
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed one-pot oxidative amidation between methylarenes and amines†
Yuhang Yang‡
a,
Jiajia Gu‡a,
Zheng Fang*a,
Zhao Yangb,
Ping Weia and
Kai Guo *ac
*ac
aCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China. E-mail: guok@njtech.edu.cn; fzcpu@163.com; Fax: +86 25 5813 9935; Tel: +86 25 5813 9926
bSchool of Engineering, China Pharmaceutical University, No. 639 Longmian Avenue, Nanjing 211198, China
cState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China
First published on 26th April 2017
Abstract
A new method for the direct one-pot oxidative amidation between methylarenes and amines catalyzed by copper has been developed. This method integrates methylarene oxidation and amide bond formation, which are usually accomplished separately, into a single operation. In addition, the reaction provides a relatively high yield and has a wide substrate scope. Moreover, the starting reagents are abundant and available in a convenient way at a cheaper price.
Introduction
The formation of amide bond is one of the most important organic chemistry reactions because amide functional group has extensive applications in pharmaceutical compounds, natural products and polymers.1 The synthesis of amides is usually carried out through the reaction of carboxylic acids or carboxylic acid derivatives with amines in the presence of coupling reagents,2 which produce byproducts.3 Moreover, these approaches have several disadvantages, such as poor atom utilization, using hazardous reagents and generation of waste which causes environmental problems. To overcome these problems, alternative methods have been developed for amide syntheses, such as the Beckmann rearrangement,4 Staudinger reaction,5 Schmidt reaction6 and Ugi reaction.7 Other methods involving selective hydrolysis of nitrile,8 dehydrogenative amidation of alcohol,9 coupling of amine with α-halonitroalkane promoted by iodonium,10 aminocarboxylation of aromatic halides,11 direct synthesis of amides from alcohols and nitroarenes or amines,12 transamidation of amines13 and oxidative amidation of aldehydes or alcohol14 have also been reported. Among these methods, oxidative amidation has received considerable attention in terms of green chemistry and economic factors and is highly desirable as it uses cheap, abundant and less hazardous starting materials.Direct oxidative amidation has attracted much attention due to atom economy and environmentally benign issues. Toluene and other methylarenes have also been used for oxidative amidation for the synthesis of aromatic amides. Mizuno and his co-workers reported the direct aerobic oxidative amidation of methylarenes using urea as the amine source in the presence of MnO2 catalyst (Scheme 1a).15 Even though the method was novel, the drawback was predictable. At 150 °C, urea decomposes easily, thereby limiting the yield. Wu16 and Sun17 also used this strategy to synthesise amides employing N,N-dimethylformamide as the amine source in the presence of ZnBr2 catalyst (Scheme 1b). Recently, Wang et al. reported iron-catalyzed oxidative amidation of methylarenes (Scheme 1c).18 Through this method, methylarenes have performed well with primary amines; however, their yield was less when secondary amines were used. Singh19 and Sekar20have reported the oxidative amidation of methylarenes using N-chloramine as the amine source in the presence of MnO2 and Fe(OAc)2, respectively (Scheme 1d). However, methylarenes with electron-withdrawing group did not perform well with this method. Moreover, economic starting materials used in these studies would be more attractive for the chemical industry. However, rigorous reaction conditions for oxidative amidation of methylarenes and the limited substrate scope limits their applications. Herein, we report an efficient and green one-pot procedure for the direct oxidative amidation of methylarenes with N-chlorosuccinimide, which have been prepared from amines, catalyzed easily by accessible copper salts (Scheme 1e). The starting materials of the reaction are abundant, economical and easily accessible, and the method is green, efficient, simple and convenient. Moreover, the reaction has a wide range of substrate scope.
