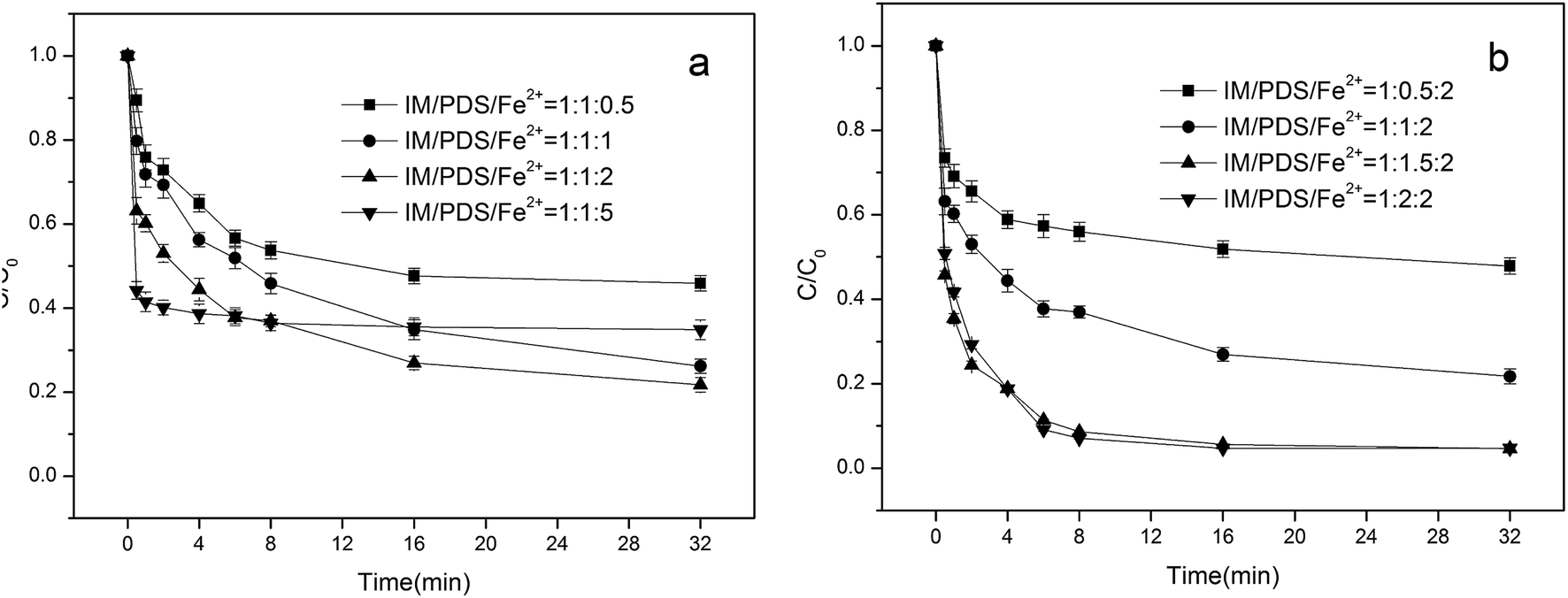
3.1.1. Effect of Fe2+ concentration in Fe2+/PDS system. Ferrous iron is a primary species that may activate peroxydisulfate to produce sulfate radicals, which can dramatically influence organic pollutant degradation efficiency. Fig. 1a depicts the influence of different initial Fe2+ concentrations on the degradation of IM. Initial concentrations of PDS were fixed at 0.02 mM. When the molar ratio of Fe2+/IM was 0.5:1, 1:1, 2:1, the degradation rate of the IM over 32 min attained 54.1%, 73.8%, and 78.3%, respectively. Under experimental conditions, the degradation rate of IM was optimal when the molar ratio of Fe2+/IM was 2:1. The higher IM degradation efficiency at higher Fe2+ doses might result from the additional production of SO4˙− in the system. However, upon further increases in the concentration of Fe2+ (to 0.1 mM), the degradation rate of IM was reduced to 63.1%; hence, excessive ferrous ions impart an inhibitory effect on the degradation of IM. This is primarily due to excessive ferrous ions, which can react with sulfate radicals to form iron ions, which consume a portion of the sulfate radicals, as represented in eqn (1) and (2) shown below.9| | |
S2O82− + Fe2+ → SO4˙− + SO42− + Fe3+
| (1) |
| | |
SO4˙− + Fe2+ → SO42− + Fe3+
| (2) |
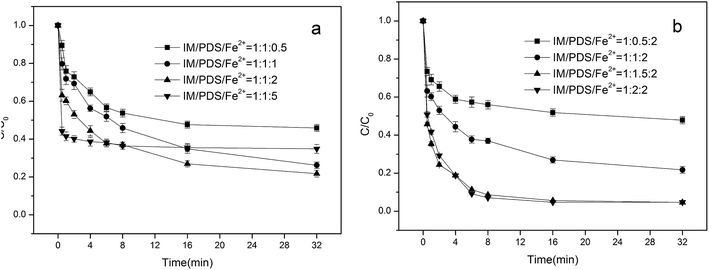 |
| | Fig. 1 Effect of Fe2+(a) and PDS (b) concentration on the degradation of IM [IM] = 0.02 mM, pH = 4.5. | |
3.1.2. Effect of PDS concentration in Fe2+/PDS system. Peroxydisulfate plays a critical role as a source of SO4˙− generation in a Fe2+ activated peroxydisulfate system. Initial concentrations of Fe2+ were fixed at 0.02 mM. The effects of peroxydisulfate concentration on the degradation of IM was examined under different peroxydisulfate dosages (from 0.01 mM to 0.04 mM), with the results shown in Fig. 1b. The degradation rate of IM increased with an increase in the initial concentration of PDS accordingly. When the initial concentration of