
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysagulide Q suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells by regulating the ROS-JAK2/Src-STAT3 signaling pathway†
Yan-Wei Yang,
Lei Yang,
Chao Zhang,
Cai-Yun Gao,
Ting Ma and
Ling-Yi Kong *
*
State Key Laboratory of Natural Medicines, Department of Natural Medicinal Chemistry, China Pharmaceutical University, 24 Tong Jia Xiang, Nanjing 210009, People's Republic of China. E-mail: cpu_lykong@126.com; Fax: +86-25-8327-1405; Tel: +86-25-8327-1405
First published on 23rd February 2017
Abstract
Physagulide Q (PQ), a new natural compound, was isolated from Physalis angulata L. in our laboratory. In this study, we observed that PQ suppressed cell proliferation and induced apoptosis in human hepatocellular carcinoma (HCC) cells. Signal transducer and activator of transcription 3 (STAT3) regulates the expression of various genes to control cell cycle progression and programmed cell death. PQ inhibited the phosphorylation of STAT3 on Tyr705 by suppressing the phosphorylation of its upstream protein kinases Janus-activated kinase 2 (JAK2) and Src. Furthermore, we found that PQ could induce production of reactive oxygen species (ROS) in human HCC cells. GSH, a ROS scavenger, abrogated the inhibitory effect of PQ on the JAK2/Src-STAT3 pathway in human HCC cells. These results suggested that PQ inhibited the STAT3 signaling pathway through the generation of ROS in human HCC cells.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancies with a high recurrence rate and mortality rate.1 Patients are often diagnosed with HCC at an advanced stage.2 The traditional approach for patients with advanced HCC is chemotherapy.3 Most chemotherapeutics have been aimed at therapeutic targets such as epidermal growth factor (EGF), vascular endothelial growth factor (VEGF) and mTOR to treat HCC. Sorafenib, a multikinase inhibitor, is the first-line treatment for HCC;4,5 however, it is also associated with drug resistance.2 Therefore, there is an urgent need to search for effective targets and chemopreventive agents for the clinical treatment of advanced HCC.Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family.6 Activation of STAT3 is often detected in many human cancers, including HCC.7 Persistent STAT3 activation, different from intermittent activation in normal cells, has anti-apoptotic, proliferative, angiogenic and metastatic effects in tumor cells.8 It has been reported that the growth of HCC cells is suppressed by blocking the activation of STAT3.7,9 Thus, STAT3 is a potential target to treat HCC. Janus-activated kinase 2 (JAK2) and Src, two important protein kinases, are the upstream regulators for STAT3 activation.10 JAK2/STAT3 is a classical pathway that is activated by interleukin 6 (IL-6), while Src/STAT3 is a receptor-independent pathway.11,12 Thus, STAT3 activation includes IL-6-induced and constitutive STAT3 activation. Phosphorylated STAT3 is the major active form of STAT3. Phosphorylated STAT3 dimerizes and then translocates into the nucleus to control the transcription of various oncogenic genes such as Bcl-2, VEGF and survivin, which regulate cell proliferation and apoptosis.13
Reactive oxygen species (ROS), which consist of reactive molecules and free radicals, induce signaling molecules that regulate cell growth, differentiation, survival and the immune response.14 ROS are very transient species that can lead to extensive damage to proteins, lipids and DNA. A high rate of ROS generation can result in oxidative stress, which is an important mechanism in the promotion of cell apoptosis.15 It has been demonstrated that the generation of ROS inhibits proliferation in human HCC cells.16
Physalis angulata L. is commonly used as a traditional Chinese medicine for antipyretic, anti-inflammatory and diuretic purposes.17 However, few studies have reported on the anti-tumor effects of Physalis angulata L.18,19 Phytochemical research has identified withanolides as the main constituents of this plant.20 We have shown previously that Physagulide M, Physagulide N and Physagulide O, withanolide compounds isolated from Physalis angulata L., have no obvious cytotoxicity in HepG2 cells.17 However, in this study we tested the cytotoxicity of another three withanolide compounds isolated from Physalis angulata L. and the cytotoxicity of PQ was higher than the other two compounds in HepG2 cells. Thus, we selected PQ to investigate its anti-tumor effect and molecular mechanism in human HCC cells.
Experimental
Materials
PQ was isolated from Physalis angulata L. in our laboratory. Samples containing 95% or higher concentrations of PQ were used in all of our experiments. PQ (40 mM) was dissolved in dimethyl sulfoxide (DMSO) to obtain a stock solution. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was obtained from Sigma-Aldrich (St. Louis, MO, USA). MTT (5 mg mL−1) was dissolved in PBS and was filter-sterilized. STAT3, phospho-STAT3 (Tyr705), JAK2, phospho-JAK2, Src, phospho-Src, Bcl-2, Bax, Cyclin B1, cdc2, phospho-cdc2, cdc25C, phospho-cdc25C, PARP, cleaved PARP, caspase-3, cleaved caspase-3, caspase-7, cleaved caspase-7, caspase-9, cleaved caspase-9, and GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).Cell lines and cultures
The human HCC cell line HepG2 and normal hepatocellular cell line L02, purchased from the Cell Bank of the Shanghai Institute of Biochemistry & Cell Biology (Shanghai, China), were maintained in DMEM medium (HyClone, Logan, USA) supplemented with 10% fetal bovine serum (Sijiqing, Hangzhou, People's Republic of China). The human HCC cell line Bel-7402, obtained from the Cell Bank of the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China), was maintained in RPMI-1640 medium (HyClone, Logan, USA) supplemented with 10% fetal bovine serum (Sijiqing, Hangzhou, People's Republic of China). All of the cell lines were cultured in a humidified atmosphere in a 5% CO2 incubator at 37 °C.Cytotoxicity assay
