
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing intrinsically photostable low band gap polymers: a smart tool combining EPR spectroscopy and DFT calculations†
Hugo Santos
Silva
a,
Isabel Fraga
Domínguez
bde,
Anthony
Perthué
bd,
Paul D.
Topham
f,
Pierre-Olivier
Bussière
cd,
Roger C.
Hiorns
ag,
Christian
Lombard
hij,
Agnès
Rivaton
bd,
Didier
Bégué
*a and
Brigitte
Pépin-Donat
*hij
aUniversité de Pau et des Pays de l'Adour, IPREM (CNRS-UMR 5254), 2 Avenue Président Angot, 64053 Pau, France. E-mail: didier.begue@univ-pau.fr
bUniversité Blaise Pascal, Institut de Chimie de Clermont-Ferrand, Équipe Photochimie, BP 10448, F-63000 Clermont-Ferrand, France
cUniversité Clermont Auvergne, SIGMA Clermont, Institut de Chimie de Clermont-Ferrand, BP 10448, F-63000 Clermont-Ferrand, France
dCNRS, UMR 6296, ICCF, Équipe Photochimie, BP 80026, F-63171 Aubière, France
eChemical Engineering and Applied Chemistry, Aston University, Birmingham, B4 7ET, UK
fAston Materials Centre, Aston University, Birmingham, B4 7ET, UK
gCNRS, IPREM (EPCP, CNRS-UMR 5254), 64053 Pau, France
hUniversité Grenoble Alpes, INAC-SPRAM, F-38000 Grenoble, France
iCNRS, INAC-SPRAM, F-38000 Grenoble, France
jCEA, INAC-SPRAM, F-38000 Grenoble, France. E-mail: brigitte.pepin-donat@cea.fr
First published on 15th September 2016
Abstract
A rapid and efficient method to identify the weak points of the complex chemical structure of low band gap (LBG) polymers, designed for efficient solar cells, when submitted to light exposure is reported. This tool combines Electron Paramagnetic Resonance (EPR) using the ‘spin trapping method’ coupled with density functional theory modelling (DFT). First, the nature of the short life-time radicals formed during the early-stages of photo-degradation processes are determined by a spin-trapping technique. Two kinds of short life-time radical (R˙ and R′O˙) are formed after ‘short-duration’ illumination in an inert atmosphere and in ambient air, respectively. Second, simulation allows the identification of the chemical structures of these radicals revealing the most probable photochemical process, namely homolytical scission between the Si atom of the conjugated skeleton and its pendent side-chains. Finally, DFT calculations confirm the homolytical cleavage observed by EPR, as well as the presence of a group that is highly susceptible to photooxidative attack. Therefore, the synergetic coupling of a spin trapping method with DFT calculations is shown to be a rapid and efficient method for providing unprecedented information on photochemical mechanisms. This approach will allow the design of LBG polymers without the need to trial the material within actual solar cell devices, an often long and costly screening procedure.
1 Introduction
Incorporating stable materials is a prerequisite in creating long-lasting, stable devices. This is particularly pertinent for materials used to make organic solar cells because polymers used in the active layer are generally unstable under light exposure. Only very few photo-induced defects formed within the polymer are required to drastically impair solar cell performance, since such defects deleteriously affect the charge generation process.1Besides photovoltaic conversion, other photophysical or photochemical processes can occur in the excited state. Among these latter processes, rearrangement, chain scission, and crosslinking all result in modification of the chemical structure of the polymer (see Scheme S1 in ESI†).2 Identifying the photodegradation mechanism(s) occurring is not easy for polymers with simple structures and even more troublesome for low band gap (LBG) polymers, which are now extensively used in efficient organic solar cells.3,4 Nevertheless, it is essential to design improved, efficient and stable LBG polymer structures to enable the fabrication of stable organic solar cells. The conjugated skeleton of LBG polymers is based on the alternation of donor and acceptor units on which alkyl side chains can be attached, where the attachments themselves may contain different types of heteroatoms. The alkyl side chains can be linear or branched, and be attached to the donor and/or acceptor units of the polymer. Based on these multiple combinations, countless types of LBG polymers can be synthesized.5,6
One of the main problems in addressing photodegradation is that there are no sufficiently sensitive tools that are able to detect at low concentrations both small structural changes in the polymer (at the molecular scale) and the primary species at the origin of the photochemical process. In fact, a dramatic decrease in organic solar cell efficiency is generally associated with only very slight changes in the ‘usual’ spectrometric parameters (Raman, UV-visible, XPS…) of the polymer.7 Furthermore, such spectroscopies cannot reveal the primary species at the origin of degradation. They only allow detection of the structural/chemical changes due to subsequent sequential steps of the degradation process. Thus, they do not give information on the mechanism behind such processes.8 It is therefore vital to develop a technique that is able to unambiguously detect, and identify, the primary species involved in photochemical processes. Electronic Paramagnetic Resonance (EPR) combines simple implementation of global tracing methods (a few milligrams of the macroscopic sample deposited on the same substrate, under the same conditions as those in real organic solar cells, are directly observed) with very high sensitivity.9 In addition, the more sophisticated ‘EPR spin trapping method’ allows the detection of primary unstable free radicals resulting from direct (intrinsic) photochemical processes. Density Functional Theory (DFT) calculations allow one to identify weak points in a chemical structure in terms of their photochemical reactivity (via the binding energies of different bonds). Herein, we exploit the remarkable synergy of EPR spectroscopy and DFT modelling to obtain information on photochemical mechanisms,10,11 thus allowing the design of push–pull polymers adapted to long-life organic solar cells without, most importantly, the necessity of testing them in real devices.
