
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow-ultrasonic synergy in click reactions for the synthesis of novel 1,2,3-triazolyl appended 4,5-unsaturated L-ascorbic acid derivatives†
Andrijana Meščića,
Anita Šalićb,
Tomislav Gregorića,
Bruno Zelićb and
Silvana Raić-Malić*a
aUniversity of Zagreb, Faculty of Chemical Engineering and Technology, Department of Organic Chemistry, Marulićev trg 20, HR-10000 Zagreb, Croatia. E-mail: sraic@fkit.hr; Fax: +385-1-4597260; Tel: +385-1-4597213
bUniversity of Zagreb, Faculty of Chemical Engineering and Technology, Department of Reaction Engineering and Catalysis, Savska cesta 16, HR-10000 Zagreb, Croatia
First published on 3rd January 2017
Abstract
A combination of flow chemistry and batch-based synthetic procedures has been successfully applied to the assembly of novel 4,5-unsaturated L-ascorbic acid series 6a–6n with diverse C-6-substituted 1,2,3-triazole moiety. We report herein the first Cu(I)-catalyzed 1,3-dipolar cycloaddition of azido functionalized L-ascorbic acid derivative and selected alkynes to provide target 1,2,3-triazolyl appended 4,5-didehydro-5,6-dideoxy-L-ascorbic acid library 6a–6n under both micro-flow and batch conditions. Implementation of ultrasound with flow chemistry accelerated hour-scale reaction conditions in batch to the minute range in micro-flow device and considerably improved the yields for the flow syntheses of 6a–6n. Moreover, the synergistic use of microreactor technology and ultrasonic irradiation highlights the sustainable eco-friendly aspect of utilized method.
Introduction
L-Ascorbic acid (AA) or vitamin C is known as a ubiquitous carbohydrate of vital importance in the living beings. Since the discovery of vitamin C, the number of its known biological functions is continually expanding.1 Thus, it plays an important role in a number of cellular processes, including collagen synthesis, cellular oxidation, and as a cofactor in several important hydroxylation reactions.2 AA is one of the most important biomolecules, which protects cellular components against oxidative damage caused by free radicals and oxidants that are involved in the development of a multitude of chronic diseases. Furthermore, some L-ascorbic acid derivatives showed to be effective antitumoral agents,3 immunostimulants4 and protecting agents against peroxidation of lipids of the biomembranes.5 In previous study we demonstrated that some pyrimidine and purine derivatives of 4,5-didehydro-5,6-dideoxy- and 6-deoxy-L-ascorbic acid exhibited selective and efficient cytostatic activities and displayed antiviral activities against varicella zoster virus (TK+VZV and TK−VZV) and cytomegalovirus (CMV).6 Structurally related γ-(triazolyl ethylidene)butenolides exhibited most potent activity against breast cancer cells and preliminary due to reactive oxygen species (ROS) generation, subsequent activation of p38, leading to apoptosis and inhibition of cancer cells.7 Furthermore, several preclinical and clinical trials on vitamin C have also supported its promising efficacy against a number of cancer types and low toxicity of high-dose vitamin C, that has been demonstrated in anticancer therapy.8On the other hand, 1,2,3-triazoles have been the nuclei of choice in recent years because of their excellent pharmacokinetic characteristics, favourable safety profile, latent ability for the formation of hydrogen bonds, moderate dipole character, rigidity and stability under in vivo conditions.9 Therefore, triazole chemistry has recently received significant impulse from the pioneering work of Sharpless and Meldal on Cu(I)-catalyzed alkyne–azide cycloaddition (CuAAC).10 The CuAAC reaction has become the cream of the crop for click chemistry and has been broadly used in many areas of modern chemistry, such as polymer and materials sciences,11 supramolecular chemistry,12 bioconjugation,13 and combinatorial chemistry.14 This reaction is also significant from the viewpoint of drug discovery,15 because 1,2,3-triazoles possess a wide range of biological properties, such as anticancer,16 antiviral,17 antibacterial18 and antifungal activities.19 Therefore, the 1,2,3-triazole moiety is extensively used in medicinal chemistry as a pharmacophore to modify known bioactive molecules and to potentiate their biological activities.9a,20 A growing effort has been devoted to the employment of click chemistry for the synthesis of functionally diverse carbohydrate derivatives as bioactive compounds and fluorescent glycoprobes.21
Although click reactions are traditionally conducted in batch process, the progress toward increased sustainability that requires novel approaches with reduced environmental impact opened up in recent years continuous-flow as a novel alternative to conventional batch-based synthesis.22 Flow chemistry technique performs a chemical reaction in a continuously flowing stream in a network of interconnecting tubes. The reagents coming from different tubes are mixed together in the reactor to start the chemical reaction. Due to its many advantages over batch reaction processes, flow chemistry has become an emerging field in organic synthesis, and has recently been employed to enhance the selectivity, safety, and efficiency of chemical syntheses, including the rapid synthesis of natural products and pharmaceuticals.23 Microreactor technology has the potential to revolutionize the pharmaceutical industry because the number of potential drug candidates that can be prepared and screened can be considerably increased, hence the likelihood of developing new drugs is considerably enhanced.24 Besides being more economical and environmentally friendly, microreactor technology (MRT) is even more advantageous than the macroscopic batch reactor method, mainly due to the high surface/volume ratio, reduced utilization of reagents, and also better control over mass as well as heat transfer.25 Many previously challenging or hazardous reactions can now be safely and efficiently conducted in flow.26 In addition, flow chemical synthesis can be performed under non-classical reaction conditions, such as applying ultrasound and microwave irradiation, to significantly improve efficiency of reactions.27 It was demonstrated that the use of ultrasound, an alternative green source of energy, to promote chemical reactions allowed a decrease in the reaction time and side reactions. On the other hand, with a well-defined inner micro-structure, a microreactor provides an ideal environment to investigate and control the acoustic cavitation process, which is the main mechanism causing sonochemical effects.28 Although the advantages of the continuous flow reactor technology for the synthesis of 1,4-disubstituted 1,2,3-triazoles over their batch reaction counterparts have been demonstrated recently29 further improvement in this direction is still mandatory. Despite the increasingly importance of flow chemistry in discovery of biologically active molecules, to our knowledge no studies are hitherto performed on the use of continuous flow to provide L-ascorbic acid and its derivatives, as molecules with unique biological properties. Realizing the benefits of flow chemistry and in continuation of our study towards the synthesis of 1,2,3-triazole-containing heterocycle pharmacophores,15b,30 our aim was to improve the practical applicability of Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC) to provide diverse C-6-substituted 1,2,3-triazolyl 4,5-unsaturated L-ascorbic acid derivatives 6a–6n using innovative enabling ultrasound (US) and continuous flow hybrid system (Fig. 1).
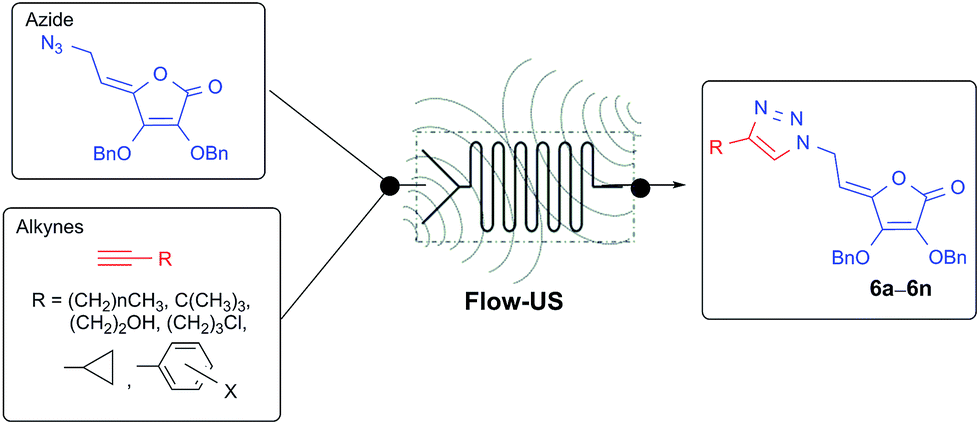 | ||
| Fig. 1 Synthesis of C-6-substituted 1,2,3-triazolyl 4,5-unsaturated L-ascorbic acid derivatives 6a–6n utilizing ultrasonic (US)-assisted continuous flow in click reactions. | ||
Herein we report classical multistep synthesis to afford azido functionalized L-ascorbic acid for clicking with terminal alkynes and investigate both continuous flow-ultrasound and batch approach for CuAAC reaction.
