
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convenient synthesis of EDOT-based dyes by CH-activation and their application as dyes in dye-sensitized solar cells†
S. M.
Abdalhadi‡
a,
A.
Connell‡
b,
X.
Zhang‡
a,
A. A.
Wiles‡
a,
M. L.
Davies
c,
P. J.
Holliman
*b and
G.
Cooke
*a
aGlasgow Centre for Physical Organic Chemistry (GCPOC), WestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: graeme.cooke@glasgow.ac.uk
bSchool of Chemistry, Bangor University, Bangor, Gwynedd LL57 2UW, UK
cSPECIFIC, Swansea University, Swansea, SA1 8EN, UK
First published on 23rd September 2016
Abstract
Precursors to three new 3,4-ethylenedioxythiophene (EDOT) incorportaing dyes have been synthesised via a one-pot C–H activation route using N,N-dimethylaniline as a donor group. We have extended this methodology to provide a convenient one-pot route to dye EDOT-Ph. The electrochemical and optical properties of the new dyes have been correlated with IV and EQE data for 1 cm2 dye-sensitized solar cell (DSSC) devices prepared using these dyes. The device data show that dye performance is strongly affected by the amount of chenodeoxycholic acid (CDCA) co-sorbent used. The best performance is for EDOT-Ph (η = 4.0%) at 10 mM CDCA compared to (η = 6.0% and η = 5.8%) for N719 and D205 control cells.
Introduction
In recent years, dye-sensitized solar cell (DSSC) devices fabricated using metal-free organic dyes have begun to overtake metal-based dyes in terms of device performance. For instance, there have been several reports of organic dyes with power conversion efficiencies (η) at 1 Sun > 12% (ref. 1–4) with the best non-verified DSSC device efficiency to date (η = 14.7%) being for a device co-sensitized with the alkoxysilyl anchor dye ADEKA-1 and a cyanoacrylate-functionalised dye LEG4.5 The interest in metal-based dyes stems from the initial report by O'Regan and Grätzel using a Ru-bipyridyl complex (N3) to sensitize TiO2 nanoparticles giving η > 7%.6 The closely related Ru-bipy dye (N719)7 increased efficiency whilst the alkyl-modified Z907 improved dye stability to water ingress.8 To date, the best Ru-bipy DSSC dye (C106) gives η = 11.4%.9 However, whilst Ru-bipy DSSC dyes are effective, their spectral response is generally restricted to <650 nm (with the exception of the related terpyridyl “Black dye”10), molar extinction coefficients (ε) are much lower than organic dyes and potential structural modifications are limited to the periphery of a relatively small number of metal-chelating ligands suitable to make stable complexes.By comparison, organic DSSC dyes can be tuned to absorb across the visible spectrum from yellow11 to red12,13 to blue,14,15 they possess much higher ε (e.g. 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for N719 (ref. 16) vs. 330000 M−1 cm−1 for SQ2 squaraine15) and they offer an almost infinite range of potential structural modifications although generally a donor–spacer–acceptor arrangement of groups in the dye is most widely utilised.17 As such, organic DSSC dyes are best grouped into dye families based on the chromophore; e.g. triphenylamines,18 indolines,19,20 coumarins21 as examples of dye which harvest light <650 nm and squaraines,14,15 phthalocyanines22,23 or cyanine24 dyes which absorb λ > 650 nm. To extend light harvesting across the visible spectrum, devices can be co-sensitized with two25 or more dyes.11,26 Another approach is to develop panchromatic dyes such as panchromatic squaraines.27 However, these dyes tend to contain many different groups which complicates synthesis and purification28 and ultimately increases dye cost which is an issue for device scaling and commercialisation.
000 M−1 cm−1 for N719 (ref. 16) vs. 330000 M−1 cm−1 for SQ2 squaraine15) and they offer an almost infinite range of potential structural modifications although generally a donor–spacer–acceptor arrangement of groups in the dye is most widely utilised.17 As such, organic DSSC dyes are best grouped into dye families based on the chromophore; e.g. triphenylamines,18 indolines,19,20 coumarins21 as examples of dye which harvest light <650 nm and squaraines,14,15 phthalocyanines22,23 or cyanine24 dyes which absorb λ > 650 nm. To extend light harvesting across the visible spectrum, devices can be co-sensitized with two25 or more dyes.11,26 Another approach is to develop panchromatic dyes such as panchromatic squaraines.27 However, these dyes tend to contain many different groups which complicates synthesis and purification28 and ultimately increases dye cost which is an issue for device scaling and commercialisation.
The majority of organic dyes for DSSCs are synthesised using standard Pd-catalysed aryl–aryl coupling reactions such as Suzuki29 and Stille30 coupling methodologies. However, the application of direct C–H activation is an attractive strategy to form new C–C bonds as it offers a largely unlimited range of commercially available substrates, and removes the necessity of preparing organoboron or toxic organotin reagents as is the case for Suzuki and Stille coupling reactions, respectively.31
This paper describes the synthesis of EDOT dyes using one-pot Pd-catalysed C–H activation protocols to afford unsymmetrical precursors to three new 3,4-ethylenedioxythiophene (EDOT)-bridged dyes EDOT-Ph, EDOT-Fu and EDOT-Th (Scheme 1). Moreover, we have extended this methodology to prepare the best performing dye (EDOT-Ph) in a one-pot reaction of EDOT and commercially available donor and acceptor residues (Scheme 2).32 Although EDOT has been shown to be a good substrate for Pd-mediated C–H activation reactions33 and has been used to prepare unsymmetrical push–pull dyes by the sequential addition of donor and acceptor units,34 our one-pot approach35 described here significantly reduces the synthesis time and associated consumables costs relating to the isolation of intermediates, allowing the rapid and convenient synthesis of EDOT-Ph in reasonable yield.