An MTT assay was conducted to examine cell viability. L02, HepG2 and Bel-7402 cells were seeded in a 96-well cell culture plate at a density of 4000 cells per well and were stabilized for 24 h. The cells were stimulated with various concentrations of PQ for 24 h. Then, 20 μL of MTT was added to every well, and the cells were incubated for an additional 4 h. The formed purple formazan crystals were dissolved in 150 μL of DMSO by gentle shaking for 10 min. The absorbance was measured using an ELISA reader (Spectra Max Plus384; Molecular Devices, Sunnyvale, CA) at a test wavelength of 570 nm and a reference wavelength of 630 nm. Cell viability and inhibition rate were calculated using the following formulas:| Cell viability = (At/As) × 100% |
| Inhibition rate = (1 − At/As) × 100% |
Colony formation assay
HepG2 and Bel-7402 cells were plated on 6-well plates at a density of eight hundred cells per well. Following incubation overnight, the cells were treated with different concentrations (0.5, 1 or 2 μM) of PQ. The medium was replaced after 24 h of incubation, and the colonies were further observed 10 days later. The plates were then stained with crystal violet solution (Sigma-Aldrich, St. Louis, MO), and photographs of the colonies were taken manually.22CFDA SE cell proliferation assay
The proliferation of HepG2 and Bel-7402 cells was investigated using a CFDA SE Cell Proliferation Assay and Tracking Kit (Beyotime, Nantong, People's Republic of China). HepG2 and Bel-7402 cells were labeled with CFDA-SE and then were plated on 6-well plates and incubated for 24 h. Subsequently, the medium was replaced with fresh medium containing PQ (1, 2, or 4 μM). After 24 h of incubation, the cells were harvested and washed twice with ice-cold PBS. The fluorescence intensity was measured by flow cytometry (BD FACS Canto, NJ, USA).Cell cycle assay
The cell cycle distribution of HepG2 and Bel-7402 cells was detected using a Cell Cycle and Apoptosis Analysis Kit (Beyotime, Nantong, People's Republic of China). After treatment with PQ (1, 2, or 4 μM) for 24 h, the cells were harvested, washed in ice-cold PBS and re-suspended in ice-cold 70% ethanol overnight. Then, the treated cells were fixed and incubated with RNAse and propidium iodide (PI) in PBS for 30 min in 37 °C in the dark. Cell cycle distribution was analyzed using a flow cytometer (BD FACS Canto, NJ, USA).JC-1 assay
The ability of PQ to depolarize the mitochondrial membrane in HepG2 cells was investigated using a JC-1 Apoptosis Detection Kit (Beyotime, Nantong, People's Republic of China). After treatment with PQ (1, 2, or 4 μM) for 24 h, the cells were harvested, washed in ice-cold PBS and incubated with JC-1 for 20 min in a humidified atmosphere in a 5% CO2 incubator at 37 °C. The fluorescence intensity was measured by flow cytometry (BD FACSCanto, NJ, USA). JC-1 staining of normal cells predominantly appears as red florescence. When cells are apoptotic or necrotic, the mitochondrial transmembrane potential (ΔΨm) is reduced, and the JC-1 stain appears as green florescence. A change in the JC-1 florescence emission from red to green indicates a depolarization of the mitochondrial transmembrane; thus, changes in ΔΨm can be measured by the ratio of red and green fluorescence.| ΔΨm% = ((Mean FL2/Mean FL1)sample/(Mean FL2/Mean FL1)control) × 100% |
Mean FL1 and Mean FL2 represent the mean value of green fluorescence intensity and the mean value of red fluorescence intensity, respectively.
Annexin V-APC/7-AAD apoptosis assay
The apoptosis of HepG2 cells was detected using fluorescein isothiocyanate Annexin-APC and 7-AAD. After treatment with PQ (1, 2, or 4 μM) for 24 h, the cells were harvested, washed in ice-cold PBS, incubated for 20 min with Annexin V-APC and 7-AAD, and analyzed using a flow cytometer (BD FACSCanto, NJ, USA). For analysis, 7-AAD-negative and Annexin V-positive cells were considered early apoptotic (lower right quadrant), while cells that were both 7-AAD- and Annexin V-positive were considered late apoptotic (higher left quadrant). In addition, cells both 7-AAD- and Annexin V-negativity were considered normal (lower left quadrant). The rate of apoptosis was the total of the early and late apoptotic cells relative to the cell population.ROS assay
ROS production in HepG2 and Bel-7402 cells was detected using a Reactive Oxygen Species Assay Kit (Beyotime, Nantong, People's Republic of China). After treatment with PQ (1, 2, or 4 μM) for 24 h, the cells were harvested, washed in ice-cold PBS, and subsequently incubated with DCFH-DA (10 μM) in serum-free medium at 37 °C for 20 min. After incubation, the DCFH fluorescence of the cells in each well was measured using flow cytometry (BD FACS Canto, NJ, USA). DCFH-DA passes through cell membranes and is cleaved to DCFH by esterase. ROS oxidizes DCFH, generating DCF, a measurable fluorescent product. Therefore, the ROS production in cells can be measured by the fluorescence intensity of DCF.Western blot assay
HepG2 and Bel-7402 cells were rinsed twice with PBS and lysed in 1 × RIPA lysis buffer (50 mM Tris–HCl, pH 7. 4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA and protease inhibitors) (Amresco, Solon, USA) for 10 min. The lysed cells were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C to collect the supernatant. The protein content in the supernatant was determined by a BCA protein kit (Beyotime, Haimen, China). The supernatant, which contained SDS sample buffer was immediately heated at 100 °C for 10 min. Equal amounts of protein from the total cell lysates (20–40 μg per lane) were separated by sodium dodecyl sulfate (8%, 10% or 12%) polyacrylamide gel electrophoresis gels (SDS-PAGE, Bio-Rad Laboratories, Hercules, CA) and wet-transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA). Nonspecific binding sites in the PVDF membrane were blocked with 5% skim milk in TBST (20 mM Tris, 166 mM NaCl, and 0.05% Tween 20, pH 7.5) for 2 h. Subsequently, the PVDF membrane was reacted first with the specific primary antibody at 4 °C overnight and with the respective horseradish peroxidase-conjugated secondary antibody at room temperature for 2 h. Finally, the PVDF membrane was washed twice with TBST. The bound immunocomplexes were detected using a ChemiDOC™ XRS + system (Bio-Rad Laboratories, Hercules, CA).23
