
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green manufacturing of metallic nanoparticles: a facile and universal approach to scaling up†
Jicheng
Feng
*a,
Xiaoai
Guo
b,
Nabil
Ramlawi
a,
Tobias V.
Pfeiffer
a,
Ruben
Geutjens
a,
Shibabrata
Basak
c,
Hermann
Nirschl
b,
George
Biskos
*ade,
Henny W.
Zandbergen
c and
Andreas
Schmidt-Ott
*a
aFaculty of Applied Sciences, Delft University of Technology, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: jic.feng@gmail.com; a.schmidt-ott@tudelft.nl
bInstitute for Mechanical Process Engineering and Mechanics, Karlsruhe Institute of Technology, Strasse am Forum 8, 76131 Karlsruhe, Germany
cKavli Institute of Nanoscience, Delft University of Technology, Lorentzweg 1, 2628 CJ, Delft, The Netherlands
dFaculty of Civil Engineering and Geosciences, Delft University of Technology, 2628 CN, Delft, The Netherlands
eEnergy Environment and Water Research Centre, The Cyprus Institute, Nicosia 2121, Cyprus. E-mail: g.biskos@cyi.ac.cy
First published on 21st June 2016
Abstract
High-yield and continuous synthesis of ultrapure inorganic nanoparticles (NPs) of well-defined size and composition has invariably been one of the major challenges in nanotechnology. Employing green techniques that avoid the use of poisonous and expensive chemicals has been realized as a necessity for manufacturing NPs on an industrial scale. In this communication, we show that a newly developed high-frequency spark (HFS) quenched by a high-purity gas yields a series of monometallic and bimetallic NPs in large quantities, with well-defined (primary) particle size (sub-10 nm) and chemical composition. The mass production rate is linearly dependent on the operating frequency, and can reach up to 1 g h−1, providing a universal and facile technology for producing multicomponent hybrid NPs. Considering also that the methodology requires neither any specialized machinery, nor any chemical reagents, product purification, or any further waste processing, it provides a green, sustainable and versatile platform for manufacturing key building blocks toward industrial scale production.
Commercial production of high-performance nanomaterial-based devices, such as batteries,1,2 sensors,3,4 and solar cells,5,6 requires scalable fabrication processes that are robust, rapid and avoid the use of expensive and toxic chemicals.7 These applications have attracted growing interest in conjunction with the synthesis of diverse inorganic nanoparticles (NPs) with sub-10 nm primary particles, a well-defined chemical composition, and high purity.8–11 Achieving high enough mass production rates of such NPs has invariably been one of the challenging tasks in nanotechnology.
Conventional wet-chemistry processes allow manipulation of the shape of NPs with homogeneous and narrow size distributions.12–14 Their scalability, however, is limited by the slow kinetics and the batch operation, which commonly results in undesirable variability of NP physicochemical properties.15 In addition, liquid-phase methods generally require a number of chemical precursors, which can lead to contamination of the resulting nanomaterial surfaces and, in some cases, hazardous wastes that have to be released to the environment.
Dry gas-phase processes, on the other hand, are continuous, rapid and environmentally friendly, and have been identified as chemical manufacturing techniques for a wide range of NPs.16 The fast kinetics of particle formation in the gas phase is not only an advantage in scaling-up; it also leads to rapid formation of fractal-like agglomerates, i.e., the primary nanoscale particles that determine their unique/novel properties. The generated aerosol NPs can be deposited and immobilized onto various (flat or porous) substrates forming, among others, hierarchically patterned coatings for new applications.6,16,17 In addition, dry gas-phase methods can also be coupled with wet-chemistry routes, which enables the synthesis of an even broader range of new building blocks.18–22
Among the numerous gas phase methods, rapidly quenched gas-phase spark ablation operating at ambient pressure provides great versatility in the synthesis of inorganic NPs consisting of a wide variety of conducting or semiconducting materials including rare earths (that are difficult to produce by traditional wet-chemistry methods), thermodynamically metastable materials,23 and alloys (e.g., steel) with virtually unlimited mixing possibilities.7,19,24–27 In addition, the method offers good control over particle size which can range from that of atomic clusters to that of singlets or agglomerates consisting of sub-10 nm primary particles.25,28,29 This technique can produce non-metallic NPs, such as semiconductors,30 oxides and hydrides,31,32 by combining appropriate gases and electrodes. As a result, recent research efforts have focused on fabricating NPs using spark ablation (at low frequencies) for a number of new applications (Table S1†).3,6,7,19,25,29,31,33–44
The carrier gases and the electrodes determine the particle composition.25 Although the manufacturing of electrodes may not be particularly green, using recycled metals allows the green manufacturing of NPs. Additionally, the energy required for the system is high (i.e., on the order of 106 J g−1, exclusively in the form of electricity), but the advantage of avoiding any solvents, toxic chemicals and wastes makes this method inherently environmentally benign. It should be pointed out that wet-chemistry manufacturing processes may have a comparable demand for energy for processing waste streams, producing precursors, and purifying nanomaterials. To make the process also sustainable with respect to its energy consumption, one could couple it with a source of renewable energy (e.g., solar panels).