Results and discussion
Synthesis of the 1,2,3-triazolyl appended 4,5-unsaturated L-ascorbic acid library under batch conditions
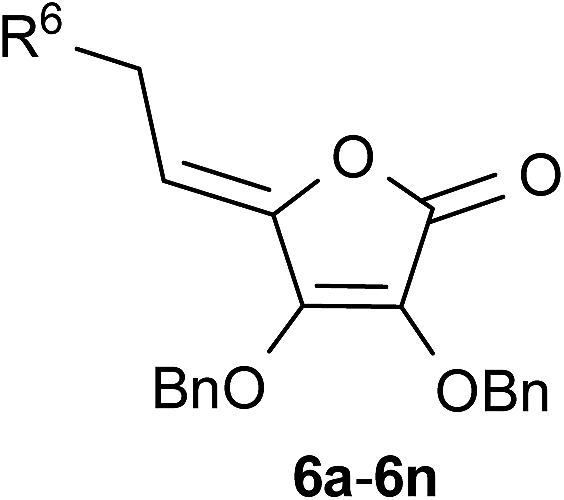
In order to synthesize C-6-substituted 1,2,3-triazolyl 4,5-didehydro-5,6-dideoxy-L-ascorbic acid derivatives, synthetic transformations of L-ascorbic acid that included selective protection and deprotection of 2,3- and 5,6-dihydroxyl groups were performed as described in the literature.31 Thus, 5,6-O,O-isopropylidene-L-ascorbic acid (1) was synthesized via reaction of L-ascorbic acid with acetyl chloride, while the remaining C-2 and C-3 hydroxyl groups of the lactone ring were protected by reaction of compound 1 with benzyl chloride in the presence of potassium carbonate to afford compound 2. To apply molecular hybridization by coupling of L-ascorbic acid derivative with 1,2,3-triazole ring, we used the strategy that involved the functionalization of L-ascorbic acid with an azide substituent at the C-6 (Scheme 1). Therefore, isopropylidene protection in 2 was removed in acidic condition to give compound 3 with free C-5 and C-6 hydroxyl functionalities, which were subsequently converted to tosylate, as a good leaving group, using p-toluenesulfonyl chloride (p-TsCl) in pyridine. The 5,6-O,O-ditosyl derivative of L-ascorbic acid (4) was used as the parent compound for the preparation of the C-6 azido derivative of 4,5-unsaturated L-ascorbic acid 5. Compound 5 was successfully synthesized from compound (4) through elimination to an exocyclic allylic tosylate which through a subsequent SN2 reaction with sodium azide (NaN3) directly afforded azide 5 in a moderate yield of 51%. 1,2,3-Triazolyl appended 4,5-unsaturated L-ascorbic acid derivatives 6a–6n with various substituents at C-8 of the triazole ring were obtained through the copper mediated 1,3-dipolar cycloaddition of C-6 azido L-ascorbic acid derivative 5 and terminal alkynes (Scheme 1). In order to assess the influence of structural modifications at C-8 of the triazole on activity, selected alkynes a–n were used in click reaction with the azide 5. The synthesis of 6a–6n was performed using Cu(OAc)2, which enabled the reduction of Cu(II) to Cu(I) ions through reaction with methanol.32 (Z)-Configuration around the C-4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-5 double bond was verified by the ROESY spectra which displayed the cross peak between the O-methylene at C-3 and methine C-5 protones. In the hypothetical (E)-configuration, where the spatial distance between C-3 and C-5 protons is too large, mentioned interactions would not be expected. These findings are in agreement with the determined (Z)-configuration of closely related pyrimidine and purine derivatives of L-ascorbic acids.33
C-5 double bond was verified by the ROESY spectra which displayed the cross peak between the O-methylene at C-3 and methine C-5 protones. In the hypothetical (E)-configuration, where the spatial distance between C-3 and C-5 protons is too large, mentioned interactions would not be expected. These findings are in agreement with the determined (Z)-configuration of closely related pyrimidine and purine derivatives of L-ascorbic acids.33
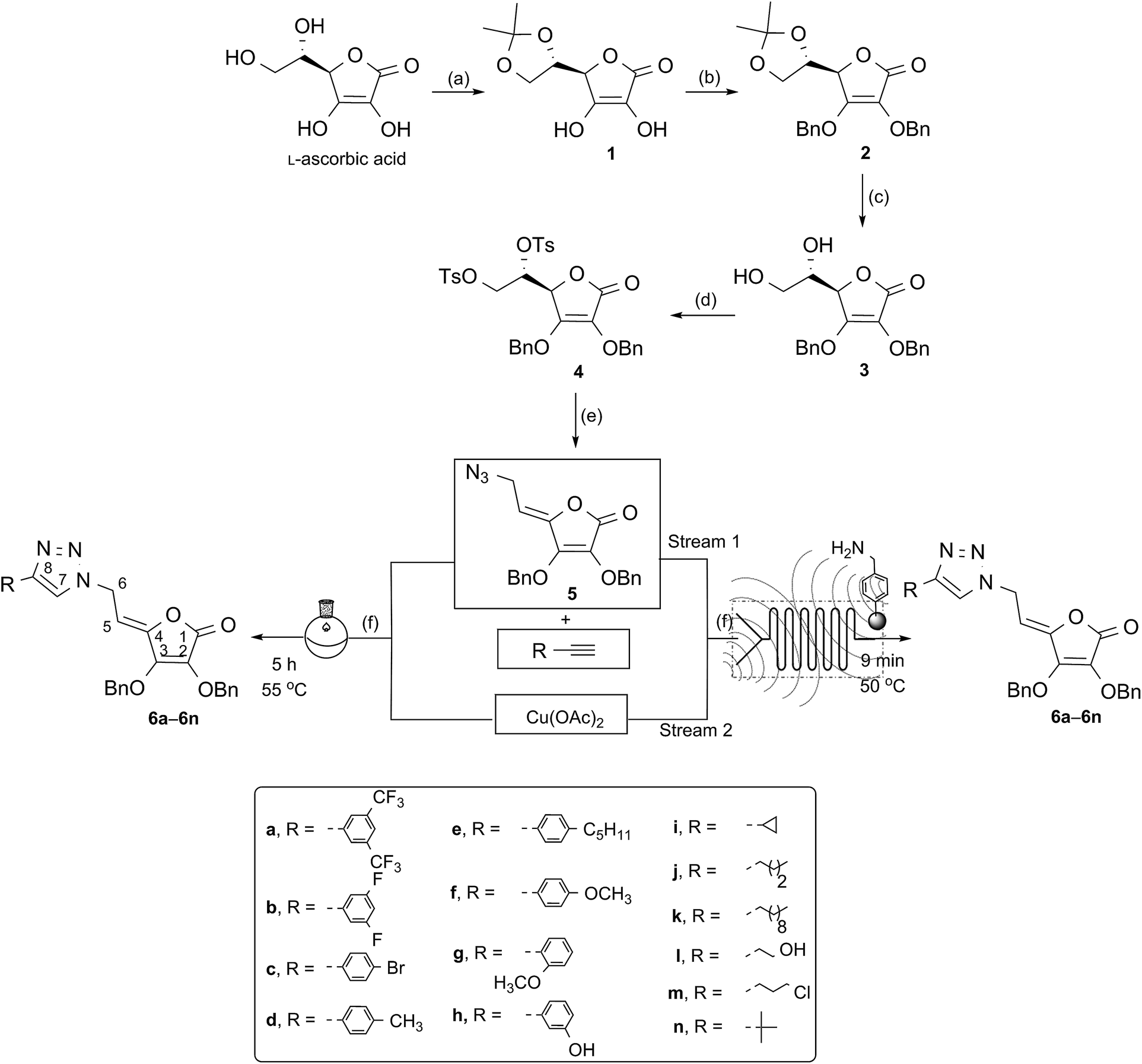 | ||
| Scheme 1 Synthesis of novel 1,4-disubstituted-1,2,3-triazole L-ascorbic acid derivatives by conventional batch and flow-ultrasound conditions. Reagents and conditions: (a) acetyl chloride, acetone; (b) benzyl chloride, K2CO3; (c) 50% acetic acid, CH3OH; (d) p-TsCl, CH2Cl2, pyridine, 0 °C; (e) NaN3, acetonitrile, 80 °C; (f) CH3OH. | ||
Synthesis of the 1,2,3-triazolyl appended 4,5-unsaturated L-ascorbic acid library under continuous flow process
In order to investigate the advantages of flow chemistry over batch methods, the Cu(I)-catalyzed click reactions of azido functionalized L-ascorbic acid derivative 5 and alkynes were performed in a microreactor system. Two enabling technologies, micro-flow chemistry and ultrasound irradiation (US) have been combined and compared to batch mode synthesis. To combine ultrasound irradiation and microfluidic system, we applied a simple and inexpensive technique, which was at the same time safe and efficient, by directly immersing the microreactor in the ultrasonic bath.For the optimization of reaction parameters, click chemistry of azido derivative of L-ascorbic acid 5 and 3,5-di(trifluoromethyl)phenylacetylene and 3-butyne-1-ol were chosen as test reactions to obtain 1,4-disubstituted 1,2,3-triazoles 6a and 6l, respectively, with the highest yield (Scheme 1 and Table 1, entries 1–4). The highest conversion of azide 5 to 1,2,3-triazole products was observed under ultrasound-assisted micro-flow using the 0.1 M solution of the starting azide and alkyne, flow rate of 0.5 μL min−1 and a slightly elevated temperature of 50 °C (Table 1, entry 3). The residence time for the compounds prepared under these reaction conditions amounted 9 min. Higher reaction temperatures typically resulted in the formation of unidentified side-products.
Higher concentration of the starting compounds and lower flow rate (0.2 μL min−1) led to the precipitation of the triazole product in the flow microreactor. However, when we attempted to decrease the residence time by increasing the flow rate to 1 μL min−1 the yields of 6a and 6l decreased.