000 rpm for 10 min at 4 °C to collect the supernatant. The protein content in the supernatant was determined by a BCA protein kit (Beyotime, Haimen, China). The supernatant, which contained SDS sample buffer was immediately heated at 100 °C for 10 min. Equal amounts of protein from the total cell lysates (20–40 μg per lane) were separated by sodium dodecyl sulfate (8%, 10% or 12%) polyacrylamide gel electrophoresis gels (SDS-PAGE, Bio-Rad Laboratories, Hercules, CA) and wet-transferred to a PVDF membrane (Bio-Rad Laboratories, Hercules, CA). Nonspecific binding sites in the PVDF membrane were blocked with 5% skim milk in TBST (20 mM Tris, 166 mM NaCl, and 0.05% Tween 20, pH 7.5) for 2 h. Subsequently, the PVDF membrane was reacted first with the specific primary antibody at 4 °C overnight and with the respective horseradish peroxidase-conjugated secondary antibody at room temperature for 2 h. Finally, the PVDF membrane was washed twice with TBST. The bound immunocomplexes were detected using a ChemiDOC™ XRS + system (Bio-Rad Laboratories, Hercules, CA).23
Electrophoretic mobility shift assay (EMSA)
STAT3-DNA binding activity was analyzed by EMSA. HepG2 and Bel-7402 cells were treated with PQ (1, 2, or 4 μM; 24 h) and then nuclear protein extracts were prepared. The sequence of the double-stranded oligonucleotide used to detect the DNA-binding activities of STAT3 was, 5′-GAT CCT TCT GGG AAT TCC TAG ATC-3′. A Biotin 3′ End DNA Labeling Kit (Thermo scientific, Rockford, U. S. A.) was used to label the double-stranded oligonucleotide. The reaction mixture for EMSA contained 1 × binding buffer, 50 ng μL−1 poly (dI-dC), 0.05% NP-40, 5 mM MgCl2, 10 mM EDTA, 2.5% glycerol and 4 μg of nuclear extract (LightShift® Chemiluminescent EMSA Kit, Thermo scientific, Rockford, U. S. A.). Unlabeled wild-type oligo-nucleotide was added to the reaction mixture and incubated for 10 min at room temperature. Biotin-labeled probe-DNA was added, and the binding reaction was allowed to proceed for another 20 min at room temperature. The mixtures were resolved on 8% polyacrylamide gels at 120 V for 3 h. Excess probe-DNA shifted to the bottom of gels due to its small molecular weight, while protein-probe-DNA stayed at the top of the gels because of its large molecular weight. Then, the gels were dried and detected using a ChemiDOC™ XRS + system (BioRad Laboratories, Hercules, CA). To ensure the credibility of this method, a positive control assay was performed with Biotin-EBNA Control DNA and EBNA extract.Statistical analysis
All of the experiments were conducted more than three times. The results were expressed as the mean ± SD and were analyzed using GraphPad Prism version 5.0 by performing one-way or two-way ANOVA (GraphPad Software, San Diego, CA, USA). Differences of P < 0.05 were considered statistically significant.Results
PQ inhibits cell proliferation in human HCC cells
We tested the cytotoxicity of PQ in the human HCC cell lines HepG2 and Bel-7402 using the MTT assay. As shown in Fig. 1A, PQ significantly inhibited the cell viability of HepG2 and Bel-7402 cells in a dose-dependent manner. Then, normal hepatocellular L02 cells were used to detect the cytotoxicity of PQ in normal human cells. Table 1 showed that the IC50 values of PQ in L02, HepG2 and Bel-7402 cells were 13.64 μM, 2.01 μM and 2.85 μM, respectively. The IC50 values in L02 cells were 5–6-fold over that of human HCC cells, indicating that PQ had less cytotoxicity in normal hepatocellular cells. We further examined the effect of PQ on proliferation in human HCC cells using a colony formation assay. Consistent with the MTT assay results, the clonogenicity of HepG2 and Bel-7402 cells was reduced in a dose-dependent manner after exposure to PQ (Fig. 1B and C), suggesting that PQ suppressed colony formation in human HCC cells. Then, a CFDA-SE assay was used to monitor the effect of PQ on cellular division in HCC cells. As shown in Fig. 1D, PQ increased the CFDA-SE fluorescence intensity in HepG2 and Bel-7402 cells, suggesting that PQ inhibited cell division and proliferation in human HCC cells. | ||
| Fig. 1 The inhibitory effects of PQ on cell proliferation in human hepatocellular carcinoma cells. (A) HepG2 and Bel-7402 cells were treated with different concentrations of PQ for 24 h. The cell viability rate was determined using the MTT assay. (B) The inhibitory effect of PQ on HepG2 and Bel-7402 cell colony formation was evaluated by a clonogenic assay. (C) Histogram represents the number of colonies. (D) HepG2 and Bel-7402 cells were treated with different concentrations (0, 1, 2 or 4 μM) of PQ for 24 h, and the CFDA-SE cell tracer assay was performed. The cells were analyzed for cell division and proliferation inhibition using flow cytometry. Histogram represents the mean value of fluorescence intensity. The data are presented as the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, compared with the control. | ||
| L02 | HepG2 | Bel-7402 | |
|---|---|---|---|
| a Values are represented as the means ± S.D. based on three independent experiments. | |||
| IC50 (μM) | 13.64 ± 0.32 | 2.01 ± 0.13 | 2.85 ± 0.12 |
PQ induces G2/M phase cell cycle arrest in human HCC cells
Cell proliferation partially depends on normal cell cycle control.24 Therefore, cell cycle analysis was used to test the effect of PQ on cell cycle progression in HepG2 and Bel-7402 cells. As shown in Fig. 2A and B, accumulation of the cell population in the G2/M phase was increased when cells were treated with PQ, suggesting that PQ induced G2/M phase arrest in HepG2 and Bel-7402 cells. Then, we examined G2/M phase-related proteins24,25 by western blotting. As shown in Fig. 2C and D, PQ up-regulated the expression of Cyclin B1 and inhibited the expression of Cdc25C and p-Cdc25C. These results revealed, at least in part, that PQ suppressed cell proliferation in human HCC cells through blocking cell cycle progress.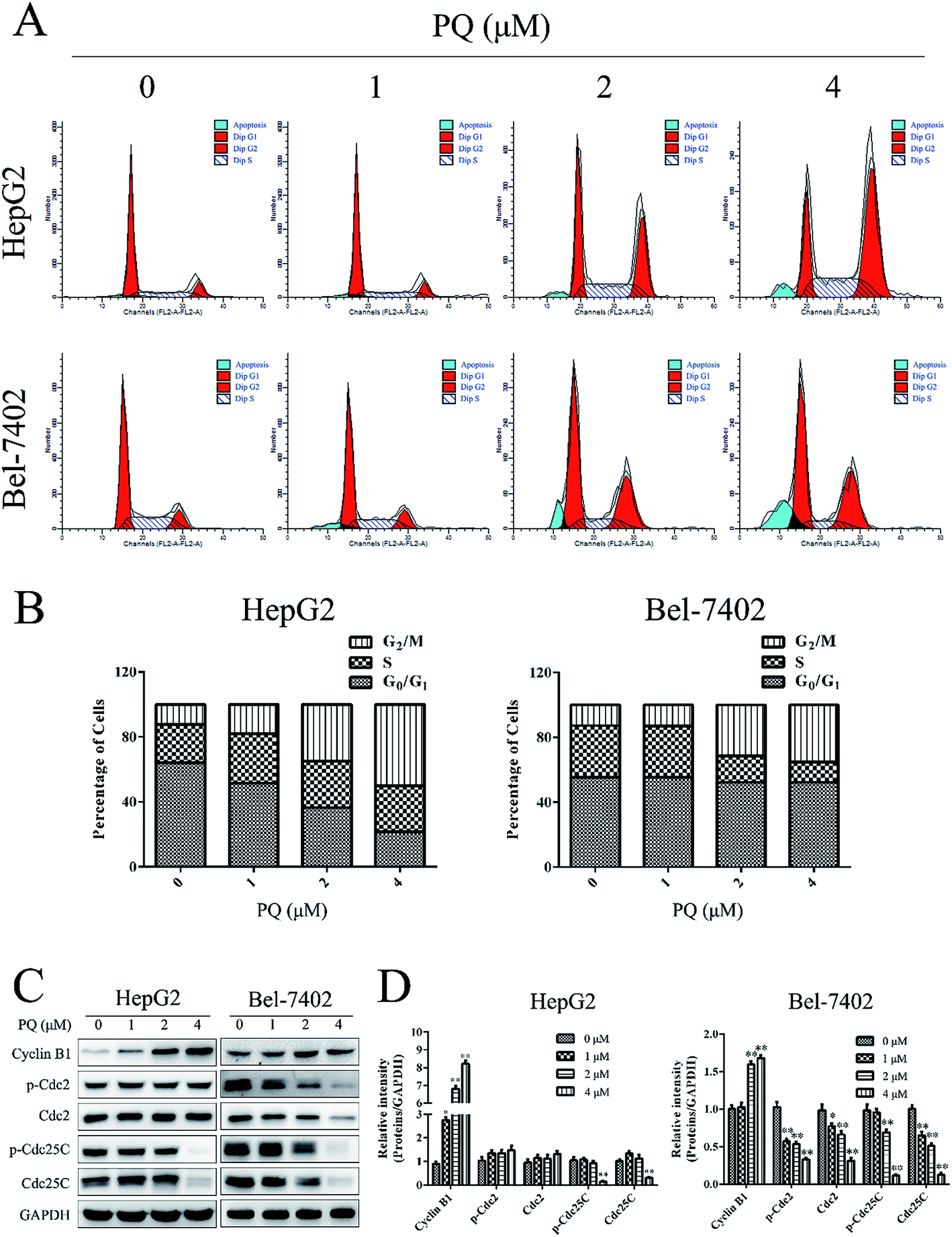 | ||