Apart from the importance of having sub-10 nm primary particles,29 achieving a high yield in NP synthesis is a vital prerequisite for numerous applications. The mass production rate of spark ablation is proportional to the spark repetition frequency and to the mass ablated per spark, which is linearly related to the spark energy as has been recently demonstrated both experimentally and theoretically.29 Spark discharges using resistance–inductance–capacitance (RLC) circuits (referred to as RLCS from this point onwards; cf. Fig. S1†)7,31 have an upper operating frequency threshold of a few hundred Hz. A continuous arc discharge develops above that threshold, which yields larger particles associated with a higher mass production rate.45 Above a certain level of the spark energy, however, undesired “splashing particles” are ejected from molten pools that are momentarily formed on the electrode surface during the spark discharges.46
Here we show that a newly developed high frequency spark (HFS; cf. Fig. S1†)7,47,48 can enormously increase the NP production rate while maintaining the small size of the primary particles in the resulting agglomerates. The core concept of the HFS is to decouple the charge and discharge cycles driven by the RLC circuit by adding a number of fast electronic switches,49 allowing spark frequencies to be controlled up to 25 kHz.7 Switching prevents the transition to the continuous arc mode and facilitates a constant low-energy per spark to be set independently of the gas characteristics (type, purity, and flow rate) and electrode gap spacing. This constant spark energy reduces the differences in the ablated mass from spark to spark and also avoids splashing, guaranteeing a consistent output when operating multiple HFSs in parallel.29 The maximum frequency of 25 kHz corresponds to a duty cycle of 25% (i.e., a typical spark duration of ca. 10 μs over a period of 40 μs for the entire cycle), thereby allowing adequate quenching between successive sparks. In contrast to continuous arc discharges, quenching is sufficient to cause a sharp temperature drop downstream of the spark zone favouring the formation of sub-10 nm primary particles.45
In this contribution we employ, for the first time, the HFS (operating frequencies ≥ 1 kHz) for high-yield synthesis of monometallic Au, Ag, Ni, Zn, Cu, and Al and bimetallic Cu–Ni (95–5 at%) NPs. The production of non-agglomerated NPs is shown to be desirable for a low particle coverage on surfaces,25,29 while agglomerates are formed by depositing particles on substrates with a high surface coverage on substrates. The size distributions of the resulting agglomerated NPs were characterized with a scanning mobility particle sizer (SMPS; cf. Section S3 in the ESI†), whereas the primary particle size of the agglomerates was analysed by transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS). High-resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy (STEM) were employed to characterize the non-agglomerated NPs. In addition, wide-angle X-ray scattering (WAXS) was used to determine the crystal phases of the resulting particles.