In the work herein, the more sophisticated ‘Spin Trap Method’ has been used in place of ‘basic’ EPR. The ‘Spin Trap Method’ consists of detecting radicals of short life-time (in this case the radicals eventually formed during the polymer photo-degradation process) by trapping them with a diamagnetic molecule to produce a stable paramagnetic adduct; it was developed in parallel in 1968 by Janzen12,13 and Lagercrantz.14 This method has been used recently to study the formation of the oxygen superoxide anion radical, which is trapped at the very beginning of illumination and assumed to be responsible for the degradation of poly(3-hexylthiophene) (P3HT) in chlorobenzene in the presence of oxygen.15 In this study, the authors did not report radical trapping arising from polymer degradation. On closer inspection of their data, and in agreement with the authors, only OH˙ radicals were trapped at the beginning of the experiment. However, after a few minutes of irradiation, a dramatic change was observed in the EPR spectrum. We ascribe this to the trapping of other radicals, such as those arising from degradation of the polymer. Therefore, the present study focuses on what happens after 15 minutes of illumination by solar simulated light in order to detect eventual radicals arising from the intrinsic polymer photochemical processes with the aim of further probing polymer photo-degradation mechanisms.
Poly[4,4′-bis(2-ethylhexyl)dithieno[3,2-b:2′,3′-d]silole]-2,6-diyl-alt-[2,5-bis(3-tetradecylthiophen-2-yl)thiazole-[5,4-d]thiazole-1,8-diyl] (PDTSTzTz) has been chosen as model LBG polymer, as its chemical structure covers most scenarios of low band gap polymers, see Fig. 1. The skeleton of PDTSTzTz comprises a ‘push’ dithenosilole (DTS) unit and a ‘pull’ thiazolothiazol (TzTz) unit; it has also two different side-chains, – one linear, one branched – with the branched side chains being attached to the conjugated backbone by an heteroatom.
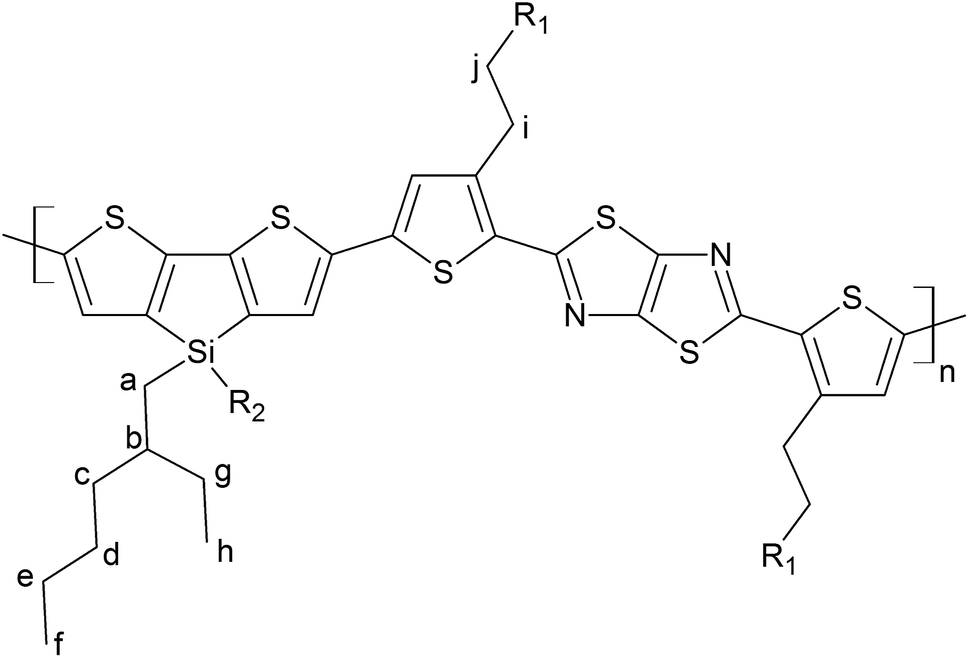 | ||
| Fig. 1 Chemical structure of PDTSTzTz, where R1 is a dodecyl linear chain and R2 is a 2-ethylhexyl chain. The calculations were performed on a monomer of this structure for which R1 is a methyl group. The hydrogen atoms used for the determination of the bond dissociation energies have been labelled a–i. | ||
2 Materials and methods
2.1 Materials
PDTSTzTz (Mn = 45 kg mol−1, Ð = 3.5) was provided by Merck Chemicals and was used without further purification (see Fig. 1 for its chemical structure). Ortho-Dichlorobenzene and para-xylene were obtained from SIGMA-ALDRICH, HPLC grade (99%). 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was obtained from SIGMA-Aldrich (≥97%). Ethanol was obtained from SIGMA-Aldrich (99.9%).2.2 Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ortho-dichlorobenzene (7:1). Films on PET/ITO substrates were drop-cast from a 1% polymer solution (w/w) in ortho-dichlorobenzene. For the purpose of the EPR experiments, polymer films on PET/ITO were introduced in EPR Quartz tubes (Ø = 3 mm) and wetted by a solution of DMPO in ethanol (50 g L−1).
:ortho-dichlorobenzene (7:1). Films on PET/ITO substrates were drop-cast from a 1% polymer solution (w/w) in ortho-dichlorobenzene. For the purpose of the EPR experiments, polymer films on PET/ITO were introduced in EPR Quartz tubes (Ø = 3 mm) and wetted by a solution of DMPO in ethanol (50 g L−1).
