
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of intercalating perfluorohexane into lipid shells on nano and microbubble stability†
Radwa H.
Abou-Saleh
 ab,
Sally A.
Peyman
a,
Benjamin R. G.
Johnson
a,
Gemma
Marston
c,
Nicola
Ingram
c,
Richard
Bushby
d,
P. Louise
Coletta
c,
Alexander F.
Markham
c and
Stephen D.
Evans
*a
ab,
Sally A.
Peyman
a,
Benjamin R. G.
Johnson
a,
Gemma
Marston
c,
Nicola
Ingram
c,
Richard
Bushby
d,
P. Louise
Coletta
c,
Alexander F.
Markham
c and
Stephen D.
Evans
*a
aMolecular and Nanoscale Physics Group, School of Physics and Astronomy, University of Leeds, LS2 9JT, UK. E-mail: s.d.evans@leeds.ac.uk
bBiophysics Group, Department of Physics, Faculty of Science, Mansoura University, Egypt
cLeeds Institute of Molecular Medicine, Wellcome Trust Brenner Building, St. James's University Hospital, Leeds, LS9 7TF, UK
dSchool of Chemistry, University of Leeds, LS2 9JT, UK
First published on 28th July 2016
Abstract
Microbubbles are potential diagnostic and therapeutic agents. In vivo stability is important as the bubbles are required to survive multiple passages through the heart and lungs to allow targeting and delivery. Here we have systematically varied key parameters affecting microbubble lifetime to significantly increase in vivo stability. Whilst shell and core composition are found to have an important role in improving microbubble stability, we show that inclusion of small quantities of C6F14 in the microbubble bolus significantly improves microbubble lifetime. Our results indicate that C6F14 inserts into the lipid shell, decreasing surface tension to 19 mN m−1, and increasing shell resistance, in addition to saturating the surrounding medium. Surface area isotherms suggest that C6F14 incorporates into the acyl chain region of the lipid at a high molar ratio, indicating ∼2 perfluorocarbon molecules per 5 lipid molecules. The resulting microbubble boluses exhibit a higher in vivo image intensity compared to commercial compositions, as well as longer lifetimes.
1 Introduction
Microbubbles (MBs) used for contrast enhanced ultrasound (CEUS) imaging are micron sized gas encapsulated spheres, stabilized with a shell made of biocompatible materials such as proteins, surfactants, phospholipids or polymers. These MBs are capable of circulating in the vasculature and their high acoustic impedance mismatch with the surrounding tissue leads to strong ultrasound scattering resulting in enhanced contrast in US imaging. Recently there has been considerable interest in the development of MBs as vehicles for drug delivery1,2 by loading them with drug-filled liposomes, attaching genes to the shell,3–5 or filling the core with a therapeutic gas.6,7 The MB complex can be targeted to the required location using antibodies, or other ligands, to provide a route for targeted, triggered delivery.8 Furthermore, it has been demonstrated that bursting the MBs using an ultrasound pulse gives rise to sonoporation of cells in the immediate vicinity of the MBs, leading to an enhanced therapeutic uptake.9,10 Their changing role requires that MBs be re-engineered to improve their functional performance, with the key MB parameters being size and dispersity, biocompatibility, shell stiffness and MB lifetime.5,11For clinical and preclinical applications MBs are typically desired to be between 1–8 μm in diameter. Control over size can be achieved by selecting a suitable production technique. Sonication and mechanical agitation are the common methods for MB production.12,13 They produce broad poly-disperse size distributions with a polydispersity index of ∼150%, with the majority of MBs being in the range of <8 μm,14 and are currently used for diagnostic imaging.15,16 Coaxial electrohydrodynamic atomisation14,17 produces bubbles which can be of a controllable size between 1 and 25 μm and with a moderate dispersity index of ∼30%. For the highest quality monodisperse MB production, the best control over the size and the dispersity index has come about through the use of flow-focus microfluidic (MF) technology.18–21 However, in this approach the MB concentration tends to be low (∼106 MB mL−1) compared to the 108 MB mL−1 used in a single bolus for in vivo imaging experiments. Recently Peyman et al. introduced a rapid pressure-drop on a chip technique, which led to the production of nano and micron sized bubbles that were typically less than 2 μm in diameter, at high concentrations (>1010 and 108 MB mL−1 respectively).22 Post production MBs are prone to fusion and dissolution unless they are stabilized with a suitable coating. Furthermore, such MBs should not only be stable in vitro but also in vivo, where they are required to survive multiple passages through the heart and lungs for successful diagnostic imaging, and possibly longer for therapeutic applications requiring the effective targeting and release of drug payloads.
Control over the in vitro and in vivo stability (or lifetime) of MBs depends upon three main considerations; (i) the conditions of the surrounding medium, such as the temperature, pressure and concentration of the dissolved gas, (ii) the MB shell composition (which controls the surface tension and the resistance to permeation) and (iii) the solubility of the encapsulated gas core in the medium.23 In 1950 Epstein and Plesset introduced a model describing the rate of growth and/or dissolution of “shell-less” or “uncoated” bubbles in aqueous media and showed them to be critically dependent on the diffusion of gas away from the bubble surface, Dw, the degree of saturation of the solution, f (=C0/Cs), and the initial bubble radius, r (Fig. 1). C0 is the concentration of dissolved gas and Cs is the concentration at saturation.24 More recently Borden and Longo suggested a modified version of the Epstein–Plesset model, as shown in eqn (1), that included the effect of the MB coating as a barrier against gas diffusion, Rshell, and a modifier of the surface tension, σ.25
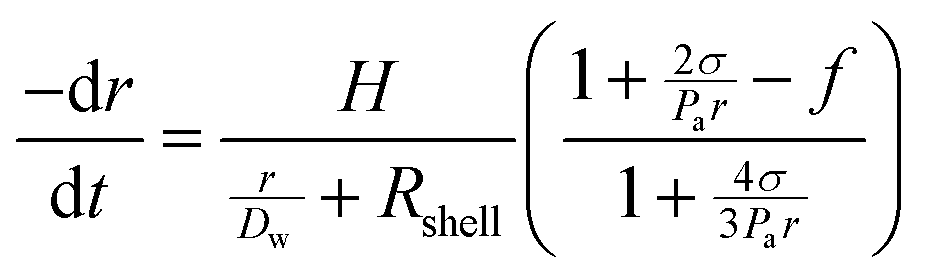 | (1) |
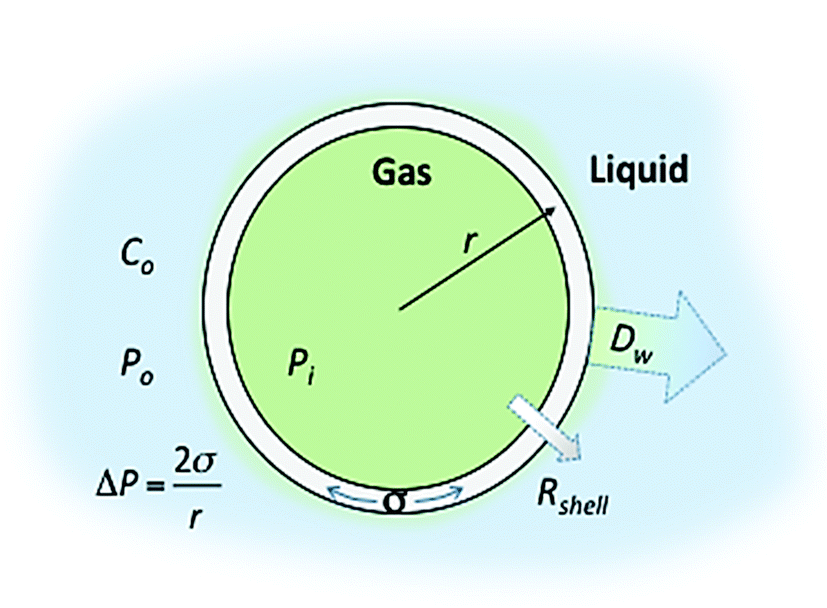 | ||
| Fig. 1 Schematic representation of a lipid shelled MB illustrating the factors that affect MB lifetime. The different terms are defined in the text. | ||