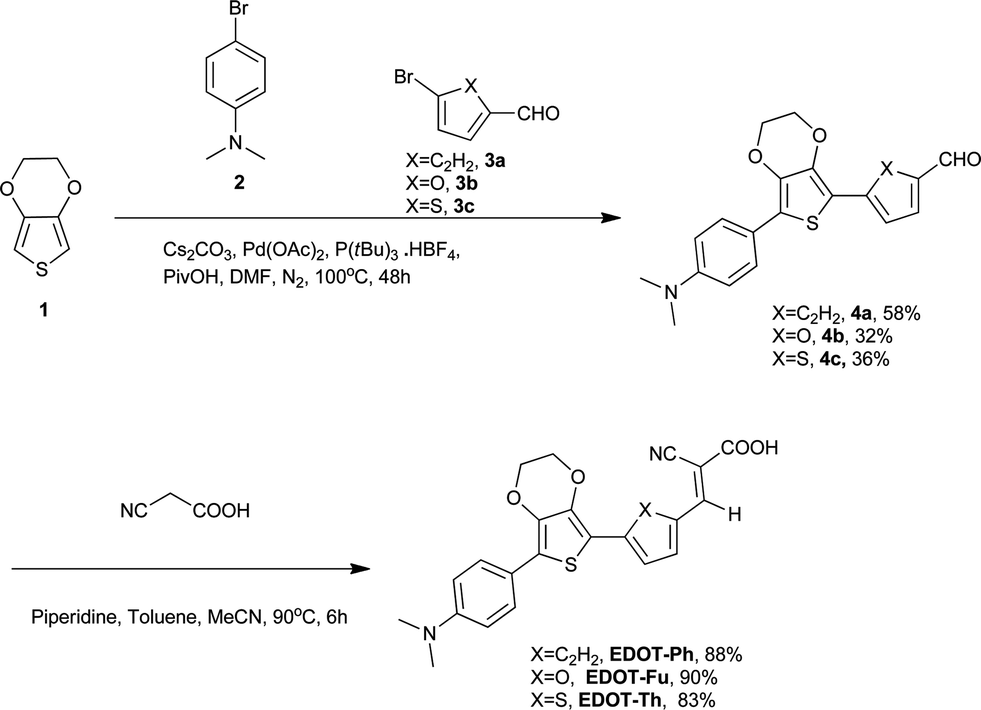 | ||
| Scheme 1 One-pot synthesis of 4a–cvia C–H activation of EDOT and their subsequent conversion to dyes EDOT-Ph, EDOT-Fu, EDOT-Th. | ||
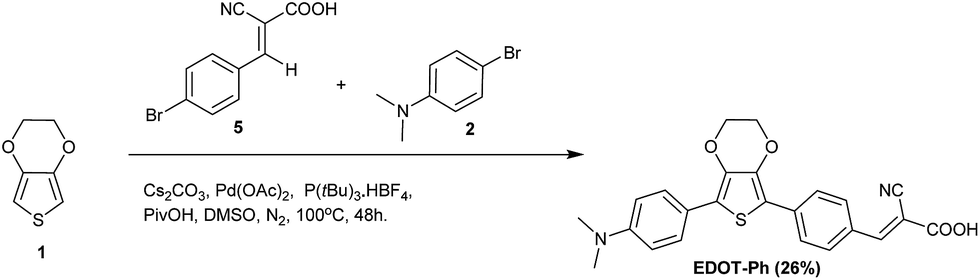 | ||
| Scheme 2 One-pot synthesis of EDOT-Ph. | ||
EDOT has been used widely in DSSC dyes as a conjugated bridge unit between the donor and acceptor residues due to its ability to facilitate co-planarity and improve light harvesting.36 Previous reports of EDOT-based DSSC dyes have typically relied on 2 or 3-step syntheses using a N,N-dimethylaniline donor bonded to EDOT with either a thiophene-acetylene linker (η = 4.5%)28 or with a triphenylamine donor bonded to EDOT with a thiophene linker (η = 6.0%)36 or a fluorinated phenyl linker (η = 8.2%).37 Furthermore, the 8.2% efficiency reported37 was achieved by modification of the acceptor moiety of the sensitizer. The new dyes and protocols described here give comparable device performance to related sensitizers,36,37 but have been synthesized using a convenient one-pot C–H activation methodology using straightforward protocols and inexpensive purification. In addition, these N,N-dimethylaniline-containing EDOT dyes give higher device efficiencies than the aforementioned sensitizers when compared to N719 in a side-by-side comparative study. Furthermore, the devices used in this study are relatively large i.e. 1 cm2cf. 0.25 cm2.28
Results and discussion
Synthesis
One-pot C–H activation was used to prepare a series of dye precursors for DSSC devices using N,N-dimethylaniline as a donor, EDOT as the π-bridge and three different aromatic/heteroaromatic aldehydes (Scheme 1). A range of conditions were investigated to maximize the yields of these reactions, and a summary is reported in the ESI.† The best conditions gave compounds 4a–c in 32–58% yield which offers convenient and rapid access to the dye precursors in a one-pot procedure. The aldehyde moiety was then reacted with cyanoacetic acid to provide three dyes EDOT-Ph, EDOT-Fu and EDOT-Th in high yields (Scheme 1). Furthermore, we were able to further exploit this methodology to prepare EDOT-Ph in a one-pot protocol in a respectable yield (Scheme 2). It is noteworthy that it was not necessary to protect the carboxylic acid unit of compound 5 which thus avoided an additional de-protection step, which is common for many DSSC dyes of this type. Whilst protection of carboxylic acids is synthetically simple to achieve, the addition and removal requires additional synthetic steps and complicates routes to high-purity dyes which perform best in devices.Optical and electrochemical properties
All three dyes were then characterised by UV-vis spectroscopy (Fig. 1) and square-wave voltammetry (Fig. S1†) and the data are summarised in Table 1. The three dyes show strong absorbance across the visible range with maxima of absorption between 444–483 nm as well as good molar extinction coefficients ranging from 13630–20790 L mol−1 cm−1. The UV-vis spectra show a red shift in the onset of absorbance on going from EDOT-Ph to EDOT-Fu and EDOT-Th suggesting a narrowing of the optical band gap (Eopt) from 2.38 eV (EDOT-Ph) to 2.20 eV (EDOT-Th).
