
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeometrically isomeric Pt(II)/Fe(II)-based heterometallo-supramolecular polymers with organometallic ligands for electrochromism and the electrochemical switching of Raman scattering†
Chanchal
Chakraborty
ab,
Rakesh K.
Pandey
a,
Utpal
Rana
a,
Miki
Kanao
a,
Satoshi
Moriyama
b and
Masayoshi
Higuchi
*a
aElectronic Functional Macromolecules Group, National Institute for Materials Science (NIMS), Tsukuba 305-0044, Japan. E-mail: HIGUCHI.Masayoshi@nims.go.jp
bInternational Center for Materials Nanoarchitectonics (MANA), NIMS, Tsukuba, Japan
First published on 12th September 2016
Abstract
Heterometallo-supramolecular polymers with Pt(II) and Fe(II) ions introduced alternately (cis-polyPtFe and trans-polyPtFe) in a precise way were prepared successfully by the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation of Fe(II) ions with cis- or trans-conformational organo-Pt(II) ligands. The conformational difference between cis- and trans-greatly changed the morphology, crystallinity, ionic conductivity, electrochromic properties, and redox-triggered fluorescence of the polymers. The cis-polyPtFe exhibited better crystallinity and low ionic conductivity, whereas trans-polyPtFe showed an amorphous nature with high ionic conductivity. Both the polymers exhibited reversible electrochromism between purple and yellow colors due to the redox of Fe(II)/(III) upon applying a potential of 0 V or +3 V. The trans-polyPtFe showed better electrochromic stability and response times compared to cis-polyPtFe. In addition, the trans-polyPtFe also showed an improved response in redox-triggered Raman scattering switching compared to cis-polyPtFe over a long time range.
:1 complexation of Fe(II) ions with cis- or trans-conformational organo-Pt(II) ligands. The conformational difference between cis- and trans-greatly changed the morphology, crystallinity, ionic conductivity, electrochromic properties, and redox-triggered fluorescence of the polymers. The cis-polyPtFe exhibited better crystallinity and low ionic conductivity, whereas trans-polyPtFe showed an amorphous nature with high ionic conductivity. Both the polymers exhibited reversible electrochromism between purple and yellow colors due to the redox of Fe(II)/(III) upon applying a potential of 0 V or +3 V. The trans-polyPtFe showed better electrochromic stability and response times compared to cis-polyPtFe. In addition, the trans-polyPtFe also showed an improved response in redox-triggered Raman scattering switching compared to cis-polyPtFe over a long time range.
Introduction
Metallo-supramolecular polymers represent a new dimension that has been growing in interest over the last ten years in the area of polymer and materials science due to their utility in a wide range of applications, including sensors, memory devices, light emitting diodes, photovoltaic devices, nanolithographic templates, stimuli responsive materials in redox-tunable photonic crystals, controlled-drug release, electrochromic displays, and molecular motors.1–7 The main reason behind this considerable interest is the great versatility of their fabrication using a very simple self-assembling process between metal cations and organic ligands.8 Using such a methodology, many one (1D), two (2D), and three (3D) dimensional structures along with different metal ions have been produced and used as smart materials for various applications.9–11 The main benefits of these metallo-supramolecular polymers over their conventional covalent counterparts originate from the presence of metallic centers, providing a wide range of properties, such as optical, electrical, redox, photochemical, magnetic, or catalytic activities.12 Due to the large range of available organic ligands and metallic cations, metallo-supramolecular polymers offer the vast potential to develop smart materials with tunable properties. In this context, if two metal ions are introduced to a metallo-supramolecular polymer chain regularly, unique properties due to the neighboring metal–metal interactions are expected to appear in the polymer.13–15 The formation of heteronuclear metal complexes using different coordination environments of ditopic ligands has been widely investigated.16–23 Constable et al. used a one-pot synthesis at different temperatures to obtain two metal ion complex structures.16 Newkome et al. reported a Sierpinski hexagonal gasket-shaped supramolecule by connecting Fe, Ru, and tris-terpyridines.24 Licciardello et al. reported the electro-optical properties of trinuclear complexes with both Ru(II) and Os(II) ions.25 Again, Ag–Cu- and Ag–Zn-containing heterometallic coordination polymers were reported by Di Sun et al. to exhibit the interesting photoluminescence property of heterometallic aggregates.26–28 However, 1D heterometallo-supramolecular polymers where metal centers are incorporated alternatively are very rarely reported, except in our previous reports,13–15 while the reaction steps are not so precise to incorporate heterometals alternately.Homometallo-supramolecular polymers were synthesized by the 1:1 complexation of metal ions and ditopic ligands (Scheme 1a),3 but the alternate introduction of heterometallic centers precisely in a polymer chain is very challenging. Scheme 1b exhibits a preparation method of a heterometallo-supramolecular polymer using an asymmetrical ligand.14 The synthetic procedure is smart, but highly selective complexation between the two metal ion species and the two coordination sites of the asymmetrical ligand are required to obtain the desired polymer structure. In order to develop divergent syntheses of heterometallo-supramolecular polymers, in the present study we focused on organometallic bonds. When ditopic ligands include metal species by organometallic bonds formation, heterometallo-supramolecular polymers are considered to form by the 1:1 complexation of the organometallic ligand and another metal ion (Scheme 1c). The obtained polymers are also expected to show unique electronic/optical properties based on the metal–metal interactions through the organometallic bonds. Alongside this, the other advantage of organometallic ligands is that it is possible to prepare two geometrical isomeric organometallic ligands (cis- and trans-) and their corresponding heterometallo-polymers from one precursor organic ligand by just changing the metal salts used for preparing the organometallic ligands (Scheme 1d).
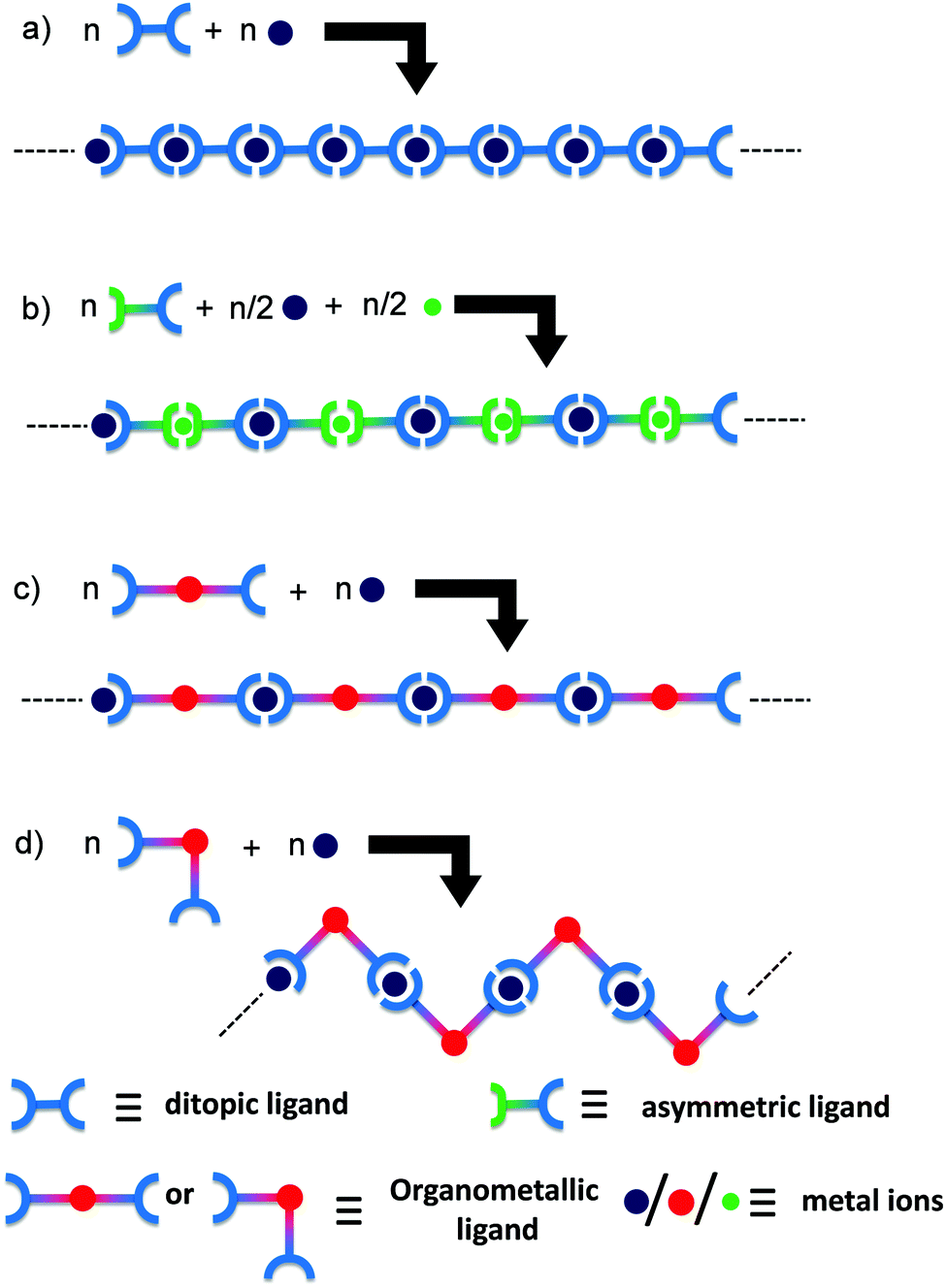 | ||
| Scheme 1 Different synthesis routes of (a) homometallo- and (b–d) heterometallo-supramolecular polymers. | ||
Recently, Fe(II)-based metallo-supramolecular polymers with bis(terpyridine) as ditopic ligands were extensively investigated as fourth generation electrochromic materials due to its redox-triggered color change from purple (Fe(II)) in the reduced state to colorless (Fe(III)) in the oxidized state.3,7,9,14,15,29–32 The electrochromic behaviors of metallo-supramolecular polymers have been widely investigated, but the correlation between the polymer structures, especially the structural characteristic, and the electrochromic activities has not been elucidated yet.