Gravimetric measurements of the electrodes were used to determine the mass production rate ṁ of the resulting particles. Fig. 1 shows the mass production rates of NPs consisting of a number of different materials, namely Au, Zn, Ag, Cu, Cu–Ni (95–5 at%), Ni and Al, produced at frequencies from 1 to 4.5 kHz. A high-purity carrier gas (Ar, 99.999%) at a total flow rate of 20 standard litres per minute (slm) is used for all the measurements (cf. Fig. S2†). A low-enough constant spark energy of 32 mJ is fixed throughout the experiments (cf. Section S3†) to avoid the formation of notable splashing particles. The red arrow (Fig. 1) indicates that the maximum frequency of RLCS corresponds to a maximum ṁ which lies below 20 × 10−3 g h−1. The measured linear relations between ṁ and f indicate that the mass ablated per spark Δm is constant and independent of frequency, and we can therefore write:
| ṁ = Δmf | (1) |
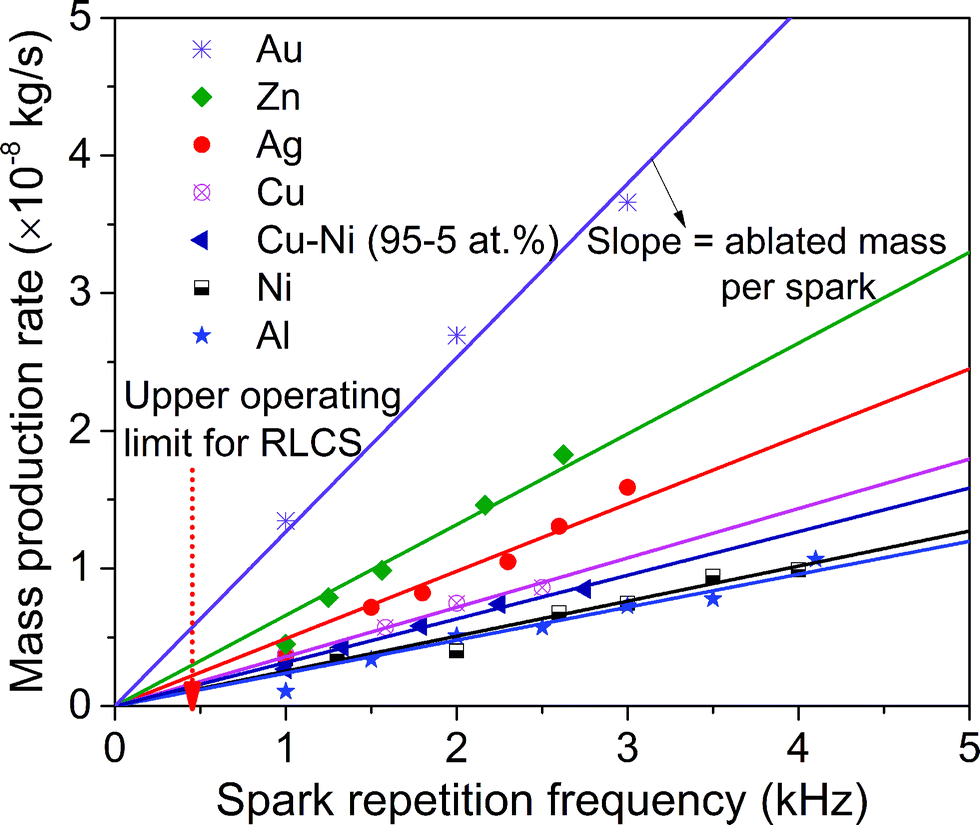 | ||
| Fig. 1 Mass production rates of HFS-produced Au, Zn, Ag, Cu, Cu–Ni (95–5 at%), Ni, and Al NPs, as a function of spark repetition frequencies. Symbols denote the gravimetric measurements of the electrodes, while the lines are fits to the measurements. The red arrow indicates the upper operating limit of the RLCS. | ||
Note that Δm is material dependent (cf. eqn (S1) and Table S2†).25
In view of these linear relations and based on the data shown in Fig. 1, the mass production rate of Au NPs can be extrapolated to ca. 1.1 g h−1 when the spark frequency is raised to 25 kHz. Operating at such high frequencies, however, would require a quenching gas flow rate higher than 100 slm in order to sufficiently cool the electrodes. However, our present system only allows flow rates of the order of a few tens of slm. The HFS could therefore be operated at 25 kHz only for a short period, and stable operation was feasible only up to 4.5 kHz at the flow rate used in this study (20 slm). A recycling flow setup capable of handling the flow rate of a few hundred slm has been designed (Fig. S3†). Continuous and stable operation at 25 kHz would also require a gap control system, since the electrodes are rapidly consumed (Section S6†).
Fig. 2 shows the non-agglomerated spherical Au and Ag NPs with high crystallinity, which were deposited (using only ca. 10 s) on a TEM grid (placed in a custom-made filter holder) at a flow rate of 20 slm and a spark repetition frequency of 1 kHz, corresponding to a production rate of ca. 60 mg h−1. Formation of these spherical NPs is driven by solid-state diffusion,29 mainly by relocation of surface atoms. However, we observe that the agglomerates inevitably form for high coverage on the surface of substrates (cf. TEM images in the ESI†). This is attributed to the incomplete coalescence of the colliding particles on substrates. Further annealing these agglomerates yields spherical particles having larger sizes than those of the primary particles within the as-deposited agglomerates. If particle growth or post-heat treatment is to be avoided, coalescence could be hindered by forming a protecting layer on the particle surface, such as coating particles by introducing a trace amount of oxygen in the carrier gas. Alternatively, decreasing the temperature below the threshold can avoid triggering coalescence. In addition, guiding the aerosol NPs to the liquid phase opens numerous possibilities to stabilize them, which, however, results in impurities and expensive/tedious washing procedures.