The Laplace pressure, ΔP, places the gas core under increased pressure due to surface tension and curvature effects and thus provides a strong driving force for MB dissolution.28 The inclusion of surfactants, in our case lipids, at the gas/liquid interface significantly reduces the surface tension and, consequently, the Laplace pressure.29,30 Such surfactants also provide a resistive barrier, Rshell, to gas leaving the MB and dissolving into the aqueous phase. Important shell properties that affect the bubble stability are surface tension, surface hardening, and resistance to gas transport.30–32 Borden and Longo25 showed that MB stability is strongly dependent on the lipid shell resistance and increases with the increasing acyl chain length of the lipids.33–36 They also showed that increasing the acyl chain length between DPPC (16) and DBPC (24) increases the lipid rigidity (Wrinkling threshold) and consequently the MB stability and circulation time. The use of higher molecular weight, less soluble gases such as SF6 and perfluorocarbons (PFCs) also significantly enhances MB lifetime and permits diagnostic and therapeutic applications.5,23,27,37 Several groups have investigated the effect of different gases, or gas mixtures, on MB stability. Sarker et al.27,38 developed a modified EP model for gas diffusion from MBs filled with air and PFC gas and showed that the dependence of shell permeability to gas type predicts a 500× increase in the time taken for MB dissolution for PFC compared to air filled bubbles. Kraft et al. used acoustical methods, to investigate the effect of the gas composition on the size and stability of shelled MBs. Using bubbles filled with nitrogen and saturated with perfluorohexane, they reported that the PFC gas increased the compressibility of the lipid monolayer resulting in more flexible and stable MBs.39,40 Kabalnov et al.26,41 presented a detailed model and in vivo experimental studies on the efficacy of different PFC gases (as osmotic agents) mixed with oxygen or nitrogen on MB dissolution in the blood stream. They showed an increase in the bubble lifetime with the increasing molecular weight of the osmotic agent when mixed with oxygen. Increasing the PFC molecular weight beyond C6F14 does not have a positive effect. Finally, Schutt et al. encapsulated a mixture of different PFCs in their gaseous state, to show that MBs containing mixtures of C4F10 and C6F14 persist for 3 min, which was longer than with either gas on its own.28,42
The combined effect of saturation of the surrounding medium, f, and different MB shells, Rshell, was investigated by Kwan and Borden in 2010.43 Sulphur hexafluoride, SF6, was encapsulated in SDS or lipid shell MBs and used in a modified perfusion chamber to observe the MB dissolution behaviour in a SF6 or air-saturated medium. When suddenly placed in an air-saturated medium, MBs initially grow (air influx) and then decrease in size as a result of SF6 efflux. Lipid coated MBs deviated from the model, as when placed in an air-saturated environment, the initial growth regime was shorter and it was followed by rapid non-uniform dissolution to the original diameter, and then a steady dissolution with a constant wall velocity and final stability at around 10 μm in diameter.
For the studies reported here, MF-MBs were produced with an average diameter of 2 μm. The stability of the population was examined in vitro, at 37 °C, and in vivo using mouse models. The aim of the study was to optimize the in vivo lifetime of the MF-MBs. Factors investigated include: shell resistance, Rshell, through control over the lipid compositions, and gas dissolution, (Dw, f, H), through control over the molecular weight of the gas and saturation of the surrounding medium. By optimising these factors we achieved a prolonged lifetime of MBs in vivo. Our optimised MBs have lifetimes of >14 minutes in vivo and also retain excellent ultrasound contrast enhancement properties.
2 Materials and methods
2.1 Materials
The lipids used throughout this study were 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-2000] (DSPE-PEG2000), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-5000] (DPPE-PEG5000) which were purchased from Avanti Polar Lipids (Alabaster, AL, USA) and used without further purification. All lipids were received in the powder form, and then dissolved in a 50/50 chloroform/methanol solution.2.2 Microbubble preparation and characterisation
The lipid composition as specified for each MB formulation was prepared as described previously.19,44 Briefly, the lipid mixture was dried under a steady stream of nitrogen gas on the vial walls. This dried film was then suspended in a solution containing 4 mg mL−1 NaCl and 1% glycerine (>99%, Sigma-Aldrich, St. Louis, MO, USA) to a final lipid concentration of 1 mg mL−1 unless otherwise specified. This solution was vortexed for 1 minute before being placed in an ultrasonic bath for 1 hour. The lipid solutions were allowed to cool down in the fridge for 5 minutes prior to use in the MF-MB maker.Polymethylmethacrylate (PMMA) MF-chips, designed in Leeds and produced by Epigem plc (Redcar, UK), were used to prepare MBs according to our previously described protocols, in which gas flow is focussed through a nozzle before being rapidly expanded to create a microspray regime.19 Two gases were investigated of different molecular weight: perfluoropropane gas (C3F8), Mw = 188 g mol−1; and perfluorobutane (C4F10), Mw = 238 g mol−1. The gas pressure was controlled using a Kukuke microprecision regulator (RS supplies, Leeds, UK). The flow rate of the liquid phase, containing the lipid products, was controlled using an Aladdin AL 2000 syringe pump (World Precision Instruments, Stevenage, UK). The flow rate was fixed for all MB preparations at 80 μL min−1. In some cases tetradecafluorohexane (C6F14, Sigma-Aldrich, St. Louis, MO, USA) was used to saturate the lipid solution (10 μL).
A typical production run produced 1 mL of MBs. For each MB population formed, a 10 μL sample (collected from the middle of the homogenous MB solution) was diluted 10-fold to facilitate counting and sizing of the bubbles. From this diluted sample 30 μL was introduced into a 50 μm chamber on a glass slide. The MBs were allowed to rise for ∼1 minute before acquiring images. An inverted microscope (Nikon, Japan) was used to image the MBs at 60× magnification. A CCD camera (DS-Fil 5 Mega pixel, Nikon, Japan) was used to capture 40 images of each sample, from which the concentration and size distribution were obtained using Image J freeware (http://rsbweb.nih.gov/ij/) and statistically analysed using Origin Pro (Version 8.5 or later).