As we can observe from Table 2, the yields of both flow-ultrasound and batch-based reactions were dependent on the C-8 substituents at 1,2,3-triazole scaffold. Interestingly, the effects of C-8 substituents in continuous micro-flow and batch syntheses were similar. Notably, all tested reactions using combination of micro-flow and ultrasound method were more successful than those under classical synthetic conditions. Among the aromatic alkynes, type of the substituents and their position on the phenyl ring were nicely tolerated in click reactions. In general, electron-donating groups gave target 1,2,3-triazolyl appended L-ascorbic acid derivatives (6e–6h) under micro-flow in excellent yields. Only exception was the click reaction with alkyne containing p-methylphenyl moiety, that afforded 4-tolyl-1,2,3-triazolyl appended L-ascorbic acid (6d) using both micro-flow and batch conditions in the lowest yields of 45% and 20%, respectively. While the position of the electron-donating substituents did not affect the CuAAC reaction in flow method, o- and m-substituted electron-donating group significantly improved the reactions (6g and 6h) in comparison to p-substituted phenyl (6e and 6f) using classical synthetic approach. From the halogen and trifluoromethyl, as electron-withdrawing substituents on the phenyl ring in selected alkynes, we can note that p-bromo substituent caused the moderate yield in the synthesis of the desired 4-(p-bromophenyl)-1,2,3-triazole appended L-ascorbic acid (6c). In contrast to that, 1,3-dipolar reaction with dipolarophile containing stronger electron-withdrawing fluorine and trifluoromethyl substituents at 3 and 5 position of the phenyl moiety proceeded under micro-flow with excellent yields (6a and 6b). From the aliphatic alkynes, we found that unbranched alkynes afforded 1,2,3-triazolyl appended L-ascorbic acid (6j–6m) with side chains at C-8 of triazole in higher yields than branched alkyne (6n). Thus, long-chain dodecyne was successfully applied, as a dipolarophile in CuAAC reaction, affording the 4-decyl-1,2,3-triazole appended L-ascorbic acid (6k) in the highest yields of 98% and 83% under micro-flow and batch conditions, respectively. Overall, it was demonstrated that the ultrasonic-assisted flow method applied in click reactions to provide target 1,2,3-triazolyl appended 4,5-unsaturated L-ascorbic acid derivatives gave improved yields and considerably shortened the reaction time of click reactions. Thereby, the applied methodology demonstrated to be a more efficient and sustainable approach than the batch-based synthesis.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Compd | R6 | Yield [%] | Entry | Compd | R6 | Yield [%] | ||
| Batcha | Flowb | Batcha | Flowb | ||||||
| a Yield was determined by HPLC chromatograms of the reaction mixture.b Yields were determined by isolation of the product. | |||||||||
| 1 | 6a | 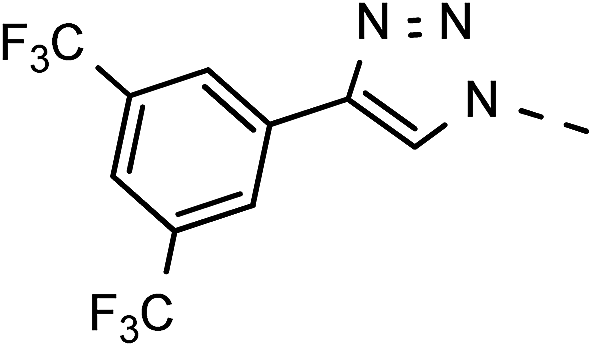 |
81 | 99 | 8 | 6h | 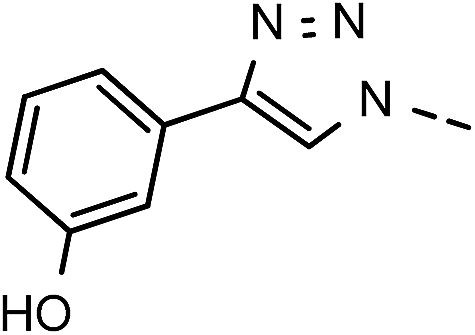 |
79 | 99 |
| 2 | 6b | 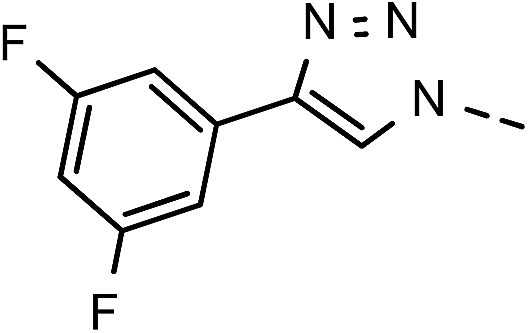 |
62 | 89 | 9 | 6i | 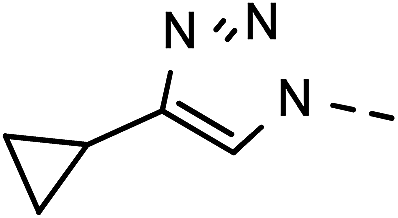 |
70 | 76 |
| 3 | 6c | 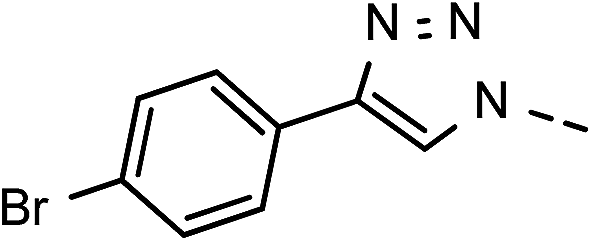 |
49 | 63 | 10 | 6j | 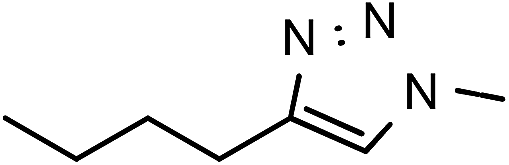 |
64 | 74 |
| 4 | 6d | 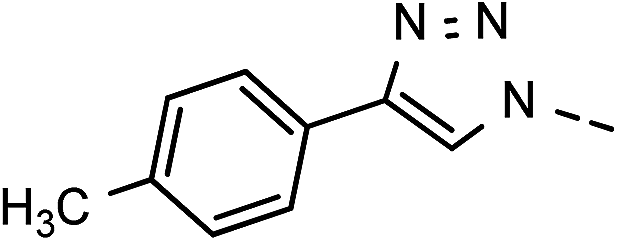 |
20 | 45 | 11 | 6k | 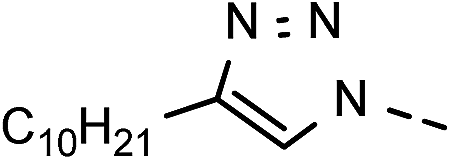 |
83 | 98 |
| 5 | 6e | 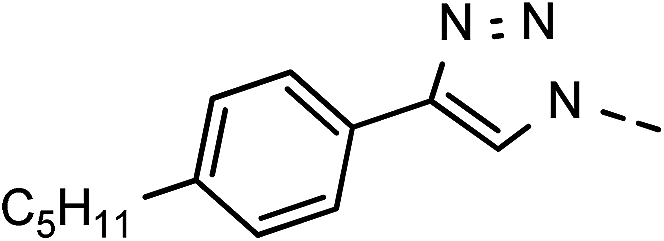 |
46 | 88 | 12 | 6l | 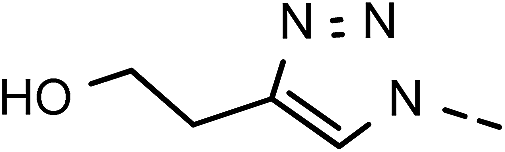 |
61 | 83 |
| 6 | 6f | 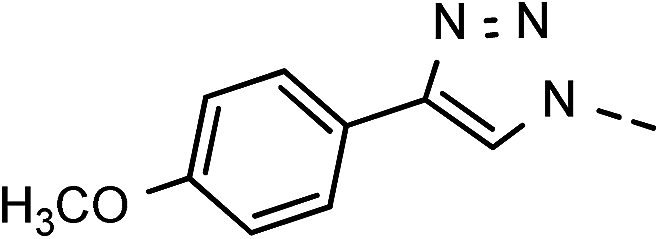 |
49 | 98 | 13 | 6m | 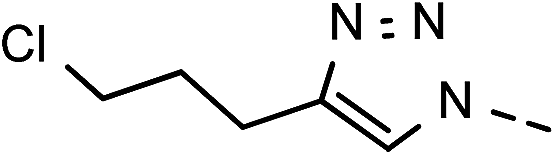 |
36 | 86 |
| 7 | 6g | 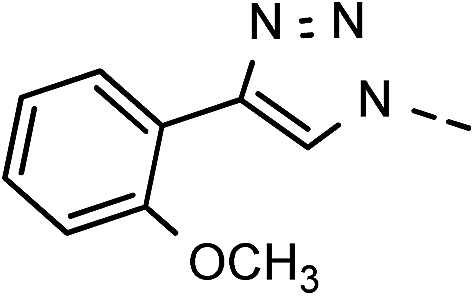 |
82 | 94 | 14 | 6n | 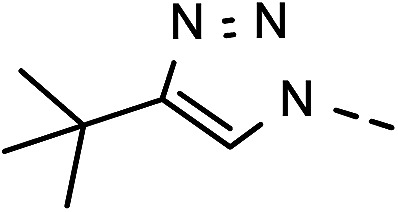 |
26 | 62 |
Conclusions
We have performed the multistep synthesis of the 6-azido derivative of L-ascorbic acid 5 as a precursor for Cu(I)-catalyzed 1,3-dipolar cycloaddition (CuAAC) with alkynes. Click reactions of the azide 5 and selected terminal alkynes were subsequently carried out in the presence of Cu(OAc)2, that was employed as a catalyst to generate the required Cu(I)-ions in situ, to provide the novel C-6-substituted 1,2,3-triazolyl 4,5-didehydro-5,6-dideoxy-L-ascorbic acid series 6a–6n. (Z)-Configuration across the C-4C-5 double bond in 6a–6n was deduced from connectivities in ROESY spectra. To evaluate the potential benefits of continuous micro-flow on CuAAC reactions, the batch conditions of click chemistry were successfully translated into a flow regime. Moreover, for the environmentally benign synthesis of 6a–6n two enabling technologies, ultrasound irradiation and flow microreactors, have been combined and compared to batch mode synthesis. The CuAAC reactions leading to triazole–L-ascorbic acid hybrids 6a–6n were greatly facilitated through the beneficial features of continuous micro-flow and ultrasound. Namely, reduced reaction time from hour-scale reaction to minute range and significantly improved yields in flow click reactions demonstrated the excellent synthetic capabilities of the applied method. Finally, we may conclude that translating batch click chemistry to ultrasound-assisted micro-flow processes in the present study highlights the successful advancement of click chemistry to generate new hybrids with L-ascorbic acid and 1,2,3-triazole units. Taking into account the potential synergy of biological activity of 1,2,3-triazole and L-ascorbic acid, we anticipate that L-ascorbic acid and 1,2,3-triazole pharmacophores if linked together would generate novel molecular entities which are likely to exhibit promising biological properties. Therefore, further antitumoral evaluations of described compounds are envisaged and will be reported on in due course.