 | ||
| Fig. 2 TEM images of Au (a, b, c) and Ag NPs (d, e, f, g; g: STEM) generated by using the HFS at 1 kHz, showing their shape and configuration and confirming their crystal structures. | ||
Besides the applications shown in Table S1,† nonagglomerated singlet particles have been shown to increase the conversion efficiency of solar cells and photocatalysts for water splitting,40,41 whereas agglomerated particles have been used to nanofinish a number of antibacterial textiles with high antibacterial activity and good washing durability.50 With respect to nanocatalysis, we found that the agglomerated NPs on the surface can be reconstructed to spheres by annealing. Even at 900 °C for 100 h, size distributions of the spherical particles remain the same, showing high stability and improved methane conversion efficiency.51
Apart from the determination of ṁ by gravimetric measurements, ṁ is derivable through the mass density of the aerosol Mag given by
| ṁ = MagQ | (2) |
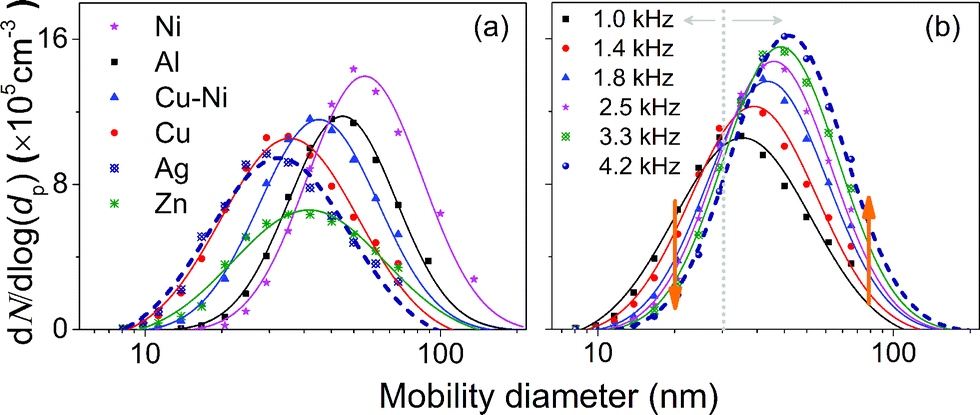 | ||
| Fig. 3 Size distributions of agglomerated NPs produced by using the HFS for (a) Ni, Al, Cu–Ni (95–5 at%), Cu, Ag, and Zn NPs produced at 1 kHz, and (b) Cu NPs produced using different spark repetition frequencies. The symbols denote the SMPS measurements and the curves are fitted log-normal distributions to the data. | ||
Fig. 3a shows the particle size distributions of Zn, Ag, Cu, Cu–Ni (95–5 at%), Ni and Al, measured by the SMPS.25 Note that the SMPS measures the agglomerate size (agglomeration is allowed here) and not the primary particle size, which can be determined by TEM imaging (Fig. S6 and S8†) and SAXS (cf.Table 2). Details of the measurements and the size distributions of different particle materials are summarized in Table S3.† In Fig. 3a, Ni shows a relatively large particle concentration and size despite its low production rate (Fig. 1). We attribute this to the small primary particle size and the tendency of magnetic materials to form chain-like agglomerates (small fractal dimension; Fig. S6†), leading to a larger mobility diameter for a given mass per agglomerate (Section S7†) and reduced losses. Fig. 2b shows the size distributions of agglomerated Cu NPs generated at frequencies ranging from 1.0 to 4.2 kHz. The size distributions shift to larger sizes as the spark frequency increases, correlating well with coagulation theory. If Q is increased linearly in accordance with f, Mag will remain unchanged for a specific material (cf.eqn (1) and (2)) as indicated by size distribution measurements discussed in Section S9 in the ESI.†
The primary particle size has greater relevance than the agglomerate size for the majority of technological applications. Concerning the invariance of the agglomerate size distribution when f/Q stays constant, we hypothesize that the process of particle formation remains similar. The TEM observations confirm that there is a strong similarity to the primary particle sizes under these conditions (cf. Fig. S8†).
To further understand this empirical result, as well as the different primary particle sizes observed for the materials investigated in the present study (cf.Table 2), it is instructive to qualitatively identify the major determinants of primary particle size. The growth of NPs in the rapidly quenched gas-phase synthesis is dominated by particle collisions and sticking. The particles formed at the early stage are liquid-like52 and therefore fully coalesce into singlets when colliding with each other until they attain a critical size, above which coalescence only partly occurs or ceases, thus forming non-spherical agglomerated particles.25 This critical size is essentially the primary particle size in the agglomerates and is temperature and material dependent. A higher surface diffusion coefficient (usually indicated by a lower melting point) yields larger primary particles. For example, the primary Zn NPs are larger than those of Ni. Together with the growth history of the particles, the temperature history is particularly influential on the primary particle size. If f/Q stays constant for a given material and specific spark energy, the particulate volume produced by each spark will be distributed over the same gas volume, suggesting a similar temperature history, as the same energy is dissipated per spark. Considering that f/Q remains unchanged, the gas impurity concentration also stays constant with respect to the particle mass concentration, thereby forming a similar surface diffusion barrier (through gas impurities reacting with the particles, usually forming an oxide layer, if non-oxide NPs are desired) hindering further coalescence. In addition, materials with a higher Δm/ρ (ρ: bulk material density) yield a greater particulate volume while maintaining identical operating conditions. This implies the tendency of forming larger primary particles, as they grow faster in the initial hot “coalescence zone”.
The aforementioned considerations highlight the difficulty in predicting the primary particle size due to the complexity of the process and due to the fact that gas impurities are unknown unless a purification system is adopted. Table 1 summarizes the criteria of controlling primary particle size, which allows us to keep the primary particle size small as we increase the production rate. The primary particle sizes of Ni, Cu, Cu–Ni, Al, and Zn determined by SAXS measurements (cf.Table 2) correlate well with the estimations using the quantity Δm/ρ (cf. Table S2†). The surface diffusion coefficient does not correspond to the melting point for alloys. In Cu–Ni (95–5 at%), the NP surface state tends to behave similarly to that of pure Ni due to segregating Ni to the surface,53 thereby forming primary particles smaller than Cu and similar to Ni (cf.Table 2). Ag NPs apparently make an exception, showing a primary particle size “too large” with respect to the rules in Table 1. We ascribe this to strong coalescence on the substrate and/or under the electron beam that we have frequently observed for noble metal NPs (Fig. S6† for Ag and Au). We believe that this is not so for most other metals because traces of reactive species exert a stabilizing effect (i.e., hindering particle coalescence).