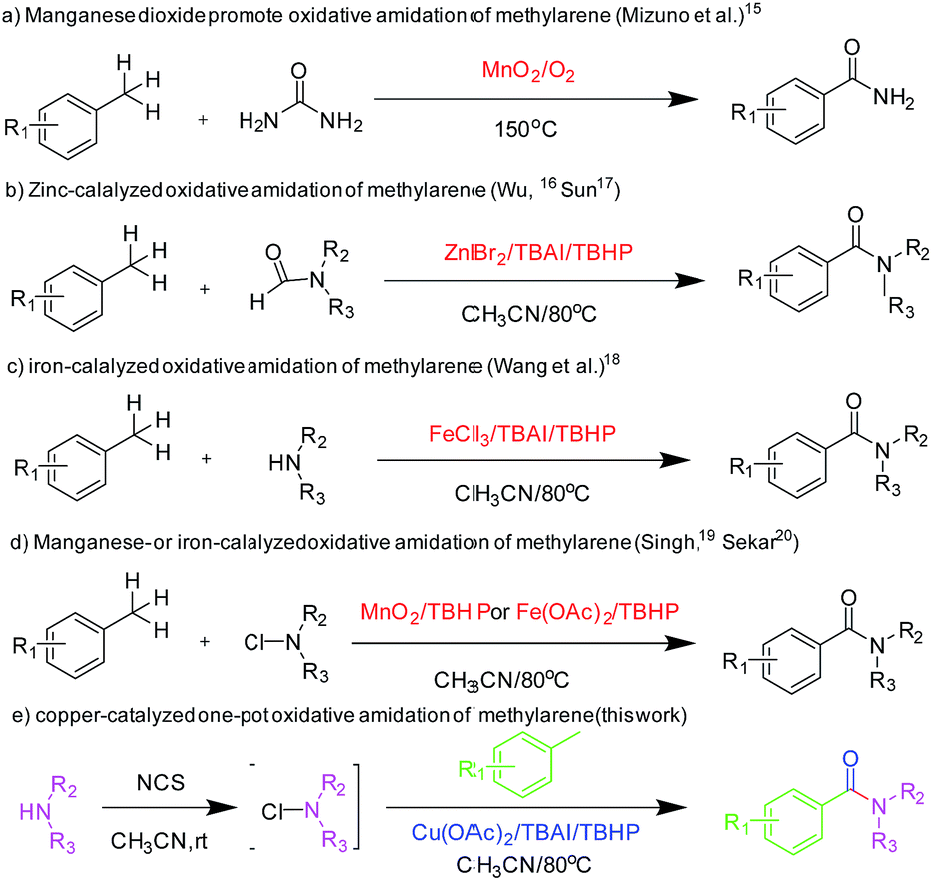 | ||
| Scheme 1 Metal-catalyzed oxidative amidation of methylarenes and amines. | ||
Results and discussion
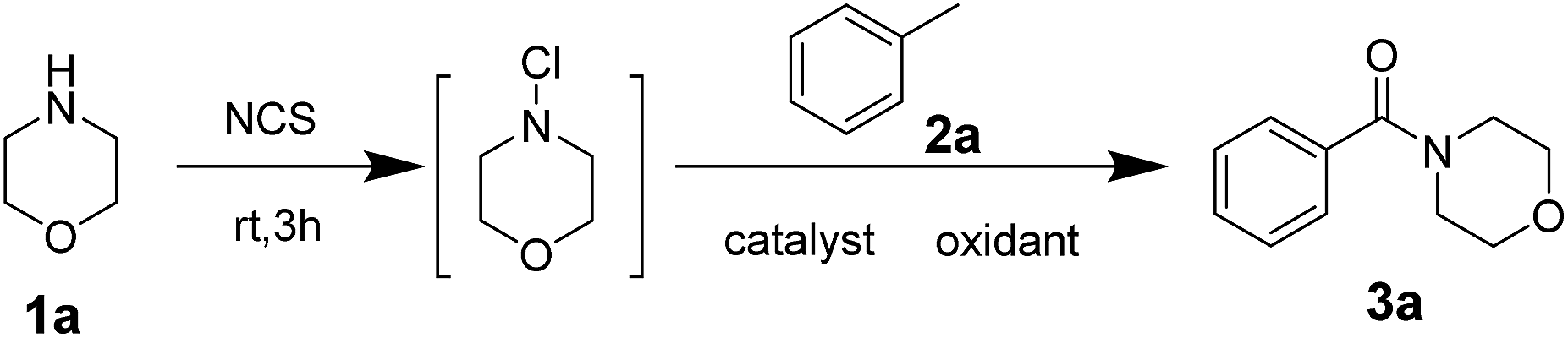
Morpholine 1a (1.0 equiv.) and methylbenzene 2a (10 equiv.) were used as starting materials to synthesise benzoyl morpholine, and the reaction was regarded as template reaction. In the beginning, morpholine was treated with N-chlorosuccinimide (NCS, 1.1 equiv.) at room temperature for 3 h, and methylbenzene, Cu(OAc)2 (10% mol) and TBHP (70% wt in water, 8.0 equiv.) were added into the reaction mixture under 80 °C to generate the amide product 3a with 48% yield (Table 1, entry 1). The yield was not good, so the reaction condition should be optimized. Different parameters such as catalysts, oxidants, and solvents were examined. In the first step, different copper salts such as Cu(OAc)2, CuSO4·5H2O, and CuCl2 were used to test the reaction and Cu(OAc)2 has shown superior activity among all of the catalysts with 48% yield (Table 1, entries 1–8). Furthermore, different oxidants involving TBPB, H2O2 and m-CPBA were used to evaluate the reaction and the best results were obtained for TBHP (Table 1, entries 9–11). Moreover, the product was obtained only in trace amounts when H2O2 and m-CPBA were used as oxidants. There was no product observed in the absence of an oxidant (Table 1, entry 12). The amounts of reactants were studied (Table 1, entries 13–15), based on which, 20% mol of catalyst and 8.0 equiv. of oxidant