Experimental part
General
Melting points (uncorrected) were determined with a Kofler micro hot-stage (Reichert, Wien, Austria). Precoated Merck (Darmstadt, Germany) silica gel 60F-254 plates were used for thin layer chromatography and the spots were detected under UV light (254 nm). Column chromatography was performed using Fluka (Buchs, Switzerland) silica gel (0.063–0.2 mm); glass columns were slurry-packed under gravity. Compounds 6a–6n where additionally filtered through a short column of Merck (Darmstadt, Germany) aluminium oxide (Al2O3, 0.063–0.2 mm) in order to scavenge the copper residues from the click reactions.All continuous flow reaction were performed using tubular glass microreactor (length: width: depth = 330 mm: 250 μm: 50 μm with the internal volume of 6 μL). Two syringe pumps (PHD 4400 Syringe Pump Series, Harvard Apparatus, USA) equipped with high pressure stainless steel syringes (8 cm3, Harvard Apparatus, USA) were used to feed substrates to the inlet streams of microreactor. Total flow velocity (Φ = 0.2–2 μL min−1) was altered and the influence on the reaction yield was monitored. When the reaction was performed in a microreactor in combination with ultrasound, microreactor was completely emerged in to an ultrasonic bath (Bandelin Sonorex, Germany) under the continuous frequency of 35 kHz and temperature of 50 °C. The samples were analyzed by high performance liquid chromatography on a Prominence HPLC system (Shimadzu, Germany) equipped with a LC-20AT pump, CBM-20A system controller, DGU-20A5 degasser, SPD-M20A photodiode array detector using an Agilent Eclipse XDB-C18 reverse-phase analytical column (4.6 × 150 mm). UV absorption was monitored at 254 nm wavelength. Water (A: 99.9% H2O, 0.1% formic acid) and acetonitrile (B: 475 mL acetonitrile, 50 mL H20, 0.5 mL formic acid) were used for elution at a flow rate of 1 mL min−1. For the analysis of the reaction mixtures 6a–6n, a gradient method starting with 40% B within 20 min, increasing to 60% B within next 25 min and then to 90% B within 25 min (total run time 70 min) and a gradient method starting with 40% B within 28 min, increasing to 60% B within next 32 min and then to 90% B within 5 min (total run time 65 min) were used. Elemental analyses were performed in the Central Analytic Service, Ruđer Bošković Institute, Zagreb. All elemental compositions were within the 0.4% of the calculated values.
1H and 13C NMR spectra were acquired on a Bruker 300 and 600 MHz NMR spectrometer (Bruker Biospin, Rheinstetten, Germany). All data were recorded in DMSO-d6 at 298 K. Chemical shifts were referenced to the residual solvent signal of DMSO at δ 2.50 ppm for 1H and δ 39.50 ppm for 13C. Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances and H–H coupling constants.
Synthetic procedures
5,6-O,O-Isopropylidene-L-ascorbic acid (1), 2,3-O,O-dibenzyl-5,6-O,O-isopropylidene-L-ascorbic acid (2), 2,3-O,O-dibenzyl-L-ascorbic acid (3), 2,3-O,O-dibenzyl-4,5-didehydro-5,6-O,O-ditosyl-L-ascorbic acid (4) were synthesized in accordance with procedures given in the literature.291H NMR (300 MHz, DMSO) δ 7.65–7.04 (m, 10H,  ), 5.53 (t, J = 7.7 Hz, 1H, H-5), 5.34 (s, 2H, OC
), 5.53 (t, J = 7.7 Hz, 1H, H-5), 5.34 (s, 2H, OC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2Ph), 5.15 (s, 2H, OC2Ph), 4.09 (d, J = 7.7 Hz, 2H, H-6) ppm. 13C NMR (151 MHz, DMSO) δ 163.6 (C-1), 147.9 (C-3), 143.6 (C-4), 135.7 (Ph-q), 135.5 (Ph-q), 128.9–127.9 (
2Ph), 5.15 (s, 2H, OC2Ph), 4.09 (d, J = 7.7 Hz, 2H, H-6) ppm. 13C NMR (151 MHz, DMSO) δ 163.6 (C-1), 147.9 (C-3), 143.6 (C-4), 135.7 (Ph-q), 135.5 (Ph-q), 128.9–127.9 ( ), 123.3 (C-2), 102.2 (C-5), 74.0 (O
), 123.3 (C-2), 102.2 (C-5), 74.0 (O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2Ph), 73.0 (OH2Ph), 44.5 (C-6) ppm.
H2Ph), 73.0 (OH2Ph), 44.5 (C-6) ppm.
General procedure for the preparation of 1,2,3-triazole-L-ascorbic acid derivatives (6a–6n) under batch conditions
The C-6 azido L-ascorbic acid derivative 5 (200–350 mg) and Cu(OAc)2 (0.05 equiv.) were dissolved in CH3OH. The corresponding alkyne (1.2 equiv.) was then added and the reaction mixture was heated for 5 h at 55 °C. The reaction mixture was stirred overnight at room temperature and the solvent was evaporated in vacuo. The product was purified with column chromatography on silica gel (CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3OH = 200:1–40:1) and Al2O3. The oily product was triturated with n-hexane in order to obtain compounds 6a–6i.
:CH3OH = 200:1–40:1) and Al2O3. The oily product was triturated with n-hexane in order to obtain compounds 6a–6i.
1H-NMR (300 MHz, DMSO) δ 9.00 (s, 1H, H-7), 8.51 (s, 2H, Ph-2′, Ph-6′), 8.07 (s, 1H, Ph-4′), 7.44–7.32 (m, 10H,  ), 5.72 (t, J = 7.7 Hz, 1H, H-5), 5.34 (2H, s, OC2Ph), 5.34 (d, J = 7.6 Hz, 2H, H-6), 5.18 (s, 2H, OC2Ph) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 143.9 (C-8), 143.7 (C-4), 135.6 (Ph-q), 135.3 (Ph-q), 133.2 (Ph-1′), 131.3; 131.1; 130.9; 130.7 (q, JCF = 33.1 Hz, Ph-3′, Ph-5′), 128.8–127.9 (
), 5.72 (t, J = 7.7 Hz, 1H, H-5), 5.34 (2H, s, OC2Ph), 5.34 (d, J = 7.6 Hz, 2H, H-6), 5.18 (s, 2H, OC2Ph) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 143.9 (C-8), 143.7 (C-4), 135.6 (Ph-q), 135.3 (Ph-q), 133.2 (Ph-1′), 131.3; 131.1; 130.9; 130.7 (q, JCF = 33.1 Hz, Ph-3′, Ph-5′), 128.8–127.9 ( ), 125.3; 125.3 (d, JCF = 3.6 Hz, Ph-2′, Ph-6′), 125.9; 124.1; 122.3; 120.5 (q, JCF = 272.7 Hz, CF), 123.5 (C-2), 123.4 (C-7), 121.2–121.0 (m, Ph-4′), 101.4 (C-5), 74.0 (OH2Ph), 73.1 (OH2Ph), 44.5 (C-6) ppm. Anal. calcd for C30H21F6N3O4: C, 59.90; H, 3.52; N, 6.99. Found: C, 59.98; H, 3.51; N, 6.99.
), 125.3; 125.3 (d, JCF = 3.6 Hz, Ph-2′, Ph-6′), 125.9; 124.1; 122.3; 120.5 (q, JCF = 272.7 Hz, CF), 123.5 (C-2), 123.4 (C-7), 121.2–121.0 (m, Ph-4′), 101.4 (C-5), 74.0 (OH2Ph), 73.1 (OH2Ph), 44.5 (C-6) ppm. Anal. calcd for C30H21F6N3O4: C, 59.90; H, 3.52; N, 6.99. Found: C, 59.98; H, 3.51; N, 6.99.
 , Ph-2′, Ph-4′, Ph-6′), 5.69 (t, J = 7.2 Hz, 1H, H-5), 5.34 (s, 2H, OC2Ph), 5.29 (d, J = 7.8 Hz, 2H, H-6), 5.18 (s, 2H, OC2Ph) ppm. 13C-NMR (75 MHz, DMSO) δ 163.5 (C-1), 165.0; 164.6; 161.3; 161.1 (dd, JCF = 246.3, 13.6 Hz, Ph-3′, Ph-5′), 147.9 (C-3), 144.6 (C-8), 143.6 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 134.3; 134.1; 133.9 (t, JCF = 11.0 Hz, Ph-1′), 128.8–127.9 (
, Ph-2′, Ph-4′, Ph-6′), 5.69 (t, J = 7.2 Hz, 1H, H-5), 5.34 (s, 2H, OC2Ph), 5.29 (d, J = 7.8 Hz, 2H, H-6), 5.18 (s, 2H, OC2Ph) ppm. 13C-NMR (75 MHz, DMSO) δ 163.5 (C-1), 165.0; 164.6; 161.3; 161.1 (dd, JCF = 246.3, 13.6 Hz, Ph-3′, Ph-5′), 147.9 (C-3), 144.6 (C-8), 143.6 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 134.3; 134.1; 133.9 (t, JCF = 11.0 Hz, Ph-1′), 128.8–127.9 ( ), 123.4 (C-2), 123.0 (C-7), 108.3; 108.1; 108.0; 107.9 (dd, JCF = 17.9, 8.6 Hz, Ph-2′, Ph-6′), 103.5; 103.1; 102.8 (t, JCF = 26.0 Hz, Ph-4′), 102.8 (C-5), 74.0(OH2Ph), 73.1 (OH2Ph), 44.4 (C-6) ppm. Anal. calcd for C28H21F2N3O4: C, 67.06; H, 4.22; N, 8.38. Found: C, 67.12; H, 4.21; N, 8.40.
), 123.4 (C-2), 123.0 (C-7), 108.3; 108.1; 108.0; 107.9 (dd, JCF = 17.9, 8.6 Hz, Ph-2′, Ph-6′), 103.5; 103.1; 102.8 (t, JCF = 26.0 Hz, Ph-4′), 102.8 (C-5), 74.0(OH2Ph), 73.1 (OH2Ph), 44.4 (C-6) ppm. Anal. calcd for C28H21F2N3O4: C, 67.06; H, 4.22; N, 8.38. Found: C, 67.12; H, 4.21; N, 8.40. ), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.27 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 145.4 (C-8), 143.4 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 131.8 (Ph), 129.9 (Ph-1′), 128.8–127.1 (
), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.27 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 145.4 (C-8), 143.4 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 131.8 (Ph), 129.9 (Ph-1′), 128.8–127.1 (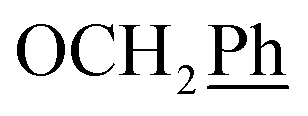 , Ph′), 123.4 (C-2), 121.9 (C-7), 120.8 (Ph-4′), 101.7 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 44.3 (C-6) ppm. Anal. calcd for C28H22BrN3O4: C, 61.77; H, 4.07; N, 7.72. Found: C, 61.83; H, 4.08; N, 7.74.