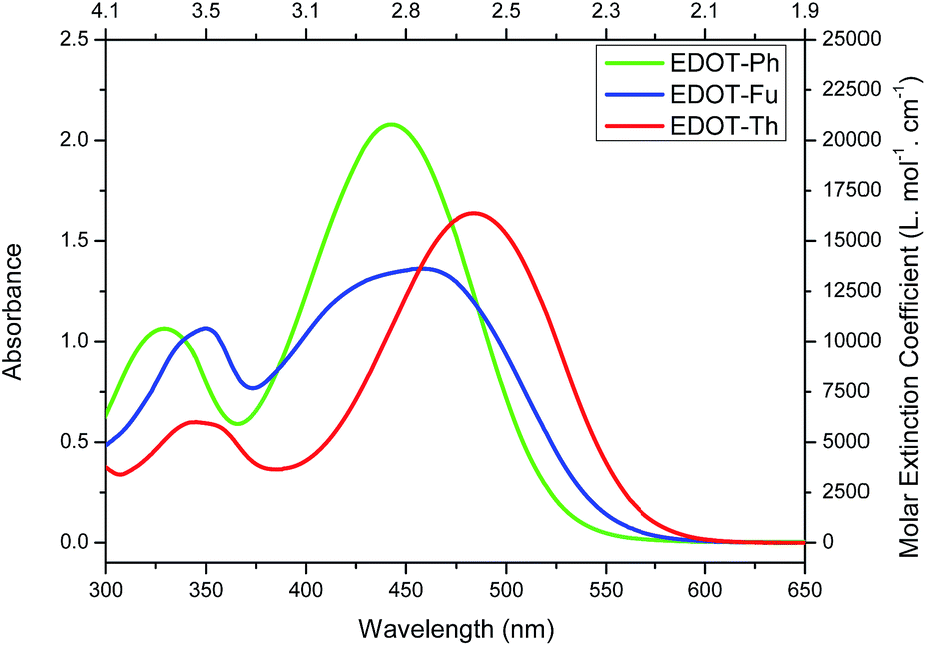 | ||
| Fig. 1 UV-vis absorbance spectra of dyes EDOT-Ph, EDOT-Fu and EDOT-Th (THF 1 × 10−4 M). | ||
| Dye | EDOT-Ph | EDOT-Fu | EDOT-Th |
|---|---|---|---|
| a Extinction coefficient is given in L mol−1 cm−1 (from A = εcl). ✗ = not calculated. Eopt is obtained from E = 1240/λmax. | |||
| Electrochemical properties | |||
| E (ox) (V) | 0.48 | 0.51 | 0.46 |
| E (red) (V) | −1.90 | ✗ | ✗ |
| IP (eV) | −5.28 | −5.31 | −5.26 |
| EA (eV) | −2.90 | ✗ | ✗ |
| E fund (eV) | 2.38 | ✗ | ✗ |
|
|||
| Optical properties | |||
| λ max (nm) | 444 (ε = 20790) |
457 (ε = 13620) |
483 (ε = 16380) |
| λ onset (nm) | 522 | 548 | 564 |
| E opt (eV) | 2.38 | 2.26 | 2.20 |
We have utilised square-wave voltammetry to determine the ionization potential (IP), electron affinity (EA) and the fundamental gap (Efund) where possible. The IP of the three dyes were relatively similar with a difference of ca. 50 mV. The reduction waves for EDOT-Ph were better resolved than the other dyes allowing the estimation of its EA (−2.90 eV). This gives an Efund of 2.38 eV for this dye which is consistent with the optically determined gap. The relatively unchanged IP and the optically measured narrowing of the energy gap suggest that the change in substituents chiefly affects the LUMO/EA of the three dyes.
Theoretical calculations
DFT calculations indicate that all dyes have essentially identical HOMO and LUMO energies (ca. −4.9 eV, −2.4 eV, respectively). The isosurface maps indicate reasonable overlap for the HOMO/LUMO, with the former being mainly located over the donor N,N-dimethylaniline and EDOT units, whereas the LUMO is largely localised over the cyanoacrylate and its adjacent aromatic linker unit (Fig. 2). The dyes are reasonably planar with EDOT-Ph providing the lowest calculated dihedral angle between the N,N-dimethylaniline and EDOT units (16.7° versus ∼17.1° for the other dyes), whilst EDOT-Th has the smallest dihedral angle between the EDOT and the aromatic linker to the cyanoacrylate residue (0.5° versus 11.1° for EDOT-Ph and 4.8° for EDOT-Fu). Interestingly, the dipole moments show some variation with EDOT-Th having the largest dipole moment of 11.6 Debyes and EDOT-Ph and EDOT-Fu possessing slightly lower values of 11.3 and 11.2 Debyes, respectively.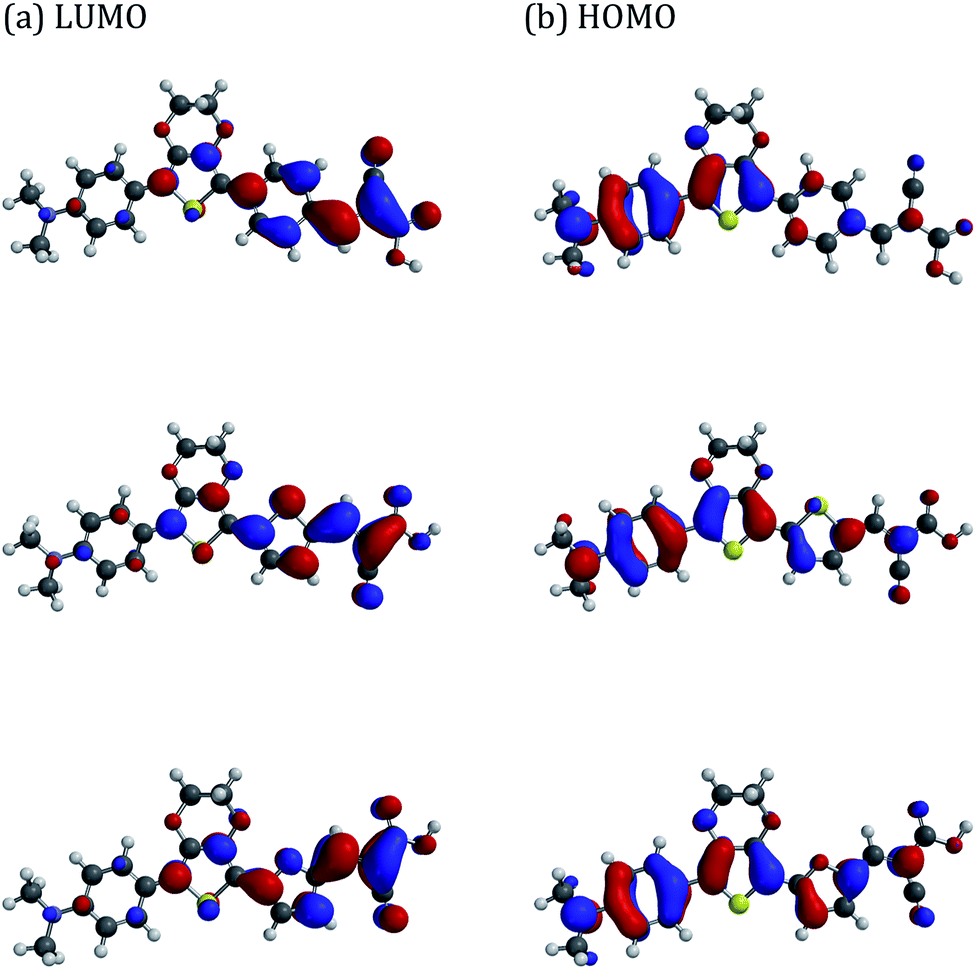 | ||