It has been reported that platinum acetylide-based organometallic oligomers and polymers are π-conjugated materials with enhanced photophysical properties33–36 and that one can tune the geometry of the Pt-complex by the proper choice of precursor Pt salt used in the complexation. It is anticipated that, if we could effectively synthesize alternatively introduced Fe(II)- and Pt(II)-containing metallo-supramolecular polymers by using a Pt(II)-based organometallic ligand, it must have an effect on the properties of the particular metal center due to the presence of hetero-metallic interaction. Thus, trans- and cis-conformational organo-Pt(II) ligands (trans-PtL and cis-PtL) were prepared by the 1:2 organometallic bond formation of Pt(II) and a terpyridyl- and ethynyl-containing compound. Heterometallo-supramolecular polymers with Pt(II) and Fe(II) ions to be introduced alternately (trans-polyPtFe and cis-polyPtFe) were precisely prepared via the 1:1 complexation of Fe(II) ions with the organo-Pt(II) ligands (Scheme 2). To the best of our knowledge, this is the first report to prepare heterometallo-supramolecular polymers using organometallic ligands. We then studied the redox-triggered optical properties, e.g., electrochromism and solid-state emission, and their effects due to the heterometallo interaction in the bimetallic Pt(II)/Fe(II) supramolecular polymers. We also investigated the geometric aspects of the trans- and cis-bimetallic polymers on their material properties, such as the electrochromism and ionic conductivity. These studies can provide a correlation between the structural aspects and the electro-optical properties, specifically the electrochromism, of metallo-supramolecular polymers in detail that has not been studied before.
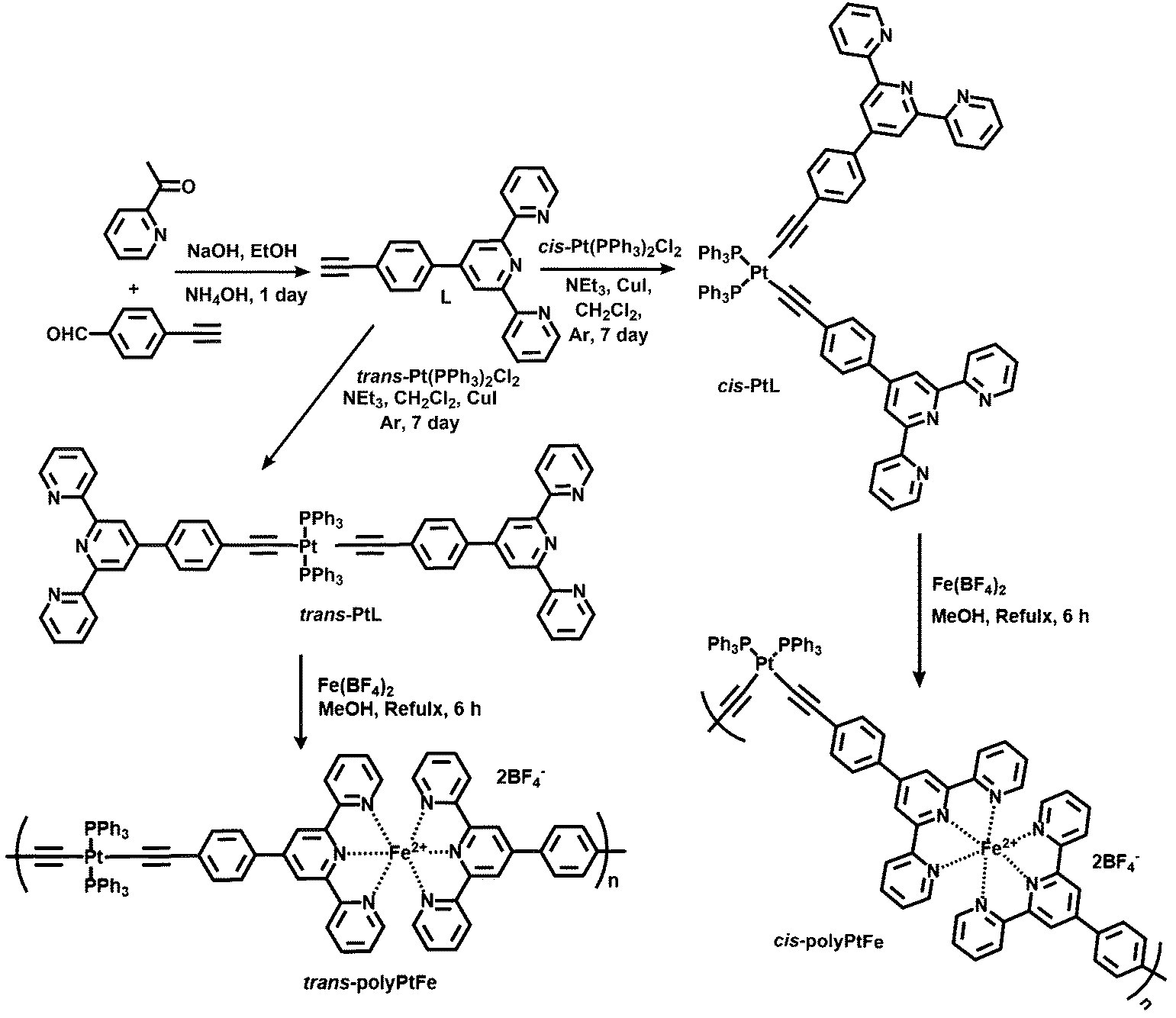 | ||
| Scheme 2 Synthesis of an unsymmetrical ligand (L) and the heterometallo-supramolecular polymers with Pt(II) and Fe(II) ions introduced alternately (cis- and trans-polyPtFe). | ||
Experimental
Materials and methods
Unless otherwise noted, all the reagents were reagent grade and were used without purification. Dehydrated ethanol, dichloromethane, and methanol were used as the reaction solvents. Spectroscopic grade methanol was used for the spectral analysis. These solvents were purchased from either Aldrich or Wako or Kanto Chemical Co. Inc. and were used as received. Deionized H2O was used in the experiments where required. 4-Ethynyl benzaldehyde, 2-acetylpyridine, sodium hydroxide, ammonia solution, cis-dichlorobis(triphenylphosphine)platinum(II), trans-dichlorobis(triphenylphosphine)platinum(II), trimethylamine, copper(I) iodide, and iron(II)tetrafluoroborate were purchased from either Sigma-Aldrich co. Ltd or Tokyo Chemical Industry Co. Ltd and used as received.Instrumentation
1H-NMR spectra were recorded at 300 MHz on a JEOL AL 300/BZ instrument using DMSO-d6 and CDCl3 as the solvent. Chemical shifts are given herein relative to TMS. DOSY NMR spectra were recorded on a JNM-ECA400 spectrometer (JEOL Resonance). MALDI mass spectra (MALDI-TOF) were measured by using the AXIMA-CFR, Shimadzu/Kratos, TOF mass spectrometer. UV-vis spectra were recorded using a Shimadzu UV-2550 UV-visible spectrophotometer. The fluorescence was measured by a Shimadzu RF-5300PC spectrofluorophotometer. The molecular weights of the polymers were determined by SEC-viscometry-RALLS (size exclusion chromatography-viscometry-right angle light scattering) on a Viscotek 270 Dual Detector instrument using polyethylene oxide PEO-22K as the standard in methanol solvent (flow rate: 1 mL min−1). Flash column chromatographic separations were performed on silica gel 60 N (neutral, 40–100 μM), Kanto Chemical Co. Inc. Wide angle XRD was done by using a RINT ULTIMA III device with Cu Kα radiation (λ = 1.54 Å), a generator voltage of 40 kV, and a current of 40 mA. Polymer powder was placed on a glass holder and scanned in a range of 2θ = 2–60° at a scan rate of 1 s per step with a step width of 0.01° at room temperature. Cyclic voltammetry (CV) and amperometric experiments were carried out by drop casting the polymer solutions onto a glassy carbon working electrode and then slowly evaporating the solution. The experiments were executed in nitrogen–saturated anhydrous acetonitrile solution containing 0.1 M of lithium perchlorate (LiClO4) as the supporting electrolyte by using an electrochemical analyzer, ALS/H CH instruments. A platinum wire was used as counter the electrode, and Ag/AgCl as the reference electrode. The scan rate for CV analysis was 10 mV s−1. For the ionic conductivity measurements, polymer powder was pressed into the form of a pellet using a press. The thicknesses of all the polymer pellets were within 0.2 to 0.4 mm. The pellet was then inserted in between the two electrodes of a sample holder. The sample holder used in the conductivity experiments was composed of two electrodes with an attached micrometer on the upper electrode and another fixed electrode with a guard ring on the electrode, which reduces the effect of stray field lines existing at the edge of the sample. The guard ring ensures that the electric field lines remain parallel all over the sample. Besides this, the ac impedance results were also normalized using the empty cell measurements by carrying out the experiment in an air-gap created with the help of the attached micrometer. This procedure eradicates the errors due to connection and equipment, etc., thus making the measurement more trustworthy. A Solartron 1260 impedance gain/phase analyzer coupled with a Solartron 1296 dielectric interface was used for the ac impedance measurements with high sensitivity. A frequency range of 50 Hz to 10 MHz was used to determine the resistance of the film under humid conditions. For comparison with various studies, polyFe was prepared according to the literature.37 Raman spectroscopy of the compounds was done with a 532 nm laser as the excitation source using a Raman 11 instrument (Nanophoton Corporation, Japan). An equimolar amount of 4′,4′′′′-(1,4-phenylene)bis(2,2′:6′,2′′-terpyridine) (Sigma-Aldrich) and Fe(BF4)2 was refluxed in methanol for 6 h. The reaction mixture was dried on a petri dish, washed several time with water and chloroform, and again dried in vacuum overnight to yield the corresponding polyFe (>95%).Synthesis of 4′-(4-ethynylphenyl)-2,2′:6′,2′′-terpyridine (L)38
2-Acetylpyridine (3.63 g, 30 mmol) and 4-ethynylbenzaldehyde (1.95 g, 15 mmol) and NaOH (1.2 g, 30 mmol) were taken in a round bottom flask. Subsequently, a solution of ethanol (200 mL) and concentrated aqueous NH3 solution (100 mL) was added to the mixture. The suspension was stirred at room temperature for 3 days. Soon after, it became a clear solution and after 3 days it formed a yellow precipitation. The formed precipitate was isolated by vacuum filtration and washed with water and ethanol. After performing flash column chromatography using chloroform in silica gel and then following recrystallization from ethanol, L was obtained as white needles (3.5 g, 70%). 1H NMR (300 MHz, DMSO-d6): 8.77 (d, J = 4.8 Hz, 2H, H6,6′′), 8.72 (s, 2H, H3′,5′), 8.69 (d, J = 9.0 Hz, 2H, H3,3′′), 8.04 (t, 2H, H4,4′′), 7.98 (d, J = 9.0, 2H, Hb), 7.70 (d, J = 9.0, 2H, Ha), 7.54 (m, 2H, H5,5′′), 4.38 (s, 1H). MALDI-TOF: [M+] calcd. for C23H15N3: 333.13, [(M + H)+] found 334.92, [(M + Na)+] found 356.79 and [(M + K)+] found 372.74.Synthesis of ligand cis-PtL