PDS was 0.01 mM, the IM degradation rate attained a 52.1% reaction after 32 min. Only when the initial concentration of PDS was increased to 0.03 mM did the IM degradation rate come close to attaining 100%. On further increases in the initial PDS concentration, the IM degradation rate was practically unchanged. This phenomenon suggested that sulfate radicals were consumed more by peroxydisulfate than with IM, as indicated in eqn (3):| | |
SO4˙− + S2O82− → S2O8˙− + SO42−
| (3) |
3.2. Influence of different factors in Fe2+/PDS system
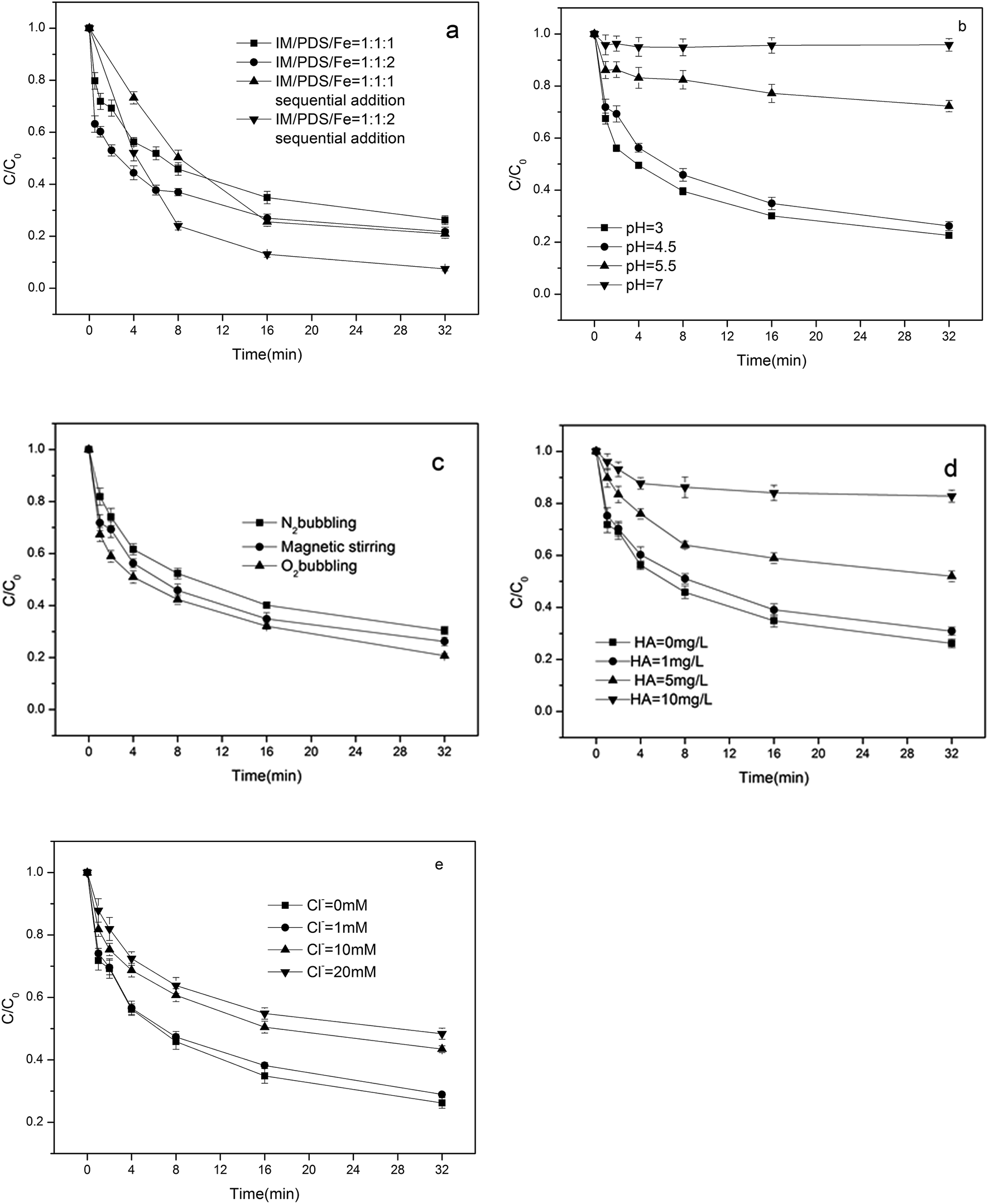
3.2.1. Influence of available Fe2+. In order to confirm the influence of available ferrous ions, experiments were conducted where ferrous ions were added sequentially as multiple equal portions to the reaction solutions, which contained a PDS/IM molar ratio of 1:1. The influence of the sequential addition of Fe2+ on IM degradation is shown in Fig. 2a. Following the successive addition of four Fe2+ doses, which were equivalent to initial Fe2+/IM molar ratios of 1:1 and 2:1, the final IM/S2O82−/Fe2+ molar ratios attained were 1:1:1, 1:1:2, and the overall IM removals were 79.1% and 92.6%, respectively. When all of the Fe2+ was added as a single dose at the same final IM/PDS/Fe2+ molar ratios as in the previous experiments, the IM removals attained were 73.8% and 78.3% for IM/PDS/Fe2+, at molar ratios of 1:1:1 and 1:1:2, respectively. It was evident that higher IM degradation efficiencies were obtained by incrementally providing the available Fe2+ to slowly generate SO4˙−, which attacked the IM with greater efficacy. This phenomenon was similar to the observations of Liang, who noted that the gradual addition of Fe2+ could minimize the oxidation of Fe2+ by SO4˙− in Fe2+/PDS systems.9 It appeared that the slower addition of Fe2+ was favorable for the production of free radicals, and that the availability of Fe2+ played an important role in controlling the ferrous ion activated peroxydisulfate reaction.