, Ph′), 123.4 (C-2), 121.9 (C-7), 120.8 (Ph-4′), 101.7 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 44.3 (C-6) ppm. Anal. calcd for C28H22BrN3O4: C, 61.77; H, 4.07; N, 7.72. Found: C, 61.83; H, 4.08; N, 7.74.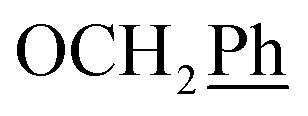 , Ph-3′, Ph-5′), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.26 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph), 2.32 (s, 3H,
, Ph-3′, Ph-5′), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.26 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph), 2.32 (s, 3H, 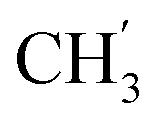 ) ppm. 13C NMR (151 MHz, DMSO) δ 163.6 (C-1), 148.0 (C-3), 146.6 (C-8), 143.3 (C-4), 137.2 (Ph-4′), 135.7 (Ph-q), 135.4 (Ph-q), 129.5–127.9 (
) ppm. 13C NMR (151 MHz, DMSO) δ 163.6 (C-1), 148.0 (C-3), 146.6 (C-8), 143.3 (C-4), 137.2 (Ph-4′), 135.7 (Ph-q), 135.4 (Ph-q), 129.5–127.9 (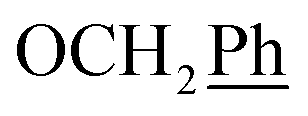 , Ph′), 127.9 (Ph-1′), 125.1 (Ph′), 123.4 (C-2), 121.2 (C-7), 102.0 (C-5), 74.1 (OH2Ph), 73.1 (OH2Ph), 44.2 (C-6), 20.8 (
, Ph′), 127.9 (Ph-1′), 125.1 (Ph′), 123.4 (C-2), 121.2 (C-7), 102.0 (C-5), 74.1 (OH2Ph), 73.1 (OH2Ph), 44.2 (C-6), 20.8 (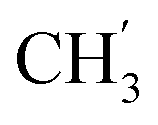 ) ppm. Anal. calcd for C29H25N3O4: C, 72.64; H, 5.25; N, 8.76. Found: C, 72.74; H, 5.26; N, 8.77.
) ppm. Anal. calcd for C29H25N3O4: C, 72.64; H, 5.25; N, 8.76. Found: C, 72.74; H, 5.26; N, 8.77.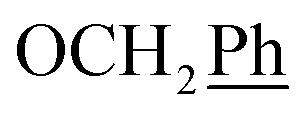 , Ph-3′, Ph-5′), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.26 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph), 2.58 (t, 2H, H-1′), 1.66–1.44 (m, 2H, H-2′), 1.32–1.15 (m, 4H, H-3′, H-4′), 0.86 (t, J = 6.8 Hz, 3H,
, Ph-3′, Ph-5′), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.26 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph), 2.58 (t, 2H, H-1′), 1.66–1.44 (m, 2H, H-2′), 1.32–1.15 (m, 4H, H-3′, H-4′), 0.86 (t, J = 6.8 Hz, 3H, 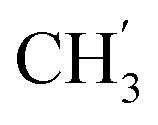 ) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 146.6 (C-8), 143.3 (C-4), 142.1 (Ph-4′), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–128.1 (
) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 146.6 (C-8), 143.3 (C-4), 142.1 (Ph-4′), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–128.1 (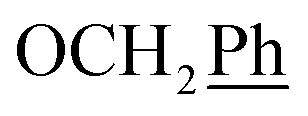 , Ph′), 127.8 (Ph-1′), 125.1 (Ph′), 123.4 (C-2), 121.1 (C-7), 101.9 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 44.2 (C-6), 34.8 (C-1′), 30.8 (
, Ph′), 127.8 (Ph-1′), 125.1 (Ph′), 123.4 (C-2), 121.1 (C-7), 101.9 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 44.2 (C-6), 34.8 (C-1′), 30.8 (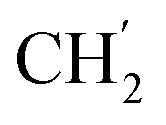 ), 30.4 (
), 30.4 (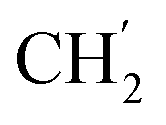 ), 21.9 (C-4′), 13.8 (
), 21.9 (C-4′), 13.8 (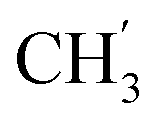 ) ppm. Anal. calcd for C33H33N3O4: C, 74.00; H, 6.21; N, 7.84. Found: C, 74.16; H, 6.20; N, 7.83.
) ppm. Anal. calcd for C33H33N3O4: C, 74.00; H, 6.21; N, 7.84. Found: C, 74.16; H, 6.20; N, 7.83.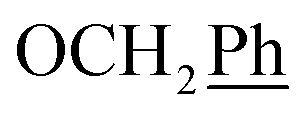 ), 7.00 (d, J = 8.8 Hz, 2H, Ph-3′, Ph-5′), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 1H, OC2Ph), 5.25 (d, J = 7.5 Hz, 1H, H-6), 5.17 (s, 1H, OC2Ph), 3.78 (s, 3H,
), 7.00 (d, J = 8.8 Hz, 2H, Ph-3′, Ph-5′), 5.69 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 1H, OC2Ph), 5.25 (d, J = 7.5 Hz, 1H, H-6), 5.17 (s, 1H, OC2Ph), 3.78 (s, 3H, 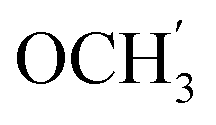 ) ppm. 13C NMR (75 MHz, DMSO) δ 163.6 (C-1), 159.0 (Ph-4′), 148.0 (C-3), 146.5 (C-8), 143.3 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–126.5 (
) ppm. 13C NMR (75 MHz, DMSO) δ 163.6 (C-1), 159.0 (Ph-4′), 148.0 (C-3), 146.5 (C-8), 143.3 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–126.5 (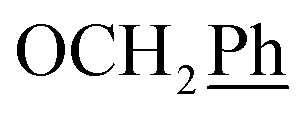 , Ph-2′, Ph-6′), 123.4 (C-2), 123.2 (Ph-1′), 120.6 (C-7), 114.3 (Ph-3′, Ph-5′), 102.0 (C-5), 74.1 (OH2Ph), 73.1 (OH2Ph), 55.1 (OCH3), 44.2 (C-6) ppm. Anal. calcd for C29H25N3O5: C, 70.29; H, 5.09; N, 8.48. Found: C, 70.31; H, 5.08; N, 8.47.
, Ph-2′, Ph-6′), 123.4 (C-2), 123.2 (Ph-1′), 120.6 (C-7), 114.3 (Ph-3′, Ph-5′), 102.0 (C-5), 74.1 (OH2Ph), 73.1 (OH2Ph), 55.1 (OCH3), 44.2 (C-6) ppm. Anal. calcd for C29H25N3O5: C, 70.29; H, 5.09; N, 8.48. Found: C, 70.31; H, 5.08; N, 8.47.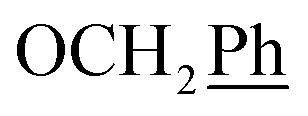 , Ph-4′), 7.13 (d, J = 8.8 Hz, 1H, Ph′), 7.08–7.01 (m, 1H, Ph′), 5.69 (t, J = 7.5 Hz, 1H), 5.33 (s, 2H, OC2Ph), 5.25 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph), 3.78 (s, 3H, OCH3) ppm. 13C NMR (151 MHz, DMSO) δ 163.6 (C-1), 155.3 (Ph-2′), 148.0 (C-3), 143.0 (C-4), 141.9 (C-8), 135.7 (Ph-q), 135.4 (Ph-q), 128.9–126.5 (
, Ph-4′), 7.13 (d, J = 8.8 Hz, 1H, Ph′), 7.08–7.01 (m, 1H, Ph′), 5.69 (t, J = 7.5 Hz, 1H), 5.33 (s, 2H, OC2Ph), 5.25 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph), 3.78 (s, 3H, OCH3) ppm. 13C NMR (151 MHz, DMSO) δ 163.6 (C-1), 155.3 (Ph-2′), 148.0 (C-3), 143.0 (C-4), 141.9 (C-8), 135.7 (Ph-q), 135.4 (Ph-q), 128.9–126.5 (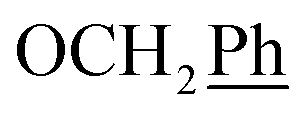 , Ph′), 124.1 (C-7), 123.3 (C-2), 120.6 (Ph-5′), 119.0 (Ph-1′), 111.5 (Ph-3′), 102.4 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 55.4 (OH3), 44.1 (C-6) ppm. Anal. calcd for C29H25N3O5: C, 70.29; H, 5.09; N, 8.48. Found: C, 70.36; H, 5.10; N, 8.50.