4′-(4-Ethynylphenyl)-2,2′:6′,2′′-terpyridine (L) (100 mg, 0.33 mmol) was added with cis-dichlorobis(triphenylphosphine)platinum(II) (118.5 mg, 0.15 mmol) under an argon environment in the presence of dry triethylamine (10 mL) and anhydrous dichloromethane (20 mL). Copper(I) iodide (6 mg, 0.075 mmol) was added to the solution and the resulting solution was stirred at room temperature for 7 days under an argon atmosphere. The yellow precipitation was filtered and successively washed several times with water and dichloromethane, respectively. The product was dried in vacuum at 50 °C, affording a greenish yellow solid (135 mg, 65%). 1H NMR (300 MHz, CDCl3): 8.71–8.59 (m, 12H), 7.86–7.77 (m, 24H), 7.53–7.40 (m, 22H). MALDI-TOF: [m/z] calcd for C82H58N6P2Pt: 1383.38, [(M + H)+] found 1384.27.Synthesis of ligand trans-PtL
4′-(4-Ethynylphenyl)-2,2′:6′,2′′-terpyridine (L) (100 mg, 0.33 mmol) was added with trans-dichlorobis(triphenylphosphine)platinum(II) (118.5 mg, 0.15 mmol) under an argon environment in the presence of dry triethylamine (10 mL) and anhydrous dichloromethane (20 mL). Copper(I) iodide (6 mg, 0.075 mmol) was added to the solution and the resulting solution was stirred at room temperature for 7 days under an argon atmosphere. The yellow precipitation was filtered and successively washed several times with water and dichloromethane, respectively. The product was dried in vacuum at 50 °C, affording a yellow solid (186 mg, 90%). 1H NMR (300 MHz, CDCl3): 8.71–8.62 (m, 12H), 8.02 (br, 4H), 7.86–7.77 (m, 20H), 7.53 (m, 4H), 7.42–7.40 (m, 18H). MALDI-TOF: [m/z] calcd for C82H58N6P2Pt: 1383.38, [(M + H)+] found 1384.24.Synthesis of cis-polyPtFe
To a 20 mL methanol solution of cis-PtL (50 mg, 0.034 mmol), a methanolic solution (10 mL) of Fe(BF4)2 (11.48 mg, 0.034 mmol) was added under inert conditions. The mixture was refluxed for 6 h with continuous stirring. The reaction mixture was dried on a petri dish, washed several time with water and chloroform, and again dried in vacuum overnight to give the corresponding cis-polyPtFe as a violet solid (46 mg, 80%). 1H NMR (300 MHz, DMSO-d6): 9.58 (br, s), 9.06 (br, s), 9.06 (br, m), 8.26 (br, m), 8.04 (br, m), 7.92–7.55 (br, m), 7.23 (br, m). Mw = 2.27 × 104 Da.Synthesis of trans-polyPtFe
The trans-polyPtFe was synthesized by the same procedure applied for the synthesis of cis-polyPtFe by using trans-PtL ligand in place of cis-PtL. The reaction mixture was dried on a petri dish, washed several times with water and chloroform, and again dried in vacuum overnight to yield the corresponding trans-polyPtFe as a violet solid (51 mg, 88%). 1H NMR (300 MHz, DMSO-d6): 9.55 (br, s), 9.04 (br, s), 8.62 (br, m), 8.22 (br, m), 8.01–7.59 (br, m), 7.20 (br, m). Mw = 2.22 × 104 Da.Preparation of a polymer film on ITO
A polyPtFe film was prepared on an ITO-coated glass by spin-coating. polyPtFe was dissolved in methanol (concentration: 2 mg mL−1). The solution (200 μL) was spin-coated (40 rpm for 250 s and 100 rpm for 600 s) onto an ITO glass (2 × 1 cm), and the prepared film was then dried at room temperature for another 20 min.Preparation of the semi-gel electrolyte
The semi-gel electrolyte was prepared by mixing 0.9 g LiClO4 and 2.1 g poly(methylmethacrylate) (PMMA) in 6 mL propylene carbonate. The mixture was added to 5 mL of acetonitrile to make a semi-gel material. The mixture was stirred at room temperature to make a well-blended semi-gel electrolyte.Preparation of the electrochromic (EC) devices and the measurements
The semi-gel electrolyte consisting of poly(methylmethacrylate) (PMMA), propylene carbonate, and lithium perchlorate in acetonitrile was put on a polymer-coated ITO. The ITO was kept for 15 min at room temperature. Finally, another ITO was put over it and it was kept at 60 °C for another 1 h for thermal curing. These devices were used for the electrochromism studies. The EC devices were oxidized or reduced by applying a voltage between the two ITO electrodes in between a potential window of +3 to 0 V by chronoamperometric studies with a 5 s interval time using an electrochemical analyzer, ALS/H CH instruments, attached to a UV-vis and fluorescence spectrophotometer.Result and discussions
A terpyridyl- and ethynyl-containing asymmetric ligand (L) was prepared by the reaction between 2-acetylpyridine and 4-ethynylbenzaldehyde in the presence of NaOH and ammonia (Scheme 2). The ligand L was characterized by MALDI-TOF and 1H NMR spectroscopy, as shown in Fig. S1 in the ESI.†L (2 equiv.) was treated with cis- or trans- dichlorobis(triphenylphosphine)platinum(II) in dichloromethane solution and catalyzed by CuI in the presence of NEt3 to prepare the corresponding cis- or trans-PtL ligands with a good yield. Both the ligands (cis- and trans-PtL) were sparsely soluble in common solvents and could be readily purified by washing with water and dichloromethane to remove the small impurities. The ligands were characterized by 1H NMR and MALDI-TOF mass spectroscopy (ESI,† Fig. S2 and S3). A comparison of the 1H NMR spectra of L and cis- and trans-PtL in Fig. S6a and b (ESI†) reveals that the characteristic singlet 1H signal of the ethynyl proton of L at 4.38 ppm completely disappeared after complexation with Pt(II). This desertion of the ethynyl proton along with no change in the terpyridine proton position of cis- and trans-PtL clearly confirmed the attachment of Pt(II) with the ethynyl binding site of L to prepare cis- and trans-PtL.The complexation behavior of cis- and trans-PtL with Fe(II) was investigated in detail by UV-vis spectroscopic measurement. The spectrum of a methanol solution of cis-PtL (6 μM) exhibited a metal-to-ligand charge transfer (MLCT) band for the complexation of Pt(II) ions with the ethynyl moiety of L at 383 nm (Fig. 1a). A methanol solution of Fe(BF4)2 was successively added to the cis-PtL solution up to a molar ratio of [Fe(BF4)2]/[cis-PtL] of 2. A new absorption at 574 nm based on the MLCT band of Fe(II) complexed with the terpyridine moieties of cis-PtL appeared during the titration. The MLCT absorption was increased linearly and was clearly saturated at a ratio of [Fe(BF4)2]/[cis-PtL] of 1 (Fig. 1a and b). The spectrum of a methanol solution of trans-PtL (6 μM) also demonstrated a MLCT band for the complexation of Pt(II) ions with the ethynyl moiety of L at 398 nm (Fig. 1c). The red-shift of the MLCT transition in trans-PtL compared to cis-PtL is due to the enhancement of the MLCT energy gap in cis-PtL rather than in trans-PtL as the π-orbitals of cis-PtL are not aligned to extend the conjugation.39 The successive addition of a methanol solution of Fe(BF4)2 to trans-PtL solution up to a molar ratio of [Fe(BF4)2]/[trans-PtL] of 2 also revealed the generation of a new absorption peak at 574 nm based on the MLCT band of Fe(II) complexed with the terpyridine moieties. Here also, the MLCT absorption was linearly increased and saturated at the ratio of [Fe(BF4)2]/[trans-PtL] of 1 (Fig. 1c and d). These spectral changes suggest the formation of a heterometallo-supramolecular polymer with Pt(II) and Fe(II) ions introduced alternately to prepare 1D linear cis- and trans-polyPtFe, according to a stepwise complexation behavior with complexation between cis- or trans-PtL and Fe(II) with a 1:1 molar ratio, as shown in Schemes 1 and 2. Furthermore, the MLCT absorption of Fe(II) did not change upon the further addition of Fe(II), indicating that the polymers were stable in solution.