| Fig. 2 PQ induced cell cycle arrest. (A) HepG2 and Bel-7402 cells were treated with various concentrations (0, 1, 2 or 4 μM) of PQ for 24 h, after which the cells were analyzed for DNA content using flow cytometry. (B) Histogram represents the percentage of cells in each phase. (C) HepG2 and Bel-7402 cells were treated with PQ (0, 1, 2 or 4 μM; 24 h). The expression of Cyclin B1, Cdc2, p-Cdc2, Cdc25C and p-Cdc25C was evaluated by western blotting. Equal loading of protein was confirmed by probing with GAPDH. (D) Histogram represents mean value of relative protein expression. The bars represent the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, compared with the control. | ||
PQ induces mitochondrial-mediated apoptosis in human HCC cells
Cell apoptosis plays a significant role in the anti-proliferative effect by external factors.26 Loss of mitochondrial transmembrane potential (ΔΨm) and activation of caspase-9 are characteristics of one of the important cell apoptotic pathways.26,27 The influence of PQ on ΔΨm in HepG2 cells was investigated by JC-1 staining. As shown in Fig. 3A and B, the red-to-green fluorescence ratio was decreased when cells were treated with PQ, indicating that PQ induced the loss of ΔΨm in a dose-dependent manner. Moreover, Bcl-2 family proteins control the ΔΨm.28 We examined the expression of Bcl-2 and Bax proteins in HepG2 and Bel-7402 cells via a western blot assay. As shown in Fig. 3C and D, PQ up-regulated the ratio of Bax/Bcl-2, consistent with the JC-1 assay result. An Annexin V/7-AAD staining assay was employed to quantify the apoptotic effect of PQ in HepG2 cells. As shown in Fig. 3E and F, treatment with PQ increased the percentage of apoptotic cells dose dependently, suggesting that PQ induced apoptosis in HepG2 cells. Caspase proteins play a central role in the mitochondrial apoptosis pathway.29 To further explore the mechanism of PQ in apoptosis, we examined the expression of caspase-related proteins. Fig. 3G and H showed a reduction in the inactive, proenzymatic form of caspase-9/7/3/PARP and a concomitant induction of cleavage of caspase-9/7/3/PARP by PQ treatments in a dose-dependent manner. These results further confirmed that the antitumor effect of PQ was mediated through activation of the apoptotic signaling pathway in human HCC cells. | ||
| Fig. 3 PQ triggered apoptosis in HepG2 and Bel-7402 cells. (A) HepG2 cells were treated with various concentrations (0, 1, 2 or 4 μM) of PQ for 24 h, and then a JC-1 assay was performed. ΔΨm was measured by flow cytometry. (B) Histogram represents the value of ΔΨm compared with control. (C) HepG2 and Bel-7402 cells were treated with PQ (0, 1, 2 or 4 μM; 24 h). The protein expression of Bcl-2 and Bax was detected using western blotting. Equal loading of proteins was confirmed by probing with GAPDH. (D) Histogram represents the mean value of relative protein expression. (E) HepG2 cells were treated with different concentrations (0, 1, 2 or 4 μM) of PQ for 24 h, and Annexin V-APC (AV) and 7-AAD were used to stain cells. The percentage of HepG2 cells undergoing apoptosis was analyzed with flow cytometry. (F) Histogram represents the relative proportion of apoptotic cells. (G) HepG2 and Bel-7402 cells were treated with PQ (0, 1, 2 or 4 μM; 24 h). The expression of caspase-9, cleaved caspase-9, caspase-3, cleaved caspase-3, caspase-7, cleaved caspase-7, PARP, and cleaved PARP protein was evaluated using western blotting. Equal loading of protein was confirmed by probing with GAPDH. (H) Histogram represents the mean value of relative protein expression. Bars represent the means ± SD of three independent experiments. *P < 0.05; **P < 0.01, compared with the control. | ||
PQ suppresses the phosphorylation of the JAK2/Src-STAT3 pathway in human HCC cells
STAT3 activation is involved in controlling programmed cell death in human HCC cells.30 To further explore the molecular mechanism of the pro-apoptotic activity of PQ in human HCC cells, we tested the effect of PQ on STAT3 activation via a western blot assay. STAT3 proteins comprise STAT3α and STAT3β forms, and both of them can be phosphorylated at tyrosine 705.31 We used a phospho-STAT3 (Tyr 705) antibody to detect the specific phosphorylation of STAT3 at tyrosine 705. There were two bands of p-STAT3 and STAT3 proteins. The upper bands were p-STAT3α and STAT3α proteins, and the lower bands were p-STAT3β and STAT3β proteins. Fig. 4A and B showed that PQ inhibited constitutive STAT3 phosphorylation at tyrosine 705 in HepG2 and Bel-7402 cells in a dose-dependent manner. In addition, IL-6 is a growth factor that can induce STAT3 activation. To determine the effect of PQ on IL-6-induced STAT3 activation, HepG2 and Bel-7402 cells were treated with or without PQ (4 μM) for various time points and then stimulated with or without IL-6 (10 ng mL−1) for 30 min. As shown in Fig. 4C and D, PQ decreased the level of IL-6-induced phosphorylation of STAT3 in a time-dependent manner, and the expression of p-STAT3 in the group of IL-6-treated cells reached a very low level at 6 h compared with the cells without PQ treatment. Next, HepG2 and Bel-7402 cells were treated with various concentrations of PQ for 6 h and then stimulated with IL-6 for 30 min. Fig. 4E and F showed that PQ suppressed IL-6-induced STAT3 phosphorylation in a dose-dependent manner. These results demonstrated that PQ inhibited the phosphorylation of STAT3 in human HCC cells. | ||