2.3 In vitro lifetime
The MB lifetime was measured in vitro as described previously.45 Briefly, 500 μL of MB solution was introduced into 500 μL of cell medium [RPMI from Invitrogen, Life Technologies, UK, with 10% (v/v) foetal calf serum (Sigma Aldrich, UK)] and incubated at 37 °C in a digital dryblock heater (Model D1100, Labnet International, USA). The vial containing the sample was left open exposed to air and 10 μL samples were collected every 15 min for sizing and counting.2.4 In vivo lifetime
For in vivo measurements 50 to 100 μL of the MB solution (in PBS) was injected via a syringe attached to a tail vein catheter. The injection of the MB bolus was controlled by a syringe driver at a rate of 0.6 mL min−1. A typical bolus contained ∼108 MBs in 100 μL.All animal work was performed under licence and in accordance with the UK Animals (Scientific Procedures) Act 1986 following local ethical review and procedures. Eighteen athymic CD1-nu/nu male mice (6–8 weeks old) were maintained in individual ventilated cages under specific pathogen-free conditions with free access to diet and water.
The mouse aorta was identified and imaged by pulsed wave (PW) Doppler imaging using a Vevo770 (FUJIFILM VisualSonics, Inc.) using the 40 MHz transducer. For post-processing analysis, a region of interest (ROI) was drawn within the aorta such that the ROI was maintained within the aorta for the whole video loop, ensuring that with respiration motion, the ROI was not sampling tissue outside the aorta, as indicated by the blue circle in Fig. 2A. Fig. 2B shows a snapshot of the accumulation of MBs in the aorta post injection.
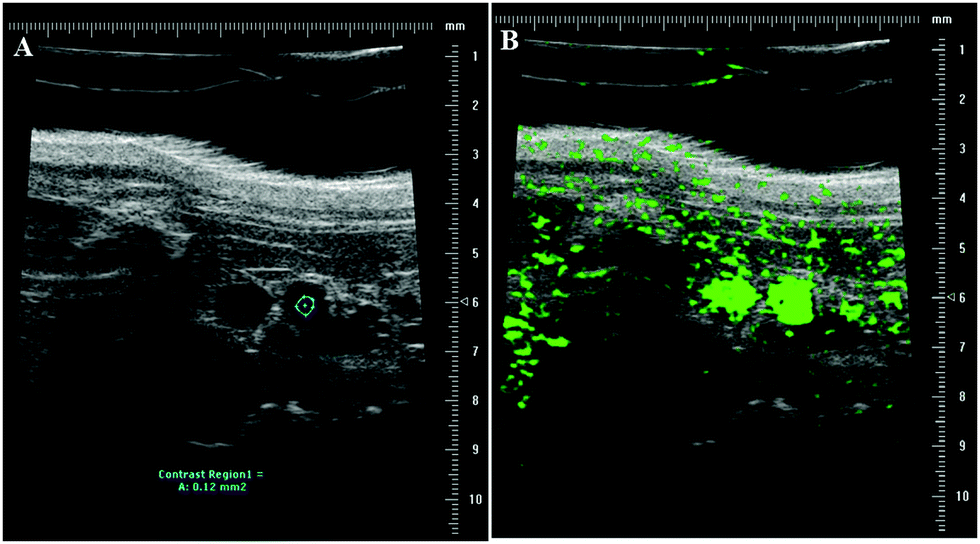 | ||
| Fig. 2 In vivo imaging of mouse aorta with high frequency ultrasound. (A) A region of interest (ROI) within the aorta was selected for post-processing analysis (blue region) to determine the contrast intensity within the aorta over time. (B) Example image showing the aorta post injection filled with MBs. | ||
2.5 Langmuir trough
A Langmuir trough (KSV Nima) was used to measure the differences in monolayer compressability and elasticity. The trough was equipped with two movable PTFE barriers to compress the monolayer symmetrically. A Wilhelmy plate tensiometer (paper method) was used to measure the surface pressure of the monolayer. This experiment was performed for the same lipid monolayer composition found to form the most stable MBs. 20 μL of a 1 mg mL−1 solution of (DPPC + 5% DSPE-PEG2000), in chloroform, was spread on the surface of a subphase of water in the presence and absence of C6F14. Pressure–area isotherms were made with the compression rate of the barriers set to 5 mm min−1 and the surface pressure was not allowed to rise beyond 30 mN m−1; this ensured the reversibility of the isotherms and allowed us to observe multiple compression/expansion cycles, which is important for the determination of whether any material is lost from the monolayer during compression.3 Results
3.1 Microbubble production and characterisation
MBs were prepared in the spray regime of a specially designed MF-chip19 shown schematically in Fig. 3A. The image (inset) in Fig. 3B shows a typical MB population, with an average MB diameter of 1.7 ± 1.1 μm and concentrations of ∼2 × 109 MB mL−1. No bubbles were produced with a diameter >7 μm. | ||
| Fig. 3 Bubble production and characterisation. (A) A schematic of the microfluidic chip design emphasizing the 3D expansion region for the production of MBs in the spray regime. (B) Graph displaying the size distribution for one of the bubble populations. The inset shows a bright field image representing the produced MBs (sample is 10-fold diluted). | ||
3.2 MB lifetime
In vitro MB lifetime was initially determined by measuring the MB concentration, using optical microscopy, every 30 min in a closed vial at room temperature and atmospheric pressure. MB concentration versus time (in vitro results) for MBs prepared with shells of DPPC (or DSPC) with 10% DSPE-PEG2000 encapsulating C3F8 gas was tested (see S1, ESI†). The data showed that the MBs were relatively long lived in both cases, i.e. for both lipid types, with no significant difference in their stability over a period of ∼3 h following production. In contrast, the in vivo lifetime in the mouse aorta, as determined from time–intensity curves (TICs), showed that both MBs decayed within 2 minutes in vivo. A slightly higher acoustic intensity signal was observed for the DPPC MBs compared to the DSPC ones, which is consistent with previous observations for such MBs.46 These initial results showed that the in vitro study of MBs, at room temperature and atmospheric pressure, is not a good indicator of the in vivo behaviour and that physiological conditions (increased temperature, pressure and potential for gas exchange around the MBs) are likely to significantly affect the MB stability. Thus, in order to better test our MBs under more realistic conditions the in vitro experiment was modified to use a cell medium (RPMI with 10% (v/v) foetal calf serum) incubated at 37 °C and the sample was left in an open tube exposed to air for gaseous exchange. Based on the in vivo data (S1, ESI†), and that DPPC MBs have a more homogenous lipid distribution on the bubble shell,47 the slightly shorter DPPC (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) lipid was chosen for subsequent MB studies over the DSPC (18:0). A series of factors were investigated to detect their effect on MB stability, firstly in vitro and then in vivo. These included: (i) the degree of pegylation in the MB shell, which affects the potential for phase separation;45,48 (ii) the gas core, by switching to a heavier gas (C4F10) to reduce solubility and diffusivity in the surrounding medium; and finally (iii) factors related to the surrounding solution, such as super saturating the lipid solution with C3F8 gas prior to bubble production, and also increasing the concentration of NaCl in the lipid solution to over 0.1 M to prevent bubble fusion.49
:0) lipid was chosen for subsequent MB studies over the DSPC (18:0). A series of factors were investigated to detect their effect on MB stability, firstly in vitro and then in vivo. These included: (i) the degree of pegylation in the MB shell, which affects the potential for phase separation;45,48 (ii) the gas core, by switching to a heavier gas (C4F10) to reduce solubility and diffusivity in the surrounding medium; and finally (iii) factors related to the surrounding solution, such as super saturating the lipid solution with C3F8 gas prior to bubble production, and also increasing the concentration of NaCl in the lipid solution to over 0.1 M to prevent bubble fusion.49
Of the factors identified above only two showed a significant effect on MB stability. Summary data has been taken from a larger data set of the variables investigated, which is detailed in the ESI† (S2). The first significant effect was that on changing from the C3F8 core to the higher molecular weight C4F10 gas, the MB population displayed a doubling in lifetime from the order of one to two hours (at 37 °C) Fig. 4A. This effect is well understood and is due to the reduced solubility of the higher Mw gas in solution (the Ostwald coefficient reduces from 5.2 × 10−4 for C3F8 to 2.0 × 10−4 for C4F10), a slightly reduced diffusion rate in water (from 7.45 × 10−10 for C3F8 to 6.9 × 10−10 for C4F10) and an increase in the resistance of the shell to the increased Mw gas. These effects have been modelled in S3 in the ESI†, following the work of Sarkar et al.27,38 for MBs with different cores and assuming the surface tension of the shelled MBs to be 25 mN m−1.