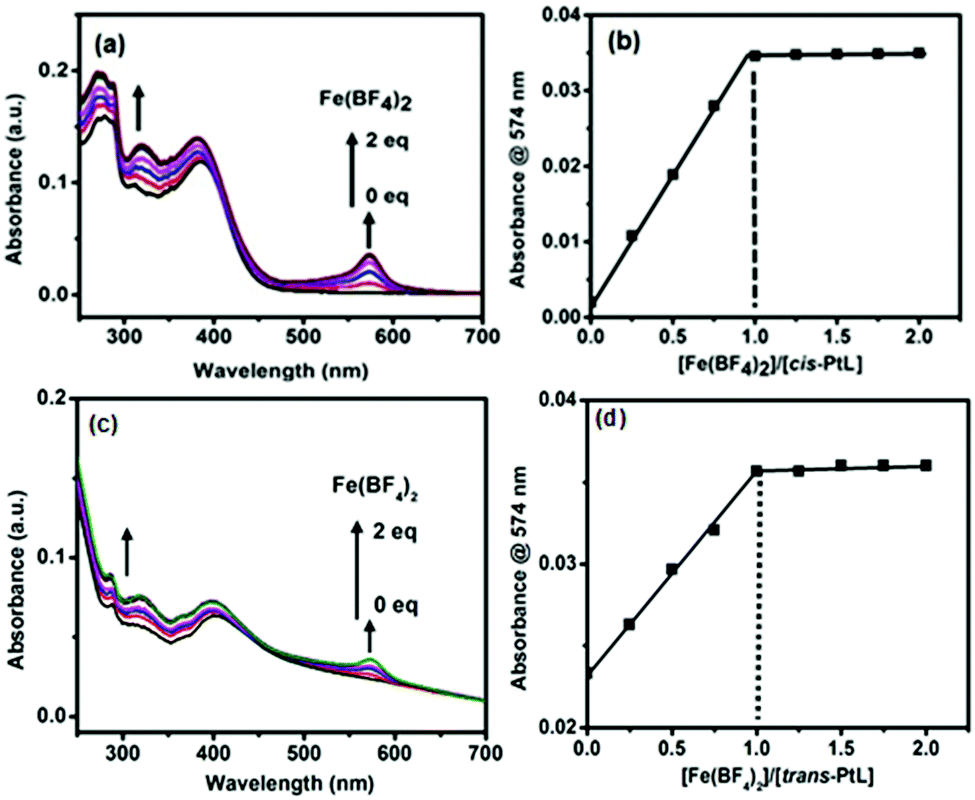 | ||
| Fig. 1 (a) UV-vis spectral change of a methanol solution of cis-PtL during titration with Fe(BF4)2 at room temperature. (b) The absorption change at 574 nm as a function of the [Fe(BF4)2]/[cis-PtL] ratio. (c) UV-vis spectral change of a methanol solution of trans-PtL during titration with Fe(BF4)2 at room temperature. (d) The absorption change at 574 nm as a function of the [Fe(BF4)2]/[trans-PtL] ratio. | ||
The cis- and trans-polyPtFe were obtained by refluxing the corresponding Pt(II)-containing ligand with Fe(BF4)2 in methanol as a violet solid in a satisfactorily high yield (>80%), as shown in Scheme 2. The polymers were soluble in alcohols, such as methanol, ethanol, and ethylene glycol, and polar solvents, such as DMSO and DMF, but sparsely soluble in the common organic solvents, such as n-hexane, chloroform, dichloromethane, and tetrahydrofuran. The molecular weights of the polymers were determined by the SEC-Viscometry/RALLS method using PEO as the standard (for cis-polyPtFe: Mw = 2.27 × 104 Da; and for trans-polyPtFe: Mw = 2.22 × 104 Da), which strongly indicated that both cis- and trans-polyPtFe form a polymer structure with moderate polydispersity. The 1H NMR spectrum of the polymers are shown in ESI,† Fig. S4 and S5. The comparison of the 1H NMR spectra of L and the cis- and trans-PtL ligands and the corresponding cis- and trans-polyPtFe in the ESI† Fig. S6a and b also revealed a broadening of the proton signals along with some downfield shifting of the terpyridyl protons after polymerization of the cis- or trans-PtL ligands by Fe(BF4)2. To gain a clearer idea about the formation of cis- and trans-metallo-supramolecular polymers, we performed two dimensional Diffusion-Ordered NMR Spectroscopy (DOSY) analysis. Generally, 1H DOSY NMR can distinguish between cis- and trans-polymers as their diffusion is different, and hence their diffusion coefficients are also different. The 1H DOSY NMR study of the cis- and trans-polyPtFe (ESI,† Fig. S7 and S8) revealed average diffusion coefficients of about 9.14 × 10−11 m2 s−1 and 1.7 × 10−10 m2 s−1, respectively, for the two polymers. The diffusion coefficient in the trans-polymer is quite a bit higher due to the better mobility in the trans-polymer chains than in the cis-configuration. The diffusion coefficient of cis-polyPtFe is one order lower due to its aggregated structure (which was further confirmed by the XRD study also), such that the diffusion is slower.
The UV-vis spectra of both cis- and trans-polyPtFe exhibited strong absorptions based on the MLCT (Fig. 2a). After polymerization with Fe(II) metal ions, the MLCT band for complexation of the Pt(II) ions with the ethynyl moiety of the two organometallic ligands were further red-shifted to 407 nm (Fig. 2a). Upon the formation of the polymer by cis- and trans-PtL with Fe(II) through terpyridyl chelation, apart from the occurrence of a new intense absorption at ca. 574 nm from the MLCT for terpyridine–Fe(II) complexation, the absorption bands due to the Pt(II)–metal-perturbed π → π* (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) and/or MLCT transitions were red-shifted ca. 24 nm for the cis-PtL and 9 nm for the trans-PtL organometallic ligands. The terpyridyl chelating to Fe(II) lowers the π* orbital energy in the CCtpy and reduces the energy gap between the HOMO (d) and LUMO (π*), thus inducing an obvious red-shift of the Pt–metal-perturbed π → π* (CC) and/or MLCT absorption.39 The red-shift was quite a bit lesser in trans-polyPtFe as the MLCT energy gap was already reduced in the trans-PtL ligand compared to its cis-counterpart. Again, the MLCT absorption due to Fe(II) complexation with terpyridine in the polyPtFe polymers is meagerly blue-shifted compared to that of the bisterpyridine ligand containing the Fe(II) metallo-supramolecular polymer polyFe (MLCT band at 583 nm). This small but significant blue-shift indicates the lowest unoccupied molecular orbital (LUMO) potential of the Pt(II)-containing ligand in the heterometallo polymer was increased rather than in the normal 4′,4′′′′-(1,4-phenylene)bis(2,2′:6′,2′′-terpyridine) ligand in polyFe due to the incorporation of Pt(II) in the ligand structure. The cyclic voltammogram of polyFe in Fig. 2b exhibits a reversible redox wave of the Fe(II)/Fe(III) couple (E1/2 = 0.77 V). The redox potential of the reversible Fe(II)/Fe(III) couple in both trans- and cis-polyPtFe showed similar values of E1/2 (0.73 V and 0.74 V for trans- and cis-polyPtFe, respectively) with a smaller negative shift of the E1/2 of the Fe(II)/Fe(III) couple than that of polyFe. This negative shift of the E1/2 of the Fe(II)/Fe(III) couple clearly supports the intramolecular metal–metal interactions between the neighboring Pt and Fe ions.13 Again, the ΔE of the Fe(II)/Fe(III) couple in polyFe (0.13 V) is slightly higher than the ΔE of the Fe(II)/Fe(III) couple in the cis- and trans-polyPtFe (0.09 V and 0.08 V for trans- and cis-polyPtFe, respectively) indicating the good electronic communication in the film of Fe/Pt-containing polymers.