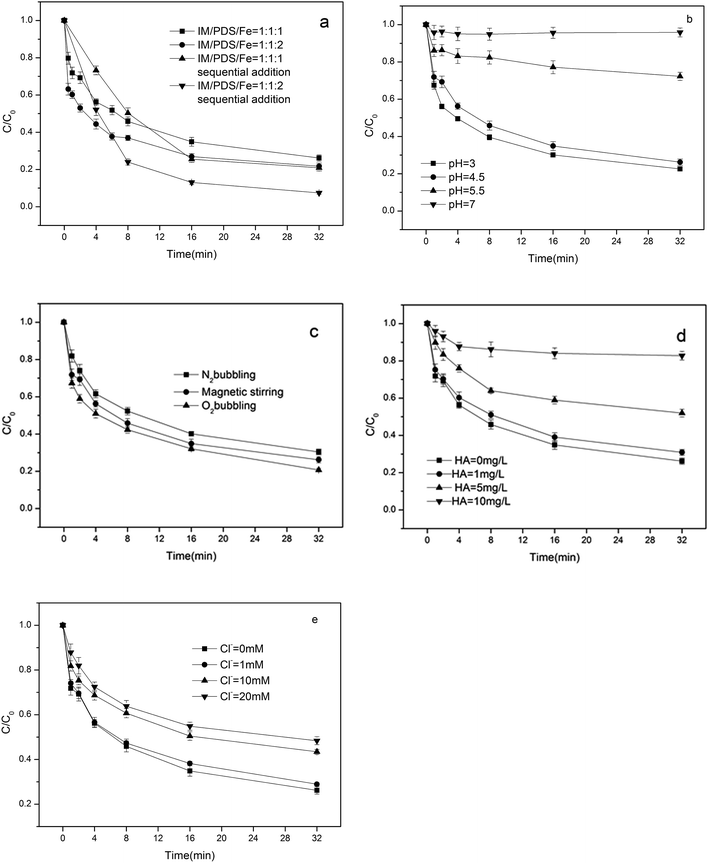 |
| | Fig. 2 Effect of different factors on the degradation of IM: (a) sequential addition of Fe2+ (b) pH (c) N2 and O2 aeration (d) HA (e) Cl−. [IM] = 0.02 mM, [PDS]:[Fe2+] = 1:1. | |
3.2.2. Influence of pH. The pH value plays a critical role in the degradation of organic pollutants. Initial concentrations of PDS and Fe2+ were fixed at 0.02 mM. The degradation of IM in the ferrous activated peroxydisulfate system was initially carried out at pH 3, 4.5, 5.5, and 7 respectively. The IM degradation rates under different pH are depicted in Fig. 2b. It may be seen that the degradation efficiency of IM decreased with increased pH, indicating that acidic pH was more favorable for the degradation of IM than neutral pH. The decreased IM degradation rate under neutral pH might be attributed to the removal of soluble iron catalysts and the self-dissociation of PDS through non-radical pathways. Further, these researchers observed that the efficiency of the Fe2+/H2O2, Fe2+/PDS reagent decreased dramatically with increased pH due to iron precipitation.18 Moreover, the quantity of soluble Fe2+ was decreased due to the formation of Fe2+ complexes, which inhibited the further reaction of Fe2+ with peroxydisulfate. In addition, the Fe3+ hydroxides exhibited low efficiencies in the activation of peroxydisulfate to produce sulfate radicals. The formation of Fe3+ hydroxides are shown in the following eqn (7).19| | |
Fe3+ + xH2O → FeOHx3−x + xH+
| (4) |
Therefore, the low activity of ferrous iron generated fewer sulfate radicals, which accounted for the lower IM degradation efficiency.