were chosen as the best mole ratio. It was found that increasing the amount of oxidant unlimitedly and decreasing the amount of oxidant did not yield good results. Finally, some additives were used to improve the yield of the reaction (Table 1, entries 16–19). After using some additives, yield of the product improved significantly, and TBAI performed better than others. In addition, using TBAI as additive without catalyst decreased the yield to a great extent (Table 1, entry 20).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Oxidant | Additive | Yieldb (%) |
| a Reaction conditions: morpholine (1.5 mmol), NCS (1.65 mmol, 1.1 equiv.), in 10 mL acetonitrile, stirring at room temperature for 3 h. To this reaction mixture, added toluene (15 mmol, 10 equiv.), catalyst (10 mol%) and oxidant (12 mmol, 8 equiv.), stirring at 80 °C for 24 h.b Isolated yield.c Reaction performed using 20 mol% of Cu(OAc)2.d Reaction performed using 50 mol% of Cu(OAc)2.e Reaction performed using 20 mol% of Cu(OAc)2 and 3 equiv. of TBHP.f Reaction performed using 20 mol% of Cu(OAc)2 and 5 equiv. of TBHP.g Reaction performed using 20 mol% of Cu(OAc)2 and 10 equiv. of TBHP.h Reaction performed using 20 mol% of Cu(OAc)2 and 20 mol% of I2.i Reaction performed using 20 mol% of Cu(OAc)2 and 20 mol% of KI.j Reaction performed using 20 mol% of Cu(OAc)2 and 20 mol% of TBAI.k Reaction performed using 20 mol% of Cu(OAc)2 and 20 mol% of NIS. | ||||
| 1 | Cu(OAc)2 | TBHP | — | 48 |
| 2 | Cu(OAc)2·H2O | TBHP | — | 44 |