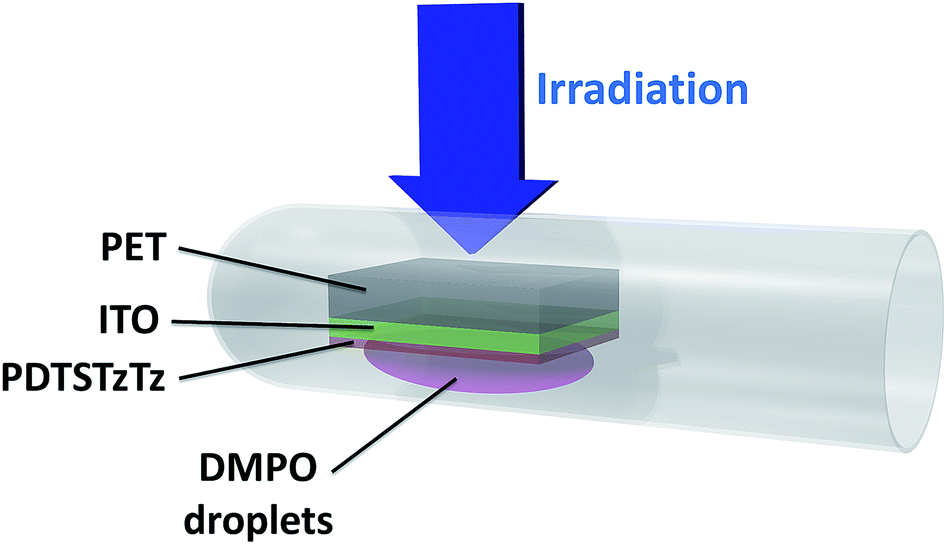 | ||
| Fig. 2 Experimental set-up related to sample irradiation for EPR spin-trapping analysis. | ||
In the configuration shown in Fig. 2, the sample was exposed to 15 min AM 1.5 light soaking and the substrate was firstly irradiated before the deposited polymer was wetted by the DMPO solution in order to minimize photo-degradation of the DMPO. EPR spectra were recorded under Ar atmosphere after air exchange inside a glovebox in order to avoid broadening of the line widths due to the presence of oxygen.
3 Results and discussion
3.1 Results
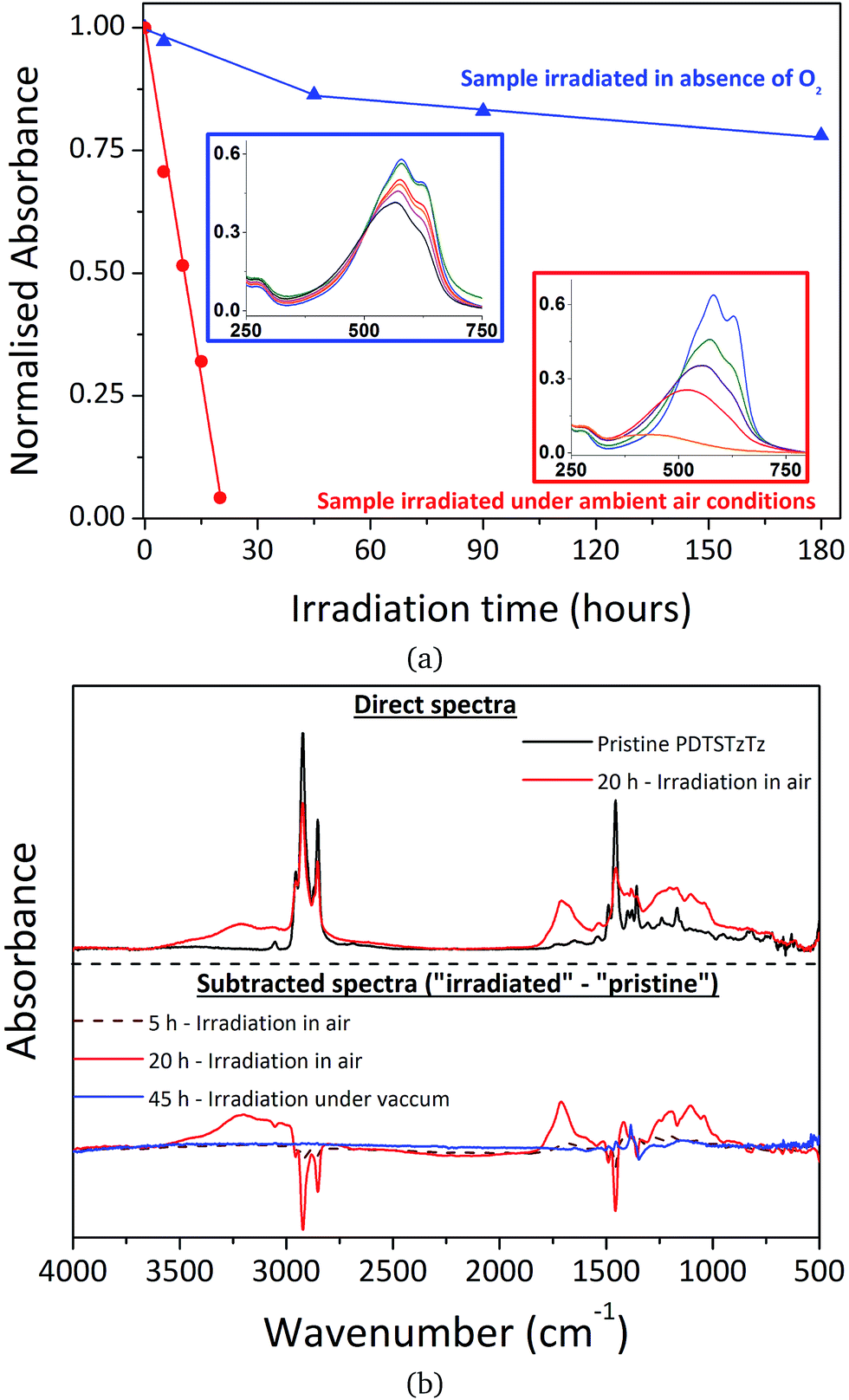 | ||
| Fig. 3 Changes in (a) UV-visible and (b) IR spectra of PDTSTzTz thin films during irradiation in the SUNTEST device under ambient conditions (red) and in the absence of oxygen (blue). | ||
It is also clear that when irradiation is performed in the absence of oxygen, the effect of photodegradation on the UV-visible absorbance is significantly reduced compared to irradiation in ambient air, as previously evidenced for MDMO-PPV, P3HT and PCDTBT.26 In parallel, IR spectroscopy revealed a decrease of the polymer bands and the formation of oxidation products upon irradiation in the presence of air, whereas no new bands were observed when samples were irradiated in glass tubes sealed under vacuum, confirming that the tubes were fully degassed (see Fig. 3b).