3.2.3. Influence of dissolved oxygen. The results of the influence of N2 bubbling on the degradation of IM is shown in Fig. 2c. Bubbling with N2 was found to decrease the removal of IM, in contrast to the condition without gas bubbling, while bubbling with O2 improved IM degradation. As the Log Kow and Henry's law constants of IM are low, bubbling with gas was not able to remove IM. The promoting effects of dissolved oxygen (DO) on the photodecomposition of ibuprofen have been reported,20 while Liu et al. found that the presence of DO exhibited inhibitory effects on the degradation of atenolol by UV/peroxymonosulfate.21The roles of DO might involve the formation of oxygen centered radicals,22 similar to the ˙OH, SO4˙− that is generated by ferrous iron activated peroxydisulfate. It can react with O2 to form reactive oxygen species such as O2˙−. Further, as shown in eqn (5),23 O2˙− may also transform to HO2˙ at acid pH, which may accelerate the IM degradation rate. In addition, the OH adducts of the substrate can react with oxygen, to produce peroxyl transients, which are rather unstable and could readily go through hydrolysis toward the formation of various by-products.24 The points mentioned above, indicating that increased DO concentrations should enhance IM degradation, were in accordance with the present results.
3.2.4. Influence of HA and Cl−. As shown in Fig. 2d, the addition of HA inhibited the IM and SO4˙− reactions significantly. This is because the HA surface contains rich organic functional groups, such as hydroxyl, phenolic hydroxyl, amino, carboxyl, carbonyl, etc. When HA with these groups coexist with IM, not only can IM be oxidized, but HA can also be attacked by SO4˙−.25 In the PDS/Fe2+ system, the presence of HA and IM tends to form a competitive mechanism, which competes with SO4˙− in the reaction. It was evident that IM decomposition dramatically decreased with increasing HA concentrations. Fig. 2e illustrates the effect of Cl− on the degradation of IM in Fe2+/PDS system. The results show that a low concentration of Cl− (1 mM) had no significant effect on the degradation of IM. This is mainly because chlorine radicals generated by the reaction of SO4˙− with Cl− did not participate in the degradation of IM due to excessive SO4˙−. Since SO4˙− was more reactive than Cl˙, the majority of IM was degraded by SO4˙−. When the concentration of Cl− is elevated (i.e. 10 mM and 20 mM), Cl− behaved primary as SO4˙− scavenger, as Cl− can react with SO4˙− to produce relatively weaker species such as Cl˙, Cl2˙−, Cl2, and HOCl according to eqn (6)–(9).26–28 reducing the degradation efficiency of IM.| | |
SO4˙− + Cl− ↔ SO42− + Cl˙
| (6) |
| | |
Cl2˙− + Cl2˙− → Cl2 + 2Cl−
| (8) |
| | |
Cl2 + H2O → HOCl + H+ + Cl−
| (9) |
3.3. Oxidative mechanism in the Fe2+/PDS system
It is widely accepted that alcohols containing alpha hydrogen, such as ethanol, readily react with hydroxyl radicals and sulfate radicals, while alcohols with no alpha hydrogen, such as tert-butanol, are effective quenching agents for hydroxyl radicals only. Therefore, ethanol is widely employed as hydroxyl and sulfate radical scavenger, whereas tert-butanol is used to quench hydroxyl radicals. Consequently, we investigated the effects of ethanol and tert-butanol on the degradation of IM in order to evaluate the reaction mechanism and the species that played major roles in the Fe2+/PDS system.
According to the literature, the rate constants for reactions of ethanol with hydroxyl radicals and sulfate radicals are 1.2–2.8 × 109 M−1 s−1 and 1.6–7.7 × 107 M−1 s−1, respectively. Although tert-butanol is an effective quenching agent for hydroxyl radicals (k = 3.8–7.6 × 108 M−1 s−1), it reacts much more slowly with sulfate radicals (k = 4.0–9.1 × 105 M−1 s−1) than with hydroxyl radicals.29,30 Thus, by comparing IM degradation rates following the addition of excess tert-butanol and ethanol in the Fe2+/PDS system may identify whether ˙OH and SO4˙− are generated.
As seen in Fig. 3, with an excess of tert-butanol and ethanol added to the reaction system, the IM degradation rate was slightly decreased; however, it could be completely degraded after 32 min, indicating that the system generated only a small amount of ˙OH, while with the addition of ethanol, the IM degradation efficiency was decreased to 62.2%. Furthermore, as tert-butanol is also a scavenger of hydroxyl radicals, the degradation of indomethacin in the presence of tert-butanol might be primarily ascribed to the formation of sulfate radical anions that are generated from reaction (1). Therefore, we believe that sulfate radical anions, as the dominant active species, are responsible for the oxidation of IM in the Fe2+/PDS system. Other radicals such as SO5˙− can also account for the degradation of IM.31 Under acidic conditions, SO4˙− is a major free radical, as shown in eqn (10) and (11);32,33 However, under alkaline conditions, the SO4˙− generated from the Fe2+/PDS system might be transformed to ˙OH via a chain reaction, as shown in eqn (12).