| Fig. 4 PQ suppressed the activation of STAT3 (Tyr705) in human HCC cells. (A) HepG2 and Bel-7402 cells were treated with PQ (0, 1, 2 or 4 μM; 24 h) and then were harvested. The expression of p-STAT3 and STAT3 was analyzed by western blotting. Equal loading of protein was confirmed by probing with GAPDH. (B) Histogram represents the mean value of p-STAT3 expression. (C) HepG2 and Bel-7402 cells were treated with 4 μM PQ for various lengths of time and then stimulated with or without IL-6 (10 ng mL−1) for 30 min. The expression of p-STAT3 and STAT3 was analyzed using western blotting. Equal loading of protein was confirmed by probing with GAPDH. (D) Histogram represents the mean value of p-STAT3 expression. (E) HepG2 and Bel-7402 cells were treated with various concentrations of PQ for 6 h and then stimulated with or without IL-6 (10 ng mL−1) for 30 min. The expression of p-STAT3 and STAT3 was analyzed by western blotting. Equal loading of protein was confirmed by probing with GAPDH. (F) Histogram represents the mean value of p-STAT3 expression. (G) HepG2 and Bel-7402 cells were treated with various concentrations of PQ (0, 1, 2 or 4 μM) for 24 h and were analyzed for nuclear STAT3-DNA binding using an EMSA. (H) HepG2 and Bel-7402 cells were treated with PQ (0, 1, 2 or 4 μM; 24 h) and then were harvested. The expression of the upstream proteins JAK2, p-JAK2, Src and p-Src was analyzed by western blotting. Equal loading of protein was confirmed by probing with GAPDH. (I) Histogram represents the mean value of relative proteins expression. The bars represent the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, compared with the control; #P < 0.05, ##P < 0.01, compared to treatment with IL-6 alone. | ||
Phosphorylated STAT3 proteins translocate from the cytoplasm into the nucleus where they bind to cis-acting sequences in the promoter of target genes involved in cell proliferation and apoptosis.8 Thus, we examined the effect of PQ on STAT3 DNA binding activity. HepG2 and Bel-7402 cells were treated with PQ, and nuclear extracts were prepared for EMSA analysis. As shown in Fig. 4G, the band labeled STAT3 is STAT3 protein that bound to the probe-DNA, and the band near the bottom is excess probe-DNA. The density of the STAT3-probe-DNA band significantly decreased when cells were treated with PQ, indicating that PQ inhibited STAT3 DNA-binding activity in a dose-dependent manner in human HCC cells.
To determine which upstream kinases are involved in PQ-mediated STAT3 inactivation, we examined the effects of PQ on the phosphorylation of JAK2 and Src in HepG2 and Bel-7402 cells. As shown in Fig. 4H and I, PQ exhibited inhibitory effects on the phosphorylation of JAK2 and Src in a dose-dependent manner. These results demonstrated that PQ inhibited STAT3 activation by inhibiting phospho-JAK2 and phospho-Src.
PQ suppresses JAK2/Src-STAT3 activation by elevating ROS production in human HCC cells
An appropriate level of intracellular ROS is important for maintaining redox balance and cell proliferation and excessive ROS production can induce cell death.32 To further explore the molecular mechanism of the anti-cancer effect of PQ, we investigated the effect of PQ on ROS production in human HCC cells. HepG2 and Bel-7402 cells were treated with PQ and the fluorescence intensity of DCF was measured by flow cytometry. As shown in Fig. 5A, the fluorescence intensity increased when cells were treated with PQ, indicating that PQ induced intracellular ROS production in HepG2 and Bel-7402 cells. Next, the cell viability was measured when HepG2 and Bel-7402 cells were incubated with GSH, a ROS scavenger, for 1 h and then treated with PQ. Fig. 5B showed that pretreatment with GSH rescued cells from the cytotoxic effects of PQ, suggesting that the anti-proliferative effect of PQ in human HCC cells was ROS-dependent. Furthermore, the expression of caspase proteins was detected after HepG2 and Bel-7402 cells were incubated with GSH for 1 h and then treated with PQ. As shown in Fig. 5C and D, GSH decreased the PQ-induced activation of caspase-9/7, suggesting that PQ induced ROS production to induce apoptosis in human HCC cells. | ||
| Fig. 5 PQ induced ROS production to inhibit the phosphorylation of the JAK/Src-STAT3 pathway. (A) HepG2 and Bel-7402 cells were treated with PQ (0, 1, 2 or 4 μM; 24 h) and labeled with the fluorescent dye DCFH-DA prior to analysis of ROS production using flow cytometry at an Ex/Em of 488 nm/525 nm. Histogram represents the mean value of fluorescence intensity. (B) HepG2 and Bel-7402 cells were treated with or without GSH (5 mM) for 1 h and then with different concentrations of PQ for 24 h. The cell viability was determined by the MTT assay. (C) HepG2 and Bel-7402 cells were treated with or without GSH (5 mM) for 1 h and then with or without PQ (4 μM) for 24 h. The expression of caspase-9, cleaved caspase-9, caspase-7 and cleaved caspase-7 was evaluated using western blotting. Equal loading of protein was confirmed by probing with GAPDH. (D) Histogram represents the mean value of relative proteins expression. (E) HepG2 and Bel-7402 cells were treated with or without GSH (5 mM) for 1 h and then with or without PQ (4 μM) for 24 h. The expression of p-JAK2, JAK2, p-Src, Src, p-STAT3 and STAT3 was analyzed by western blotting. Equal loading of protein was confirmed by probing with GAPDH. (F) Histogram represents the mean value of relative proteins expression. The bars represent the means ± SD of three independent experiments. *P < 0.05, **P < 0.01, compared with the control; #P < 0.05, ##P < 0.01, related to treatment with PQ alone. | ||
Since STAT3 plays a key role in cell proliferation and apoptosis.8 we next examined the relationship between PQ-induced ROS production and PQ-induced STAT3 inactivation in human HCC cells. HepG2 and Bel-7402 cells were incubated with GSH for 1 h and then treated with PQ. GSH decreased the inhibitory effect of PQ on phosphorylated JAK2, Src and STAT3 (Fig. 5E and F), indicating that PQ inhibited the phosphorylation of the JAK/Src-STAT3 pathway by inducing ROS production.