C) and/or MLCT transitions were red-shifted ca. 24 nm for the cis-PtL and 9 nm for the trans-PtL organometallic ligands. The terpyridyl chelating to Fe(II) lowers the π* orbital energy in the CCtpy and reduces the energy gap between the HOMO (d) and LUMO (π*), thus inducing an obvious red-shift of the Pt–metal-perturbed π → π* (CC) and/or MLCT absorption.39 The red-shift was quite a bit lesser in trans-polyPtFe as the MLCT energy gap was already reduced in the trans-PtL ligand compared to its cis-counterpart. Again, the MLCT absorption due to Fe(II) complexation with terpyridine in the polyPtFe polymers is meagerly blue-shifted compared to that of the bisterpyridine ligand containing the Fe(II) metallo-supramolecular polymer polyFe (MLCT band at 583 nm). This small but significant blue-shift indicates the lowest unoccupied molecular orbital (LUMO) potential of the Pt(II)-containing ligand in the heterometallo polymer was increased rather than in the normal 4′,4′′′′-(1,4-phenylene)bis(2,2′:6′,2′′-terpyridine) ligand in polyFe due to the incorporation of Pt(II) in the ligand structure. The cyclic voltammogram of polyFe in Fig. 2b exhibits a reversible redox wave of the Fe(II)/Fe(III) couple (E1/2 = 0.77 V). The redox potential of the reversible Fe(II)/Fe(III) couple in both trans- and cis-polyPtFe showed similar values of E1/2 (0.73 V and 0.74 V for trans- and cis-polyPtFe, respectively) with a smaller negative shift of the E1/2 of the Fe(II)/Fe(III) couple than that of polyFe. This negative shift of the E1/2 of the Fe(II)/Fe(III) couple clearly supports the intramolecular metal–metal interactions between the neighboring Pt and Fe ions.13 Again, the ΔE of the Fe(II)/Fe(III) couple in polyFe (0.13 V) is slightly higher than the ΔE of the Fe(II)/Fe(III) couple in the cis- and trans-polyPtFe (0.09 V and 0.08 V for trans- and cis-polyPtFe, respectively) indicating the good electronic communication in the film of Fe/Pt-containing polymers.
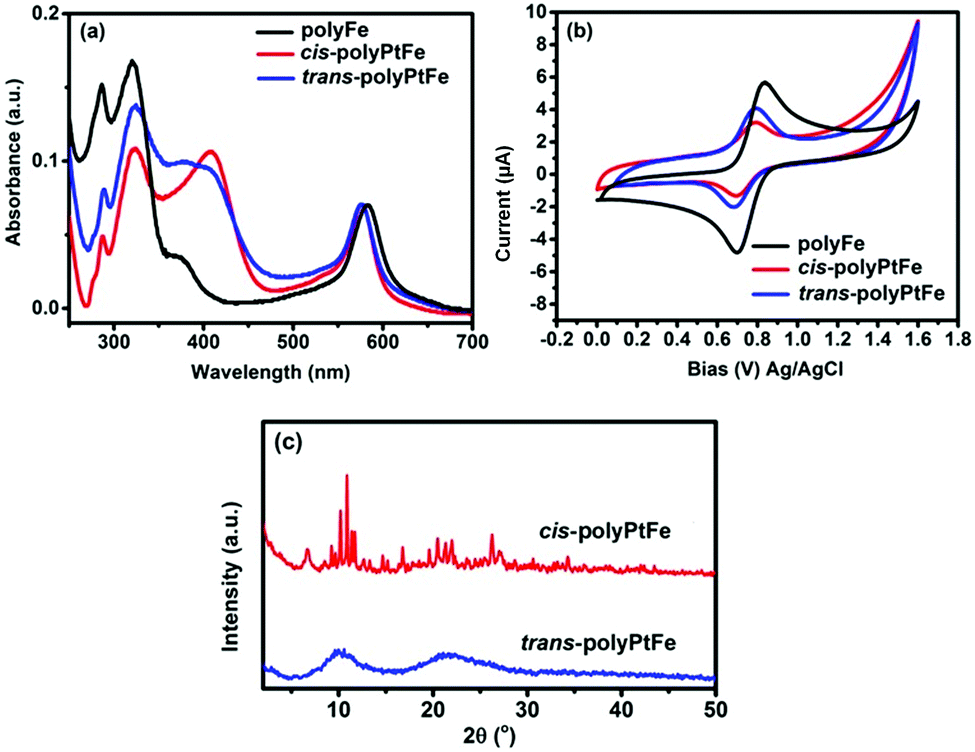 | ||
| Fig. 2 (a) UV-vis spectra of polyFe and the cis- and trans-polyPtFe polymers in methanol solution and (b) cyclic voltammograms for polyFe and the cis- and trans-polyPtFe films coated on a glassy carbon electrode in nitrogen–saturated anhydrous acetonitrile (electrolyte: 0.1 M LiClO4; counter electrode: Pt wire; reference electrode: Ag/Ag+; scan rate: 10 mV s−1). (c) Powder XRD of cis- and trans-polyPtFe. | ||
The comparison of the powder XRD study of cis- and trans-polyPtFe revealed very sharp peaks from the cis-polyPtFe compared to from the trans-polyPtFe (Fig. 2c). From this study, we can assume that cis-polyPtFe possessed a better crystallinity, whereas trans-polyPtFe was amorphous in nature. In the cis-polymer structure, we can assume a polarization occurred as the individual Pt-complex centers have some dipole moment due to its angular shape (Fig. S9, ESI†); whereas the trans-polymer is linear in shape and the individual Pt-complex centers do not possess any dipole moment as it is cancelled out due to their linear shape. As the polarization and dipole moment are higher in individual Pt centers in the cis-polymer, we can assume there is a better crystallinity in the cis-polymer.40,41
The morphological characteristics of the two polymers were studied by a SEM image study (Fig. 3a–d). The SEM images revealed an agglomerated polymer network in cis-polyPtFe (Fig. 3a and b) whereas, in trans-polyPtFe, the polymer network was very well defined to produce high-aspect-ratio fibers (Fig. 3c and d). In the trans-polymer, self-assembly between the polymer chains is reasonably easier due to its linear structure, which gives more structural preference rather than in the cis-polymer to produce a well-defined polymer network.
 | ||
| Fig. 3 SEM images of cis-polyPtFe (a and b) and trans-polyPtFe (c and d) film drop-casted over glass from methanol solution.. | ||
Electrochromic materials (ECMs) have received increasing interest for their use in optoelectronic applications, such as smart windows and electronic papers.3,42,43 Recently, it has been revealed that metallo-supramolecular polymers synthesized by the 1:1 complexation of transition metals, such as Fe(II)/Ru(III) and Fe(II)/Cu(I) ions with some ditopic ligands, are an excellent ECM with both high durability and ample color variation.13,15 These polymers display a specific color based on MLCT absorption and can show electrochromic behavior based on the disappearance/reappearance of MLCT absorption, which is triggered by the electrochemical redox reaction of the metal ions. A solid-state device (2 × 1 cm, Fig. 4a) was fabricated using a cis- or trans-polyPtFe film on ITO and a semi-gel electrolyte layer including a lithium perchlorate salt was sandwiched between the ITO-containing polymer film and another ITO (ESI,† Fig. S10). Both devices showed reversible electrochromic behavior (from purple to blue to yellow) when the applied voltage was changed from 0 to +3 V (Fig. 4b and d). This electrochromic color change could be clearly monitored by UV-vis spectroscopy (Fig. 4c and e). For the device with cis-polyPtFe, the UV-vis spectrum showed two absorptions around 467 and 580 nm based on the MLCT absorptions of the Pt(II)–ethynyl and Fe(II)–terpyridyl complex moieties, respectively (Fig. 4c). Generally the λmax of the polymer in the solid state is bathochromically shifted when compared to the solution spectra, due to the major conformational order, resulting in a different energy levels distribution. So, the MLCT peak for the Pt(II)–ethynyl complex at 407 nm in methanol solution in Fig. 2a is red-shifted to 467 nm in the solid film in EC devices in Fig. 4c and e. Again, the MLCT transitions are typically extremely sensitive to the solvent polarity because they give rise to a strong change in the molecular dipole moment in the excited state. Using a polar solvent, like methanol, can enhance the energy gap between the HOMO (d) and LUMO (π*), thus inducing the obvious blue-shift to 407 nm in methanol solution in Fig. 2a from 467 nm in the solid film in EC devices in Fig. 4c and e.