| 3 | CuSO4·5H2O | TBHP | — | 32 |
| 4 | CuCl2 | TBHP | — | 26 |
| 5 | CuO | TBHP | — | 33 |
| 6 | CuBr | TBHP | — | 42 |
| 7 | CuI | TBHP | — | 38 |
| 8 | Cu2O | TBHP | — | 21 |
| 9 | Cu(OAc)2 | TBPB | — | 39 |
| 10 | Cu(OAc)2 | H2O2 | — | Trace |
| 11 | Cu(OAc)2 | m-CPBA | — | Trace |
| 12 | Cu(OAc)2 | — | — | — |
| 13c | Cu(OAc)2 | TBHP | — | 57 |
| 14d | Cu(OAc)2 | TBHP | — | 59 |
| 15e | Cu(OAc)2 | TBHP | — | 34 |
| 16f | Cu(OAc)2 | TBHP | — | 42 |
| 17g | Cu(OAc)2 | TBHP | — | 58 |
| 18h | Cu(OAc)2 | TBHP | I2 | 72 |
| 19i | Cu(OAc)2 | TBHP | KI | 79 |
| 20j | Cu(OAc)2 | TBHP | TBAI | 82 |
| 21k | Cu(OAc)2 | TBHP | NIS | 74 |
| 22 | — | TBHP | TBAI | 29 |
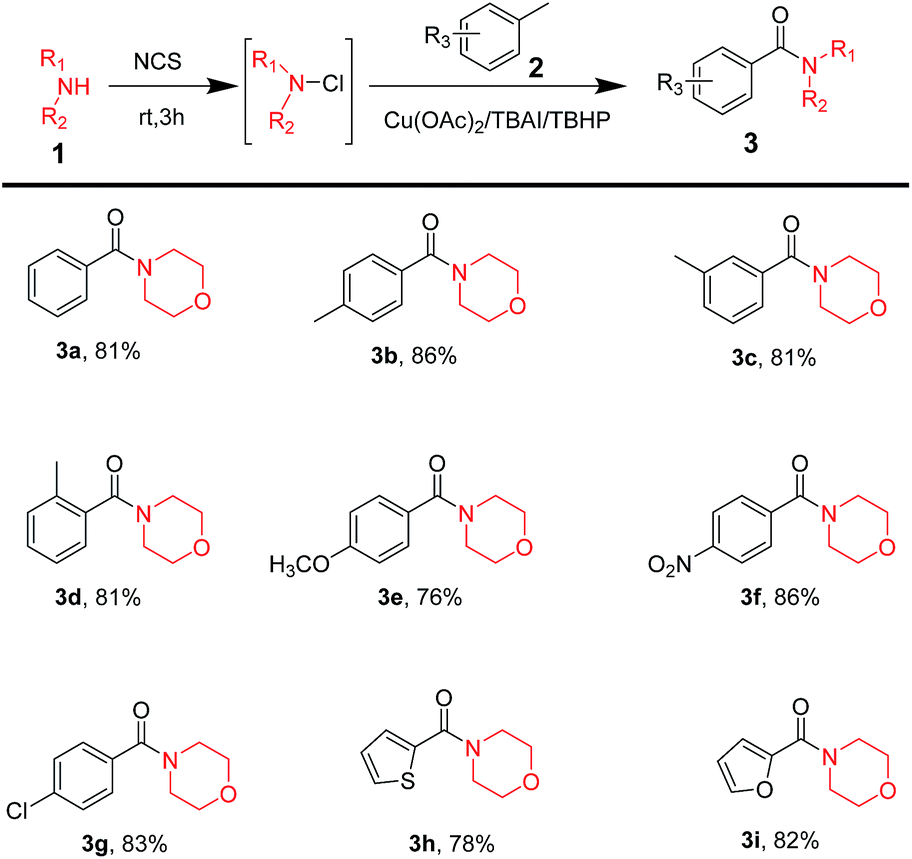
After figuring out optimum reaction conditions, a large range of commercially available methylarenes was used to test the substrate scope for this reaction; the result has been displayed in Table 2. It was surprising to note that methylarenes with electron-donating groups, such as CH3 (Table 2, 3b–3d), OCH3 (Table 2, 3e), or electron-withdrawing groups, like NO2 (Table 2, 3f), Cl (Table 2, 3g), resulted in corresponding amide products with excellent yield. Moreover, it was notable that the reaction possessed excellent chemo-selectivity when amines reacted with p-xylene (Table 2, 3b), m-xylene (Table 2, 3c) and o-xylene (Table 2, 3d) to obtain mono-amide products. The reaction could even tolerate some heterocycles as substrates, such as thiophene (Table 2, 3h) or oxole (Table 2, 3i) and they performed well.