It is noteworthy that photooxidative processes occurring with irradiation in ambient air, resulting in easily identifiable oxygen fixation on the chemical structure, does not exclude the involvement of pure photochemical processes that are unaffected by atmospheric conditions. Furthermore, if an efficient encapsulation barrier may suppress, or at least drastically reduce, oxidation processes, pure photochemical processes have to be regarded as the intrinsic fragility of the polymer. Therefore, characterizing exclusively light-induced degradation processes is crucial if one wishes to develop intrinsically more stable materials, which will then allow significant improvements in device lifetime to be made. The spectroscopic analysis of PDTSTzTz deposits submitted to long duration irradiations in the total absence of oxygen reveals the presence of intrinsic photochemical processes that lead to some modification of the PDTSTzTz backbone structure. In this respect, previous work devoted to other polymers, such as P3HT and PCDTBT, suggested that irradiation in the absence of oxygen provoked homolytical scission between the side chain and conjugated backbone. Successive saturation of the P3HT conjugated skeleton by the alkyl fragments would induce a reduction of the conjugation length of the π-conjugated system, thus explaining the decrease in absorbance.27 Macroradical recombination was proposed in the case of PCDTBT, with parallel formation of low molecular weight alkyl products.9 Moreover, to our knowledge, scientific literature reports no direct photochemical process which would uniquely involve the building blocks of the conjugated skeleton leading to the disruption of π-conjugation. The side-chain cleavage hypotheses presented in the literature for the degradation of P3HT and PCDTBT cannot, however, be validated for PDTSTzTz uniquely on the basis of UV-vis and IR analyses presented herein.
In order to clarify the mechanisms occurring in the early stages of polymer degradation and investigate the possible side-chain cleavage processes, intermediate short-lived free radicals formed during the irradiation process have been studied herein by performing spin trapping experiments.
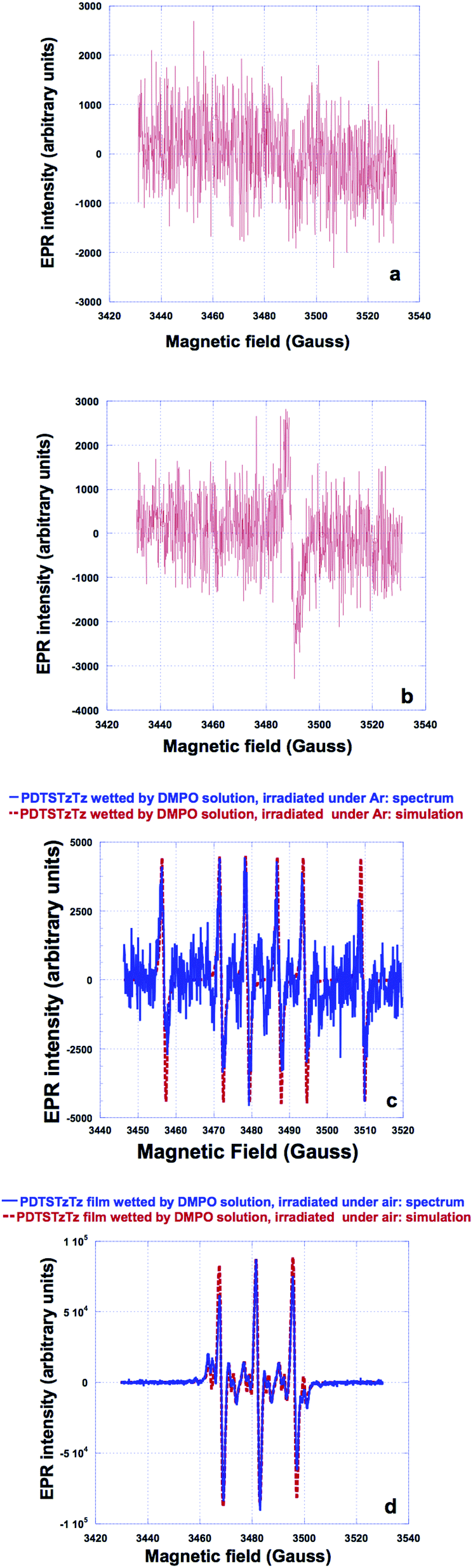 | ||
| Fig. 4 EPR spectra of PDTSTzTz not wetted by DMPO solution, and irradiated with the solar simulator (a) under argon and; (b) in the presence of air (g = 2.0023). EPR spectra of PDTSTzTz wetted by DMPO solution and irradiated (c) under argon; (d) in presence of air. Parts (c) and (d) show the experimental spectra (solid blue line) together with their simulations (dotted red line). | ||
Accordingly, the spectrum of PDTSTzTz wetted by the DMPO solution and illuminated under argon was then recorded. Fig. 4c presents the recorded spectrum together with its simulation created with a single radical species using three hyperfine couplings: An = 15.2 G; AH1 = 22 G and AH2 = 0.5 G. These hyperfine couplings are very similar to those observed for an alkyl radical of R˙ type (for example CH3CH2˙ in water gives rise to hyperfine couplings of AN = 16.3 G, AH1 = 23.5 G).28‡ The fact that hyperfine couplings observed in our case are slightly lower is attributed to the difference in polarity