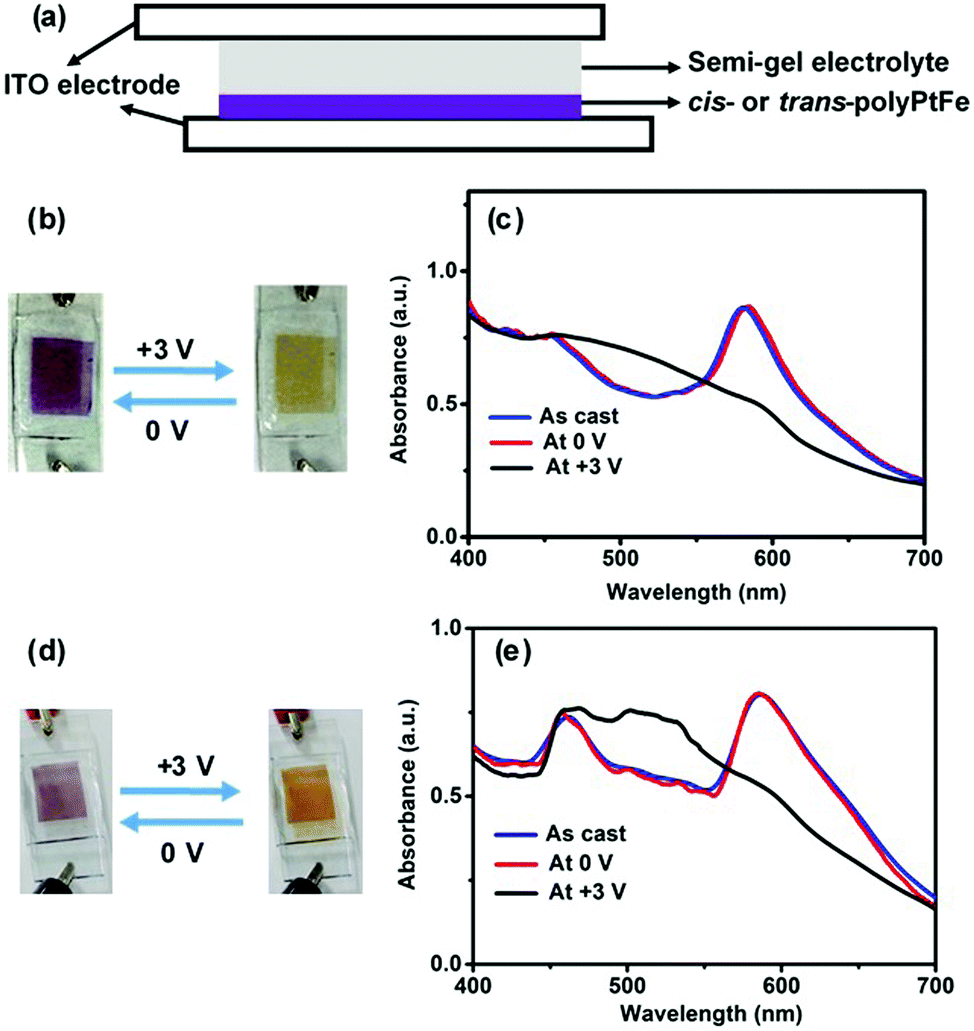 | ||
| Fig. 4 (a) Electrochromic device structure with a heterometallo-supramolecular polymer film and a semi-gel electrolyte layer. (b) Color change of the cis-polyPtFe polymer device during the electrochemical studies and (c) that for the cis-polyPtFe polymer device during the electrochemical studies. (d) UV-Vis spectral change of the EC device of the trans-polyPtFe polymer by applying a potential (from 0 to +3 V) and (e) that for the EC device of the trans-polyPtFe polymer by applying a potential (from 0 to +3 V). | ||
Interestingly, the absorption around 580 nm for the MLCT absorption of the Fe(II)–terpyridyl complex disappeared when the device was exposed at +3 V, because of the electrochemical oxidation of Fe(II) to Fe(III). This phenomenon was also reflected in the solid-state color change of the devices from purple to yellow in Fig. 4b. The yellow color is prominent due to only the MLCT of the Pt(II)–ethynyl complex. The spectral change was reversible; the original spectrum could be re-obtained by applying the opposite voltage (0 V) to the device (Fig. 4b). As Fe(III) centers were reduced to Fe(II) at 0 V, the prominent purple color of the MLCT absorption of the Fe(II)–terpyridyl complex reappeared. We observed the same phenomenon with the device made of trans-polyPtFe (Fig. 4d and e). It also showed same reversible electrochromic behavior by the electrochemical redox of Fe ions, whereby the MLCT absorption at 580 nm of the Fe(II)–terpyridine complex disappeared to give a color change from purple to yellow, upon the oxidation of Fe(II) to Fe(III) by exposing the film at +3 V and then reappeared upon the reduction of Fe(III) to Fe(II) at 0 V.
In order to confirm the device durability in these EC changes, a long-term redox-switching repeatability study was done by applying two voltages (+3 and 0 V) alternately (interval time: 5 s for each step) to both the devices for up to 200 s, and the MLCT absorption at 580 nm was monitored as a function of time. Fig. 5a and b show the switching behavior of the MLCT absorptions from 0 to 200 s for cis- and trans-polyPtFe, respectively. Both the polymer devices revealed good switching stability with the applied potential; however, the device with the cis-polyPtFe polymer film showed some decrement of the absorbance difference of the Fe–terpyridine MLCT between the reduced and oxidized states with time (Fig. 5a). We calculated the response times for the disappearance (bleaching time tb) and reappearance (coloring time tc) of the MLCT absorption at 580 nm, and for the cis-polyPtFe polymer device the values were 4.23 and 2.17 s, respectively (Fig. 5c); whereas, the response times for the disappearance (tb) and reappearance (tc) of the MLCT absorption at 580 nm were 0.93 and 1.08 s, respectively, for the trans-polyPtFe device (Fig. 5d).
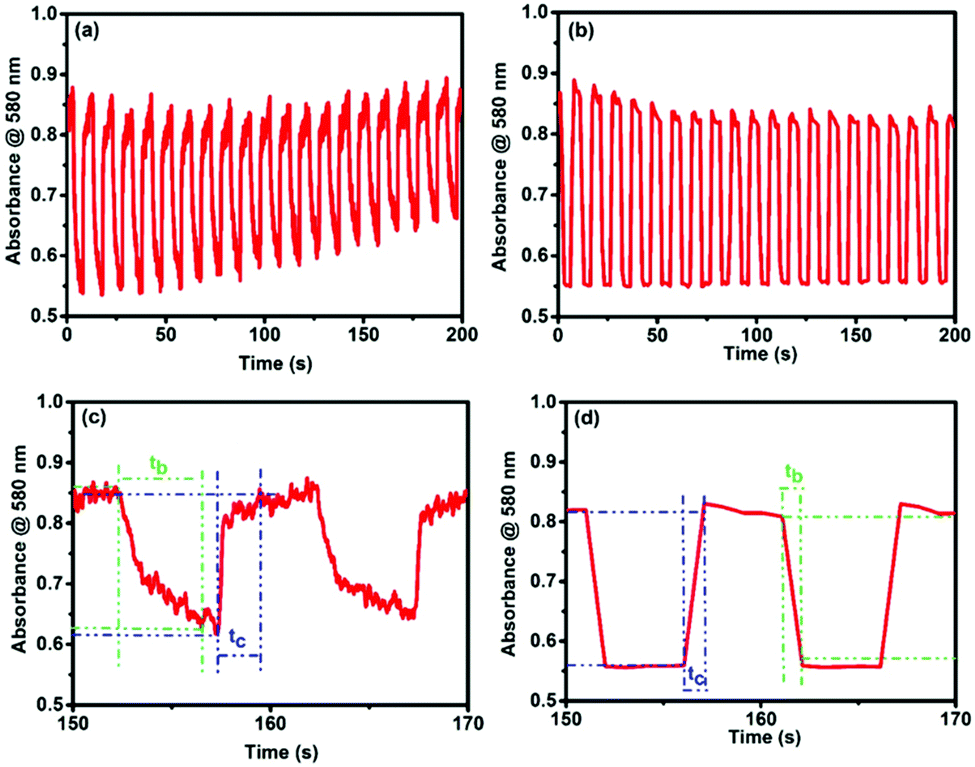 | ||
| Fig. 5 Absorbance change in the absorption at 580 nm during the electrochromic switching of the EC device (applied voltages: 0 and +3 V; interval time for each step: 5 s) for up to 200 s of (a) cis-polyPtFe and (b) trans-polyPtFe. (c and d) Are the absorbance change of cis-polyPtFe and trans-polyPtFe, respectively, during two cycles (150–170 s). The times for coloring and bleaching (tc and tb) were defined as the time taken for 95% of the Δ absorbance to change. | ||
Again, in the fluorescence spectroscopy both polymers revealed a narrow bandwidth peak around 503 nm (19880 cm−1) when excited at 467 nm (21413 cm−1), which is characteristic of the MLCT absorption of the Pt–ethynyl unit (Fig. 6a and b). However, this narrow bandwidth peak at 503 nm cannot be the emission peak for the organo-Pt moiety as the bandwidth is too small and the peak position is far more blue-shifted than usual for organo-Pt lumophores.44 To evaluate the origin of this narrow bandwidth peak, such as whether it comes from an emission from the organo-Pt moiety or terpyridine–ethynyl ligand itself, we also performed an emission study of the pristine ligand L in the solid film state. The ligand L has only one absorption peak for π–π* transition at 323 nm (ESI,† Fig. S11). The emission study of L with 323 nm excitation exhibited only one emission band, with its maxima at 384 nm (ESI,† Fig. S12). However, the emission study of L with excitation at 467 nm (where no absorption is present from L) revealed an identical fluorescence pattern (λmax at 504 nm, 19841 cm−1) as for polyPtFe polymers (ESI,† Fig. S13). This experiment clearly indicated that the narrow peak around 503 nm in both the polymers is not from the emission of organo-Pt moieties but may be due to Raman scattering from the ligand. To further investigate the peak and whether it came from the Raman scattering or not, we studied the fluorescence spectra of L at two other arbitrary excitations, such as 480 and 488 nm, and in both cases we obtained the same pattern of spectra with only a change in the peak positions (519 and 529 nm, respectively) (ESI,† Fig. S13). However, if the peak is due to the emission, then it should not change with changing the excitation wavelengths. The energy gap between the excitation wavenumber and the emission peak was about ∼1570 cm−1, which is a typical value for the first Stokes line of Raman scattering by a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond and so on. In polymers, the energy gap between the excitation wavenumber (21413 cm−1) and the emission peak (19880 cm−1) is also about ∼1533 cm−1. So, we can assume that the narrow emission peak at 503 nm is solely due to Raman scattering of the ligand L. To gain further insight, we studied the Raman scattering of ligand L and both the polymers, which revealed a strong peak with a Raman shift of ∼1530–1580 cm−1 for typical CC stretching (ESI,† Fig. S14). We assume that when the emission was measured, this peak interfered and exhibited a narrow bandwidth peak at 503 nm in both the polymers. Again, an enhancement of this Raman scattering band was observed upon the electrochemical oxidation of Fe(II) ions to Fe(III) at +3 V (Fig. 6a and b). The Raman scattering intensity was reversibly weakened again by the electrochemical reduction of Fe(III) to Fe(II). We also performed a repeatability study of this redox-triggered-scattering switching behavior of these two devices within a potential range of +3 and 0 V (Fig. 6c and d). Initially, both polymers showed very nice redox-triggered-scattering switching; however, in cis-polyPtFe, the intensity difference between the two states decreased with time (Fig. 6c). The Raman scattering intensity enhancement after electrochemical oxidation to Fe(II) to Fe(III) in both polymers is due to electrochromic bleaching, which can increase the optical transmittance of the polymer film.45 When the polymer film was reduced, the optical transmittance of the film again was reduced to exhibit an overall lower intensity of Raman scattering.