| a Reaction conditions: morpholine (1.5 mmol), NCS (1.65 mmol, 1.1 equiv.), in 10 mL acetonitrile, stirring at room temperature for 3 h. To this reaction mixture, added methylarenes (15 mmol, 10 equiv.), catalyst (20 mol%), additive (20 mol%) and oxidant (12 mmol, 8 equiv.), stirring at 80 °C for 24 h. |
|---|
 |
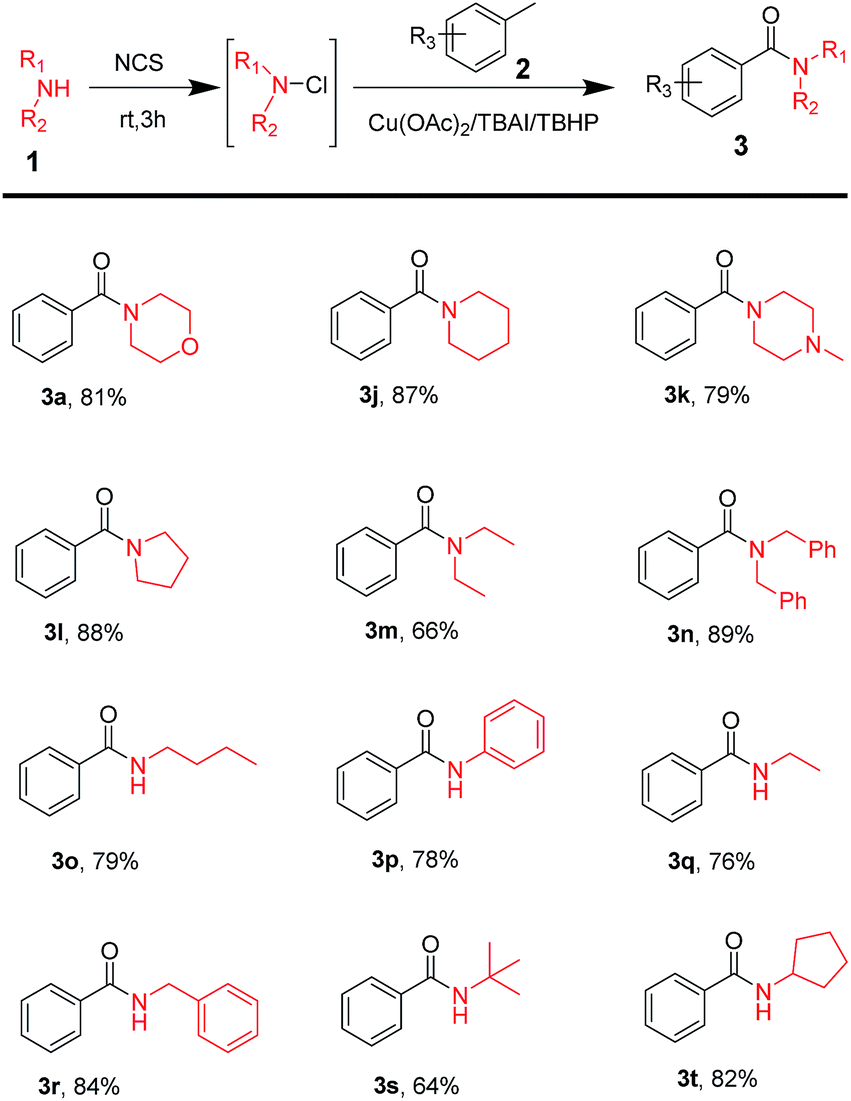
Moreover, commercially available amines were used to test the substrate scope of this reaction. The results of the test are shown in Table 3. The table shows that disubstituted amines, such as cyclic (Table 3, 3j–3l) and acyclic amines (Table 3, 3m–3n) played a great role in the reaction to obtain the corresponding amide products in good yields. Monosubstituted amines (Table 3, 3o–3r) have also shown excellent tolerance. On using amines with steric hindrance (Table 3, 3s–3t), results were good.
| a Reaction conditions: amines (1.5 mmol), NCS (1.65 mmol, 1.1 equiv.), in 10 mL acetonitrile, stirring at room temperature for 3 h. To this reaction mixture, added toluene (15 mmol, 10 equiv.), catalyst (20 mol%), additive (20 mol%) and oxidant (12 mmol, 8 equiv.), stirring at 80 °C for 24 h. |
|---|
 |
Aiming to exhibit the practical applications of the technology described above, we used copper-catalyzed one-pot oxidative amidation as a key step to synthesise moclobemide, a type of monoamine oxidase inhibitor antidepressant. On beginning with commercially available methylarenes, parachlorotoluene, we were able to obtain the corresponding product with 69% yield (Scheme 2).