| Increasing the below factors | Increase (↑) or decrease (↓) |
|---|---|
| Quenching gas volume flow rate Q | ↓ |
| Spark repetition frequency f | ↑ |
| Particulate volume Δm/ρ | ↑ |
| Surface diffusion coefficient | ↑ |
| Reactivity with gas impurities | ↓ |
| NPs | SAXS | WAXS |
|---|---|---|
| d pp (nm) | Crystal phase | |
| a Occurrence of oxide crystal phases (e.g. CuO and ZnO) is probably due to the partial oxidation of NPs during the transport and handling as well as the presence of trace amounts of oxygen and/or water in the carrier gas during NP production. | ||
| Ni | 7.13 | Ni |
| Cu | 8.58 | Cu, CuOa |
| Al | 11.53 | Al |
| Zn | 14.70 | Zn, ZnOa |
| Ag | 26.51 | Ag |
| CuNi | 7.28 | Cu, Ni, CuOa |
In the SAXS/WAXS observations, the scattering intensity of deposited particles is measured as a function of scattering angle 2θ or scattering vector q = (4π/λ)sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ (the wavelength of the X-ray beam λ = 0.154 nm). Fig. 4 shows the SAXS/WAXS results of Ni NPs generated at 3 kHz, whereas the data for other NPs are summarized in Table 2. The SAXS curve in a log–log plot shows two power-law regimes, and the values of their exponents P can be determined from the slopes of the linear parts (black and green lines). Due to the growth mechanism, the particle samples have fractal surfaces and form a mass fractal structure. The mass fractal describes the 3D network structure of agglomerates, whereas the surface fractal describes the roughness of the particle surface. In general, for surface fractals (with exponents 3 < P < 4) the surface fractal dimension ds is given by ds = 6 − P. For the two-phase structure with sharp boundaries and smooth surfaces, P is equal to four. For mass fractals (with 1 < P < 3) the mass-fractal dimension dmf is equal to P, which describes the mass-fractal scaling inside the aggregates or agglomerates.54,55 Ni agglomerates have a dmf of 2.12 and a smooth surface (ds = 2.0). A “shoulder” appears between two power-law regimes, as marked by a red dashed curve, yielding the gyration radius of the particles (Rg = 2.75 nm), thereby determining dpp ≃ 7 nm
θ (the wavelength of the X-ray beam λ = 0.154 nm). Fig. 4 shows the SAXS/WAXS results of Ni NPs generated at 3 kHz, whereas the data for other NPs are summarized in Table 2. The SAXS curve in a log–log plot shows two power-law regimes, and the values of their exponents P can be determined from the slopes of the linear parts (black and green lines). Due to the growth mechanism, the particle samples have fractal surfaces and form a mass fractal structure. The mass fractal describes the 3D network structure of agglomerates, whereas the surface fractal describes the roughness of the particle surface. In general, for surface fractals (with exponents 3 < P < 4) the surface fractal dimension ds is given by ds = 6 − P. For the two-phase structure with sharp boundaries and smooth surfaces, P is equal to four. For mass fractals (with 1 < P < 3) the mass-fractal dimension dmf is equal to P, which describes the mass-fractal scaling inside the aggregates or agglomerates.54,55 Ni agglomerates have a dmf of 2.12 and a smooth surface (ds = 2.0). A “shoulder” appears between two power-law regimes, as marked by a red dashed curve, yielding the gyration radius of the particles (Rg = 2.75 nm), thereby determining dpp ≃ 7 nm 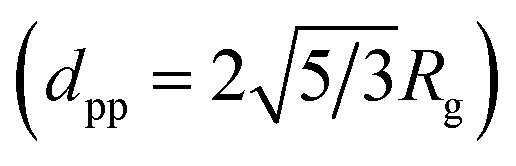 .54,55 This estimation is in line with the TEM observations (cf. inset in Fig. 4a). Simultaneous WAXS measurements identify the crystal phases of the resulting Ni NPs (cf.Fig. 4b) by comparing them with the background peaks. The mean primary particle sizes dpp and crystal phases of Cu, Al, Zn, Ag and Cu–Ni NPs generated at 3 kHz are reported in Table 2 (see details of the SAXS/WAXS measurements in Section S11 in the ESI†).
.54,55 This estimation is in line with the TEM observations (cf. inset in Fig. 4a). Simultaneous WAXS measurements identify the crystal phases of the resulting Ni NPs (cf.Fig. 4b) by comparing them with the background peaks. The mean primary particle sizes dpp and crystal phases of Cu, Al, Zn, Ag and Cu–Ni NPs generated at 3 kHz are reported in Table 2 (see details of the SAXS/WAXS measurements in Section S11 in the ESI†).