, Ph′), 124.1 (C-7), 123.3 (C-2), 120.6 (Ph-5′), 119.0 (Ph-1′), 111.5 (Ph-3′), 102.4 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 55.4 (OH3), 44.1 (C-6) ppm. Anal. calcd for C29H25N3O5: C, 70.29; H, 5.09; N, 8.48. Found: C, 70.36; H, 5.10; N, 8.50. , Ph′), 6.74–6.71 (m, 1H, Ph-4′), 5.70 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.26 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph) ppm. 13C NMR (151 MHz, DMSO) δ 163.7 (C-1), 157.8 (Ph-3′), 148.1 (C-3), 146.7 (C-8), 143.4 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 131.9 (Ph-1′), 130.0–128.0 (
, Ph′), 6.74–6.71 (m, 1H, Ph-4′), 5.70 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.26 (d, J = 7.5 Hz, 2H, H-6), 5.17 (s, 2H, OC2Ph) ppm. 13C NMR (151 MHz, DMSO) δ 163.7 (C-1), 157.8 (Ph-3′), 148.1 (C-3), 146.7 (C-8), 143.4 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 131.9 (Ph-1′), 130.0–128.0 ( , Ph′), 123.4 (C-2), 121.6 (C-7), 116.1 (Ph′), 115.0 (Ph′), 111.9 (Ph′), 102.1 (C-5), 74.1 (OH2Ph), 73.1 (OH2Ph), 44.3 (C-5) ppm. Anal. calcd for C28H23N3O5: C, 69.84; H, 4.81; N, 8.73. Found: C, 69.70; H, 4.81; N, 8.73.
, Ph′), 123.4 (C-2), 121.6 (C-7), 116.1 (Ph′), 115.0 (Ph′), 111.9 (Ph′), 102.1 (C-5), 74.1 (OH2Ph), 73.1 (OH2Ph), 44.3 (C-5) ppm. Anal. calcd for C28H23N3O5: C, 69.84; H, 4.81; N, 8.73. Found: C, 69.70; H, 4.81; N, 8.73. ), 5.60 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.14 (s, 2H, OC2Ph), 5.12 (s, 2H, H-6), 1.96–1.87 (m, 1H, H-1′), 0.91–0.84 (m, 2H,
), 5.60 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.14 (s, 2H, OC2Ph), 5.12 (s, 2H, H-6), 1.96–1.87 (m, 1H, H-1′), 0.91–0.84 (m, 2H, 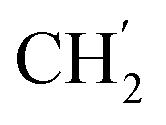 ), 0.71–0.66 (m, 2H,
), 0.71–0.66 (m, 2H, 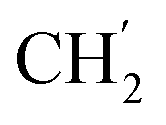 ) ppm. 13C-NMR (151 MHz, DMSO) δ 163.5 (C-1), 149.2 (C-8), 147.9 (C-3), 143.1 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–127.9 (
) ppm. 13C-NMR (151 MHz, DMSO) δ 163.5 (C-1), 149.2 (C-8), 147.9 (C-3), 143.1 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–127.9 (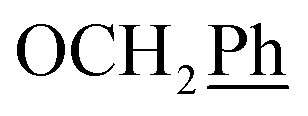 ), 123.3 (C-2), 120.8 (C-7), 102.2 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.9 (C-6), 7.5 (C-2′, C-3′), 6.5 (C-1′) ppm. Anal. calcd for C25H23N3O4: C, 69.92; H, 5.40; N, 9.78. Found: C, 70.04; H, 5.41; N, 9.77.
), 123.3 (C-2), 120.8 (C-7), 102.2 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.9 (C-6), 7.5 (C-2′, C-3′), 6.5 (C-1′) ppm. Anal. calcd for C25H23N3O4: C, 69.92; H, 5.40; N, 9.78. Found: C, 70.04; H, 5.41; N, 9.77.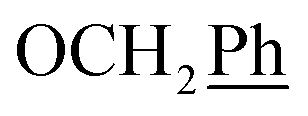 ), 5.61 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.22–5.03 (m, 4H, OC2Ph, H-6), 2.59 (t, J = 7.5 Hz, 2H, H-1′), 1.63–1.50 (m, 2H, H-2′), 1.41–1.14 (m, 2H, H-3′), 0.88 (t, J = 7.3 Hz, 3H,
), 5.61 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.22–5.03 (m, 4H, OC2Ph, H-6), 2.59 (t, J = 7.5 Hz, 2H, H-1′), 1.63–1.50 (m, 2H, H-2′), 1.41–1.14 (m, 2H, H-3′), 0.88 (t, J = 7.3 Hz, 3H, 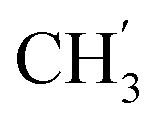 ) ppm. 13C NMR (75 MHz, DMSO) δ 163.6 (C-1), 148.0 (C-3), 147.1 (C-8), 143.0 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–127.9 (
) ppm. 13C NMR (75 MHz, DMSO) δ 163.6 (C-1), 148.0 (C-3), 147.1 (C-8), 143.0 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–127.9 (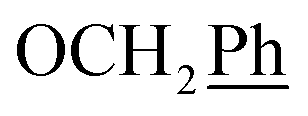 ), 123.3 (C-2), 121.9 (C-7), 102.3 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.9 (C-6), 31.0 (C-1′), 24.6 (
), 123.3 (C-2), 121.9 (C-7), 102.3 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.9 (C-6), 31.0 (C-1′), 24.6 (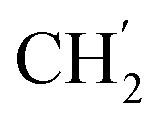 ), 21.6, (
), 21.6, (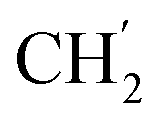 ), 13.7 (
), 13.7 (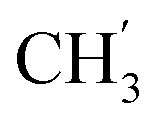 ) ppm. Anal. calcd for C26H27N3O4: C, 70.09; H, 6.11; N, 9.43. Found: C, 69.99; H, 6.08; N, 9.40.
) ppm. Anal. calcd for C26H27N3O4: C, 70.09; H, 6.11; N, 9.43. Found: C, 69.99; H, 6.08; N, 9.40.1H-NMR (300 MHz, DMSO) δ 7.87 (s, 1H, H-7), 7.44–7.31 (m, 10H, 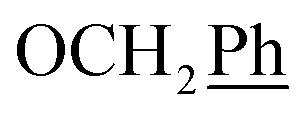 ), 5.61 (t, J = 7.2 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.19–5.15 (m, 4H, OC2Ph, H-6), 2.57 (t, J = 7.5 Hz, 2H, H-1′), 1.60–1.51 (m, 2H, H-2′), 1.23 (s, 14H,
), 5.61 (t, J = 7.2 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.19–5.15 (m, 4H, OC2Ph, H-6), 2.57 (t, J = 7.5 Hz, 2H, H-1′), 1.60–1.51 (m, 2H, H-2′), 1.23 (s, 14H, 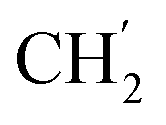 × 7), 0.84 (t, J = 6.9 Hz, 3H,
× 7), 0.84 (t, J = 6.9 Hz, 3H, 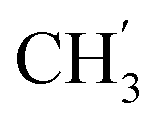 ) ppm. 13C-NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 147.2 (C-8), 143.0 (C-3), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–127.9 (
) ppm. 13C-NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 147.2 (C-8), 143.0 (C-3), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–127.9 (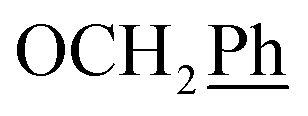 ), 123.3 (C-2), 121.8 (C-7), 102.2 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.8 (C-6), 31.2 (C-1′), 28.9 (
), 123.3 (C-2), 121.8 (C-7), 102.2 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.8 (C-6), 31.2 (C-1′), 28.9 (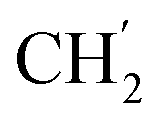 ), 28.9 (
), 28.9 (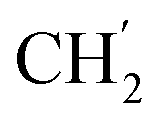 ), 28.9 (
), 28.9 (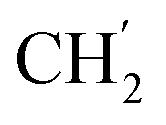 ), 28.7 (
), 28.7 (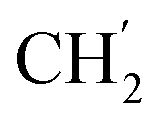 ), 28.6 (
), 28.6 (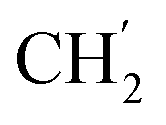 ), 28.5 (
), 28.5 (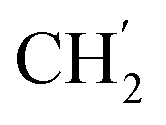 ), 24.9 (
), 24.9 (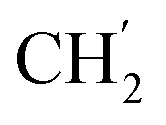 ), 22.1 (
), 22.1 (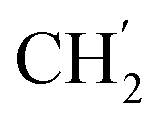 ), 13.9 (
), 13.9 (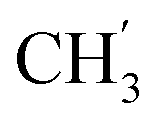 ) ppm. Anal. calcd for C32H39N3O4: C, 72.56; H, 7.42; N, 7.93. Found: C, 72.69; H, 7.43; N, 7.95.
) ppm. Anal. calcd for C32H39N3O4: C, 72.56; H, 7.42; N, 7.93. Found: C, 72.69; H, 7.43; N, 7.95.
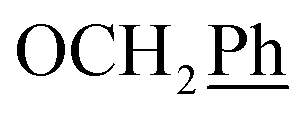 ), 5.61 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.24–5.09 (m, 4H, OC2Ph, H-6), 4.67 (t, J = 5.0 Hz, 1H, OH), 3.61 (dd, J = 11.6, 6.7 Hz, 2H, H-2′), 2.75 (t, J = 6.9 Hz, 1H, H-1′) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 144.7 (C-8), 143.1 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–127.9 (
), 5.61 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.24–5.09 (m, 4H, OC2Ph, H-6), 4.67 (t, J = 5.0 Hz, 1H, OH), 3.61 (dd, J = 11.6, 6.7 Hz, 2H, H-2′), 2.75 (t, J = 6.9 Hz, 1H, H-1′) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 144.7 (C-8), 143.1 (C-4), 135.7 (Ph-q), 135.4 (Ph-q), 128.8–127.9 (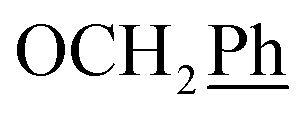 ), 123.3 (C-2), 122.5 (C-7), 102.2 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 60.3 (C-2′), 43.8 (C-6), 29.1 (C-1′) ppm. Anal. calcd for C24H23N3O5: C, 66.50; H, 5.35; N, 9.69. Found: C, 66.74; H, 5.36; N, 9.69.