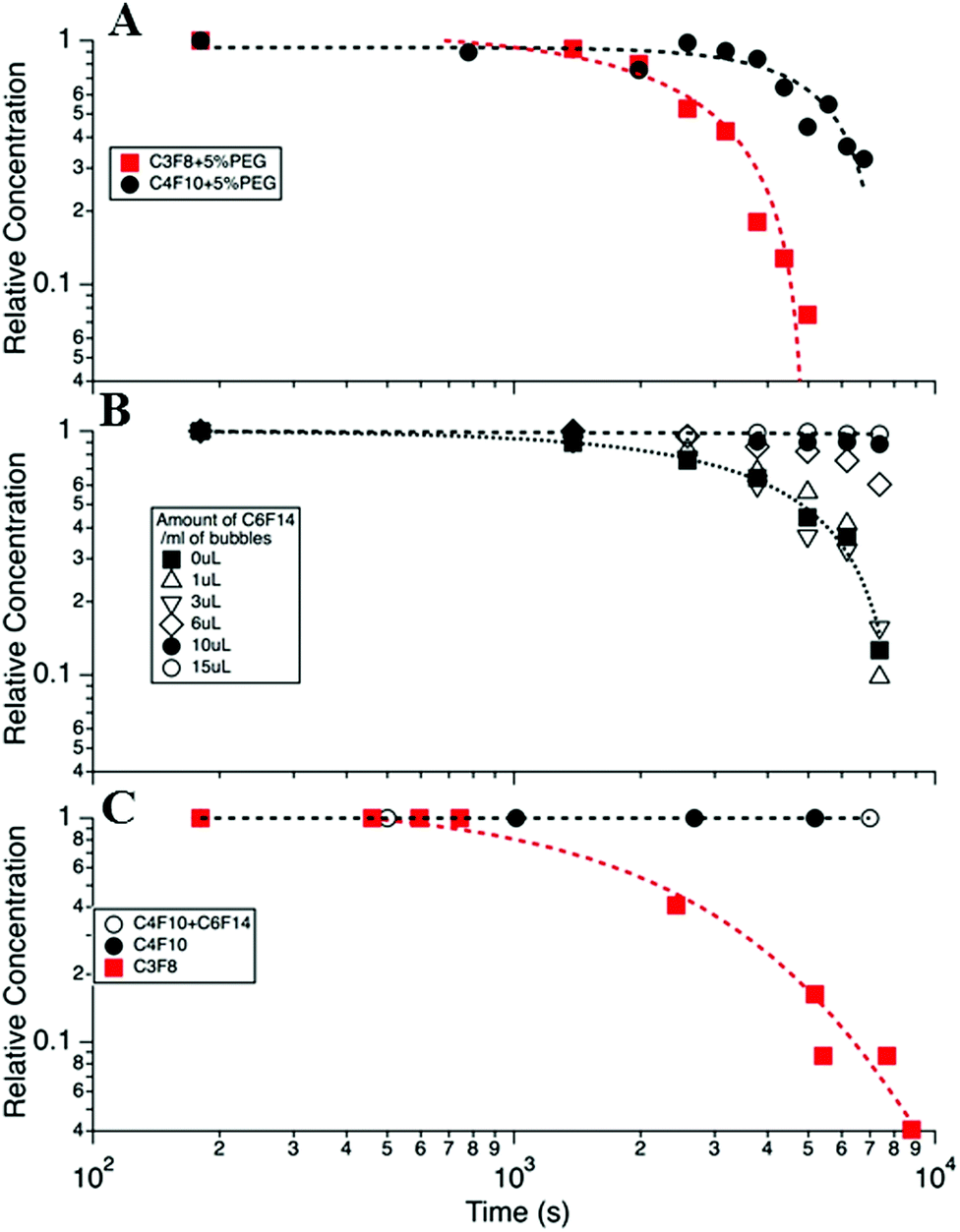 | ||
| Fig. 4 (A) MB concentration versus time for DPPC + 5% PEG DSPE MBs with C3F8 and C4F10 gas cores. (B) The effect of adding liquid C6F14 to the MB bolus, in increasing amounts from 0 to 15 μL mL−1, on MB lifetime. (C) Calculated lifetime expected for the MB distribution profile shown in Fig. 3 for 3 cases, (i) with a C3F8 core, (ii) with a C4F10 core and (iii) C4F10 + C6F14 (open circles). The parameters for calculations were taken from Sarkar27 and given in detail in the ESI† (S4). All data was taken at 37 °C in cell media and at atmospheric pressure. | ||
Decreasing the PEG concentration from 10% to 5% led to a modest increase in the average lifetime (S4, ESI†). However, in spite of this significant improvement in the in vitro lifetime, at body temperature, these formulations showed negligible improvement in the in vivo MB lifetime, as indicated by the TIC curves (S4, ESI†).
To control the saturation, f, or solubility of the gas in the solution, H, the medium containing the MBs was saturated with C6F14, which is a liquid at room temperature. The solubility of C6F14 in water, at 25 °C, is 2.7 × 10−4 mol L−1, which is equivalent to ∼0.05 μL for 1 mL of the lipid solution.11,50 The volume of C6F14 per mL of MB was varied between 0 and 15 μL mL−1 in the lipid solution (DPPC + 5% DSPE-PEG2000) to determine the optimum concentration at which the MBs have the longest lifetime. Fig. 4B shows the fraction of bubbles remaining in the medium at 37 °C, as a function of time for different volumes of C6F14 added to a 1 mL MB bolus. The data shows that MB lifetime was significantly increased upon the addition of C6F14 for concentrations ≥6 μL mL−1, with the 15 μL mL−1 sample producing the most stable MB population. However, the addition of such volumes of C6F14 adversely affected the MB production leading to a 10-fold reduction in the concentration of MBs produced. Thus the addition of 10 μL of C6F14 was selected as being the optimal amount. At this concentration MBs were still produced at a high concentration of ∼1 × 109 MB mL−1 whilst also increasing MBs stability. The MB decay rate was reduced to only 0.07% over the 2 h period.