Discussion
PQ is a new withanolide compound that was isolated from Physalis angulata L. Some withanolide compounds isolated from Physalis angulata L. such as Physangulidine A33 and Physalin F18 have been reported to show anti-cancer activity. Physangulidine A induces G2/M phase cell cycle arrest and apoptosis in prostate cancer cells,33 while Physalin F induces apoptosis in human renal carcinoma cells.18 However, the molecular mechanism of the anti-cancer effects of withanolides remains largely unknown. In this study, we first reported the anti-cancer activity of PQ and explored its potential molecular mechanism in human HCC cells.Unlimited proliferation is an important feature of cancer cells, and compounds that exhibit anti-tumor effects always inhibit cell proliferation.30,34 There are reports that evodiamine inhibited tumor growth in a subcutaneous HepG2 xenografts model by decreasing the proliferative ability of cells.30 In this study, we found that PQ inhibited cell proliferation in human HCC cells. Cell cycle plays an important role in cell proliferation.24 Previously, we found that Physagulide I, a structural analogues of PQ, inhibited cell proliferation partially by inducing G2/M phase arrest in human osteosarcoma cells.35 In our study, we found that PQ induced G2/M phase cell cycle arrest in human HCC cells. Moreover, the progression of cells from G2 to M phase is controlled by several key regulatory proteins including Cyclin B1, Cdc2, and Cdc25C.35 We also demonstrated that PQ down-regulated the expression of Cdc25C and p-Cdc25C and up-regulated the expression of Cyclin B1, which was in good agreement with previous reports that Physagulide I induced G2/M phase cell cycle arrest by modulating related proteins.35 Thus, we provided a novel finding that PQ, a new withanolide compound, suppressed proliferation in human HCC cells partially by inducing G2/M phase cell cycle arrest.
Increasing evidence suggests that withanolides exert an anti-cancer effect by inducing apoptosis in cancer cells.18,33,36 The mitochondria-mediated apoptosis pathway is frequently impaired in human cancers and loss of ΔΨm plays a pivotal role in the mitochondrial apoptosis pathway.28,37 ΔΨm is tightly controlled by various factors including the Bcl-2 family.28 The members of the Bcl-2 protein family can act as either pro-apoptotic (e.g., Bax, Bid) or anti-apoptotic (e.g., Bcl-2, Bcl-XL) factors.26 An increase in the ratio of Bax/Bcl-2 protein expression has been reported to induce the loss of ΔΨm and cause mitochondrial-mediated apoptosis.38,39 In our previous research, we demonstrated that physapubenolide, a structural analogue of PQ, triggered the mitochondrial apoptosis in HCC cells.21 In this work, we found that PQ also induced the loss of ΔΨm and triggered apoptosis in human HCC cells. Moreover, the expression of cleaved caspase proteins also play an important role in mitochondria-mediated apoptosis.40 Previous research has demonstrated that withaferin A, a structural analogue of PQ, induced mitochondria-mediated apoptosis by up-regulating the ratio of Bax/Bcl-2 and cleaved PARP in human breast cancer cells.36 In our research, we also confirmed that PQ up-regulated the ratio of Bax/Bcl-2 and cleaved caspase 9/3/7/PARP to induce mitochondrial-mediated apoptosis in human HCC cells. We are the first time to report that PQ induces mitochondria-apoptosis in human HCC cells.
Herein, we verified the anti-cancer activity of PQ, but the molecular mechanisms of the anti-proliferative and pro-apoptotic effects of PQ in human HCC cells were not well elucidated. STAT3, a nuclear transcription factor belonging to the STAT family, plays an important role in tumor growth.41 Phosphorylated STAT3 has been reported to have anti-apoptotic, proliferative, angiogenic and metastatic effects in tumor cells.8 Many STAT3 inhibitors have been reported to have promising anti-cancer activity by inhibiting STAT3 phosphorylation, including peptides, peptidomimetics and natural products.10 For example, Stattic, a small-molecule inhibitor of STAT3, induced apoptosis in breast cancer and HNSCC cells and inhibited the growth of orthotopic HNSCC tumor xenografts.10,42 Evodiamine is a natural product that inhibited STAT3 phosphorylation and suppressed tumor growth in a subcutaneous HepG2 xenograft model.30 Here, we found that PQ inhibited STAT3 phosphorylation at tyrosine 705, a site that regulates STAT3 dimer formation, nuclear translocation and DNA binding.43 Subsequently, an EMSA assay demonstrated that STAT3-DNA binding was inhibited by PQ, suggesting that PQ induced apoptosis partially by inhibiting the phosphorylation of STAT3 in human HCC cells. Furthermore, the phosphorylation of STAT3 at tyrosine 705 is mediated by upstream kinases such as JAK2 (ref. 44) and Src.12 It has been reported that withaferin A inhibited STAT3 phosphorylation by suppressing JAK2 activation but not Src activation in human renal carcinoma Caki cells.6 In this study, we demonstrated that PQ inhibited the phosphorylation of both JAK2 and Src to suppress STAT3 phosphorylation in human HCC cells. Thus, we provided evidence that PQ inhibited the phosphorylation of the JAK2/Src-STAT3 signaling pathway to exhibit an anti-proliferation effect in human HCC cells.