), 123.3 (C-2), 122.5 (C-7), 102.2 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 60.3 (C-2′), 43.8 (C-6), 29.1 (C-1′) ppm. Anal. calcd for C24H23N3O5: C, 66.50; H, 5.35; N, 9.69. Found: C, 66.74; H, 5.36; N, 9.69.1H NMR (300 MHz, DMSO) δ 7.94 (s, 1H, H-7), 7.54–7.17 (m, 10H, 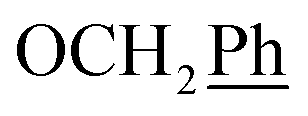 ), 5.62 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.25–5.06 (m, 4H, OC2Ph, H-6), 3.67 (t, J = 6.5 Hz, 2H, H-3′), 2.74 (t, J = 7.4 Hz, 2H, H-1′), 2.14–1.88 (m, 2H, H-2′) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 145.7 (C-8), 143.1 (C-3), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–127.9 (
), 5.62 (t, J = 7.5 Hz, 1H, H-5), 5.32 (s, 2H, OC2Ph), 5.25–5.06 (m, 4H, OC2Ph, H-6), 3.67 (t, J = 6.5 Hz, 2H, H-3′), 2.74 (t, J = 7.4 Hz, 2H, H-1′), 2.14–1.88 (m, 2H, H-2′) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 147.9 (C-3), 145.7 (C-8), 143.1 (C-3), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–127.9 (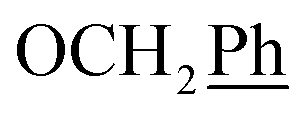 ), 123.3 (C-2), 122.2 (C-7), 102.1 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 44.6 (C-3′), 43.9 (C-6), 31.7 (C-1′), 22.2 (C-2′) ppm. Anal. calcd for C25H24ClN3O4: C, 64.44; H, 5.19; N, 9.02. Found: C, 64.58; H, 5.20; N, 9.04.
), 123.3 (C-2), 122.2 (C-7), 102.1 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 44.6 (C-3′), 43.9 (C-6), 31.7 (C-1′), 22.2 (C-2′) ppm. Anal. calcd for C25H24ClN3O4: C, 64.44; H, 5.19; N, 9.02. Found: C, 64.58; H, 5.20; N, 9.04.
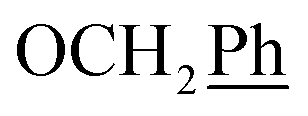 ), 5.62 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.23–5.09 (m, 4H, OC2Ph, H-6), 1.25 (s, 9H, 3 ×
), 5.62 (t, J = 7.5 Hz, 1H, H-5), 5.33 (s, 2H, OC2Ph), 5.23–5.09 (m, 4H, OC2Ph, H-6), 1.25 (s, 9H, 3 × 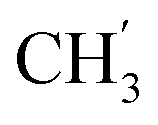 ) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 156.4 (C-8), 147.9 (C-3), 143.0 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–127.9 (
) ppm. 13C NMR (151 MHz, DMSO) δ 163.5 (C-1), 156.4 (C-8), 147.9 (C-3), 143.0 (C-4), 135.7 (Ph-q), 135.3 (Ph-q), 128.8–127.9 (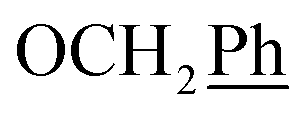 ), 123.3 (C-2), 119.9 (C-7), 102.3 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.9 (C-6), 30.5 (C-1′), 30.2 (
), 123.3 (C-2), 119.9 (C-7), 102.3 (C-5), 74.0 (OH2Ph), 73.0 (OH2Ph), 43.9 (C-6), 30.5 (C-1′), 30.2 (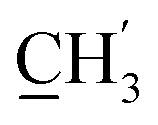 ) ppm. Anal. calcd for C26H27N3O4: C, 70.09; H, 6.11; N, 9.43. Found: C, 69.97; H, 6.11; N, 9.44.
) ppm. Anal. calcd for C26H27N3O4: C, 70.09; H, 6.11; N, 9.43. Found: C, 69.97; H, 6.11; N, 9.44.General flow procedure for the synthesis of 1,2,3-triazolyl L-ascorbic acid derivatives 6a–6n in a microreactor
Solution 1: C-6 azido L-ascorbic acid derivative (5) (0.1 M) and terminal alkyne (1 equiv.) were diluted in CH3OH.Solution 2: Cu(OAc)2 was diluted in CH3OH (0.005 M) and homogenized by sonication.
Solution 1 and 2 were loaded into an injection loop and injected into the micro-flow reactor set at 50 °C at a rate of 0.5 μL min−1 (9 min residence time). The microreactor was positioned into an ultrasound bath. Outflows from the microreactors were collected in vials placed on ice and filled with QuadraPure BZA copper-scavenger resin (polymer supported benzylamine) to stop the reaction. Reaction mixture was filtered via cotton. Prior to the HPLC analysis the sample (10 μL) was diluted in H2O (300 μL) and acetonitrile (400 μL). Conversion of compound 5 in flow reactions was determined using a prepared calibration curve of compound 5.
Acknowledgements
We greatly appreciate the financial support of the Croatian Science Foundation (project No. IP-2013-11-5596).Notes and references
- (a) J. L. Svirbely and A. Szent-Györgyi, Biochem. J., 1933, 27, 279–285 CrossRef CAS PubMed; (b) J. Dua, J. J. Cullena and G. R. Buettner, Biochim. Biophys. Acta, 2012, 1826, 443–457 Search PubMed.
- G. Grosso, R. Bei, A. Mistretta, S. Marventano, G. Calabrese, L. Masuelli, M. G. Giganti, A. Modesti, F. Galvano and D. Gazzol, Front. Biosci., Landmark Ed., 2013, 18, 1017–1029 CrossRef CAS.
- (a) J. Yun, E. Mullarky, C. Lu, K. N. Bosch, A. Kavalier, K. Rivera, J. Roper, I. I. Chio, E. G. Giannopoulou, C. Rago, A. Muley, J. M. Asara, J. Paik, O. Elemento, Z. Chen, D. J. Pappin, L. E. Dow, N. Papadopoulos, S. S. Gross and L. C. Cantley, Science, 2015, 350, 1391–1396 CrossRef CAS PubMed; (b) K. Blaschke, K. T. Ebata, M. M. Karimi, J. A. Zepeda-Martínez, P. Goyal, S. Mahapatra, A. Tam, D. J. Laird, M. Hirst, A. Rao, M. C. Lorincz and M. Ramalho-Santos, Nature, 2013, 500, 222–226 CrossRef CAS PubMed; (c) H. E. Sauberlich, Annu. Rev. Nutr., 1994, 14, 371–385 CrossRef CAS PubMed; (d) M. Osmak, I. Kovacek, I. Ljubenkov, R. Spaventi and M. Eckert-Maksic, Neoplasma, 1997, 44, 101–107 CAS; (e) H. Sakagami, M. Takeda, A. Utsumi, S. Fujinaga, A. Tsunoda, N. Yasuda, M. Shibusawa, T. Koike, H. Ota, K. Kazama, D. Shiokawa, S. Tanuma and M. Kochi, Anticancer Res., 1996, 13, 65–71 Search PubMed.
- (a) H. Sakagami, M. Takeda, A. Utsumi, S. Fujinaga, A. Tsunoda, N. Yasuda, M. Shibusawa, T. Koike, H. Ota, K. Kazama, D. Shiokawa, S. Tanuma and M. Kochi, Anticancer Res., 1996, 13, 65–71 Search PubMed; (b) P. E. Maxim and R. W. Veltri, FASEB J., 1988, 2, A370 Search PubMed.
- (a) Y. Nihro, H. Miyataka, T. Sudo, H. Matsumoto and T. Satoh, J. Med. Chem., 1991, 34, 2152–2157 CrossRef CAS PubMed; (b) R. P. Tripathi, B. Singh, S. S. Bisht and J. Pandey, Curr. Org. Chem., 2009, 13, 99–122 CrossRef CAS.
- (a) S. Raić-Malić, A. Hergold-Brundić, A. Nagl, M. Grdiša, K. Pavelić, E. De Clercq and M. Mintas, J. Med. Chem., 1999, 42, 2673–2678 CrossRef PubMed; (b) S. Raić-Malić, D. Svedruzić, T. Gazivoda, A. Marunović, A. Hergold-Brundić, A. Nagl, J. Balzarini, E. De Clercq and M. Mintas, J. Med. Chem., 2000, 43, 4806–4811 CrossRef; (c) T. Gazivoda, M. Šokčević, M. Kralj, L. Šuman, K. Pavelić, E. De Clercq, G. Andrei, R. Snoeck, J. Balzarini, M. Mintas and S. Raić-Malić, J. Med. Chem., 2007, 50, 4105–4112 CrossRef CAS PubMed.
- N. Devender, K. K. G. Ramakrishna, Hamidullah, S. K. Shukla, R. Konwar and R. P. Tripathi, Eur. J. Med. Chem., 2014, 82, 106–119 CrossRef CAS PubMed.
- (a) J. J. Casciari, N. H. Riordan, T. L. Schmidt, X. L. Meng, J. A. Jackson and H. D. Riordan, Br. J. Cancer, 2001, 84, 1544–1550 CrossRef CAS PubMed; (b) S. W. Hong, D. H. Jin, E. S. Hahm, S. H. Yim, J. S. Lim, K. I. Kim, Y. Yang, S. S. Lee, J. S. Kang, W. J. Lee, K. W. Lee and M. S. Lee, Oncol. Rep., 2007, 18, 811–815 CAS; (c) P. Chen, J. Yu, B. Chalmers, J. Drisko, J. Yang, B. Li and Q. Chen, Anti-Cancer Drugs, 2012, 23, 437–444 CrossRef CAS PubMed; (d) H. Fritz, G. Flower, L. Weeks, K. Cooley, M. Callachan, J. McGowan, B. Skidmore, L. Kirchner and D. Seely, Integr. Cancer Ther., 2014, 13, 280–300 CrossRef CAS PubMed; (e) C. Vollbracht, B. Schneider, V. Leendert, G. Weiss, L. Auerbach and J. Beuth, In Vivo, 2011, 25, 983–990 CAS.