between water and ethanol. Finally, the spectrum of PDTSTzTz, wetted by the DMPO solution, and irradiated under oxygen was recorded; Fig. 4d shows the experimental spectrum together with its simulation/fit. The experimental spectrum is satisfactorily fitted with two radicals: the first radical (of low intensity) gives rise to hyperfine couplings of AN = 13.4 G, AH1 = 7.5 G and AH2 = 1.8 G (six pairs of lines), while the second radical (of higher intensity) gives rise to only one hyperfine coupling of AN = 14 G. The hyperfine couplings of the first radical species are very similar to those observed for an alkoxy radical of the type R′O˙ (for example CH3CH2O˙ in ethanol exhibits AN = 13.5 G, AH1 = 7.4 G and AH2 = 1.7 G).28 The hyperfine AN coupling of the second radical fits well with that of the oxidative degradation product of DMPO.28 Both species present a g-factor in the order of 2.006. The EPR data herein are interpreted as follows: first, the stable spectrum (g = 2.0023) observed for the polymer alone (without DMPO) when illuminated under oxygen is ascribed to the formation of the cation radical of the polymer associated with the superoxide anion radical, as previously reported for other polymers.29 Second, when irradiation of the polymer with DMPO is performed, the observed R˙ signal (Fig. 4b) is attributed to a ‘small’ alkyl molecule and not to a macroradical. In fact, if this signal would correspond to a polymeric structure, the lines would be broader due to the rotational diffusion correlation time, especially the one located at high external magnetic field.30,31 Thirdly, when irradiation of the polymer with DMPO is performed under oxygen, the observed R′O˙ (Fig. 4b) results from the oxidation of alkyl radicals.
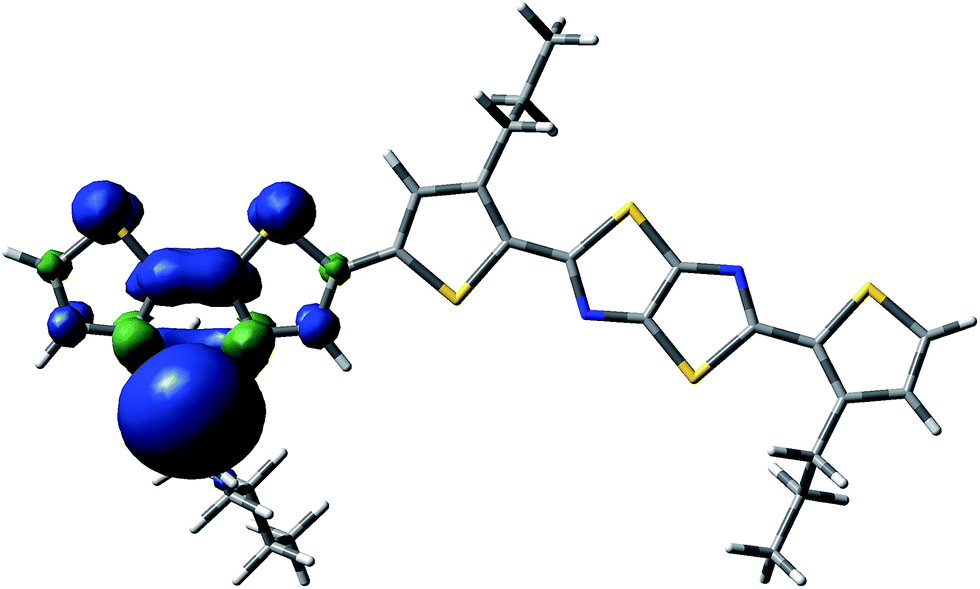 | ||
| Fig. 5 Spin delocalization over the PDTSTzTz repeat unit which has lost the lateral chain of the Si atom moiety. | ||
In addition to the homolytic scissions, irradiation of the sample in the presence of oxygen would also lead to photooxidative processes, commonly starting by abstraction of labile hydrogen atoms of the polymer as reported for PCDTBT9 and PVK.32,33 Using DFT calculations, the most labile hydrogen atoms of PDTSTzTz have been identified. The bond dissociation energies (BDEs) were calculated for the labelled protons in Fig. 1 and the values are given in Table 1.
| Label | BDE (kcal mol−1) |
|---|---|
| a | 100.18 |
| b | 97.43 |
| c | 101.45 |
| d | 103.92 |
| e | 104.63 |
| f | 108.76 |
| g | 101.26 |
| h | 106.98 |
| i | 89.93 |
| j | 104.42 |
From the data shown in Table 1, one can see that the hydrogen atom in the alpha position to the thiophene ring is the most labile one, and can therefore be more easily abstracted from the chain, even more so than the hydrogen on the tertiary carbon of the 2-ethyl-hexyl chain, in line with Santos Silva et al.23 Furthermore, the spin delocalization over the polymer repeat unit which has lost a H radical in the alpha-position of the thiophene units takes place over a large amount of neighbouring double bonds as presented in Fig. 6. Therefore, although the energy of dissociation of the C–H bond is larger than that of Si–C bond, this radical is more delocalized, thus, more stable.