Cell proliferation and apoptosis are closely linked to the level of intracellular ROS in cancer cells.32 Appropriate intracellular ROS production is helpful for cell proliferation, but excessive ROS can induce cell death.39 Existing studies have shown that compounds that lead to ROS accumulation can induce cell apoptosis, and this effect is associated with apoptosis-related proteins such as caspase 9/7/3.18,45 In this study, we demonstrated that PQ increased ROS production to induce apoptosis in human HCC cells. Moreover, recent studies have established a relationship between ROS generation and the phosphorylation of proteins in the JAK2/Src-STAT3 signaling pathway in cancer cells.39 Previous reports have shown that apoptosis-inducing compounds could inhibit the phosphorylation of STAT3 signaling pathway through ROS production.46,47 In our study, we found that PQ induced inactivation of the JAK2/Src-STAT3 pathway via generation of ROS in HCC cells. In addition, there are close links between the structures of compounds and their biological activity. Natural and synthetic compounds containing electrophilic Michael acceptor pharmacophores may display promising anti-cancer activity.48,49 This is because Michael acceptors display a high electrophilicity and are associated with the redox balance.49 The molecular structure of PQ contains two Michael acceptor moieties, which may contribute to ROS production. Moreover, cancer cells are more sensitive to rapid increases in ROS levels than normal cells.50 Accordingly, this can partially explain why, in our research, the IC50 values of L02 cells was 5–6-fold over that of human HCC cells. Taken together, we provide a novel finding that PQ triggers apoptosis and exerts anti-proliferation effects in human HCC cells by regulating the ROS-JAK2/Src-STAT3 signaling pathway (Fig. 6).
 | ||
| Fig. 6 The proposed signaling pathways of PQ-induced anti-proliferation and apoptosis of HCC cells. | ||
Conclusions
Our study firstly demonstrated that PQ obviously suppressed proliferation and induced apoptosis in human HCC cells. The molecular mechanism of PQ on anti-cancer activity in HCC cells was associated with the ROS-JAK2/Src-STAT3 signaling pathway. These findings expanded the research scope of Physalis angulata L. and withanolide compounds and suggested that PQ can be considered as a potential anti-cancer candidate for the therapy of HCC.Abbreviations
| HCC | Human hepatocellular carcinoma |
| STAT3 | Signal transducer and activator of transcription 3 |
| JAK2 | Janus-activated kinase 2 |
| ROS | Reactive oxygen species |
| MTT | (3-4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide |
| ΔΨm | Mitochondrial transmembrane potential |
| PARP | Poly-(ADP-ribose)-polymerase |
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NO. 81673554 and 81503211); the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R63).Notes and references
- Y. Cui, P. Lu, G. Song, Q. Liu, D. Zhu and X. Liu, Food Chem. Toxicol., 2016, 92, 26–37 CrossRef CAS PubMed.
- T. Y. Zhou, L. H. Zhuang, Y. Hu, Y. L. Zhou, W. K. Lin, D. D. Wang, Z. Q. Wan, L. L. Chang, Y. Chen, M. D. Ying, Z. B. Chen, S. Ye, J. S. Lou, Q. J. He, H. Zhu and B. Yang, Sci. Rep., 2016, 6, 30483 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, X. Chi, Z. Zhao, D. Huang, J. Jin and J. Gao, Sci. Rep., 2016, 6, 31009 CrossRef CAS PubMed.
- Y. Xiao, Y. Liu, S. Yang, B. Zhang, T. Wang, D. Jiang, J. Zhang, D. Yu and N. Zhang, Colloids Surf., B, 2016, 141, 83–92 CrossRef CAS PubMed.
- R. Lencioni, J. M. Llovet, G. Han, W. Y. Tak, J. Yang, A. Guglielmi, S. W. Paik, M. Reig, Y. Kim do, G. Y. Chau, A. Luca, L. R. del Arbol, M. A. Leberre, W. Niu, K. Nicholson, G. Meinhardt and J. Bruix, J. Hepatol., 2016, 64, 1090–1098 CrossRef CAS PubMed.
- H. J. Um, K. J. Min, D. E. Kim and T. K. Kwon, Biochem. Biophys. Res. Commun., 2012, 427, 24–29 CrossRef CAS PubMed.
- L. Lin, Z. Yao, K. Bhuvaneshwar, Y. Gusev, B. Kallakury, S. Yang, K. Shetty and A. R. He, Carcinogenesis, 2014, 35, 2393–2403 CrossRef CAS PubMed.
- K. S. Siveen, S. Sikka, R. Surana, X. Dai, J. Zhang, A. P. Kumar, B. K. Tan, G. Sethi and A. Bishayee, Biochim. Biophys. Acta, Gen. Subj., 2014, 1845, 136–154 CAS.
- Y. Hong, L. Zhou, H. Xie, W. Wang and S. Zheng, Biochem. Biophys. Res. Commun., 2015, 461, 513–518 CrossRef CAS PubMed.
- G. Miklossy, T. S. Hilliard and J. Turkson, Nat. Rev. Drug Discovery, 2013, 12, 611–629 CrossRef CAS PubMed.
- H. Zhao, Y. Guo, S. Li, R. Han, J. Ying, H. Zhu, Y. Wang, L. Yin, Y. Han, L. Sun, Z. Wang, Q. Lin, X. Bi, Y. Jiao, H. Jia, J. Zhao, Z. Huang, Z. Li, J. Zhou, W. Song, K. Meng and J. Cai, Oncotarget, 2016, 6, 31927–31943 Search PubMed.
- Q. Tan, H. Wang, Y. Hu, M. Hu, X. Li, Aodengqimuge, Y. Ma, C. Wei and L. Song, Cancer Sci., 2015, 106, 1023–1032 CrossRef CAS PubMed.
- W. Yu, H. Xiao, J. Lin and C. Li, J. Med. Chem., 2013, 56, 4402–4412 CrossRef CAS PubMed.
- M. O. Yu, K. J. Park, D. H. Park, Y. G. Chung, S. G. Chi and S. H. Kang, J. Neuro-Oncol., 2015, 125, 55–63 CrossRef CAS PubMed.
- H. Li, H. He, Z. Wang, J. Cai, B. Sun, Q. Wu, Y. Zhang, G. Zhou and L. Yang, Gene, 2016, 585, 256–264 CrossRef CAS PubMed.
- H. S. Seol, S. E. Lee, J. S. Song, H. Y. Lee, S. Park, I. Kim, S. R. Singh, S. Chang and S. J. Jang, Cancer Lett., 2016, 382, 157–165 CrossRef CAS PubMed.
- C.-Y. Gao, T. Ma, J. Luo and L.-Y. Kong, Nat. Prod. Commun., 2015, 10, 2059–2062 Search PubMed.
- S. Y. Wu, Y. L. Leu, Y. L. Chang, T. S. Wu, P. C. Kuo, Y. R. Liao, C. M. Teng and S. L. Pan, PLoS One, 2012, 7, e40727 CAS.