- (a) J. Hou, X. Liu, J. Shen, G. Zhao and P. G. Wang, Expert Opin. Drug Discovery, 2012, 7, 489–501 CrossRef CAS PubMed; (b) N. V. Sokolova and V. G. Nenajdenko, RSC Adv., 2013, 3, 16212–16242 RSC.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2001, 40, 2004–2021 CrossRef CAS; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- (a) K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–197 RSC; (b) R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- (a) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC; (b) A. C. Fahrenbach and J. F. Stoddart, Chem.–Asian J., 2011, 6, 2660–2669 CrossRef CAS PubMed.
- A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC.
- (a) K. A. Kalesh, P. Y. Yang, R. Srinivasan and S. Q. Yao, QSAR Comb. Sci., 2007, 26, 1135–1144 CrossRef CAS; (b) C. M. Salisbury and B. F. Cravatt, QSAR Comb. Sci., 2007, 26, 1229–1238 CrossRef CAS.
- (a) J. L. Hou, X. F. Liu, J. Shen, G. L. Zhao and P. G. Wang, Expert Opin. Drug Discovery, 2012, 7, 489–501 CrossRef CAS PubMed; (b) S. Raić-Malić and A. Meščić, Curr. Med. Chem., 2015, 22, 1462–1499 CrossRef.
- (a) P. S. Rao, C. Kurumurthy, B. Veeraswamy, G. S. Kumar, Y. Poornachandra, G. C. Kumar, S. B. Vasamsetti, S. Kotamraju and B. Narsaiah, Eur. J. Med. Chem., 2014, 80, 184–191 CrossRef PubMed; (b) Y. C. Duan, Y. C. Zheng, X. C. Li, M. M. Wang, X. W. Ye, Y. Y. Guan, G. Z. Liu, J. X. Zheng and H. M. Liu, Eur. J. Med. Chem., 2013, 64, 99–110 CrossRef CAS PubMed; (c) H. Elamari, R. Slimi, G. G. Chabot, L. Quentin, D. Scherman and C. Girard, Eur. J. Med. Chem., 2013, 60, 360–364 CrossRef CAS PubMed.
- (a) A. Krajczyk, K. Kulinska, T. Kulinski, B. L. Hurst, C. W. Day, D. F. Smee, T. Ostrowski, P. Januszczyk and J. Zeidler, Antiviral Chem. Chemother., 2014, 23, 161–171 CrossRef PubMed; (b) Y. W. He, C. Z. Dong, J. Y. Zhao, L. L. Ma, Y. H. Li and H. A. Aisa, Eur. J. Med. Chem., 2014, 76, 245–255 CrossRef CAS PubMed; (c) H. Cheng, J. Wan, M. I. Lin, Y. Liu, X. Lu, J. Liu, Y. Xu, J. Chen, Z. Tu, Y. S. Cheng and K. Ding, J. Med. Chem., 2012, 55, 2144–2153 CrossRef CAS PubMed.
- (a) H. Behbehani, H. M. Ibrahim, S. Makhseed and H. Mahmoud, Eur. J. Med. Chem., 2011, 46, 1813–1820 CrossRef CAS PubMed; (b) N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, M. V. Deshpande, S. Kadreppa, S. Chattopadhyay and R. G. Gonnade, Org. Biomol. Chem., 2008, 6, 3823–3830 RSC.
- (a) X. L. Wang, K. Wan and C. H. Zhou, Eur. J. Med. Chem., 2010, 45, 4631–4639 CrossRef CAS PubMed; (b) P. M. Chaudhary, S. R. Chavan, F. Shirazi, M. Razdan, P. Nimkar, S. P. Maybhate, A. P. Likhite, R. Gonnade, B. G. Hazara, M. V. Deshpande and S. R. Deshpande, Bioorg. Med. Chem., 2009, 17, 2433–2440 CrossRef CAS PubMed.
- S. B. Ötvös, I. M. Mándity, L. Kiss and F. Fülöp, Chem.–Asian J., 2013, 8, 800–808 CrossRef PubMed.
- (a) X. P. He, Y. Zang, T. D. James, J. Li, G. R. Chen and J. Xie, Chem. Commun., 2017, 53, 82–90 RSC; (b) X. P. He, Y. L. Zeng, Y. Zang, J. Li, R. A. Field and G. R. Chen, Carbohydr. Res., 2016, 429, 1–22 CrossRef CAS PubMed.
- (a) A. R. Bogdan and K. James, Chem.–Eur. J., 2010, 16, 14506–14512 CrossRef CAS PubMed; (b) A. R. Bogdan and K. James, Org. Lett., 2011, 13, 4060–4063 CrossRef CAS PubMed; (c) A. R. Bogdan, S. V. Jerome, K. N. Houk and K. James, J. Am. Chem. Soc., 2012, 134, 2127–2138 CrossRef CAS PubMed; (d) L. Vaccaro, D. Lanari, A. Marrocchi and G. Strappaveccia, Green Chem., 2014, 16, 3680–3704 RSC.
- (a) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS; (b) J. Bao and G. K. Tranmer, Chem. Commun., 2015, 51, 3037–3044 RSC; (c) T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 5750–5758 CrossRef CAS PubMed; (d) I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 24, 2566–2568 RSC; (e) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC; (f) C. Wiles and P. Watts, Future Med. Chem., 2009, 1, 1593–1612 CrossRef CAS PubMed; (g) L. Matel-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef PubMed; (h) R. L. Hartman and K. F. Jensen, Lab Chip, 2009, 9, 2495–2507 RSC.
- (a) P. Watts and S. J. Haswell, Drug Discovery Today, 2003, 8, 586–593 CrossRef CAS PubMed; (b) D. M. Roberage, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–322 CrossRef.
- (a) P. Watts and C. Wiles, Org. Biomol. Chem., 2007, 5, 727–732 RSC; (b) C. Wiles and P. Watts, Chem. Commun., 2011, 47, 6512–6535 RSC; (c) W. Ehrfeld, V. Hessel and H. Löwe, Microreactors: New technology for modern chemistry, Wiley-VCH, Weinheim, 2000, pp. 1–12 Search PubMed; (d) G. N. Doku, D. N. Verboom, D. N. Reinhoudt and A. van den Berg, Tetrahedron, 2005, 61, 2733–2742 CrossRef CAS; (e) A. Šalić and B. Zelić, RSC Adv., 2014, 4, 41714–41721 RSC.
- (a) D. H. DeWitt, Curr. Opin. Chem. Biol., 1999, 3, 350–356 CrossRef PubMed; (b) Y. Zhang, T. F. Jamison, S. Patel and N. Mainolfi, Org. Lett., 2011, 13, 280–283 CrossRef CAS PubMed.
- (a) Z. Dong, C. Yao, X. Zhang, J. Xu, G. Chen, Y. Zhao and Q. Yuan, Lab Chip, 2015, 15, 1145–1152 RSC; (b) S. Hübner, S. Kressirer, D. Kralisch, C. Bludszuweit-Philipp, K. Lukow, I. Jänich, A. Schilling, H. Hieronymus, C. Liebner and K. Jähnisch, ChemSusChem, 2012, 5, 279–288 CrossRef PubMed; (c) G. Cravotto and P. Cintas, Chem. Soc. Rev., 2006, 35, 180–196 RSC; (d) D. F. Rivas, P. Cintas and H. J. G. E. Gardeniers, Chem. Commun., 2012, 48, 10935–10947 RSC.
- (a) D. F. Rivas, M. Ashokkumar, T. Leong, K. Yasui, T. Tuziuti, S. Kentish, D. Lohse and H. J. G. E. Gardeniers, Ultrason. Sonochem., 2012, 19, 1252–1259 CrossRef PubMed; (b) T. Tandiono, S. W. Ohl, D. S. W. Ow, E. Klaseboer, V. V. Wong, R. Dumkec and C. D. Ohl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5996–5998 CrossRef PubMed; (c) T. Tandiono, S. W. Ohl, D. S. W. Ow, E. Klaseboer, V. V. Wong, A. Camattari and C. D. Ohl, Lab Chip, 2010, 10, 1848–1855 RSC; (d) J. Rooze, E. V. Rebrov, J. C. Schouten and J. T. F. Keurentjes, Ultrason. Sonochem., 2013, 20, 1–11 CrossRef CAS PubMed.
- (a) C. O. Kappe and E. Van der Eycken, Chem. Soc. Rev., 2010, 39, 1280–1290 RSC; (b) S. B. Ötvös and F. Fülöp, Catal. Sci. Technol., 2015, 5, 4926–4941 RSC.
- (a) S. Maračić, T. Gazivoda Kraljević, H. Čipčić Paljetak, M. Perić, M. Matijašić, D. Verbanac, M. Cetina and S. Raić-Malić, Bioorg. Med. Chem., 2015, 23, 7448–7463 CrossRef PubMed; (b) T. Gazivoda Kraljević, A. Harej, M. Sedić, S. Kraljević Pavelić, V. Stepanić, D. Drenjančević, J. Talapko and S. Raić-Malić, Eur. J. Med. Chem., 2016, 124, 794–808 CrossRef PubMed.
- (a) F. Von Dallacker and J. Sanders, Chem.-Ztg., 1985, 109, 197–202 Search PubMed; (b) T. Gazivoda, K. Wittine, I. Lovrić, D. Makuc, J. Plavec, M. Cetina, D. Mrvoš-Sermek, L. Šuman, M. Kralj, K. Pavelić, M. Mintas and S. Raić-Malić, Carbohydr. Res., 2006, 4, 433–442 CrossRef PubMed.
- G. C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, J. Am. Chem. Soc., 2011, 133, 13984–14001 CrossRef CAS PubMed.
- T. Gazivoda, M. Plevnik, J. Plavec, S. Kraljević, M. Kralj, K. Pavelić, J. Balzarini, E. De Clercq, M. Mintas and S. Raić-Malić, Bioorg. Med. Chem., 2005, 13, 131–139 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of 6a–6n, and 2D NMR spectra of 6a and 6n, HPLC chromatograms for micro-flow reactions of 6a–6n. See DOI: 10.1039/c6ra25244c |
| This journal is © The Royal Society of Chemistry 2017 |