Fig. 4C shows the calculated MB population profile expected based on the calculation of a typical MB distribution, given in Fig. 3B, and that the MBs decay in accordance with the modified Epstein–Plesset model proposed by Borden25 and Sarkar.27 In these models the surface tension was assumed to be 25 mN m−1 and the saturation, f, was taken as unity, and all other parameters are given in the ESI.† We note that in this model MBs of a diameter <0.5 μm were considered to not be observable optically and so bubbles were removed from the MB population when their size decreased below 0.5 μm. Essentially we see the expected trend that C4F10 > C3F8, however we note that the model predicts longer lifetimes than were experimentally observed, probably indicating that the MB surface tension, shell resistance and/or the degree of saturation are not accurately modelled in our system.
Fig. 5A shows the in vivo TIC for four different MB variants, comparing both the effect of the presence of C6F14 in the bolus and the concentration of the lipid. Comparison of the area under the curves for the cases with and without C6F14 clearly indicates that the presence of C6F14 significantly enhances MB persistence in the bloodstream. Furthermore, by doubling the lipid concentration, from 1 mg mL−1 to 2 mg mL−1, during the production of the MBs, the concentration of the MBs was doubled from 1 × 109 to 2 × 109 MB mL−1, and the lifetime was modestly enhanced, leading to a larger area beneath the TIC curves. Fig. 5B shows the TIC curves for our best in vivo MB in comparison to those obtained using commercially available MBs (Micromarker (MM), Definity).
 | ||
| Fig. 5 Ultrasound time intensity curves in mice aorta. (A) shows the effect of increasing the final lipid concentration, and saturating the lipid solution with C6F14 on MB lifetime for MBs made with DPPC + 5% PEG2000 and encapsulating C4F10 gas. (B) compares TIC curves for our improved in-house MBs and commercial MBs. | ||
For each MB population the intensity versus time curves were collected from 5 mice, and from each curve 6 different parameters were extracted, as shown in Fig. 6A. These can facilitate the comparison of the different MBs and aid the determination of the one with the best properties for in vivo imaging. These parameters are presented with the codes as follows: (1) rate of decay. This can be calculated from the slope of the curve after the peak point. The smaller the decay rate the longer the MB lifetime. (2) Peak enhancement, which is the point at the highest contrast intensity. This mainly depends on the MB shell properties and concentration. (3) Area under the curve, which is proportional to both the MB concentration and persistence time. A greater area indicates better quality and stability of MBs. (4) Time to peak, which is the time to reach the maximum intensity at the ROI. It is better if this is short. (5) Peak enhancement duration, which is the time at which there was the highest contrast intensity after injection. (6) Time to half peak contrast. The longer this parameter is, the more prolonged the lifetime of the MBs is at the ROI.
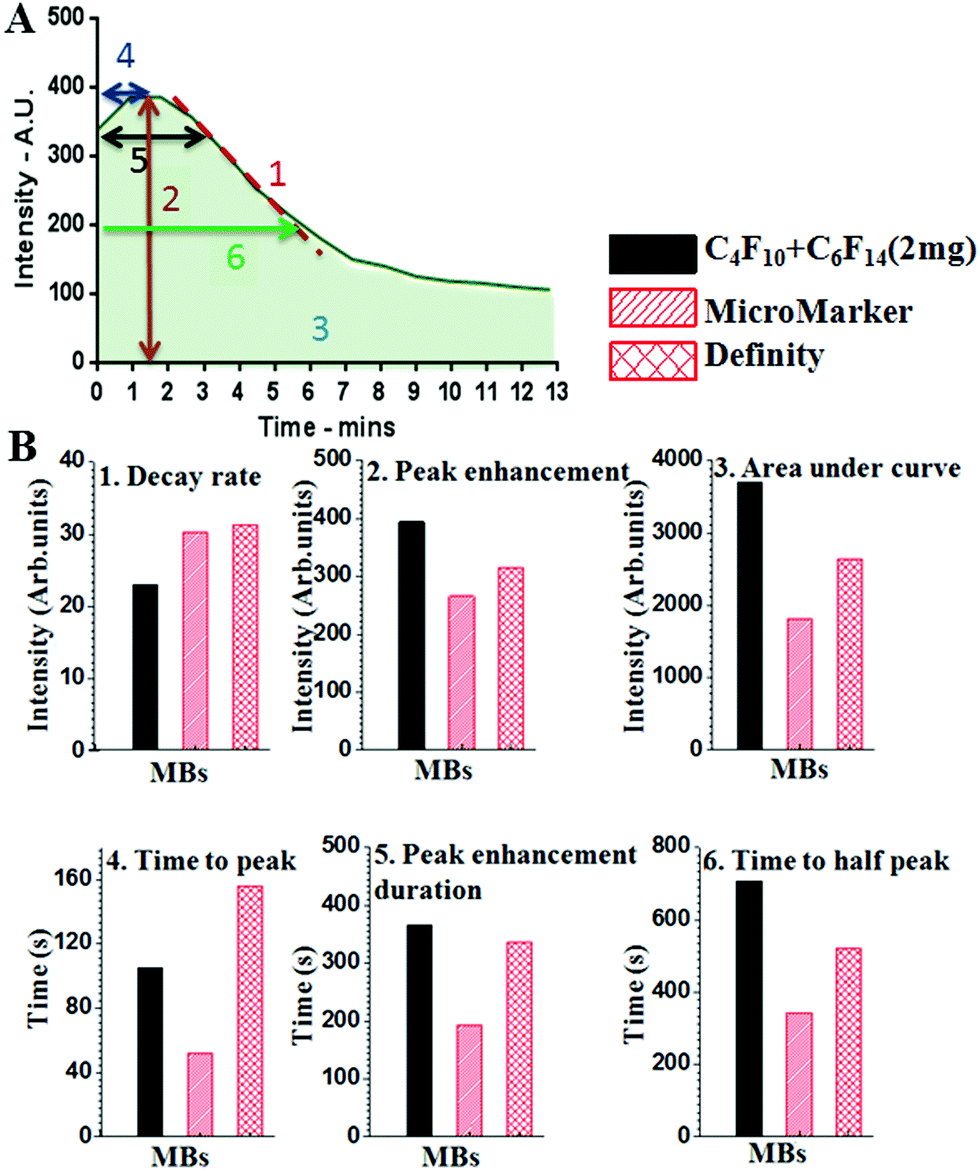 | ||
| Fig. 6 In vivo TIC analysis. (A) shows an example time–intensity curve (TIC) for MB flow in a mouse aorta and defines six key parameters for defining MB lifetime: (1) is the decay rate calculated from the slope of the curve after the peak intensity, the slower the decay rate the greater the MB stability; (2) is the peak enhancement, which is the maximum intensity signal, and is due to the MB shell properties as well as the gas used; (3) is the area under the curve, which indicates the signal from the total MB population present and therefore the concentration of MBs; (4) is the time to peak, which is the time to reach the peak signal intensity, it should be short enough to enable imaging within a realistic time-frame; (5) is the peak enhancement duration, which is the length of time the MB signal persists, this needs to be of sufficient duration to allow a useful imaging session following MB injection; (6) is the time to half peak, which is the time it takes the signal to reduce to half the peak intensity, which is a further measure of the duration of the signal. (B) The six parameters for our in-house MBs and two commercial MB formulations. In-house MBs showed (1) slower decay rate, (2) improved peak enhancement signal and (5) duration, (3) larger area under the curve, (4) shorter time after injection to reach the maximum intensity, and (6) significantly longer time to half peak, indicating better in vivo stability, better contrast enhancement and longer imaging time. | ||
Fig. 6B shows the data analysis for the TIC curves comparing the six parameters between our in-house MF-MBs and the commercially available MBs. The data suggests that the MF-MBs produced in-house display an improved peak enhancement and area under the curve, and a significantly longer time to half peak.