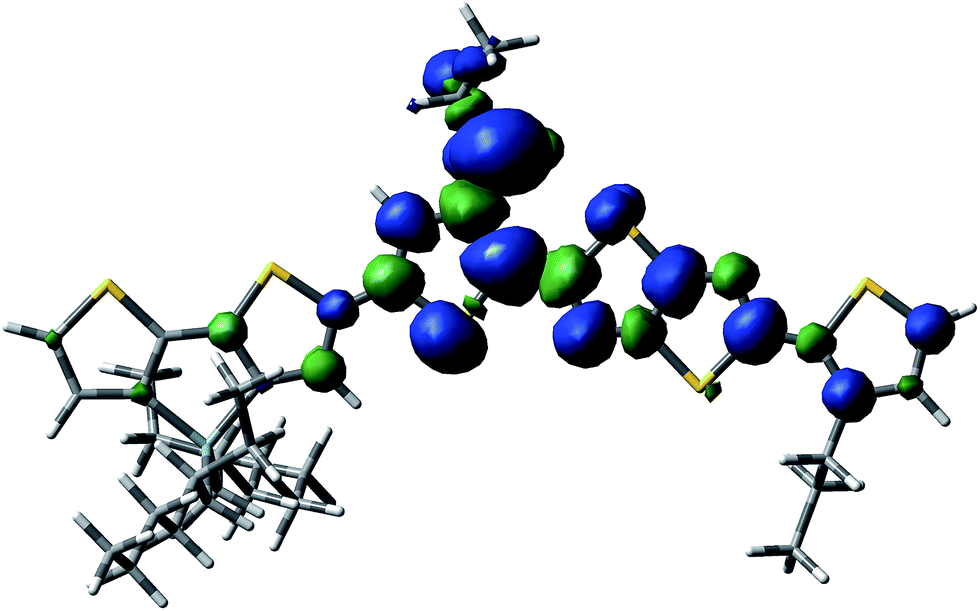 | ||
| Fig. 6 Spin delocalization over the PDTSTzTz repeat unit which has lost a H radical in the alpha-position of the thiophene units. | ||
3.2 Discussion
The UV-vis absorbance decay of PDTSTzTz films indicate a higher degree of degradation for samples irradiated in air. Thus, whilst pure photolytic processes can happen both in the presence and absence of oxygen, photooxidative processes are those predominantly leading to the degradation of the polymer. As for photolytic processes (i.e. processes occurring upon irradiation under inert atmosphere), DFT calculations indicate that the Si–R bond can be homolitically broken. Such photochemical homolysis would lead to the formation of two types of radicals: a macro-Si˙ radical and a ˙C–R radical, with the latter being unstable as delocalization is not possible (see Fig. 7). This result leads one to assume that the ˙C–R trapped by DMPO under illumination without oxygen, observed by EPR, is likely to originate from the homolysis of the alkyl chain linked to the Si atom, the so-called according to the labelling in Fig. 1; with the calculated g-value for one DMPO adduct being in full agreement with that observed by EPR (g = 2.006). No spectrum corresponding to the macro-Si˙ radical was observed from the present spin trapping experiments. This is not surprising since, to our knowledge, no adduct of DMPO with Si˙ has ever been reported in the literature. The so-formed alkyl radicals could saturate the π-conjugated skeleton of the polymer (as suggested for P3HT)27 and the macroradicals formed on the Si atom are also susceptible to combination, thus provoking the crosslinking of the irradiated material (as suggested for PCDTBT9 and PVK)32,33. Both processes could be responsible for the change in chemical structure of the polymer leading to a decrease in the UV-vis absorption of the polymer.
according to the labelling in Fig. 1; with the calculated g-value for one DMPO adduct being in full agreement with that observed by EPR (g = 2.006). No spectrum corresponding to the macro-Si˙ radical was observed from the present spin trapping experiments. This is not surprising since, to our knowledge, no adduct of DMPO with Si˙ has ever been reported in the literature. The so-formed alkyl radicals could saturate the π-conjugated skeleton of the polymer (as suggested for P3HT)27 and the macroradicals formed on the Si atom are also susceptible to combination, thus provoking the crosslinking of the irradiated material (as suggested for PCDTBT9 and PVK)32,33. Both processes could be responsible for the change in chemical structure of the polymer leading to a decrease in the UV-vis absorption of the polymer.