| Fig. 2 HOMO/LUMO maps of dyes predicted by DFT calculations. Top: EDOT-Ph, middle: EDOT-Fu, bottom: EDOT-Th. | ||
Device testing
Table 2 and Fig. 3 show I–V data for the devices prepared using the EDOT dyes with and without 10 mM chenodeoxycholic acid (CDCA) and/or 0.05 mM LiI. The data show that working DSSC devices are produced from all variables with comparable performance from EDOT-Ph and EDOT-Fu without any CDCA or LiI (η = 1.0% and 1.2%, respectively). However, adding CDCA has a profound influence on device efficiencies for all the dyes improving η by 2–5 times to 3.9%, 2.4% and 2.7% for EDOT-Ph, EDOT-Fu and EDOT-Th, respectively.| Dye | LiI | CDCA | FF | V oc (V) | J sc (mA cm−2) | η (%) |
|---|---|---|---|---|---|---|
| EDOT-Ph | N | N | 0.62 | 0.59 | 2.70 | 1.0 |
| N | Y | 0.64 | 0.75 | 8.20 | 3.9 | |
| Y | Y | 0.62 | 0.62 | 9.80 | 4.0 | |
| EDOT-Fu | N | N | 0.63 | 0.61 | 3.10 | 1.2 |
| N | Y | 0.63 | 0.70 | 5.40 | 2.4 | |
| Y | Y | 0.62 | 0.61 | 7.70 | 2.9 | |
| EDOT-Th | N | N | 0.34 | 0.54 | 2.50 | 0.5 |
| N | Y | 0.62 | 0.70 | 6.50 | 2.7 | |
| Y | Y | 0.67 | 0.70 | 6.00 | 2.8 | |
| N719 | 0.67 | 0.73 | 12.30 | 6.0 | ||
| D205 | 0.68 | 0.74 | 11.40 | 5.8 |
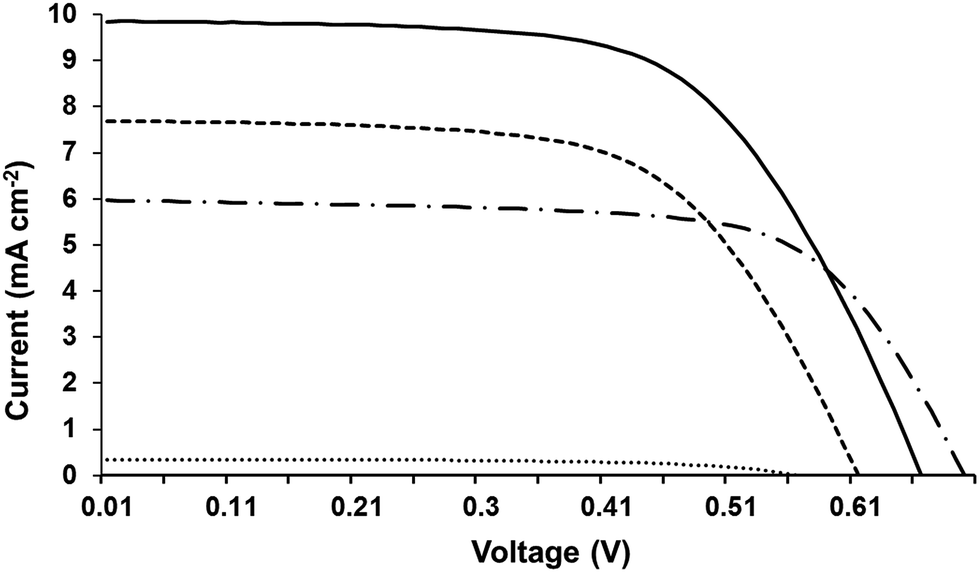 | ||
| Fig. 3 I–V curves for EDOT-Ph (solid line), EDOT-Fu (dashed line), EDOT-Th (dot-dash line) and dark current for EDOT-Ph (dotted line). | ||
We have observed the positive effect of CDCA on device performance previously but not to this extent.11–13,25 For all the dyes, the improvements reflected increased Voc and Jsc. For instance, adding 10 mM CDCA to EDOT-Ph increased Voc from 0.59 to 0.75 V and Jsc from 2.7 to 8.2 mA cm−2. These changes support previous reports that CDCA improves the balance of electron injection over recombination by shifting the position of the conduction band.38,39 Also, because these dyes are small, unbranched and highly conjugated they may aggregate on the TiO2 surface which is detrimental to device performance. However, CDCA suppresses dye aggregation38–40 which also improves device efficiencies.