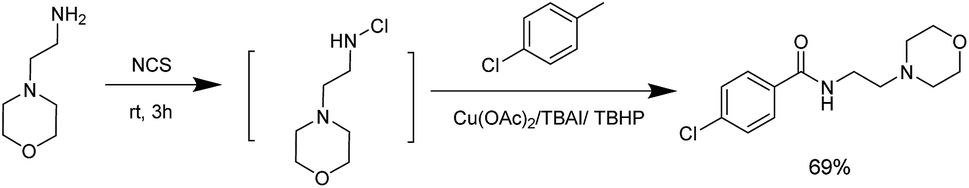 | ||
| Scheme 2 Synthesis of moclobemide. | ||
Based on previously reported studies, we considered a possible mechanism for this reaction and it is shown in Scheme 3. In the first step, TBAI was oxidized to IO− (or IO2−) by TBHP, and TBHP transformed into t-BuOH. Then, IO− (or IO2−) acted as real oxidizing agent to oxidize toluene to benzyl alcohol and benzyl alcohol was further oxidized to benzyl aldehyde. Copper played an important role in the reaction. Cu(II) reacted with TBHP in such a way that TBHP transformed into tert-butylperoxy radical and H+. Cu(II) also transformed into Cu(I). Tert-butylperoxy radical captured a hydrogen from an aldehyde, thereby transforming the aldehyde into acyl group. With redox reaction, the protonated N-chloramine was transformed into amino radical, as reported by Minisci.21,22 In the last step, amino radical and the acyl groups were coupled to yield corresponding amide products. To verify the conjecture, acyl radical, generated from toluene under the abovementioned reaction conditions, was trapped with 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) resulting in the formation of TEMPO adduct, which was detected by HRMS (Scheme 4).
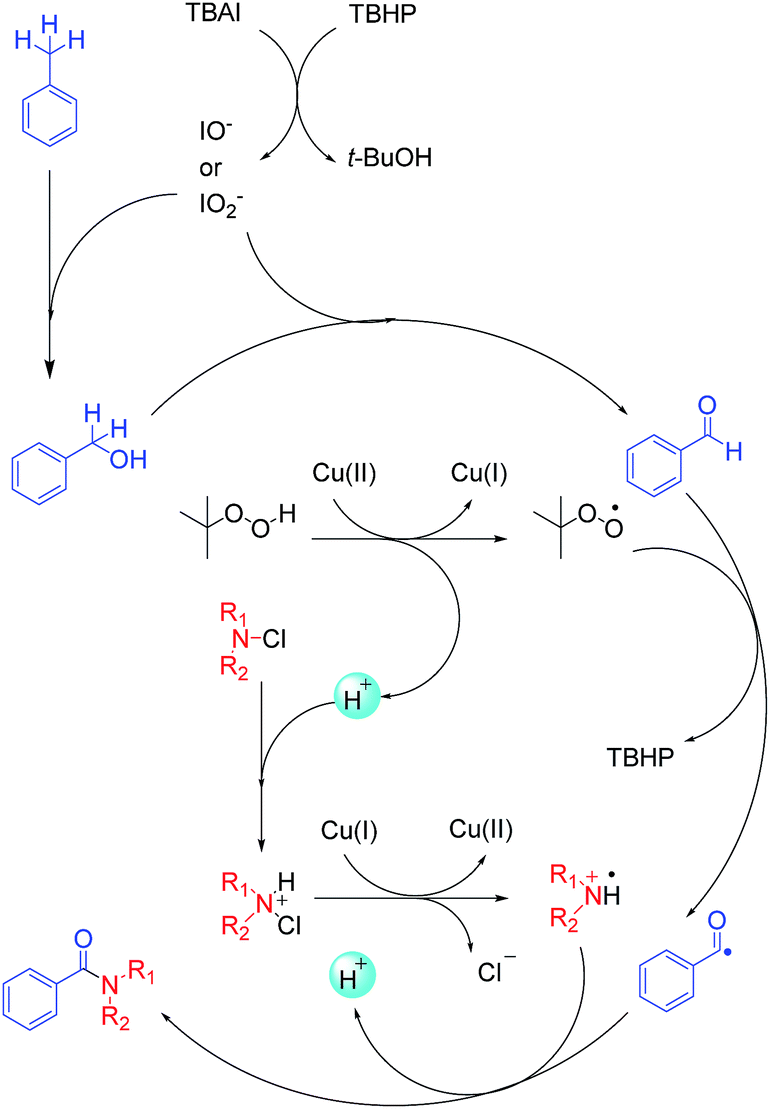 | ||
| Scheme 3 Proposed mechanism for amide synthesis. | ||
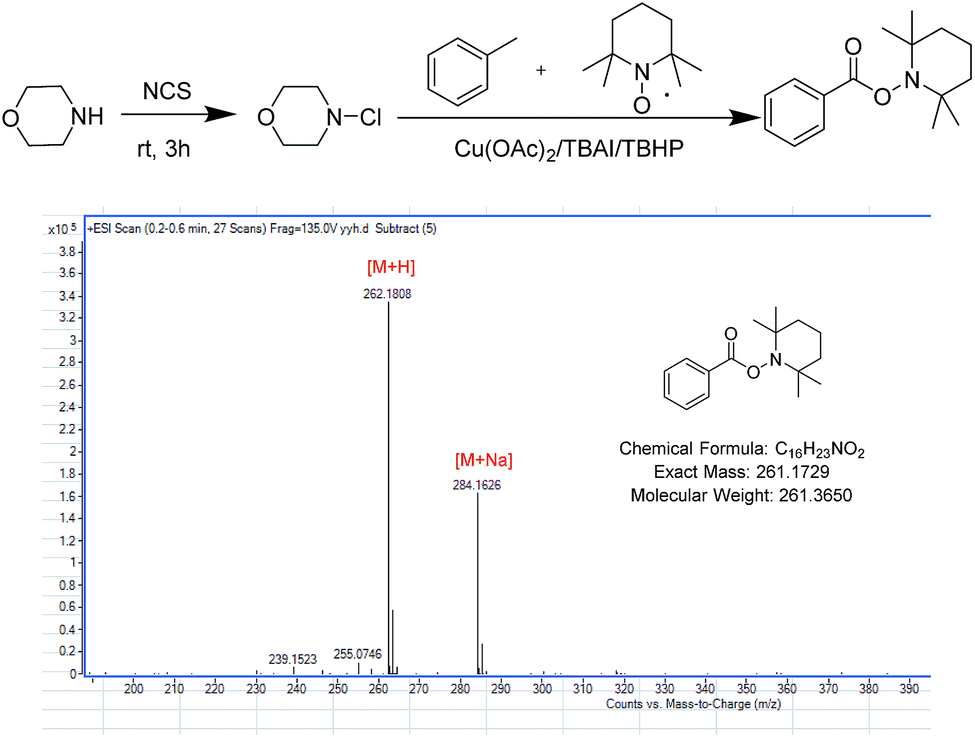 | ||
| Scheme 4 Trapping of the acyl radical. | ||
Conclusions
In summary, we developed an effective and environmentally friendly method for the synthesis of amides via copper-catalyzed one-pot direct oxidative amidation between methylarenes and amines. This method has combined methylarenes oxidation and amide bond formation into one procedure, which