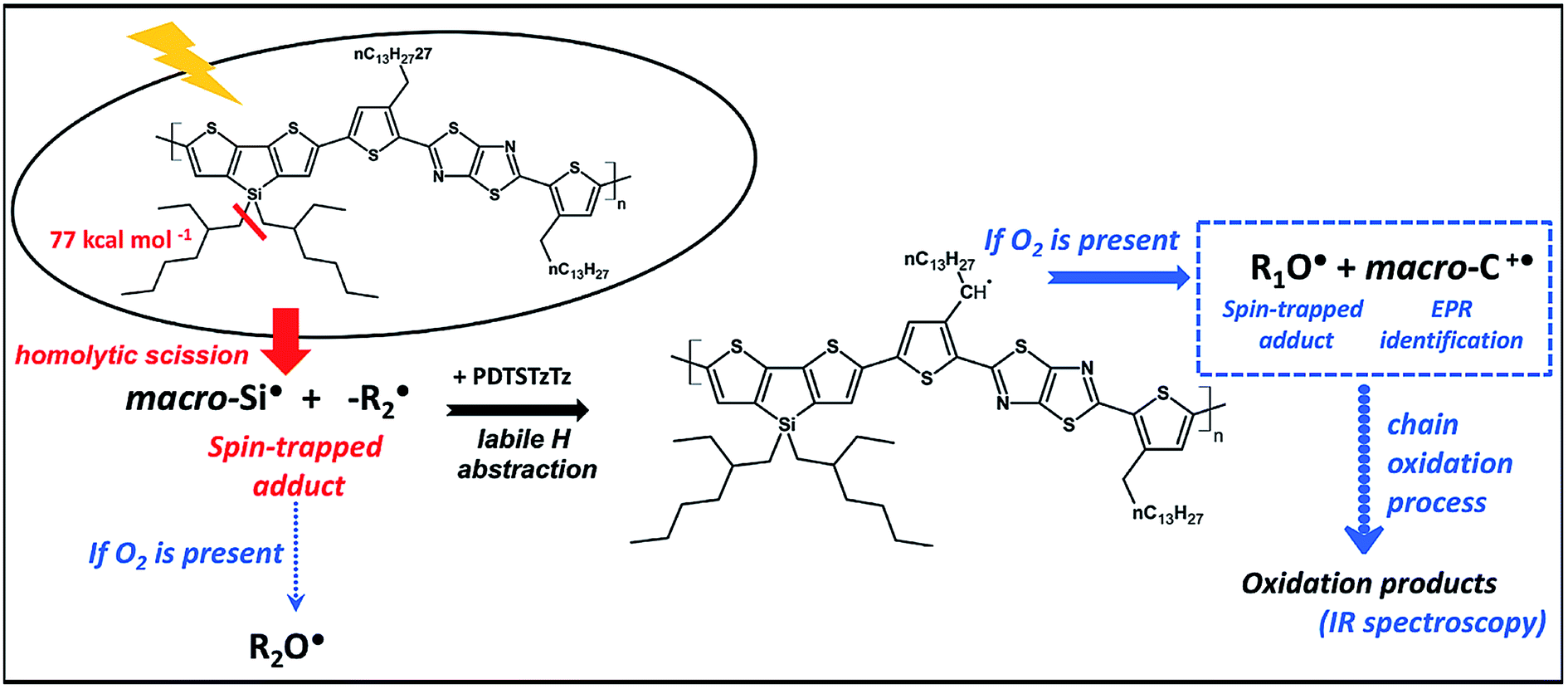 | ||
| Fig. 7 Schematic representation of the early stages of the light-induced degradation of the PDTSTzTz polymer. | ||
Under photooxidative conditions, radicals (such as those created by solely homolytic processes) are able to propagate the chain oxidation process by preferentially abstracting the most labile hydrogen atoms, i.e. those in the alpha position to the thiophene ring according to the data obtained by DFT calculations (Table 1). Irradiation of PDTSTzTz films in air led to EPR identification of the positive polaron (macro-C+˙ in Fig. 7) and of alkoxy radicals, in good accordance with these calculations. As depicted in Fig. 7, such alkoxy radicals can stem from the oxidation of the R1 radical (R1O˙) or the aforementioned 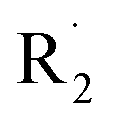 , generated by homolytical scission. Although the EPR spectrum in Fig. 4d has signals ascribed to both R1O˙ and R2O˙ (their adducts with DMPO giving similar EPR spectra), evolution and kinetics obtained by UV-vis and IR analysis clearly show that oxidative processes are predominant over intrinsic photochemical processes. Therefore, one can assume that RO˙ are primarily related to R1 moieties.
, generated by homolytical scission. Although the EPR spectrum in Fig. 4d has signals ascribed to both R1O˙ and R2O˙ (their adducts with DMPO giving similar EPR spectra), evolution and kinetics obtained by UV-vis and IR analysis clearly show that oxidative processes are predominant over intrinsic photochemical processes. Therefore, one can assume that RO˙ are primarily related to R1 moieties.
In this work, spin-trapping EPR spectroscopy is shown as a very sensitive tool, even after very short irradiation times (only 15 min AM 1.5 exposure), to identify the radical species (stable and unstable) resulting from photochemical processes. In combination with DFT calculations, the most likely photochemical processes occurring at these early stages of degradation are summarised in Fig. 7. According to the results herein presented, the absorption of UV-vis light by PDTSTzTz leads to (at least in part) the homolysis of the bond between the Si atom and its side-chain. Interestingly, previous investigations have indicated that replacing the C atom by Si, as a bridging atom to the alkyl-chains, actually enhances the stability of the attached side-chain against photooxidation processes.11 However, according to the study herein presented, the presence of a Si atom bearing a side-chain does represent the Achilles' heel of the chemical structure of PDTSTzTz; on the one hand, direct degradation can occur at this point under inert conditions via light-induced homolysis of this bond. On the other hand, if oxygen is present, the radicals produced are able to propagate oxidation, thus further degrading the polymer. More generally, irradiation in presence of oxygen is identified as the most destructive for the polymer. DFT calculations demonstrate that the primary steps of this oxidation process occur at the side chain attached to the thiophene ring, characterised by a low C–H binding energy (see Fig. 7). In summary, the work presented identifies the combination between EPR and DFT calculations as a smart tool to identify the species at the origin of degradation. Furthermore, our findings are highly relevant for the organic electronic and photovoltaic communities as for designing polymers with more stable chemical structures.