For the best dye (EDOT-Ph), the further addition of 0.05 mM LiI improves η very slightly (4.0% vs. 3.9%). Underlying this are larger changes; e.g. Jsc increases from 8.2 to 9.8 mA cm−2 whilst Voc drops from 0.75 to 0.62 V. This reflects Li+ ions adsorbing on the TiO2 surface and shifting the conduction band as reported previously.41,42 This improves electron injection but at the expense of voltage. Adding LiI has little effect on EDOT-Th devices but it does improve the performance of EDOT-Fu from 2.4 to 2.9% for similar reasons to EDOT-Ph but the Voc drops less. In addition, the dark currents for the three EDOT dyes are all very similar (Fig. S4). This suggests that the dyes exhibit similar rates of recombination at the electrolyte–electrode interface and hence infers that a different factor is influencing device performance for these dyes.
Despite the 3 dyes all exhibiting the similar structural motifs and N,N-dimethylaniline donor groups, EDOT-Ph gives the best performance. The addition of a hetero-atom (O or S) into the linker unit does not increase device efficiency with the highest Jsc (Table 2) and EQE (Fig. 4) recorded for the EDOT-Ph dye containing a phenyl linker group. Interestingly, the EQE data show spectral response from 350 to 600 nm for all 3 dyes indicating that the heteroatoms in EDOT-Fu and EDOT-Th do not directly influence the λ range of light harvesting. However, UV-visible data for the dyes adsorbed onto TiO2 films (Fig. S2d†) show that light absorption is linked to Jsc with the highest Jsc and absorbance for EDOT-Ph (9.80 mA cm−2 and Abs = 2.86) and the lowest Jsc and absorbance for EDOT-Th (6.00 mA cm−2 and Abs = 1.58). Therefore, light harvesting appears to be strongly correlated with dye loading.
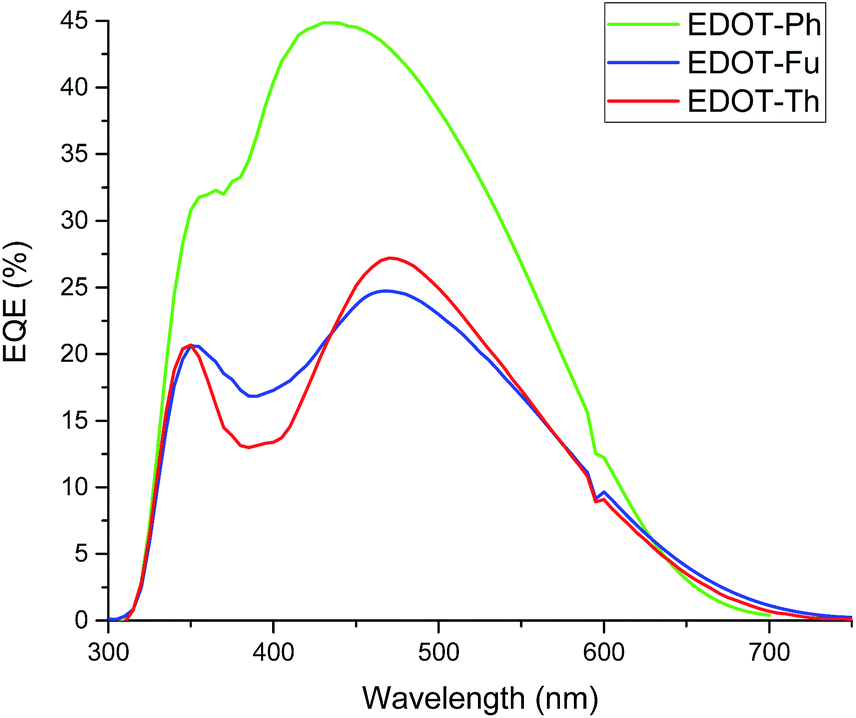 | ||
| Fig. 4 EQE data for EDOT-Ph (green line), EDOT-Fu (red line) and EDOT-Th (blue line) each with 10 mM CDCA and 0.05 mM LiI (1 cm2 devices). | ||
Electronic spectra of adsorbed dyes
UV-vis spectra (Fig. S2†) have been studied for the three EDOT dyes adsorbed from 0.5 mM THF solutions by 18 h passive dyeing onto transparent, mesoporous TiO2 films produced by doctor blading 1 layer of DSL18NR-T colloid onto glass (film thickness ca. 7 μm). The absorbance data have been measured in the presence of varying concentrations of CDCA to study the influence of this additive on dye aggregation. The data show λmax at ca. 440 nm for EDOT-Ph and EDOT-Fu but a red-shift for EDOT-Th to ca. 550 nm reflecting the addition of the sulphur heteroatom in the thiophene moiety of this dye. In addition, each of the 3 dyes exhibit similar trends of narrowing of the main absorption peak as the CDCA concentration increases. Taking the EDOT-Fu data as an example (Fig. S2†), this shows the broadest absorption in the absence of any CDCA; e.g. Abs > 0.9 stretches from 360 to 550 nm. However, even the addition of 1 mM CDCA results in a drastic narrowing of the absorption peak (Abs > 0.9 is 400 to 505 nm) whilst at ≥ 15 mM CDCA Abs > 0.9 narrows to 420 to 470 nm. This effect is ascribed to dye aggregation which is at its greatest when there are only EDOT molecules present because the intermolecular dye interactions are also at their strongest which causes a significant broadening of the main absorption peak. However, when 1 mM CDCA is added to the dye solution, there are 2 CDCA molecules for every EDOT dye and this significantly reduces the inter-dye interactions. This leads to fewer aggregated dye molecules when they adsorb on the TiO2 surface. This is important because dye aggregation has previously been shown to reduce device efficiency (e.g. for half-squarylium12,13 and squaraine dyes).38,43 So, at first sight, the broader light absorption without CDCA might seem beneficial but the poorer electron injection and dye regeneration combined with increased inter-dye recombination between aggregated dye molecules are believed to outweigh the enhanced light harvesting. When comparing the different dyes, CDCA has the greatest influence on EDOT-Fu which suggests greater aggregation for this dye. This may be due to the oxygen of the furan moiety causing greater polarisation within this dye and add to the π–π stacking interactions which are likely between these small, unbranched but highly conjugated dye molecules.Experimental
General synthetic, analytical and computational procedures
All synthetic transformations were performed under a nitrogen atmosphere. Solvents were purified using a PureSolv solvent purifier system. Melting points are uncorrected. 1H NMR and 13C NMR spectroscopy were recorded on a Bruker AV III (500 MHz) spectrometer, operating at 500 MHz and 125 MHz, respectively. Chemical shifts are reported in ppm and are relative to TMS. UV-vis spectra were recorded using a Perkin Elmer Lambda 25 spectrophotometer. Square wave voltammetry was recorded at room temperature under nitrogen on a CH-Instruments 440A potentiostat using a three-electrode cell with a Pt working electrode, a Pt wire counter electrode and an Ag wire pseudo-reference electrode. Bu4NPF6 (0.1 M) was used as the supporting electrolyte in DMF. All voltages are referenced to Fc/Fc+ redox couple and are adjusted to 0 V. DFT calculations were performed using Spartan '14 (64 bit).44 The structures were first optimized semi-empirically (AM1) and then re-optimized using DFT (B3LYP/6-31G*). The resulting structures were local minima, as none of the vibrational frequencies generated imaginary frequencies.Synthesis
:DCM; 1:2).