| | |
S2O82− + H+ → HS2O8−
| (10) |
| | |
HS2O8− → SO4˙− + HSO4−
| (11) |
| | |
SO4˙− + OH− → SO42− + ˙OH
| (12) |
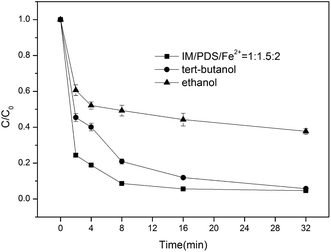 |
| | Fig. 3 Effects of the addition of tert-butanol and ethanol on IM degradation. [IM] = 0.02 mM, [tert-butanol] = [ethanol] = 20 mM. | |
3.4. Mineralization of indomethacin
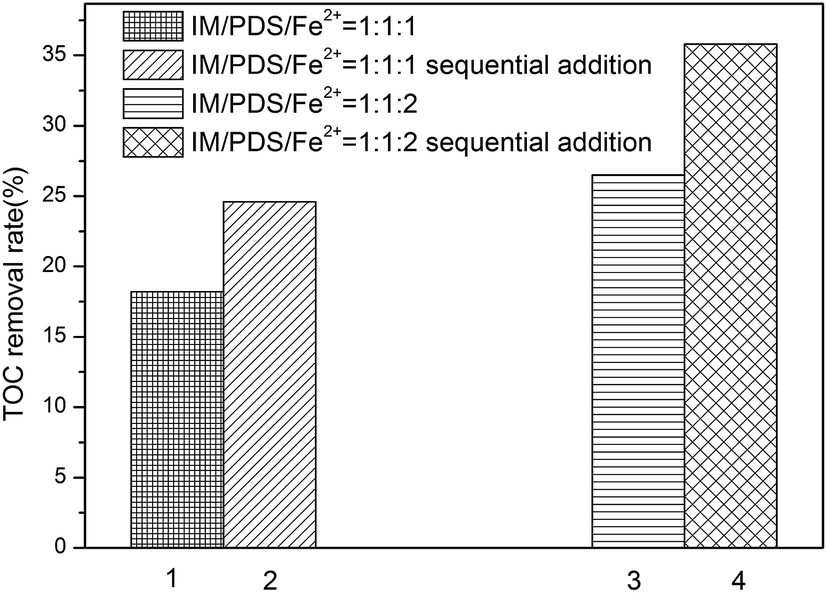
Mineralization of IM in the Fe2+-peroxydisulfate was evaluated by measuring the removal of TOC. According to Fig. 2a, it can be seen that the removal of IM under the molar ratio of IM:PDS:Fe2+ at 1:1:1 and 1:1:2 was found to be efficient. However, as shown in Fig. 4, it was found inefficient for TOC removal, while only 18.2% and 26.5% TOC was removed, respectively. Higher TOC removal rate was obtained with higher total amount of Fe2+ employed. Otherwise, the sequential addition of Fe2+ may seem to be an important factor in the mineralization of IM. As described previously, the total TOC of sequential addition of Fe2+ in small increments, the final IM/PDS/Fe2+ molar ratios attained were 1:1:1, 1:1:2, increased to 24.6% and 35.8%, which is higher than that of one-off dosing process. This may be due to the involvement of secondary reaction, which is harmful for SO4˙− to react with IM and its transformation products. We can conclude that the way of Fe2+ addition is not only critical for the oxidation of IM, but also the mineralization. It's important to maintain the Fe2+ level to ensure effective and higher extent of mineralization.
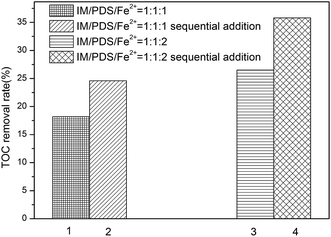 |
| | Fig. 4 TOC removal efficiencies of IM after 24 min reaction. [IM = 0.02 mM]. | |
3.5. Identification of transformation products
Fig. SM-1† represents the total ion chromatograms (TIC) of indomethacin oxidized by ferrous iron-activated peroxydisulfate. Indomethacin was degraded into a variety of transformation products, as demonstrated by UHPLC-MS/MS analysis (Fig. SM-2†). The chromatograms in negative ionization mode presented in Fig. SM-1† reveal the detected transformation products of indomethacin in the Fe2+/PDS system. Five products from the degradation of indomethacin by ferrous iron-activated peroxydisulfate were identified using MS2 scan mode for preliminary screening, and using product ion scan mode for confirmation. To elucidate the chemical structures of indomethacin by UHPLC-MS/MS, the MS2 spectra obtained from the product ion scan mode of indomethacin and Fig. SM-2† provided the basis for the interpretation of the transformation products.
P I: The MS spectrum of product I in Fig. SM-2a† shows a deprotonated molecular ion at m/z 155, and one major fragment ion at m/z 111, which was due to the loss of CO2 in the ionization source.34
P II: The spectrum of productS in Fig. SM-2b† did not show the carboxylic acid neutral losses, indicating that it was a decarboxylated product, the cations C7H4ClO− and C6H4Cl− are observed, which indicated that the chlorinated ring remained unaltered.