otherwise would follow two distinct steps. Using copper as a catalyst and TBHP as the sole oxidant proved to be economical too. What's more, the starting materials of the reaction are abundant, economical and easily accessible, and the reaction has a wide range of substrate scope.Acknowledgements
This research has been supported by the National Natural Science Foundation of China (Grant No. 21522604, U1463201 and 21402240); the youth in Jiangsu Province Natural Science Fund (Grant No. BK20150031 and BY2014005-03); a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243 CrossRef CAS PubMed.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827 CrossRef CAS.
- Selected recent examples: (a) Y. J. Kang, H. A. Chung, J. J. Kim and Y. J. Yoon, Synthesis, 2002, 733 CrossRef CAS; (b) I. Azumaya, T. Okamoto, F. Imabeppu and H. Takayanagi, Tetrahedron Lett., 2003, 59, 2325 CrossRef CAS; (c) A. Teichert, K. Jantos, K. Harms and A. Studer, Org. Lett., 2004, 6, 3477 CrossRef CAS PubMed; (d) D. M. Shendage, R. Froehlich and G. Haufe, Org. Lett., 2004, 6, 3675 CrossRef CAS PubMed; (e) D. A. Black and B. A. Arndtsen, Org. Lett., 2006, 8, 1991 CrossRef CAS PubMed; (f) A. R. Katritzky, C. Cai and S. K. Singh, J. Org. Chem., 2006, 71, 3375 CrossRef CAS PubMed; (g) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed.
- (a) E. Beckmann, Ber. Dtsch. Chem. Ges., 1886, 89, 988 CrossRef; (b) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599 CrossRef CAS PubMed.
- (a) Y. G. Gololobov and L. F. Kasukhin, Tetrahedron, 1992, 48, 1353 CrossRef CAS; (b) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007 CrossRef CAS PubMed; (c) F. Damkaci and P. DeShong, J. Am. Chem. Soc., 2003, 125, 4408 CrossRef CAS PubMed.