4 Conclusions
Most polymers are photo-instable, and the identification of their Achilles' heel is particularly difficult when focusing on complex chemical structures such as LBG polymers. To this end we have demonstrated in the present study that the judicious combination of EPR spectroscopy and DFT calculations is a smart and efficient approach to unambiguously determine radical processes occurring in the early stages of photodegradation of low band gap polymers. Implementing this tool on PDTSTzTz, as a model polymer, we have revealed that it is unstable under light irradiation, as most polymers are, to identify the chemical structure of transient radicals formed during the photodegradation process and consequently to point out the Achilles' heel of the polymer structure. As follow up to this work, DFT modelling could allow one to tune the effect of structural modifications (for instance, carbon and germanium-analogue structures) to design a more photostable material. More generally, the smart combined approach demonstrated herein is a way to intuitively expand the tool box of building blocks available to design photostable low band gap polymers, which is a prerequisite for materials which will be submitted to UV-vis light irradiation over the lifetime of the organic solar cell.Acknowledgements
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2011) under Grant Agreement ESTABLIS No. 290022.References
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- Y.-W. Su, S.-C. Lan and K.-H. Wei, Mater. Today, 2012, 15, 554–562 CrossRef CAS.
- P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2010, 23, 456–469 CrossRef.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- H. Van Mullekom, J. Vekemans, E. Havinga and E. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- S. Karuthedath, T. Sauermann, H.-J. Egelhaaf, R. Wannemacher, C. J. Brabec and L. Lüer, J. Mater. Chem. A, 2015, 3, 3399–3408 CAS.
- L. Müller-Meskamp, J. Fahlteich and F. Krebs, Stab. Degrad. Org. Polym. Sol. Cells, 2012, 269–329 Search PubMed.
- A. Tournebize, A. Rivaton, J.-L. Gardette, C. Lombard, B. Pépin-Donat, S. Beaupré and M. Leclerc, Adv. Energy Mater., 2014, 4, 1301530 CrossRef.
- A. Tournebize, J.-L. Gardette, C. Taviot-Guého, D. Bégué, M. A. Arnaud, C. Dagron-Lartigau, H. Medlej, R. C. Hiorns, S. Beaupré and M. Leclerc, et al. , Polym. Degrad. Stab., 2015, 112, 175–184 CrossRef CAS.
- I. Fraga Domínguez, P. D. Topham, P.-O. Bussière, D. Bégué and A. Rivaton, J. Phys. Chem. C, 2015, 119, 2166–2176 Search PubMed.
- E. G. Janzen and B. J. Blackburn, J. Am. Chem. Soc., 1968, 90, 5909–5910 CrossRef CAS.
- E. G. Janzen and B. J. Blackburn, J. Am. Chem. Soc., 1969, 91, 4481–4490 CrossRef CAS.
- C. Lagercrantz and S. Forshult, Trapping of free radicals formed by γ-irradiation of organic compounds, Univ. of goteborg technical report, 1968 Search PubMed.
- L. Chen, J. Mizukado, Y. Suzuki, S. Kutsuna, Y. Aoyama, Y. Yoshida and H. Suda, Chem. Phys. Lett., 2014, 605, 98–102 CrossRef.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- P. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- K. Kim and K. Jordan, J. Phys. Chem., 1994, 98, 10089–10094 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- H. S. Silva, A. Tournebize, D. Bégué, H. Peisert, T. Chassé, J.-L. Gardette, S. Therias, A. Rivaton and R. Hiorns, RSC Adv., 2014, 4, 54919–54923 RSC.
- W. Kutzelnigg, U. Fleischer and M. Schindler, Deuterium and shift calculation, Springer, 1990, pp. 165–262 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. Rivaton, A. Tournebize, J. Gaume, P.-O. Bussière, J.-L. Gardette and S. Therias, Polym. Int., 2014, 63, 1335–1345 CrossRef CAS.
- M. Manceau, A. Rivaton, J.-L. Gardette, S. Guillerez and N. Lemaître, Sol. Energy Mater. Sol. Cells, 2011, 95, 1315–1325 CrossRef CAS.
- G. R. Buettner, Free Radicals Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- Y. Aoyama, T. Yamanari, T. N. Murakami, T. Nagamori, K. Marumoto, H. Tachikawa, J. Mizukado, H. Suda and Y. Yoshida, Polym. J., 2015, 47, 26–30 CrossRef CAS.
- A. Jeunet, B. Nickel and A. Rassat, Nouv. J. Chim., 1986, 10, 123–132 CAS.
- Y. Xia, Y. Li, A. O. Burts, M. F. Ottaviani, D. A. Tirrell, J. A. Johnson, N. J. Turro and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 19953–19959 CrossRef CAS PubMed.
- A. Tournebize, P.-O. Bussière, P. Wong-Wah-Chung, S. Thérias, A. Rivaton, J.-L. Gardette, S. Beaupré and M. Leclerc, Adv. Energy Mater., 2013, 3, 478–487 CrossRef CAS.
- A. Rivaton, B. Mailhot, G. Derderian, P. Bussiere and J.-L. Gardette, Macromolecules, 2003, 36, 5815–5824 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta05455b |
| ‡ And references therein. |
| This journal is © The Royal Society of Chemistry 2016 |