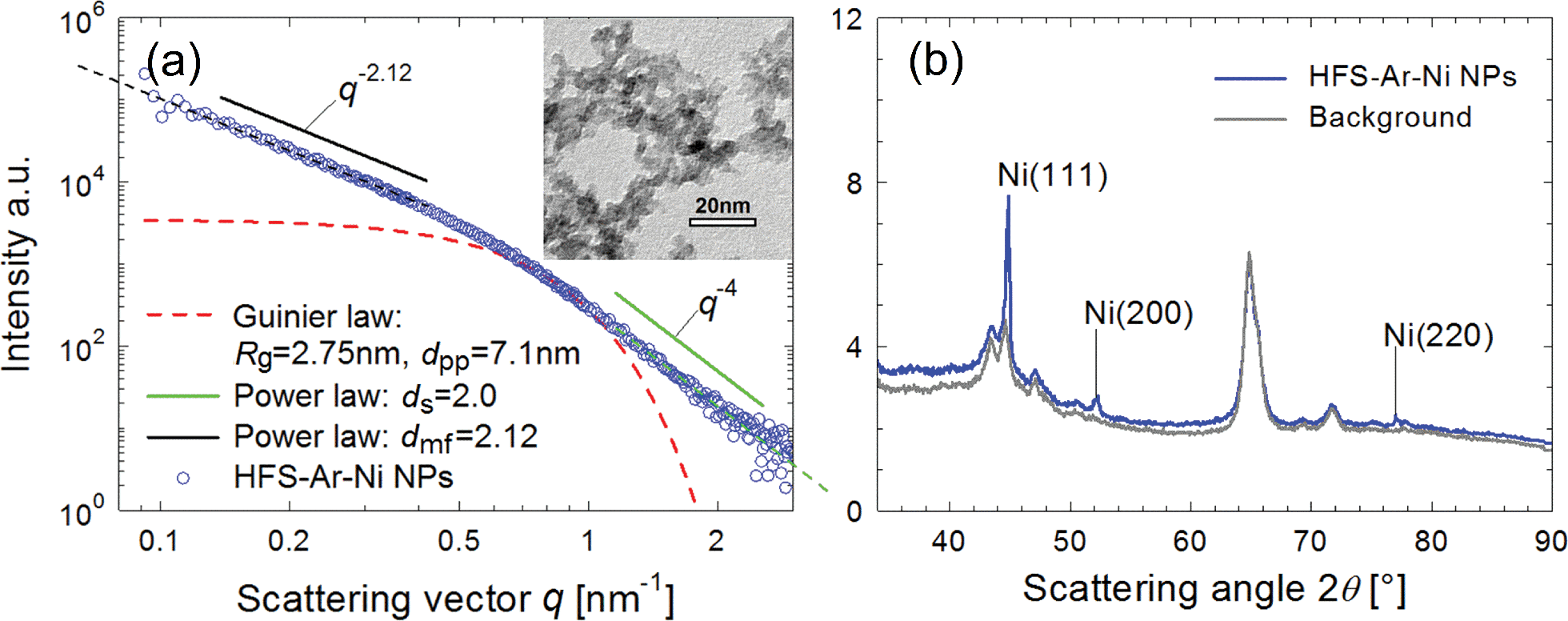 | ||
| Fig. 4 SAXS/WAXS and TEM measurements of Ni NPs generated at 3 kHz. (a) SAXS curve and the TEM image (inset), which provides small primary particles of a few nm in size. (b) WAXS curves. | ||
The direct deposition of NPs onto substrates normally enhances the product properties,40,41,50 albeit a low surface coverage (in the order of 10–1000 μg m−2). Note that the mass production rate demonstrated here (in the order of 0.1 g h−1) exceeds such deposition capacity. The direct incorporation of NP deposition onto substrates or process lines (e.g. textile nanofinishing,50 methane reforming) not only avoids the use of any capping agent that is detrimental to most applications, but also minimizes their associated risks, such as their pyrophoricity and potential release during transport and handling.
Conclusions
In summary, we have shown that a newly developed HFS allows the preparation of an extensive variety of inorganic NPs of well-defined (primary) particle size (both agglomerated and non-agglomerated particles have been produced by controlling their coverage on the surfaces of substrates) and composition at yields of up to 130 × 10−3 g h−1 (operated at 3 kHz). Such yields of small NPs have never been achieved before by evaporation–condensation processes that do not involve liquid precursors. The mass production rates for the NPs consisting of different materials increase linearly with spark frequency. As a result, the HFS can achieve NP mass production rates of the order of 1 g h−1 as extrapolated from the results presented here. Considering that the generic process is easily scalable by parallelizing, the high frequency spark (HFS) can achieve mass production rates to meet industrial demands. Besides the production capacity, the HFS also maintains consistency in product quality, attributed to good control over the kinetics of the process, in a continuous manner. In addition, the fast quenching in principle makes this method feasible to produce thermodynamically metastable materials and to achieve efficient mixing on the atomic or nanometre scale. In contrast to chemical routes, our system avoids using any precursor solutions, thereby allowing single-step synthesis of complex multicomponent inorganic nanomaterials with high purity in a predictable and green fashion. Our findings pave the way for upscaling an extensive variety of nanomaterial syntheses, providing a green and versatile approach to nanofabrication. The wide variety of NPs produced with this technique will be of interest in diverse fields such as materials science, chemistry, and physics as well as in industrial applications.Acknowledgements
This work was funded by the European Union's Seventh Framework Program (EU FP7) under Grant Agreement No. 280765 (BUONAPART-E). The authors are grateful to Dr. A. Maisser's discussion and D. Butterman-Dorey's proofreading.Notes and references
- C. Wang, M. Lan, Y. Zhang, H. Bian, M.-F. Yuen, K. Ken Ostrikov, J. Jiang, W. Zhang, Y. Y. Li and J. Lu, Green Chem., 2016, 18, 3029–3039 RSC.