P III: The spectrum of product III in Fig. SM-2c† was similar to that of P II, and a loss of CH3O (from the ether) was observed. The ion at m/z 155 was assigned to 4-hydroxybenzoate, which resulted from the migration of an OH group.
P IV: The spectrum of product IV in Fig. SM-2d† shows the loss of CO2, CH3, and CO, and an ion at m/z = 137 was observed, indicating that the Cl had been substituted by OH.
P V: The MS spectrum of product V in Fig. SM-2e† shows the sequential losses of H2O and CO2 (typical of carboxylic), CO (corresponding to carbonyl groups), and CH3, yielding the ions at m/z = 372, 346, 318, 303. The loss of H2O was attributed to the ˙OH, which was attacked on the pyrrole ring. The loss of CO2 confirmed the presence of the carboxylic group. The ion at m/z 155 was assigned to 4-hydroxybenzoate, resulting from the migration of an OH group, and the ion at m/z 111 confirmed that the chlorinated ring did not undergo transformation.
3.6. Reaction mechanisms and pathways
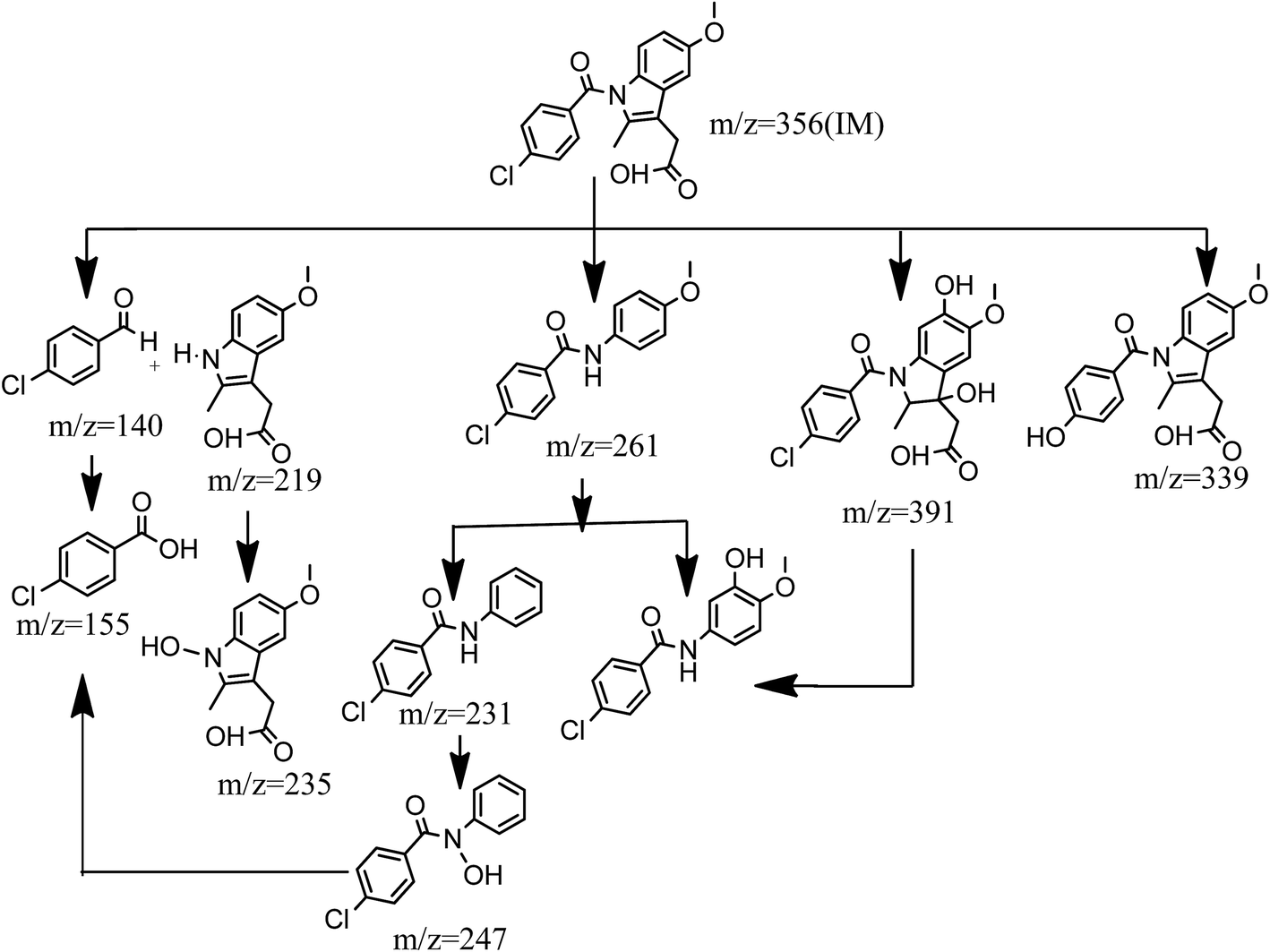
Transformation pathways of indomethacin in the Fe2+ activated peroxydisulfate system are proposed in Fig. 5. When SO4˙− and ˙OH attack organic compounds, SO4˙− preferentially reacts through an electron transfer process, while ˙OH participates by the way of hydrogen abstraction or addition reactions.35 It may be seen that there are four primary pathways. Pathway I is the cleavage of the carbonyl carbon on the benzene ring, and nitrogen on the indole group, which is attributed to the attack of SO˙− or ˙OH. The cleavage of carbonyl carbon on the benzene ring, and nitrogen on indole group, produces 4-chloro-benzaldehyde m/z 140 and m/z 219, where 4-chloro-benzaldehyde is further oxidized to form 4-chloro-benzoic acid. The N on the m/z 219 is inclined to be attacked by SO4˙− to yield a N-centered radical through one electron transfer, which can react with H2O to form m/z 235.36 Pathway II is subject to the cleavage of C–C and C–N bonds in the indole group, and produce m/z 261. Subsequently, m/z 261 goes through an ˙OH addition on the aromatic ring and the drop of methyl group on the side chain of the aromatic ring to produce m/z 277 and m/z 247, respectively. Pathway III is attributed to a one electron-transfer process that occurs to form an IM radical cation (IM˙+), which reacts quickly with H2O by way of hydroxyl abstraction, or addition reaction, to generate hydroxylated IM, which may be explained by the reactions shown in eqn (13) and (14).37 The aromatic ring and double bond in the indole group may serve as the attack point of SO4˙−, which leads to the formation of m/z 391. The interesting thing is that m/z 391 may initiate the cleavage of C–C and C–N bonds in the indole group to generate the same m/z 277 products of pathway II. Pathway IV is initiated by the substitution of chlorine by a hydroxyl group on the aromatic ring, which causes the formation of m/z 339. These results are consistent with previous research reported by Ji et al.38