In order to understand the role of C6F14 in enhancing MB lifetime, a Langmuir trough was used to plot the relationship of the surface pressure, Π (mN m−1), versus the average molecular area, for a 1 mg mL−1 (DPPC + 5% PEG2000) solution spread at the air/water interface in the presence and absence of C6F14 in the sub-phase. Fig. 7 shows the compression isotherms of the monolayer. All conditions are fixed for both cases with the only difference being the sub-phase; either just with MilliQ water or MilliQ water plus C6F14 at the same concentration used during MB production (10 μL mL−1).
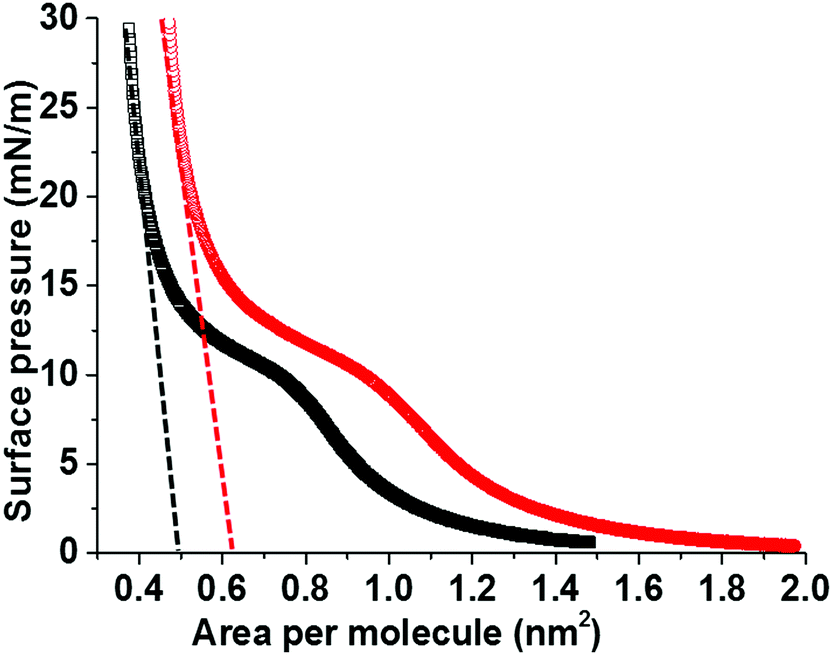 | ||
| Fig. 7 Langmuir isotherms of DPPC + 5% DSPE-PEG2000 monolayers, on water sub-phase (black) and water with 10 μL mL−1 C6F14 added to the sub-phase (red). Dashed lines show the extrapolated average molecular area for the DPPC/DSPE-PEG2000 phase. | ||
The mean molecular area was found to increase from 0.5 nm2 to 0.63 nm2 upon the inclusion of C6F14 in the sub-phase, which indicates the incorporation of the C6F14 molecules within the lipid layer. The fractional component of C6F14 in the MB shell was calculated to be 0.4, which means an estimation of 2 PFC molecules for every 5 lipid molecules. As a consequence of incorporating C6F14 (surface tension 11 mN m−151) at this proportion in the lipid layer, the surface tension of the monolayer is reduced from 25 mN m−127,38 to 19 mN m−1. Consequently, this leads to a reduction in the Laplace pressure from 50 × 103 N m−1 in the absence of C6F14, to 38 × 103 N m−1 in the presence of C6F14, which leads to an improved estimate in the MB lifetime. The hollow circles in Fig. 4C indicate the predicted enhancement in MB lifetime due to the reduction in surface tension. This is shown more clearly in S3 (ESI†).
The isothermal compressibility, C, and the compression modulus (elasticity), K, of the lipid modified interface are calculated from the extrapolated lines shown on the isotherm (Fig. 7). These calculations showed that the existence of PFC in the surrounding solution increased the compressibility (C) from 1.0 × 10−2 to 1.2 × 10−2 m mN−1, which corresponds to a decrease in the elasticity (K) from 97 mN m−1 to 88 mN m−1 in the presence of PFC molecules. These results are in good agreement with the previously published work by Krafft39 on the effect of saturating the surrounding air with different PFC gases on the compressibility and elasticity of lipid monolayers.
4 Discussion and conclusion
MB lifetime stability has been investigated by changing different factors that are known to have an effect on MB lifetime. The MB architectures have a shell comprising a DPPC/DSPE-PEG2000 binary system, the components of which are completely miscible below 15% PEG2000 concentrations forming a single condensed phase with low permeability.52 We have previously shown that reducing the PEG2000 concentration from 10 to 5% helped increase the MB lifetime.45 MB lifetime was also improved by changing the encapsulated gas from C3F8 to one with a larger molecular weight, i.e. C4F10. This decreases the coefficient of diffusivity from 7.45 × 10−10 to 6.9 × 10−10 m2 s−1.27 Combining the changes outlined above has resulted in a considerable improvement in the in vitro lifetime of the MBs, as discussed in the Results section above.Recently, C6F14 has been encapsulated together with Dox or gold nano-rods in nanoparticles and locally activated at the site of a tumour to form MBs, and C6F14 had been shown to enhance the acoustic imaging, cavitation and therapeutic delivery effects.53,54 In this work, saturating the medium surrounding the MBs with liquid C6F14 and using C4F10 in the gas core are considered key to the improvements in MB lifetime. The presence of C6F14 in the surrounding medium increased the stability and lifetime of the MBs in vitro and showed an improvement of up to 14 min in vivo (Fig. 5). We believe this improvement in stability arises for two reasons (Fig. 8); firstly, the addition of C6F14 effectively results in the saturation of the surrounding medium, reducing the ability for C4F10 to partition into the aqueous phase. Secondly, the incorporation of C6F14 molecules into the lipid shell of the MB, as concluded from Fig. 7, leads to a 25% reduction in the surface tension of the membrane, and hence also a reduction in the Laplace driving force for dissolution. This also modifies the mechanical properties of the shell, where a 17% increase in the compressibility is observed accompanied by a 10% decrease in the shell elasticity. The effect of saturating the gas medium with PFC gases on the lipid monolayers has been studied by Gerber et al.,55,56 who saturated the atmosphere above the Langmuir monolayer of DPPC with vapours of C6F14. They reported that saturating the gaseous medium with PFC gas has a fluidizing effect on the DPPC monolayer adsorption at the gas/water interface, as it prevents the liquid condensed phase formation and hinders lipid crystallisation. Krafft et al.39 also demonstrated that DMPC lipid shell MBs showed a 20% increase in compressibility of the membrane and a 26% decrease in the surface tension upon saturating the air with C6F14 in the gas medium. Krafft and Fainerman et al.57,58 theoretically modelled and practically showed that in case of DPPC monolayers, saturating the gas phase with C6F14− led to C6F14 molecules being adsorbed at the lipid surface, causing a reduction in the energy of attraction between the DPPC molecules, which leads to a fluidization of the monolayer. From the data shown here it appears that the surface tension alone is not a significant enough change to account for the increased MB stability and thus we also believe that the inclusion of the C6F14 molecules into the lipid chain must also increase the shell resistance.