C double bond and so on. In polymers, the energy gap between the excitation wavenumber (21413 cm−1) and the emission peak (19880 cm−1) is also about ∼1533 cm−1. So, we can assume that the narrow emission peak at 503 nm is solely due to Raman scattering of the ligand L. To gain further insight, we studied the Raman scattering of ligand L and both the polymers, which revealed a strong peak with a Raman shift of ∼1530–1580 cm−1 for typical CC stretching (ESI,† Fig. S14). We assume that when the emission was measured, this peak interfered and exhibited a narrow bandwidth peak at 503 nm in both the polymers. Again, an enhancement of this Raman scattering band was observed upon the electrochemical oxidation of Fe(II) ions to Fe(III) at +3 V (Fig. 6a and b). The Raman scattering intensity was reversibly weakened again by the electrochemical reduction of Fe(III) to Fe(II). We also performed a repeatability study of this redox-triggered-scattering switching behavior of these two devices within a potential range of +3 and 0 V (Fig. 6c and d). Initially, both polymers showed very nice redox-triggered-scattering switching; however, in cis-polyPtFe, the intensity difference between the two states decreased with time (Fig. 6c). The Raman scattering intensity enhancement after electrochemical oxidation to Fe(II) to Fe(III) in both polymers is due to electrochromic bleaching, which can increase the optical transmittance of the polymer film.45 When the polymer film was reduced, the optical transmittance of the film again was reduced to exhibit an overall lower intensity of Raman scattering.
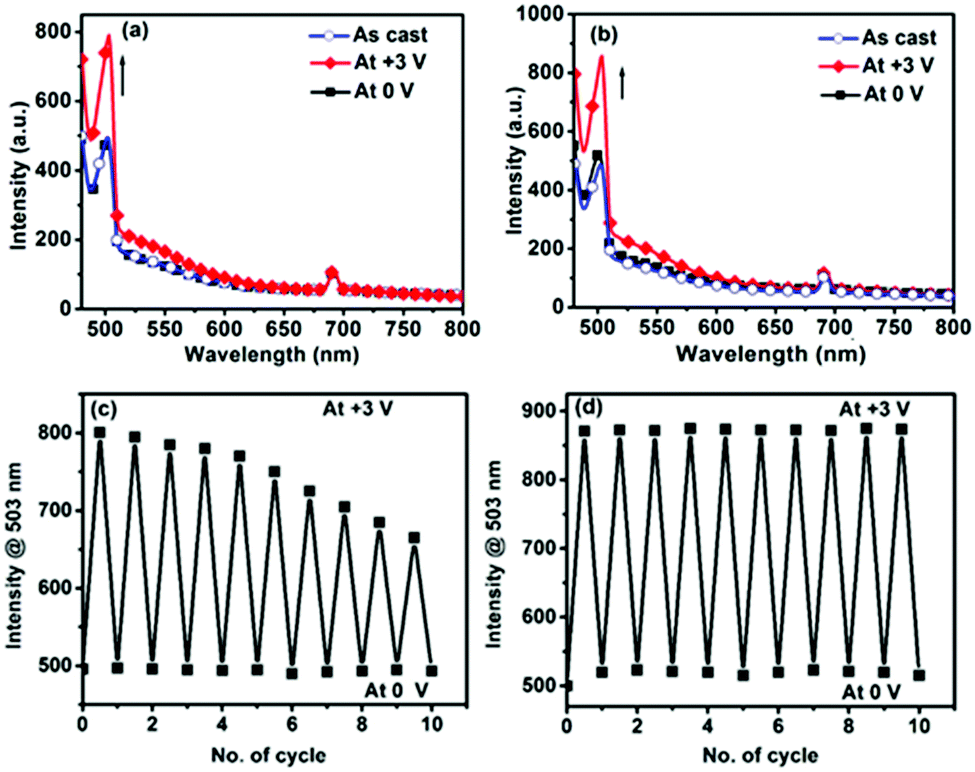 | ||
| Fig. 6 The fluorescence spectra of EC devices of (a) cis-polyPtFe and (b) trans-polyPtFe at potential of as cast, +3.0 and 0 V. Corresponding intensity change of the devices of (c) cis-polyPtFe and (d) trans-polyPtFe at +3.0 and 0 V exited at 467 nm. | ||
To gain an insight into the reason for the better electrochromic- and redox-triggered Raman scattering switching behavior in the trans-polyPtFe compared to in the cis-polyPtFe device, we measured the ionic conductivity of the polymers by impedance measurement. Fig. 7 shows the impedance response for the trans- and cis-polyPtFe. The conductivities of the polymers are 3.0 × 10−5 mS cm−1 and 0.9 × 10−5 mS cm−1, respectively. The more than three times higher conductivity in the trans-polyPtFe compared to that of the cis-polyPtFe could be assigned to the structural preference in the trans-polyPtFe as it contains a straight chain type structure, which facilitates a swift ion transfer process in the polymer. From the SEM images (Fig. 3), it also evident that the counter anion mobility is easier in trans-polyPtFe as it contains a well-defined nanofiber structure, which can produce a channel to transport the counter anions. As the ionic conductivity is higher in the trans-polyPtFe, it showed better electrochromic behavior over its cis-counterpart. Again, from the XRD study in Fig. 2c, it is evident that the cis-polyPtFe possessed higher crystallinity in the film rather than the trans-polyPtFe, which has a higher amorphous content. Owing to its higher crystalline nature in film, the cis-polyPtFe showed poor electrochromic performance, as electrochemical reactions are harder in a rigid structure with higher crystallinity. In the cis-polymer, the interchange and diffusion of the counter anion is difficult than in the trans-polymer due to its more crystalline nature. As a result, the cis-polymer showed a weaker and slow response rather than its trans-counterpart.