- W.-N. Zhang and W.-Y. Tong, Chem. Biodiversity, 2016, 13, 48–65 Search PubMed.
- A. G. Schirmer Pigatto, L. A. Mentz and G. L. Gonçalves Soares, Biochem. Syst. Ecol., 2014, 54, 40–47 CrossRef CAS.
- T. Ma, B. Y. Fan, C. Zhang, H. J. Zhao, C. Han, C. Y. Gao, J. G. Luo and L. Y. Kong, Sci. Rep., 2016, 6, 29926 CrossRef PubMed.
- K. Shenoy, Y. Wu and S. Pervaiz, Cancer Res., 2009, 69, 1941–1950 CrossRef CAS PubMed.
- Y. D. Geng, L. Yang, C. Zhang and L. Y. Kong, Food Chem. Toxicol., 2014, 73, 7–16 CrossRef CAS PubMed.
- Y. C. Ma, N. Su, X. J. Shi, W. Zhao, Y. Ke, X. Zi, N. M. Zhao, Y. H. Qin, H. W. Zhao and H. M. Liu, Toxicol. Appl. Pharmacol., 2015, 282, 227–236 CrossRef CAS PubMed.
- J. Zhang, H. Li, Z. Huang, Y. He, X. Zhou, T. Huang, P. Dai, D. Duan, X. Ma, Q. Yin, X. Wang, H. Liu, S. Chen, F. Zou and X. Chen, Cell Stress Chaperones, 2016, 21, 339–348 CrossRef CAS PubMed.
- Y. Kiraz, A. Adan, M. Kartal Yandim and Y. Baran, Tumour Biol., 2016, 37, 8471–8486 CrossRef CAS PubMed.
- Q. Tang, G. Li, X. Wei, J. Zhang, J. F. Chiu, D. Hasenmayer, D. Zhang and H. Zhang, Cancer Lett., 2013, 336, 325–337 CrossRef CAS PubMed.
- S. Fulda, Semin. Cancer Biol., 2015, 31, 84–88 CrossRef CAS PubMed.
- W. Ye, Y. Chen, H. Li, W. Zhang, H. Liu, Z. Sun, T. Liu and S. Li, Molecules, 2016, 21, 781 CrossRef PubMed.
- J. Yang, X. Cai, W. Lu, C. Hu, X. Xu, Q. Yu and P. Cao, Cancer Lett., 2013, 328, 243–251 CrossRef CAS PubMed.
- S. Biethahn, F. Alves, S. Wilde, W. Hiddemann and K. Spiekermann, Exp. Hematol., 1999, 27, 885–894 CrossRef CAS PubMed.
- K. R. Martin and J. C. Barrett, Hum. Exp. Toxicol., 2002, 21, 71–75 CAS.
- E. M. Reyes-Reyes, Z. Jin, A. J. Vaisberg, G. B. Hammond and P. J. Bates, J. Nat. Prod., 2013, 76, 2–7 CrossRef CAS PubMed.
- C. T. Kuo and W. S. Lee, Cancer Lett., 2016, 378, 104–110 CrossRef CAS PubMed.
- T. Ma, W.-N. Zhang, L. Yang, C. Zhang, R. Lin, S.-M. Shan, M.-D. Zhu, J.-G. Luo and L.-Y. Kong, RSC Adv., 2016, 6, 53089–53100 RSC.
- S. D. Stan, E. R. Hahm, R. Warin and S. V. Singh, Cancer Res., 2008, 68, 7661–7669 CrossRef CAS PubMed.
- Y. D. Geng, C. Zhang, Y. M. Shi, Y. Z. Xia, C. Guo, L. Yang and L. Y. Kong, Cancer Lett., 2015, 366, 19–31 CrossRef CAS PubMed.
- C. Y. Cheng and C. C. Su, Mol. Med. Rep., 2010, 3, 645–650 CAS.
- D. H. Kim, K. W. Park, I. G. Chae, J. Kundu, E. H. Kim, J. K. Kundu and K. S. Chun, Mol. Carcinog., 2016, 55, 1096–1110 CrossRef CAS PubMed.
- M. A. Rahman, K. Bishayee, K. Habib, A. Sadra and S. O. Huh, Biochem. Pharmacol., 2016, 117, 97–112 CrossRef CAS PubMed.
- J. Diao, J. Wei, R. Yan, X. Liu, Q. Li, L. Lin, Y. Zhu and H. Li, Biochem. Biophys. Res. Commun., 2016, 477, 1024–1030 CrossRef CAS PubMed.
- J. Schust, B. Sperl, A. Hollis, T. U. Mayer and T. Berg, Chem. Biol., 2006, 13, 1235–1242 CrossRef CAS PubMed.
- J. Lee, E. R. Hahm and S. V. Singh, Carcinogenesis, 2010, 31, 1991–1998 CrossRef CAS PubMed.
- P. Rajendran, F. Li, M. K. Shanmugam, R. Kannaiyan, J. N. Goh, K. F. Wong, W. Wang, E. Khin, V. Tergaonkar, A. P. Kumar, J. M. Luk and G. Sethi, Cancer Prev. Res., 2012, 5, 631–643 CrossRef CAS PubMed.
- Y. Q. Hou, Y. Yao, Y. L. Bao, Z. B. Song, C. Yang, X. L. Gao, W. J. Zhang, L. G. Sun, C. L. Yu, Y. X. Huang, G. N. Wang and Y. X. Li, Oxid. Med. Cell. Longevity, 2016, 2016, 4941623 Search PubMed.
- D. Dixit, V. Sharma, S. Ghosh, N. Koul, P. K. Mishra and E. Sen, Free Radical Biol. Med., 2009, 47, 364–374 CrossRef CAS PubMed.
- Y. Liu, L. Wang, Y. Wu, C. Lv, X. Li, X. Cao, M. Yang, D. Feng and Z. Luo, Toxicology, 2013, 304, 120–131 CrossRef CAS PubMed.
- F. F. Gan, K. K. Kaminska, H. Yang, C. Y. Liew, P. C. Leow, C. L. So, L. N. Tu, A. Roy, C. W. Yap, T. S. Kang, W. K. Chui and E. H. Chew, Antioxid. Redox Signaling, 2013, 19, 1149–1165 CrossRef CAS PubMed.
- P. Zou, J. Zhang, Y. Xia, K. Kanchana, G. Guo, W. Chen, Y. Huang, Z. Wang, S. Yang and G. Liang, Oncotarget, 2015, 6, 5860–5876 CrossRef PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25032g |
| This journal is © The Royal Society of Chemistry 2017 |