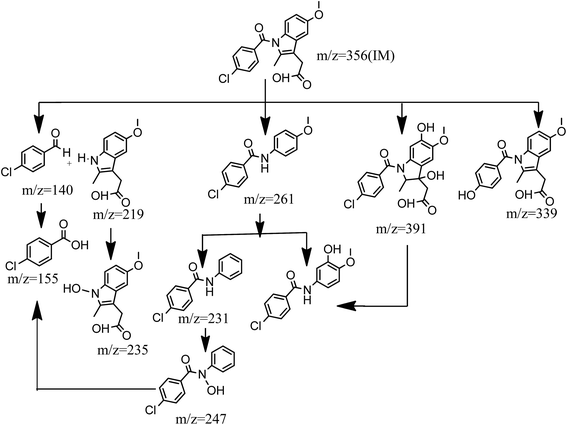 |
| | Fig. 5 Possible degradation pathways of IM in Fe2+-peroxydisulfate system. | |
The dominant path in the degradation of indomethacin by Fe2+ activated peroxydisulfate is unclear; however, the intensity of the iron peak in the total ion chromatography demonstrated that pathway III is relatively important. It should be emphasized that most of the transformation products were chlorinated compounds, which raised our concerns to assess their potential risks to humans, ecosystems, and the environment at large.
| | |
IM + SO4˙− → IM+ + SO42−
| (13) |
| | |
IM+ + H2O → (OH)IM + H+
| (14) |
4. Conclusions
This study revealed that initial PDS and Fe2+ concentrations had an important influence on the degradation of IM. With the increase of initial PDS concentrations, the IM removal efficiency was improved. A further enhancement both in oxidation and mineralization was obtained with the sequential addition of Fe2+. Increasing the solution pH acted to decrease the degradation rate of IM. The decrease of DO caused by N2 aeration slowed down the degradation efficiency, while IM degradation was inhibited by humic acid. The predominant oxidizing species responsible for IM degradation was identified as SO4˙− via radical scavenger tests.
It was demonstrated that a sustainable supply of Fe2+ introduced at a slow rate might make good use of generated SO4˙− for the degradation of IM. The primary degradation pathways of IM included the hydroxylation of the aromatic ring and the indole group, cleavage of C–N bonds, and the substitution of chlorine by hydroxyl radicals. The present study provides important information for the potential practical application of Fe2+/PDS.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21377031 and 21677040), the Innovative Team Program of High Education of Guangdong Province (2015KCXTD007)
References
- S. Mompelat, B. L. Bot and O. Thomas, Environ. Int., 2009, 35, 803–814 CrossRef CAS PubMed.
- R. Rosal, A. Rodríguez, J. A. Perdigón-Melón, A. Petre, E. García-Calvo, M. J. Gómez, A. Agüera and A. R. Fernández-Alba, Water Res., 2010, 44, 578–588 CrossRef CAS PubMed.
- Q. Sui, J. Huang, S. Deng, G. Yu, Q. Fan, T. Ternes and U. V. Gunten, Water Res., 2010, 44, 417–426 CrossRef CAS PubMed.
- J. T. Yu, E. J. Bouwer and M. Coelhan, Agr. Water. Manag., 2006, 86, 72–80 CrossRef.