- S. Kondrat, P. Wu, R. Qiao and A. A. Kornyshev, Nat. Mater., 2014, 13, 387–393 CrossRef CAS PubMed.
- N. A. Isaac, P. Ngene, R. J. Westerwaal, J. Gaury, B. Dam, A. Schmidt-Ott and G. Biskos, Sens. Actuators, B, 2015, 221, 290–296 CrossRef CAS.
- S. Tanwar, M.-C. Chuang, K. S. Prasad and J. A. Ho, Green Chem., 2012, 14, 799 RSC.
- S. M. Kang, S. Jang, J.-K. Lee, J. Yoon, D.-E. Yoo, J.-W. Lee, M. Choi and N.-G. Park, Small, 2016, 12, 2443–2449 CrossRef CAS PubMed.
- S. Jang, J. Yoon, K. Ha, M. Kim, D. H. Kim, S. M. Kim, S. M. Kang, S. J. Park, H. S. Jung and M. Choi, Nano Energy, 2016, 22, 499–506 CrossRef CAS.
- T. V. Pfeiffer, J. Feng and A. Schmidt-Ott, Adv. Powder Technol., 2014, 25, 56–70 CrossRef CAS.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- N. a. Frey, S. Peng, K. Cheng and S. Sun, Chem. Soc. Rev., 2009, 38, 2532–2542 RSC.
- J. Chang, X. Huang, G. Zhou, S. Cui, P. B. Hallac, J. Jiang, P. T. Hurley and J. Chen, Adv. Mater., 2014, 26, 758–764 CrossRef CAS PubMed.
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580–587 CrossRef CAS PubMed.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- G. H. Jeong, Y. W. Lee, M. Kim and S. W. Han, J. Colloid Interface Sci., 2009, 329, 97–102 CrossRef CAS PubMed.
- M. R. Buck and R. E. Schaak, Angew. Chem., Int. Ed., 2013, 52, 6154–6178 CrossRef CAS PubMed.
- J.-M. Lim, A. Swami, L. M. Gilson, S. Chopra, S. Choi, J. Wu, R. Langer, R. Karnik and O. C. Farokhzad, ACS Nano, 2014, 8, 6056–6065 CrossRef CAS PubMed.
- A. Tricoli, M. Righettoni and A. Teleki, Angew. Chem., Int. Ed. Engl., 2010, 49, 7632–7659 CrossRef CAS PubMed.
- A. Gutsch, H. Mühlenweg and M. Krämer, Small, 2005, 1, 30–46 CrossRef CAS PubMed.
- Z. Jin, M. Xiao, Z. Bao, P. Wang and J. Wang, Angew. Chem., Int. Ed. Engl., 2012, 51, 6406–6410 CrossRef CAS PubMed.
- J. H. Byeon and Y.-W. Kim, Nanoscale, 2012, 4, 6726–6729 RSC.
- R. G. Sanedrin, D. G. Georganopoulou, S. Park and C. A. Mirkin, Adv. Mater., 2005, 17, 1027–1031 CrossRef CAS.
- J. M. Tour, Nature, 2014, 512, 30–31 CrossRef CAS PubMed.
- C. Boissiere, D. Grosso, A. Chaumonnot, L. Nicole and C. Sanchez, Adv. Mater., 2011, 23, 599–623 CrossRef CAS PubMed.
- V. Sebastian, M. Arruebo and J. Santamaria, Small, 2014, 10, 835–853 CrossRef CAS PubMed.
- J. Feng, N. Ramlawi, G. Biskos and A. Schmidt-Ott, RSC Adv. Search PubMed, submitted.
- J. Feng, G. Biskos and A. Schmidt-Ott, Sci. Rep., 2015, 5, 15788 CrossRef CAS PubMed.
- J. H. Byeon, J. H. Park, K. Y. Yoon and J. Hwang, Nanoscale, 2009, 1, 339–343 RSC.
- J. H. Byeon, J. H. Park and J. Hwang, J. Aerosol Sci., 2008, 39, 888–896 CrossRef CAS.
- A. Maisser, K. Barmpounis, M. B. Attoui, G. Biskos and A. Schmidt-Ott, Aerosol Sci. Technol., 2015, 49, 886–894 CrossRef CAS.
- J. Feng, L. Huang, L. Ludvigsson, M. E. Messing, A. Maisser, G. Biskos and A. Schmidt-Ott, J. Phys. Chem. C, 2016, 120, 621–630 CAS.