- (a) T. Ribelin, C. E. Katz, D. G. English, S. Smith, A. K. Manukyan, V. W. Day, B. Neusenswander, J. L. Poutsma and J. Aube, Angew. Chem., 2008, 120, 6329 (Angew. Chem., Int. Ed., 2008, 47, 6233) CrossRef; (b) S. Lang and J. A. Murphy, Chem. Soc. Rev., 2006, 35, 146 RSC.
- I. Ugi, Angew. Chem., 1962, 74, 9 (Angew. Chem., Int. Ed. Engl., 1962, 1, 8) CrossRef CAS.
- (a) P. M. Bendale and B. M. Khadilkar, Synth. Commun., 2000, 30, 1715 Search PubMed; (b) Z. Gordi and H. Eshghi, J. Korean Chem. Soc., 2011, 55, 716 Search PubMed.
- (a) S. Cho, E. Yoo, I. Bae and S. Chang, J. Am. Chem. Soc., 2005, 127, 16046 CrossRef CAS PubMed; (b) Z. W. Chen, H. F. Jiang, X. Y. Pan and Z. J. He, Tetrahedron, 2011, 67, 5920 CrossRef CAS.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027 CrossRef CAS PubMed.
- J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed.
- C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20 CAS.
- (a) M. Tamura, T. Tonomura, K. i. Shimizu and A. Satsuma, Green Chem., 2012, 14, 717 RSC; (b) T. A. Dineen, M. A. Zajac and A. G. Myers, J. Am. Chem. Soc., 2006, 128, 16406 CrossRef CAS PubMed; (c) M. Zhang, S. Imm, S. Baehn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 3905 CrossRef CAS PubMed.
- (a) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992 CrossRef PubMed; (b) S. C. Ghosh, J. S. Y. Ngiam, C. L. L. Chai, A. M. Seayad, D. T. Tuan and A. Chen, Adv. Synth. Catal., 2012, 354, 1407 CrossRef CAS; (c) C. G. Subhash, S. Y. N. Joyce, A. M. Seayad, T. T. Dang, C. L. L. Chai and A. Q. Chen, J. Org. Chem., 2012, 77, 8007 CrossRef PubMed; (d) C. Roberta, P. Andrea, G. Giampaolo and L. D. Luca, Org. Lett., 2012, 14, 5014 CrossRef PubMed; (e) J. J. Gu, Z. Fang, C. K. Liu, X. Li, P. Wei and K. Guo, RSC Adv., 2016, 6, 72121 RSC; (f) J. J. Gu, Z. Fang, Y. H. Yang, Z. Yang, X. Li, L. Wan, P. Wei and K. Guo, RSC Adv., 2016, 6, 89413 RSC; (g) J. J. Gu, Z. Fang, C. K. Liu, Z. Yang, X. Li, P. Wei and K. Guo, RSC Adv., 2015, 5, 95014 RSC; (h) K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544 CrossRef CAS PubMed; (i) K. Xu, Y. Hu, S. Z. Zha and Z. Wang, Chem.–Eur. J., 2012, 18, 9793 CrossRef CAS PubMed.
- Y. Wang, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7250 CrossRef CAS PubMed.
- J. B. Feng, D. Wei, J. L. Gong, X. Qi and X. F. Wu, Tetrahedron Lett., 2014, 55, 5082 CrossRef CAS.
- B. Du and P. Sun, Sci. China: Chem., 2014, 57, 1176 CrossRef CAS.
- T. Wang, L. Yuan, Z. G. Zhao, A. L. Shao, M. Gao, Y. F. Huang, F. Xiong, H. L. Zhang and J. F. Zhao, Green Chem., 2015, 17, 2741 RSC.
- R. Vanjari, T. Guntreddi and K. N. Singh, Org. Lett., 2013, 15, 4908 CrossRef CAS PubMed.
- S. S. Kotha, S. Badigenchala and G. Sekar, Adv. Synth. Catal., 2015, 357, 1437 CrossRef.
- F. Minisci, F. Fontana, S. Araneo, F. Recupero, S. Ban and S. Quici, J. Am. Chem. Soc., 1995, 117, 226 CrossRef CAS.
- F. Minisci, Synthesis, 1973, 1 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General information and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c7ra02942j |
| ‡ These authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2017 |