Compound 4a was prepared using aldehyde 3a and was isolated as a red solid (341.6 mg, 58%). Mp 155–156 °C; 1H NMR (CDCl3, 500 MHz, ppm) δH 9.95 (1H, s, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) O), 7.88 (2H, d, J 8.5, C-Ar), 7.84 (2H, d, J 8.5, C-Ar), 7.65 (2H, d, J 9.0, C-Ar), 6.74 (2H, d, J 9.0, C-Ar), 4.39–4.41 (2H, m, C2), 4.34–4.36 (2H, m, C2), 3.00 (6H, s, 2 × C3); 13C NMR (CDCl3, 125 MHz, ppm) δC 191.5 (
O), 7.88 (2H, d, J 8.5, C-Ar), 7.84 (2H, d, J 8.5, C-Ar), 7.65 (2H, d, J 9.0, C-Ar), 6.74 (2H, d, J 9.0, C-Ar), 4.39–4.41 (2H, m, C2), 4.34–4.36 (2H, m, C2), 3.00 (6H, s, 2 × C3); 13C NMR (CDCl3, 125 MHz, ppm) δC 191.5 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HO), 149.7 (–NH2), 141.0 (–OCH2), 139.6 (–OCH2), 137.1 (–CHO), 133.4 (), 130.2 (H-Ar), 127.4 (H-Ar), 125.5 (H-Ar), 120.7 (), 119.6 (), 112.3 (H-Ar), 111.7 (), 64.9 (H2), 64.4 (H2), 40.5 (2 × H3); HRMS m/z (ESI+) [M]+ 365.1065 (requires 365.1080 for C21H19NO3S).
HO), 149.7 (–NH2), 141.0 (–OCH2), 139.6 (–OCH2), 137.1 (–CHO), 133.4 (), 130.2 (H-Ar), 127.4 (H-Ar), 125.5 (H-Ar), 120.7 (), 119.6 (), 112.3 (H-Ar), 111.7 (), 64.9 (H2), 64.4 (H2), 40.5 (2 × H3); HRMS m/z (ESI+) [M]+ 365.1065 (requires 365.1080 for C21H19NO3S).
Compound 4b was prepared using aldehyde 3b and was isolated as a red solid (181 mg, 32%). Mp 146–147 °C; 1H NMR (CDCl3, 500 MHz, ppm) δH 9.55 (1H, s, CO), 7.63 (2H, d, J 9.0, 2 × C-Ar), 7.31 (1H, d, J 3.8, C-Fu), 6.74 (3H, m, 2 × C-Ar and C-Fu), 4.42 (2H, dt, J 3.9, 2.0, C2), 4.36 (2H, dt, J 3.9, 2.0, C2), 3.01 (6H, s, 2 × H3); 13C NMR (CDCl3, 125 MHz, ppm) δC 176.4 (HO), 154.4 (), 150.5 (), 150.2 (), 141.9 (), 136.8 (), 127.8 (H-Ar), 127.8 (H-Fu), 121.8 (), 120.8 (), 112.7 (H-Ar), 108.3 (H-Fu), 102.6 (), 65.5 (H2), 64.8 (H2), 40.8 (2 × H3); HRMS m/z (ESI+) [M + Na]+ 378.0754 (requires 378.0770 for C19H17NNaO4S).
Compound 4c was prepared using aldehyde 3c and was isolated as a red solid (215 mg, 36%). Mp 140–141 °C; 1H NMR (CDCl3, 500 MHz, ppm) δH 9.84 (1H, s, CO), 7.65 (1H, d, J 4.1, C–Th), 7.62 (2H, d, J 9.1, C-Ar), 7.23 (1H, d, J 4.1, C–Th), 6.73 (2H, d, J 9.1, C-Ar), 4.42–4.44 (2H, m, C2), 4.34–4.37 (2H, m, C2), 3.00 (6H, s, 2 × C3); 13C NMR (CDCl3, 125 MHz, ppm) δC 182.7 (HO), 150.2 (), 145.9 (), 141.4 (), 140.4 (), 137.4 (H–Th), 137.0 (), 127.8 (H-Ar), 122.5 (H–Th), 120.7 (), 120.3 (), 112.7 (H-Ar), 107.4 (), 65.6 (H2), 64.9 (H2), 40.8 (H3); HRMS m/z (ESI+) [M + Na]+ 394.0559 (requires 394.0548 for C19H17NNaO3S2).
:1 mixture of toluene and acetonitrile (10 mL + 10 mL) in the presence of piperidine (1.2 mL, 21.0 mmol), and then heated to 85–90 °C for 6 h. The mixture was then cooled to room temperature, and solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (DCM:MeOH; 4:1).
EDOT-Ph was isolated as a red powder (520 mg, 88%). Mp > 250 °C; 1H NMR (DMSO-d6, 400 MHz, ppm) δH 7.93 (1H, s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 7.90 (2H, d, J 8.5, 2 × C-Ar), 7.77 (2H, d, J 8.5, 2 × C-Ar), 7.56 (2H, d, J 9.0, 2 × C-Ar), 6.77 (2H, d, J 9.0, 2 × C-Ar), 4.43 (2H, dd, J 5.8, 2.1, C2), 4.38 (2H, dd, J 5.8, 2.1, C2), 2.94 (6H, s, 2 × C3); HRMS m/z (ESI−) [M − H]− 431.1061 (requires 431.1071 for C24H19N2O4S).