- V. A. Vons, L. C. P. M. de Smet, D. Munao, A. Evirgen, E. M. Kelder and A. Schmidt-Ott, J. Nanopart. Res., 2011, 13, 4867–4879 CrossRef CAS.
- A. Anastasopol, T. V. Pfeiffer, J. Middelkoop, U. Lafont, R. J. Canales-Perez, A. Schmidt-Ott, F. M. Mulder and S. W. H. Eijt, J. Am. Chem. Soc., 2013, 135, 7891–7900 CrossRef CAS PubMed.
- V. A. Vons, A. Anastasopol, W. J. Legerstee, F. M. Mulder, S. W. H. Eijt and A. Schmidt-Ott, Acta Mater., 2011, 59, 3070–3080 CrossRef CAS.
- M. E. Messing, R. Westerström, B. O. Meuller, S. Blomberg, J. Gustafson, J. N. Andersen, E. Lundgren, R. van Rijn, O. Balmes, H. Bluhm and K. Deppert, J. Phys. Chem. C, 2010, 114, 9257–9263 CAS.
- J. H. Byeon and J.-W. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 3105–3110 CAS.
- Y. H. Joe, W. Ju, J. H. Park, Y. H. Yoon and H. Jungho, Aerosol Air Qual. Res., 2013, 13, 1009–1018 CAS.
- J. Byeon, D. Park and J. Kim, Nanoscale, 2015, 7, 2271–2275 RSC.
- J. H. Byeon and J.-W. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 947–951 CAS.
- D. Z. Pai, K. Ken Ostrikov, S. Kumar, D. A. Lacoste, I. Levchenko and C. O. Laux, Sci. Rep., 2013, 3, 1221 Search PubMed.
- T. Pfeiffer, P. Kedia, M. E. Messing, M. Valvo and A. Schmidt-Ott, Materials, 2015, 8, 1027–1042 CrossRef.
- T. V. Pfeiffer, J. Ortiz-Gonzalez, R. Santbergen, H. Tan, A. Schmidt-Ott, M. Zeman and A. H. M. Smets, Energy Procedia, 2014, 60, 3–12 CrossRef CAS.
- M. Valenti, D. Dolat, G. Biskos, A. Schmidt-ott and W. A. Smith, J. Phys. Chem. C, 2015, 119, 2096–2104 CAS.
- V. A. Vons, H. Leegwater, W. J. Legerstee, S. W. H. Eijt and A. Schmidt-Ott, Int. J. Hydrogen Energy, 2010, 35, 5479–5489 CrossRef CAS.
- S. K. Sengar, B. R. Mehta, R. Kumar and V. Singh, Sci. Rep., 2013, 3, 2814 Search PubMed.
- J. H. Park, K. Y. Yoon, H. Na, Y. S. Kim, J. Hwang, J. Kim and Y. H. Yoon, Sci. Total Environ., 2011, 409, 4132–4138 CrossRef CAS PubMed.
- E. Hontañón, J. M. Palomares, M. Stein, X. Guo, R. Engeln, H. Nirschl and F. E. Kruis, J. Nanopart. Res., 2013, 15, 1957 CrossRef.
- N. S. Tabrizi, M. Ullmann, V. A. Vons, U. Lafont and A. Schmidt-Ott, J. Nanopart. Res., 2009, 11, 315–332 CrossRef CAS.
- A. Schmidt-Ott and T. V. Pfeiffer, WO Pat., 2013/115644, 2013.
- T. V. Pfeiffer, PhD thesis, Delft University of Technology, 2014.
- V. Vons, PhD thesis, Delft University of Technology, 2008.
- J. Feng, E. Hontañón, M. Blanes, J. Meyer, X. Guo, L. Santos, L. Paltrinier, N. Ramlawi, L. C. P. M. de Smet, H. Nirschl, F. E. Kruis, A. Schmidt-Ott and G. Biskos, ACS Appl. Mater. Interfaces, 2016, 8, 14756–14765 Search PubMed.
- C. Denonville and H. Malerød-Fjeld, Personal communication on Ni nanoparticles as catalysis in catalytic reactors, Duisburg, January 2016 Search PubMed.
- A. B. Bourlinos, R. Herrera, N. Chalkias, D. D. Jiang, Q. Zhang, L. A. Archer and E. P. Giannelis, Adv. Mater., 2005, 17, 234–237 CrossRef CAS.
- Q. Wu, L. D. L. Duchstein, G. L. Chiarello, J. M. Christensen, C. D. Damsgaard, C. F. Elkjaer, J. B. Wagner, B. Temel, J.-D. Grunwaldt and A. D. Jensen, ChemCatChem, 2014, 6, 301–310 CrossRef CAS.
- X. Guo, A. Gutsche and H. Nirschl, J. Nanopart. Res., 2013, 15, 2058 CrossRef.
- X. Guo, M. Wagner, A. Gutsche, J. Meyer, M. Seipenbusch and H. Nirschl, J. Aerosol Sci., 2015, 85, 17–29 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta03221d |
| This journal is © The Royal Society of Chemistry 2016 |