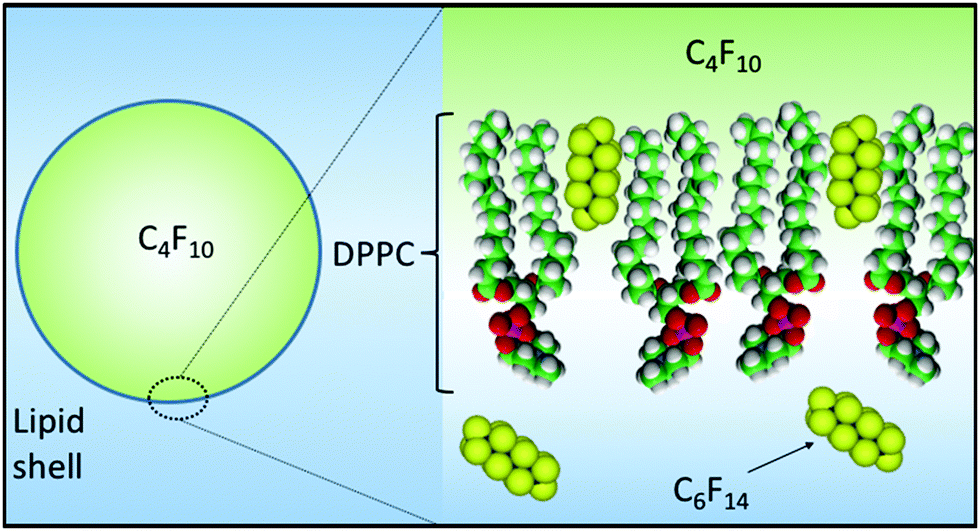 | ||
| Fig. 8 Schematic diagram showing (i) the incorporation of C6F14 into the MB shell thereby reducing surface tension and the Laplace driving force for dissolution and (ii) the saturation of the medium with C6F14 thereby reducing the solubility of C4F10 in the ambient phase. | ||
In vivo MB lifetime improved from 2 min to 14 min through a combination of changes to the lipid shell, the gas core and the addition of C6F14 to the medium. Furthermore, comparing in-house MF-MBs and commercial MBs (Fig. 5) showed that the sat-C4F10 MBs provided enhanced lifetime and contrast properties compared to their commercial counterparts.
In conclusion, control of MB properties including decay rate, intensity and enhancement duration, was achieved by changing the phospholipid shell composition, the encapsulated gas and, most significantly, the degree of saturation of the surrounding medium with C6F14. Our data suggests the C6F14 molecules are incorporated in the lipid shell reducing the surface tension and also increasing the mechanical compressibility and collapse pressure. MBs for drug delivery applications are likely to require longer lifetimes to allow for accumulation at the ROI for optimal dose delivery and will thus benefit greatly from such improvements.
Acknowledgements
The authors would like to acknowledge the EPSRC for funding (EP/I000623, EP/K023845) and thank the Microbubble Consortium (http://www.microbubbles.leeds.ac.uk/) at the University of Leeds for useful discussions. The data presented in this article are openly available from the University of Leeds Data Repository http://doi.org/10.5518/68.References
- S. B. Feinstein, Am. J. Physiol.: Heart Circ. Physiol., 2004, 287, H450–457 CrossRef CAS PubMed.
- J. R. Lindner, Nat. Rev. Drug Discovery, 2004, 3, 527–532 CrossRef CAS PubMed.
- B. Geers, I. Lentacker, N. N. Sanders, J. Demeester, S. Meairs and S. C. De Smedt, J. Controlled Release, 2011, 152, 249–256 CrossRef CAS PubMed.
- J. R. McLaughlan, N. Ingram, R. H. Abou-Saleh, S. Harput, T. Evans, S. D. Evans, L. Coletta and S. Freear, High-Frequency Subharmonic Imaging of Liposome-Loaded Microbubbles, Prague, 2013 Search PubMed.
- E. C. Unger, T. Porter, W. Culp, R. LaBell, T. Matsunaga and R. Zutshi, Adv. Drug Delivery Rev., 2004, 56, 1291–1314 CrossRef CAS PubMed.
- J. Zhang, Y. Luan, Z. Lyu, L. Wang, L. Xu, K. Yuan, F. Pan, M. Lai, Z. Liu and W. Chen, Nanoscale, 2015, 7, 14881–14888 RSC.
- F. Cavalieri, I. Finelli, M. Tortora, P. Mozetic, E. Chiessi, F. Polizio, T. B. Brismar and G. Paradossi, Chem. Mater., 2008, 20, 3254–3258 CrossRef CAS.
- B. Geers, I. Lentacker, N. N. Sanders, J. Demeester, S. Meairs and S. C. De Smedt, J. Controlled Release, 2011, 152, 249–256 CrossRef CAS PubMed.
- R. Chen, Y. Luan, Z. Liu, W. Song, L. Wu, M. Li, J. Yang, X. Liu, T. Wang, J. Liu and Z. Ye, BioMed Res. Int., 2015, 2015, 514234 Search PubMed.
- Y. Luan, Z. Chen, G. Xie, J. Chen, A. Lu, C. Li, H. Fu, Z. Ma and J. Wang, J. Nanosci. Nanotechnol., 2015, 15, 1357–1361 CrossRef CAS PubMed.
- E. G. Schutt, D. H. Klein, R. M. Mattrey and J. G. Riess, Angew. Chem., Int. Ed., 2003, 42, 3218–3235 CrossRef CAS PubMed.
- C. Christiansen, H. Kryvi, P. C. Sontum and T. Skotland, Biotechnol. Appl. Biochem., 1994, 19(pt 3), 307–320 CAS.
- G. Pu, M. A. Borden and M. L. Longo, Langmuir, 2006, 22, 2993–2999 CrossRef CAS PubMed.
- E. Stride and M. Edirisinghe, Med. Biol. Eng. Comput., 2009, 47, 883–892 Search PubMed.
- J. R. Lindner, M. P. Coggins, S. Kaul, A. L. Klibanov, G. H. Brandenburger and K. Ley, Circulation, 2000, 101, 668–675 CrossRef CAS PubMed.
- J. R. Lindner, P. A. Dayton, M. P. Coggins, K. Ley, J. Song, K. Ferrara and S. Kaul, Circulation, 2000, 102, 531–538 CrossRef CAS PubMed.
- U. Farook, H. B. Zhang, M. J. Edirisinghe, E. Stride and N. Saffari, J. Biomed. Eng., 2007, 29, 749–754 CAS.