- M. M. Sein, M. Zedda, J. Tuerk, T. C. Schmidt, A. Golloch and C. V. Sonntag, Environ. Sci. Technol., 2008, 42, 6656–6662 CrossRef CAS PubMed.
- T. Rodriguez-Alvarez, R. Rodil, J. B. Quintana, S. Trinanes and R. Cela, Water Res., 2013, 47, 3220–3230 CrossRef CAS PubMed.
- J. B. Quintana, R. Rodil, P. Lópezmahía, S. Muniateguilorenzo and D. Pradarodríguez, Water Res., 2010, 44, 243–255 CrossRef CAS PubMed.
- I. González-Mariño, J. B. Quintana, I. Rodríguez and R. Cela, Water Res., 2011, 45, 6770–6780 CrossRef PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1213–1223 CrossRef CAS PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1225 CrossRef CAS PubMed.
- H. Hori, A. Yamamoto, E. Hayakawa, S. Taniyasu, N. Yamashita, S. Kutsuna, H. Kiatagawa and R. Arakawa, Environ. Sci. Technol., 2005, 39, 2383 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS PubMed.
- C. Qi, X. Liu, C. Lin, X. Zhang, J. Ma, H. Tan and W. Ye, Chem. Eng. J., 2014, 249, 6–14 CrossRef CAS.
- C. Qi, X. Liu, C. Lin, H. Zhang, X. Li and J. Ma, Chem. Eng. J., 2017, 315, 201–209 CrossRef CAS.
- P. Neta, V. Madhavan, H. Zemel and R. W. Fessenden, J. Am. Chem. Soc., 1977, 99, 1 CrossRef.
- G. R. Peyton, Mar. Chem., 1993, 41, 91–103 CrossRef CAS.
- P. Neta, R. E. Huie and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 1027–1284 CrossRef CAS.
- A. Rastogi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 85, 171–179 CrossRef CAS.
- X. Xiong, B. Sun, J. Zhang, N. Gao, J. Shen, J. Li and X. Guan, Water Res., 2014, 62, 53–62 CrossRef CAS PubMed.
- R. K. Szabó, C. Megyeri, E. Illés, K. Gajda-Schrantz, P. Mazellier and A. Dombi, Chemosphere, 2011, 84, 1658–1663 CrossRef PubMed.
- X. Liu, T. Zhang, Y. Zhou, L. Fang and Y. Shao, Chemosphere, 2013, 93, 2717–2724 CrossRef CAS PubMed.
- I. Kim and H. Tanaka, Environ. Int., 2009, 35, 793–802 CrossRef CAS PubMed.
- X. Xu, G. Pliego, J. A. Zazo, J. A. Casas and J. J. Rodriguez, J. Hazard. Mater., 2016, 318, 355–362 CrossRef CAS PubMed.
- B. Lee and M. Lee, Environ. Sci. Technol., 2005, 39, 9278–9285 CrossRef CAS PubMed.
- C. S. Wang, S. F. Kang, H. J. Yang, S. Y. Pa and H. W. Chen, Environ. Technol., 2002, 23, 1415 CrossRef CAS PubMed.
- Y. Ji, C. Dong, D. Kong, J. Lu and Q. Zhou, Chem. Eng. J., 2015, 263, 45–54 CrossRef CAS.
- R. Yuan, Z. Wang, Y. Hu, B. Wang and S. Gao, Chemosphere, 2014, 109, 106–112 CrossRef CAS PubMed.
- X. Lou, D. Xiao, C. Fang, Z. Wang, J. Liu, Y. Guo and S. Lu, Environ. Sci. Pollut. Res., 2016, 23, 4778–4785 CrossRef CAS PubMed.
- E. Hayon, A. Treinin and J. Wilf, J. Am. Chem. Soc., 1972, 94, 47–57 CrossRef CAS.
- G. Wu, Y. Katsumura and G. Chu, Phys. Chem. Chem. Phys., 2000, 2, 5602–5605 RSC.
- Y. Guo, X. Lou, C. Fang, D. Xiao, Z. Wang and J. Liu, Environ. Sci. Technol., 2013, 47, 11174–11181 CrossRef CAS PubMed.
- K. L. Tim, W. Chu and N. J. D. Graham, Environ. Sci. Technol., 2007, 41, 613–619 CrossRef.
- B. H. Hameed and T. W. Lee, J. Hazard. Mater., 2009, 164, 468 CrossRef CAS PubMed.
- X. Hu, J. Yang and J. Zhang, J. Hazard. Mater., 2011, 196, 220–227 CrossRef CAS PubMed.
- F. Minisci, A. Citterio and C. Giordano, Acc. Chem. Res., 1983, 16, 27–32 CrossRef CAS.
- M. Mahdi Ahmed, S. Barbati, P. Doumenq and S. Chiron, Chem. Eng. J., 2012, 197, 440–447 CrossRef CAS.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- Y. Ji, C. Ferronato, A. Salvador, X. Yang and J. M. Chovelon, Sci. Total Environ., 2014, 472, 800–808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra03364h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Fuhua Lic and
Wenying Lva
*a,
Fuhua Lic and
Wenying Lva