C), 7.90 (2H, d, J 8.5, 2 × C-Ar), 7.77 (2H, d, J 8.5, 2 × C-Ar), 7.56 (2H, d, J 9.0, 2 × C-Ar), 6.77 (2H, d, J 9.0, 2 × C-Ar), 4.43 (2H, dd, J 5.8, 2.1, C2), 4.38 (2H, dd, J 5.8, 2.1, C2), 2.94 (6H, s, 2 × C3); HRMS m/z (ESI−) [M − H]− 431.1061 (requires 431.1071 for C24H19N2O4S).
EDOT-Fu was isolated as a red powder (532 mg, 90%). Mp > 250 °C; 1H NMR (DMSO-d6, 500 MHz, ppm) δH 7.69 (1H, s, CC), 7.54 (2H, d, J 9.1, 2 × C-Ar), 7.25 (1H, d, J 3.7, C-Fu), 6.78 (2H, d, J 9.1, 2 × C-Ar), 6.76 (1H, d, J 3.7, C-Fu), 4.43–4.45 (2H, m, C2), 4.38–4.40 (2H, m, C2), 2.95 (6H, s, 2 × C3); HRMS m/z (ESI−) [M − H]− 421.0875 (requires 421.0864 for C22H17N2O5S).
EDOT-Th as isolated as a red powder (490 mg, 83%). Mp 240 °C (dec); 1H NMR (DMSO-d6, 500 MHz, ppm) δH 8.13 (1H, d, J 0.4, CC), 7.65 (1H, d, J 4.1, C–Th), 7.56 (2H, d, J 9.1, 2 × C-Ar), 7.28 (1H, d, J 4.1, C–Th), 6.77 (2H, d, J 9.1, 2 × C-Ar), 4.47 (2H, dt, J 3.8, 2.1, C2), 4.40 (2H, dt, J 3.8, 2.1, C2), 2.95 (6H, s, 2 × C3); HRMS m/z (ESI−) [M − H]− 437.0626 (requires 437.0635 for C22H17N2O4S2).
:MeOH; 5:1) afforded EDOT-Ph as a red solid (0.31 g, 26%).
Conclusions
Our one-pot, three component C–H activation protocols have allowed the rapid synthesis of precursors for three EDOT-containing dyes. Moreover, we have extended this methodology to produce EDOT-Ph in a respectable yield following facile purification using column chromatography. In the longer term, our protocols could be further extended to provide a combinatorial-like approach to allow the rapid screening of the structure–property relationships of diverse libraries of dyes.35,45Interestingly, a comparison of device performance data of the three dyes shows that the N,N-dimethyaniline moiety produces devices with comparable efficiency to analogous sensitizers containing a N,N-dimethylaniline donor unit attached to the EDOT unit via an acetylene bridge or dyes containing more complicated indoline or triphenylamine donor units reported in the literature.26,28,37 A substantial improvement in device performance (8.2%) was reported for a triphenylamine containing EDOT sensitizer but this is due to a modification of the acceptor unit in the dye.37 Therefore, a comparison of the EDOT dyes reported in the literature with the EDOT dyes reported in this work would suggest that the N,N-dimethylaniline moiety is not a significantly poorer donor group than triphenylamine. However, we do observe unusually high device efficiency improvements on addition of CDCA. For example, optimised CDCA concentrations required for the sensitizers reported in this study are 10 mM by comparison to 1 mM and 6 mM for sensitizers reported in other studies38,39 respectively. This suggests that the bulkier indoline and triphenylamine groups may reduce dye aggregation more effectively than the N,N-dimethylaniline unit. At the other end of the dye molecules, the phenyl-linked dye EDOT-Ph gives rise to better device performance than the heterocyclic variants despite the latter extending their light harvesting to longer wavelengths.
Acknowledgements
We gratefully acknowledge funding from Welsh Government for Sêr Cymru (PJH) and EPSRC (EP/M015254/1 (AC)) and EP/E036244/1. S. M. A. acknowledges the Ministry of Higher Education and Scientific Research in Iraq and Kufa University for funding and support.Notes and references
- Z. Yao, M. Zhang, R. Li, L. Yang, Y. Qiao and P. Wang, Angew. Chem., Int. Ed., 2015, 54, 5994 CrossRef CAS PubMed.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799 CrossRef CAS PubMed.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973 CrossRef CAS PubMed.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, T. Kyomen and M. Hanaya, Chem. Commun., 2015, 51, 6315 RSC.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894 RSC.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402 CrossRef CAS PubMed.
- Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CAS.
- M. K. Nazeeruddin, P. Péchy and M. Grätzel, Chem. Commun., 1997, 1705 RSC.
- P. J. Holliman, M. Mohsen, A. Connell, M. L. Davies, K. Al-Salihi, M. B. Pitak, G. J. Tizzard, S. J. Coles, R. W. Harrington, W. Clegg, C. Serpa, O. H. Fontes, C. Charbonneau and M. J. Carnie, J. Mater. Chem., 2012, 22, 13318 RSC.
- A. Connell, P. J. Holliman, M. L. Davies, C. D. Gwenin, S. Weiss, M. B. Pitak, P. N. Horton, S. J. Coles and G. Cooke, J. Mater. Chem. A, 2014, 2, 4055 CAS.
- A. Connell, P. J. Holliman, E. W. Jones, L. Furnell, C. Kershaw, M. L. Davies, C. D. Gwenin, M. B. Pitak, S. J. Coles and G. Cooke, J. Mater. Chem. A, 2015, 3, 2883 CAS.
- A. Burke, L. Schmidt-Mende, S. Ito and M. Grätzel, Chem. Commun., 2007, 234 RSC.
- T. Geiger, S. Kuster, J.-H. Yum, S.-J. Moon, M. K. Nazeeruddin, M. Grätzel and F. Nüesch, Adv. Funct. Mater., 2009, 19, 2720 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopolous and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734 RSC.
- S. Hwang, J.-H. Lee, C. Park, H. Lee, C. Kim, C. Park, M.-H. Lee, W. Lee, J. Lee, K. Kim and C. Kim, Chem. Commun., 2007, 4887 RSC.