- M. Hashimoto and G. M. Whitesides, Small, 2010, 6, 1051–1059 CrossRef CAS PubMed.
- S. A. Peyman, R. H. Abou-Saleh, J. R. McLaughlan, N. Ingram, B. R. G. Johnson, K. Critchley, S. Freear, J. A. Evans, A. F. Markham, P. L. Coletta and S. D. Evans, Lab Chip, 2012, 12, 4544–4552 RSC.
- E. Talu, K. Hettiarachchi, R. L. Powell, A. P. Lee, P. A. Dayton and M. L. Longo, Langmuir, 2008, 24, 1745–1749 CrossRef CAS PubMed.
- E. Talu, K. Hettiarachchi, S. Zhao, R. L. Powell, A. P. Lee, M. L. Longo and P. A. Dayton, Mol. Imaging, 2007, 6, 384–392 Search PubMed.
- S. A. Peyman, J. R. McLaughlan, R. H. Abou-Saleh, G. Marston, B. R. Johnson, S. Freear, P. L. Coletta, A. F. Markham and S. D. Evans, Lab Chip, 2016, 16, 679–687 RSC.
- J. J. Kwan and M. A. Borden, Adv. Colloid Interface Sci., 2012, 183, 82–99 CrossRef PubMed.
- P. S. Epstein and M. S. Plesset, Adv. Colloid Interface Sci., 1950, 183, 82–99 Search PubMed.
- M. A. Borden and M. L. Longo, Langmuir, 2002, 18, 9225–9233 CrossRef CAS.
- A. Kabalnov, D. Klein, T. Pelura, E. Schutt and J. Weers, Ultrasound Med. Biol., 1998, 24, 739–749 CrossRef CAS PubMed.
- K. Sarkar, A. Katiyar and P. Jain, Ultrasound Med. Biol., 2009, 35, 1385–1396 CrossRef PubMed.
- E. G. Schutt, D. P. Evitts, C. D. Anderson and J. G. Weers, US Pat., US 6953569 B2, 2005 Search PubMed.
- H. D. Van Liew and M. E. Burkard, Adv. Exp. Med. Biol., 1997, 411, 395–401 CrossRef CAS PubMed.
- D. E. Yount, Aviat., Space Environ. Med., 1979, 50, 44–50 CAS.
- H. D. Van Liew and S. Raychaudhuri, J. Appl. Physiol., 1997, 82, 2045–2053 CAS.
- C. Li, J. Chen, J. Wang, Z. Ma, P. Han, Y. Luan and A. Lu, Sci. Total Environ., 2015, 521–522, 101–107 CrossRef CAS PubMed.
- B. A. Lewis and D. M. Engelman, J. Mol. Biol., 1983, 166, 211–217 CrossRef CAS PubMed.
- K. H. Kim, S. Q. Choi, J. A. Zasadzinski and T. M. Squires, Soft Matter, 2011, 7, 7782–7789 RSC.
- D. H. Kim, M. J. Costello, P. B. Duncan and D. Needham, Langmuir, 2003, 19, 8455–8466 CrossRef CAS.
- J. J. Kwan and M. A. Borden, Soft Matter, 2012, 8, 4756–4766 RSC.
- K. Ferrara, R. Pollard and M. Borden, Annu. Rev. Biomed. Eng., 2007, 9, 415–447 CrossRef CAS PubMed.
- A. Katiyar and K. Sarkar, J. Colloid Interface Sci., 2010, 343, 42–47 CrossRef CAS PubMed.
- C. Szijjarto, S. Rossi, G. Waton and M. P. Krafft, Langmuir, 2012, 28, 1182–1189 CrossRef CAS PubMed.
- S. Rossi, G. Waton and M. P. Krafft, Langmuir, 2010, 26, 1649–1655 CrossRef CAS PubMed.
- A. Kabalnov, J. Bradley, S. Flaim, D. Klein, T. Pelura, B. Peters, S. Otto, J. Reynolds, E. Schutt and J. Weers, Ultrasound Med. Biol., 1998, 24, 751–760 CrossRef CAS PubMed.
- E. G. Schutt, C. D. Anderson and D. P. Evitts, US Pat., 5605673, 1997 Search PubMed.
- J. J. Kwan and M. A. Borden, Langmuir, 2010, 26, 6542–6548 CrossRef CAS PubMed.
- R. H. Abou-Saleh, S. A. Peyman, K. Critchley, S. D. Evans and N. H. Thomson, Langmuir, 2013, 29, 4096–4103 CrossRef CAS PubMed.
- R. H. Abou-Saleh, S. D. Evans and N. H. Thomson, Langmuir, 2014, 30, 5557–5563 CrossRef CAS PubMed.
- T. V. Rooij, Y. Luan, G. Renaud, A. F. W. van der Steen, M. Versluis, N. De Jong and K. Kooiman, Ultrasound Med. Biol., 2015, 41, 1432–1445 CrossRef PubMed.
- K. Kooiman, T. J. A. Kokhuis, T. V. Rooij, I. Skachkov, A. Nigg, J. G. Bosch, A. F. W. van der Steen, W. A. van Cappellen and N. De Jong, Eur. J. Lipid Sci. Technol., 2014, 116, 1217–1227 CrossRef CAS.
- A. K. Kenworthy, S. A. Simon and T. J. McIntosh, Biophys. J., 1995, 68, 1903–1920 CrossRef CAS PubMed.
- B. W. Ninham and P. Lo Nostro, Molecular forces and self assambly: in colloid, nano science and biology, Cambridge University Press, USA, 2010, ch. 8, pp. 232–249 Search PubMed.
- A. S. Kabalnov, K. N. Makarov and O. V. Scherbakova, J. Fluorine Chem., 1990, 50, 271–284 CrossRef CAS.
- Y. Luan, J. Xu, B. Huang, X. Liu, Y. Liu, L. Chen and X. Chu, Zhonghua Xueyexue Zazhi, 2015, 36, 286–290 CAS.
- M. M. Lozano and M. L. Longo, Soft Matter, 2009, 5, 1822–1834 RSC.
- J. Zhong, S. Yang, L. Wen and D. Xing, J. Controlled Release, 2016, 226, 77–87 CrossRef CAS PubMed.
- N. Zhang, X. Cai, W. Gao, R. Wang, C. Xu, Y. Yao, L. Hao, D. Sheng, H. Chen, Z. Wang and Y. Zheng, Theranostics, 2016, 6, 404–417 CrossRef CAS PubMed.
- F. Gerber, M. P. Krafft, T. F. Vandamme, M. Goldmann and P. Fontaine, Angew. Chem., Int. Ed., 2005, 44, 2749–2752 CrossRef CAS PubMed.
- F. Gerber, M. P. Krafft, T. F. Vandamme, M. Goldmann and P. Fontaine, Biophys. J., 2006, 90, 3184–3192 CrossRef CAS PubMed.
- M. P. Krafft, V. B. Fainerman and R. Miller, Colloid Polym. Sci., 2015, 293, 3091–3097 CAS.
- V. B. Fainerman, E. V. Aksenenko and R. Miller, Adv. Colloid Interface Sci., 2015 DOI:10.1016/j.cis.2015.11.004.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sm00956e |
| This journal is © The Royal Society of Chemistry 2016 |