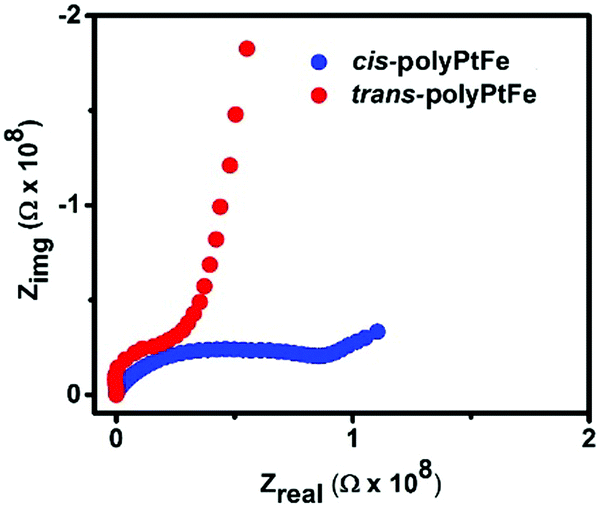 | ||
| Fig. 7 The Nyquist plots for cis- and trans-polyPtFe at 25 °C and 98% RH. | ||
Conclusions
cis- and trans-conformational heterometallo-supramolecular polymers with Pt(II) and Fe(II) ions introduced alternately (cis- and trans-polyPtFe) were synthesized by the 1:1 complexation of Fe(II) ions with organo-Pt(II) ligands. Both polymers exhibited similar molecular weights, but their crystallinity and the ionic conductivity of their solid films were different due to their different spatial arrangements. An electronic interaction between the two hetero-metal ions through the π-conjugated spacer unit of the ligand was observed in the cyclic voltammetry measurements. Both the polymers exhibited reversible electrochromism between purple and yellow based on the electrochemical redox of Fe(II) to Fe(III). The trans-polyPtFe showed better electrochromic stability and response times compared to the cis-polyPtFe owing to its better ionic conductivity and higher amorphous nature of the film. The bimetallic polymers also revealed reversible Raman scattering switching by triggering the redox of the Fe(II) ions in the solid thin film. The trans-polyPtFe also showed improved response in redox-triggered Raman scattering switching compared to the cis-polyPtFe over the long-time range. The studies have tremendous impact for understanding the correlation between the structural characteristic and ionic conductivity and the electrochromic behavior of metallo-supramolecular polymers with different geometries. As we successfully prepared two precise heterometallo-supramolecular polymers with Pt(II) and Fe(II) ions introduced alternately via stepwise complexation, these polymers can also be used as a precursor to fabricate patterning of FePt nanoparticles.
Acknowledgements
The authors thank the CREST Project, Japan Science and Technology Agency (JST) for financial support. Authors also acknowledge Dr Miharu Eguchi for her help regarding photophysical discussion.Notes and references
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS PubMed.
- J.-C. Eloi, L. Chabanne, G. R. Whittell and I. Manners, Mater. Today, 2008, 11, 28 CrossRef CAS.
- M. Higuchi, J. Mater. Chem. C, 2014, 2, 9331 RSC.
- A. Bandyopadhyay, S. Sahu and M. Higuchi, J. Am. Chem. Soc., 2011, 133, 1168 CrossRef CAS PubMed.
- T. Sato and M. Higuchi, Chem. Commun., 2012, 48, 4947 RSC.
- R. I. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14 CrossRef CAS PubMed.
- F. S. Han, M. Higuchi and D. G. Kurth, Adv. Mater., 2007, 19, 3928 CrossRef CAS.
- D. G. Kurth and M. Higuchi, Soft Matter, 2006, 2, 915 RSC.
- C.-W. Hu, T. Sato, J. Zhang, S. Moriyama and M. Higuchi, ACS Appl. Mater. Interfaces, 2014, 6, 9118 CAS.
- S. B. Clendenning, S. Fournier-Bidoz, A. Pietrangelo, G. Yang, S. Han, P. M. Brodersen, C. M. Yip, Z.-H. Lu, G. A. Ozin and I. Manners, J. Mater. Chem., 2004, 14, 1686 RSC.
- C. Chakraborty, R. K. Pandey, M. D. Hossain, Z. Futera, S. Moriyama and M. Higuchi, ACS Appl. Mater. Interfaces, 2015, 7, 19034 CAS.
- J. Lombard, D. A. Jose, C. E. Castillo, R. Pansu, J. Chauvin, A. Deronzier and M.-N. Collomb, J. Mater. Chem. C, 2014, 2, 9824 RSC.
- M. D. Hossain, J. Zhang, R. K. Pandey, T. Sato and M. Higuchi, Eur. J. Inorg. Chem., 2014, 3763 CrossRef CAS.
- T. Sato and M. Higuchi, Chem. Commun., 2013, 49, 5256 RSC.
- C.-W. Hu, T. Sato, J. Zhang, S. Moriyama and M. Higuchi, J. Mater. Chem. C, 2013, 1, 3408 RSC.
- E. C. Constable, A. J. Edwards, P. R. Raithby and J. K. Walker, Angew. Chem., Int. Ed. Engl., 1993, 32, 1465 CrossRef.
- E. C. Constable and A. M. W. C. Thompson, J. Chem. Soc., Dalton Trans., 1995, 1615 RSC.
- C. D. Buchecker, B. Colasson, M. Fujita, A. Hori, N. Geum, S. Sakamoto, K. Yamaguchi and J. P. Sauvage, J. Am. Chem. Soc., 2003, 125, 5717 CrossRef PubMed.
- D. Imbert, M. Cantuel, J. C. G. Bunzli, G. Bernardinelli and C. Piguet, J. Am. Chem. Soc., 2003, 125, 15698 CrossRef CAS PubMed.
- C. Piguet, G. Hopfgartner, B. Bocquet, O. Schaad and A. F. Williams, J. Am. Chem. Soc., 1994, 116, 9092 CrossRef CAS.
- R. J. Abergel and K. N. Raymond, Inorg. Chem., 2006, 45, 3622 CrossRef CAS PubMed.
- A. R. Stefankiewicz, J. Harrowfield, A. Madalan, K. Rissanen, A. N. Sobolev and J. M. Lehn, Dalton Trans., 2011, 40, 12320 RSC.
- H. Fenton, I. S. Tidmarsh and M. D. Ward, Dalton Trans., 2010, 39, 3805 RSC.
- G. R. Newkome, P. S. Wang, C. N. Moorefield, T. J. Cho, P. P. Mohapatra, S. N. Li, S. H. Hwang, O. Lukoyanova, L. Echegoyen, J. A. Palagallo, V. Iancu and S. W. Hla, Science, 2006, 312, 1782 CrossRef CAS PubMed.
- F. Puntoriero, S. Serroni, A. Licciardello, M. Venturi, A. Juris, V. Ricevuto and S. Campagna, J. Chem. Soc., Dalton Trans., 2001, 1035 RSC.
- D. Sun, L. Zhang, H. Lu, S. Feng and D. Sun, Dalton Trans., 2013, 42, 3528 RSC.
- D. Sun, L. Zhang, Z. Yan and D. Sun, Chem. – Asian J., 2012, 7, 1558 CrossRef CAS PubMed.
- D. Sun, D.-F. Wang, X.-G. Han, N. Zhang, R.-B. Huang and L.-S. Zheng, Chem. Commun., 2011, 47, 746 RSC.
- M. Higuchi, Y. Akasaka, T. Ikeda, A. Hayashi and D. G. Kurth, J. Inorg. Organomet. Polym. Mater., 2009, 19, 74 CrossRef CAS.
- M. Higuchi, Polym. J., 2009, 41, 511 CrossRef CAS.
- F. Han, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2008, 130, 2073 CrossRef CAS PubMed.
- A. Bandyopadhyay and M. Higuchi, Eur. Polym. J., 2013, 49, 1688 CrossRef CAS.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong and K.-K. Cheung, Organometallics, 2001, 20, 4476 CrossRef CAS.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447 CrossRef CAS PubMed.
- W.-Y. Wong, J. Inorg. Organomet. Polym. Mater., 2005, 15, 197 CrossRef CAS.
- Y. Liu, S. Jiang, K. Glusac, D. H. Powell, D. F. Anderson and K. S. Schanze, J. Am. Chem. Soc., 2002, 124, 12412 CrossRef CAS PubMed.
- M. Higuchi and D. G. Kurth, Chem. Rec., 2007, 7, 203 CrossRef CAS PubMed.
- A. Wild, A. Teichler, C.-L. Ho, X.-Z. Wang, H. Zhan, F. Schlütter, A. Winter, M. D. Hager, W.-Y. Wong and U. S. Schubert, J. Mater. Chem. C, 2013, 1, 1812 RSC.
- H.-B. Xu, J. Ni, K.-J. Chen, L.-Y. Zhang and Z.-N. Chen, Organometallics, 2008, 27, 5665 CrossRef CAS.
- V. Bharti, H. S. Xu, G. Shanthi and Q. M. Zhang, J. Appl. Phys., 2000, 87, 452 CrossRef CAS.
- A. Dey and G. R. Desiraju, Chem. Commun., 2005, 2486 RSC.
- R. J. Mortimer, Annu. Rev. Mater. Res., 2011, 41, 241 CrossRef CAS.
- S. Shankar, M. Lahav and M. E. van der Boom, J. Am. Chem. Soc., 2015, 137, 4050–4053 CrossRef CAS PubMed.
- E. Glimsdal, M. Carlsson, B. Eliasson, B. Minaev and M. Lindgren, J. Phys. Chem. A, 2007, 111, 244 CrossRef CAS PubMed.
- S.-H. Lee, H. M. Cheong, N.-G. Park, C. E. Tracy, A. Mascarenhas, D. K. Benson and S. K. Deb, Solid State Ionics, 2001, 140, 135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterizations of ligands and cis- and trans-polyPtFe by different 1H and DOSY NMR techniques, electrochromic device fabrication for electrochromic measurements, emission study and Raman spectroscopy. See DOI: 10.1039/c6tc02929a |
| This journal is © The Royal Society of Chemistry 2016 |