- S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Pechy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202 CrossRef CAS.
- Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Grätzel and W. Zhu, Energy Environ. Sci., 2012, 5, 8261 CAS.
- K. Hara, Z. S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. D. Oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476 CrossRef CAS PubMed.
- J.-H. Yum, S. Jang, R. Humphry-Baker, M. Grätzel, J.-J. Cid, T. Torres and M. K. Nazeeruddin, Langmuir, 2008, 24, 5636 CrossRef CAS PubMed.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. Wei-Guang Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646 CrossRef CAS PubMed.
- T. Ono, T. Yamaguchi and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2009, 93, 831 CrossRef CAS.
- P. J. Holliman, M. L. Davies, A. Connell, B. Vaca Velasco and T. M. Watson, Chem. Commun., 2010, 46, 7256 RSC.
- P. J. Holliman, K. J. Al-Salihi, A. Connell, M. L. Davies, E. W. Jones and D. A. Worsley, RSC Adv., 2014, 4, 2515 RSC.
- S. Pack, H. Choi, C. Kim, N. Cho, S. So, K. Song, M. K. Nazeeruddin and J. Ko, Chem. Commun., 2011, 47, 2874 RSC.
- M. Al-Eid, S. H. Lim, K.-W. Park, B. Fitzpatrick, C.-H. Han, K. Kwak, J. Hong and G. Cooke, Dyes Pigm., 2014, 104, 197 CrossRef CAS.
- A. K. Mohanakrishnan, A. Hucke, M. A. Lyon, M. V. Laksmikantham and M. P. Cava, Tetrahedron, 1999, 55, 11745 CrossRef CAS.
- S. S. Zhu and T. M. Swager, J. Am. Chem. Soc., 1997, 119, 12568 CrossRef CAS.
- J. Schatz and I. Hoffmann, Top. Heterocycl. Chem., 2015, 39, 109 CAS.
- Although compound 5 is commercially available, we have synthesised this compound for this article. For an alternative synthesis see: E. Obrador, M. Castro, J. Tamariz, G. Zepeda, R. Miranda and F. Delgado, Synth. Commun., 1998, 28, 4649 CrossRef CAS.
- For representative examples see: (a) A. K. Mohanakrishnan, P. Amaladass and J. A. Clement, Tetrahedron Lett., 2007, 48, 539 CrossRef CAS; (b) G. Trippé-Allard and J.-C. Lacroix, Tetrahedron, 2013, 69, 861 CrossRef; (c) C.-Y. Liu, H. Zhao and H.-h. Yu, Org. Lett., 2011, 13, 4068 CrossRef CAS PubMed; (d) H. Chong, H.-A. Lin, M.-Y. Shen, C.-Y. Lu, H. Zhao and H.-h. Yu, Org. Lett., 2015, 17, 3198 CrossRef CAS PubMed.
- P. Amaladass, J. A. Clement and A. K. Mohanakrishnan, Tetrahedron, 2007, 63, 10363 CrossRef CAS.
- For examples of one-pot approaches to dyes for DSSCs see: (a) S. Fuse, S. Sugiyama, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, R. Katoh, T. Kaiho and T. Takahashi, Chem.–Eur. J., 2014, 20, 10685 CrossRef CAS PubMed; (b) K. Matsumura, S. Yoshizaki, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, S. Fuse, H. Tanaka and T. Takahashi, Chem.–Eur. J., 2015, 21, 9742 CrossRef CAS PubMed; (c) S. Irie, S. Fuse, M. M. Maitani, Y. Wada, Y. Ogomi, S. Hayase, T. Kaiho, H. Masui, H. Tanaka and T. Takahashi, Chem.–Eur. J., 2016, 22, 2507 CrossRef CAS PubMed.
- W. Li, B. Liu, Y. Wu, S. Zhu, Q. Zhang and W. Zhu, Dyes Pigm., 2013, 99, 176 CrossRef CAS.
- B.-S. Chen, D.-Y. Chen, C.-L. Chen, C.-W. Hsu, H.-C. Hsu, K.-L. Wu, S.-H. Liu, P.-T. Chou and Y. Chi, J. Mater. Chem., 2011, 21, 1937 RSC.
- J.-H. Yum, S. J. Moon, R. Humphry-Baker, P. Walter, T. Geiger, F. Nüesch, M. Grätzel and M. D. K. Nazeeruddin, Nanotechnology, 2008, 19, 424005 CrossRef CAS PubMed.
- T. Maeda, S. Mineta, H. Fujiwara, H. Nakao, S. Yagi and H. Nakazumi, J. Mater. Chem. A, 2013, 1, 1303 CAS.
- G. Cicero, G. Musso, A. Lambertini, B. Camino, S. Bianco, D. Pugilese, F. Risplendi, A. Sacco, N. Shahzad, A. M. Ferrari, B. Ballarin, C. Barolo, E. Tresso and G. Caputo, Phys. Chem. Chem. Phys., 2013, 15, 7198 RSC.
- R. Li, D. Liu, D. Zhou, Y. Shi, Y. Wang and P. Wang, Energy Environ. Sci., 2010, 3, 1765 CAS.
- Y. Bai, J. Zhang, Y. Wang, M. Zhang and P. Wang, Langmuir, 2011, 27, 4749 CrossRef CAS PubMed.
- J. H. Delcamp, Y. Shi, J. H. Yum, T. Sajoto, E. D. Orto, S. Barlow, M. K. Nazeeruddin, S. R. Marder and M. Grätzel, Chem.–Eur. J., 2013, 19, 1819 CrossRef CAS PubMed.
- Spartan '14 (64 bit), Wavefunction, Inc., 18401 Von Karman Ave., Suite 370, Irvine, CA, 92612 Search PubMed.
- S. Fuse, K. Matsumura, A. Wakamiya, H. Masui, H. Tanaka, S. Yoshikawa and T. Takahashi, ACS Comb. Sci., 2014, 16, 494 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic method development, square wave voltammetry, device characterization and testing, UV-vis spectra of thin films of the dyes and solution NMR spectra. See DOI: 10.1039/c6ta04483b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |