
Crystallization-driven one-dimensional self-assembly of polyethylene-b-poly(tert-butylacrylate) diblock copolymers in DMF: effects of crystallization temperature and the corona-forming block†
Bin
Fan
,
Lei
Liu
,
Jun-Huan
Li
,
Xi-Xian
Ke
,
Jun-Ting
Xu
*,
Bin-Yang
Du
and
Zhi-Qiang
Fan
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science & Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: xujt@zju.edu.cn
First published on 28th September 2015
Abstract
Crystallization-driven self-assembly of polyethylene-b-poly(tert-butylacrylate) (PE-b-PtBA) block copolymers (BCPs) in N,N-dimethyl formamide (DMF) was studied. It is found that all three PE-b-PtBA BCPs used in this work can self-assemble into one-dimensional crystalline cylindrical micelles. When the BCP solution is cooled to crystallization temperature (Tc) from 130 °C, the seed micelles may be produced via two competitive processes in the initial period: stepwise micellization/crystallization and simultaneous crystallization/micellization. Subsequently, the seed micelles can undergo growth driven by the epitaxial crystallization of the unimers. The lengths of both the seed micelles and the grown micelles are longer for the BCP with a longer PtBA block at a higher Tc. Quasi-living growth of the PE-b-PtBA crystalline cylindrical micelles is achieved at a higher Tc. A longer PtBA block evidently retards the attachment of unimers to the crystalline micelles, leading to a slower growth rate.
Introduction
Block copolymers (BCPs) can form micelles of different morphologies in selective solvents, driven by the solvent-philic/solvent-phobic force. Recently, BCP micelles with a crystalline core-forming block, such as polyferrocenyldimethylsilane (PFDMS),1–4 polyethylene (PE),5–12 poly(ethylene oxide) (PEO),13–16 poly(ε-caprolactone) (PCL),17–24 poly(L-lactide) (PLLA),25–29 polyacrylonitrile (PAN),30 poly(p-dioxanone),31,32 and polythiophene,33,34 have received considerable attention. Halperin et al. showed that the “crystalline micelles” tended to have platelet morphology due to the crystallization of the core-forming block.35 However, the repulsion force among the soluble corona-forming chains may compete with the crystallization force of the core-forming block, leading to other micellar morphologies, such as a cylinder and a sphere. As a result, the crystallization force plays a vital role in the morphology of crystalline micelles of BCPs.36 Crystallization adds lots of unique characteristics to the BCP micelles, such as “living growth” and the construction of “block co-micelles” and micelles with a complicated structure,37–41 which cannot be achieved in the amorphous BCP micelles so far.Control and regulation of micellar morphology and size are important, since they can largely determine the function and performance of the micelles.42,43 There are generally two methods to prepare crystalline micelles of BCPs with well-controlled morphology and size. Hillmyer proposed a stepwise micellization/crystallization method.12 In this protocol, the amorphous micelles were first prepared above the melting temperature of the core-forming block, and then the micellar solution was cooled to a lower temperature to allow crystallization of the micellar core. When crystallization is confined inside the core of single micelles, the overall morphology of the amorphous micelles at high temperature may be retained after crystallization. As a consequence, the morphology and aggregation parameters of the crystalline micelles can be regulated and designed based on the theory for amorphous BCPs. However, this method is only applicable to the BCP/solvent systems having a very low and nearly invariable critical micellization concentration (CMC) with temperature. Because almost all polymer chains exist in the form of micelles at higher temperature, no more micelles will be generated upon lowering temperature. If the CMC of BCPs changes greatly with temperature, micelles with a different morphology may be formed upon cooling. Moreover, if break-out crystallization occurs among different micelles, the micellar morphology will be altered as well.13,14 The second method is “self-seeding”,44,45 which imitates the way for culture of polymer single crystals. In this method, a small amount of crystalline seed micelles was first prepared, then the unimers in the solution or extra added unimers can epitaxially grow on the active end of seed micelles. The shape and size of the crystalline micelles can be readily regulated using this method. However, a high solubility of the BCPs in the solvent (i.e. a high CMC) is necessary for this method. On the one hand, no additional crystalline seed micelles will be produced when the dissolved unimers are cooled from a higher temperature or the extra unimers dissolved in a co-solvent of both blocks are added. The reason why the seed crystalline micelles can survive at the self-seeding temperature is due to the solidification effect of crystallization in spite of the large CMC. On the other hand, a large amount of unimers in the solution can grow on the active ends of the seed crystalline micelles.
Nevertheless, most of the BCP/solvent systems are between the situations required by the above-mentioned two methods, i.e. the BCP has an intermediate CMC in the selective solvent and the CMC varies with temperature. When temperature is lowered, micellization and growth of the crystalline micelles will occur simultaneously. This may lead to difficulty in control of the shape and size of crystalline micelles of BCPs. This is the reason why Winnik and Manners pointed out that, in the self-seeding method the self-nucleation of the crystals (micellization of unimers) upon cooling or addition of a large amount of co-solvent should be avoided.45,46 So far, the formation mechanism and growth of the crystalline micelles under such a complicated situation has not been extensively studied yet, though its importance is needless to say. Herein three polyethylene-b-poly(tert-butylacrylate) (PE-b-PtBA) BCPs were synthesized and their self-assembly in N,N-dimethyl formamide (DMF) was studied. The effects of the chain structure and temperature on micellization and growth of the crystalline cylindrical micelles were discussed.
Cylindrical or worm-like BCP micelles have distinguished characteristics due to their one-dimensional shape and high aspect ratio.42,43 However, PE-containing BCPs usually form platelet or disc-like micelles and cylindrical micelles are difficult to obtain,5–8 possibly due to the strong crystallizability of PE. So far cylindrical micelles have been only reported for polystyrene-b-polyethylene-b-poly(methyl methacrylate) (PS-b-PE-b-PMMA) triblock terpolymers in toluene and THF and poly(N,N-dimethylacrylamide)-b-polyethylene in water.9–12 In the present work, we found that the PE-b-PtBA BCPs with different compositions could readily self-assemble into cylindrical micelles in DMF. On the other hand, the length of the cylindrical micelles could be regulated by crystallization temperature and the chain structure of the PE-b-PtBA BCPs.
Experimental section
Materials
The PE-b-PtBA BCPs were synthesized according to the procedure reported in the literature.47 Hydroxyl-terminated PE (PE-OH) with a narrow molecular weight distribution was first prepared through polyhomologation of dimethylsulfoxonium methylide and the subsequent oxidation of trisorganoborane using trimethylamine-N-oxide dihydrate.48 The end hydroxyl group in PE-OH was esterified with 2-bromo-2-methylpropionyl bromide, which was used as a macroinitiator for atom transfer radical polymerization (ATRP) of tert-butylacrylate. Details of synthesis and characterization of PE-b-PtBA BCPs are described in the ESI.† The sample information of three PE-b-PtBA BCPs used in the present work is given in Table 1.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800
800Preparation of the micellar solutions
PE-b-PtBA was dispersed in DMF with a concentration of 0.042 mmol L−1 and the solution was stirred at 130 °C in an oil bath for 1 h. Subsequently, the homogeneous solution was transferred to another oil bath with preset crystallization temperature (Tc) and held for different times. Four Tcs, 25 °C, 50 °C, 70 °C and 90 °C, were selected. Crystallization of the core-forming block inside the micelles was confirmed by synchrotron wide angle X-ray diffraction (Fig. S3 in the ESI†).Characterization of the micelles
Dynamic laser scattering (DLS) measurements were performed on a Brookhaven Instrument BI-200SM with a laser wavelength of 636 nm at 25 °C with a scattering angle of 90°. The data were analyzed with the COUNTIN program supplied by Brookhaven. Transmission electron microscopy (TEM) characterization was carried out on a JEOL JEM-1230 electron microscope at an acceleration voltage of 80 kV. If not specifically indicated, the TEM samples were prepared by dropping 2 μL of the micellar solution onto carbon-coated copper grids and were allowed to dry in air at room temperature. Sometimes the samples for TEM observation were also prepared by freeze-drying method. The hot micellar solution was taken out by a pre-heated burette and dropped onto the carbon-coated copper grids frozen with liquid nitrogen, and then freeze-dried under vacuum. Since the coated carbon film on the copper grids is easily broken upon freeze-drying, this method is only adopted when it is necessary. The contour lengths of the cylindrical micelles were analyzed with Image-Pro Plus software. In a general procedure, we firstly draw the outline of the long cylindrical micelles in the TEM micrograph. The pixels of the micelles along the longest dimension were automatically measured by the software and then the micellar length was calculated with respect to the scale bar. Moreover, the short cylindrical micelles formed by the unimers during TEM sample preparation were omitted and screened from the images to avoid any possible interference. In order to ensure accuracy and reliability, more than 100 micelles from different images were traced for each sample and only those micelles with a clear contour were chosen for analysis.Results and discussion
Possible pathways for micellization and crystallization of PE-b-PtBA in DMF
Since it is difficult to find a co-solvent that can dissolve both PE and PtBA at room temperature, the PE-b-PtBA micelles cannot be prepared by addition of the non-solvent of PE to the solution of the BCPs in a co-solvent, which is the most frequently used method for the preparation of BCP micelles.49,50 The PE-b-PtBA micelles were prepared through a stepwise heating and quenching process. The PE-b-PtBA/DMF solution was first heated to 130 °C, which is above the melting temperature of the PE block, and held for 1 h. In order to know the starting state of PE-b-PtBA in DMF at 130 °C, TEM characterization was carried out. The samples for TEM observation were prepared by a common process and a freeze-drying method, respectively. As illustrated in Fig. 1a, one can see that, when the solution was dried at room temperature, lots of short cylinders are formed. By contrast, for the freeze-dried sample, no short cylinders but only a few spheres are observed (Fig. 1b). This shows that the sample preparation method for TEM observation is quite important. The short cylinders in Fig. 1a are an artifact resulting from crystallization and micellization of PE-b-PtBA unimers at room temperature during sample preparation, which is not the real situation in the hot solution. Crystallization of the PE block can be hindered by freeze-drying.51 This also leads to the low contrast of the TEM image in Fig. 1b. The TEM result reveals that there are only small amounts of spherical PE-b-PtBA micelles in DMF at 130 °C and most of the PE-b-PtBA chains are dissolved and exist as unimers. This implies that most of PE100 can be dissolved in DMF at 130 °C, possibly due to its low molecular weight. | ||
| Fig. 1 TEM micrographs of PE100-b-PtBA70/DMF (0.042 mmol L−1) solution after aging at 130 °C for 1 h with different TEM preparation processes. (a) Prepared by dropping the micellar solution onto carbon-coated copper grids and dried in air at room temperature; (b) prepared by a freeze-drying method. The red squares highlight the amorphous spherical micelles formed at 130 °C. | ||
After being held at 130 °C for 1 h, the PE-b-PtBA/DMF solution was cooled to a lower temperature to allow micellization and/or crystallization of PE-b-PtBA. The DSC crystallization curves of the PE-b-PtBA/DMF mixtures at a cooling rate of 1 °C min−1 are shown in Fig. S4 in the ESI.† It is found that the crystallization peak temperatures (Tcps) of PE100-b-PtBA30, PE100-b-PtBA48 and PE100-b-PtBA70 in DMF are 99.0 °C, 69.2 °C and 64.5 °C, respectively. However, PE100-b-PtBA48 and PE100-b-PtBA70 start to crystallize at around 90 °C in DMF. As a result, the highest Tc for the preparation of PE-b-PtBA micelles in DMF was selected as 90 °C in the present work. The PE-b-PtBA micelles were also prepared at other three Tcs (70 °C, 50 °C and 25 °C). Fig. 2 shows the TEM micrographs of PE100-b-PtBA70/DMF after being held at 90 °C for 1 h. When the TEM sample is prepared at room temperature, a mixture of long and short cylinders is observed. The length of the long cylinders ranges from 400 nm to 800 nm, while the short cylinders are only about 20 nm. Because of the large difference in the length, these two populations of the cylinder can be readily distinguished. The sample for TEM observation was prepared by a freeze-drying method as well. As can be seen in Fig. 2b, only long cylinders are observed and the short ones nearly completely disappear. As a result, the short cylinders are produced by the unimers at room temperature during sample preparation, which do not exist in the solution at 90 °C. By contrast, the long cylinders are produced upon being held at 90 °C.
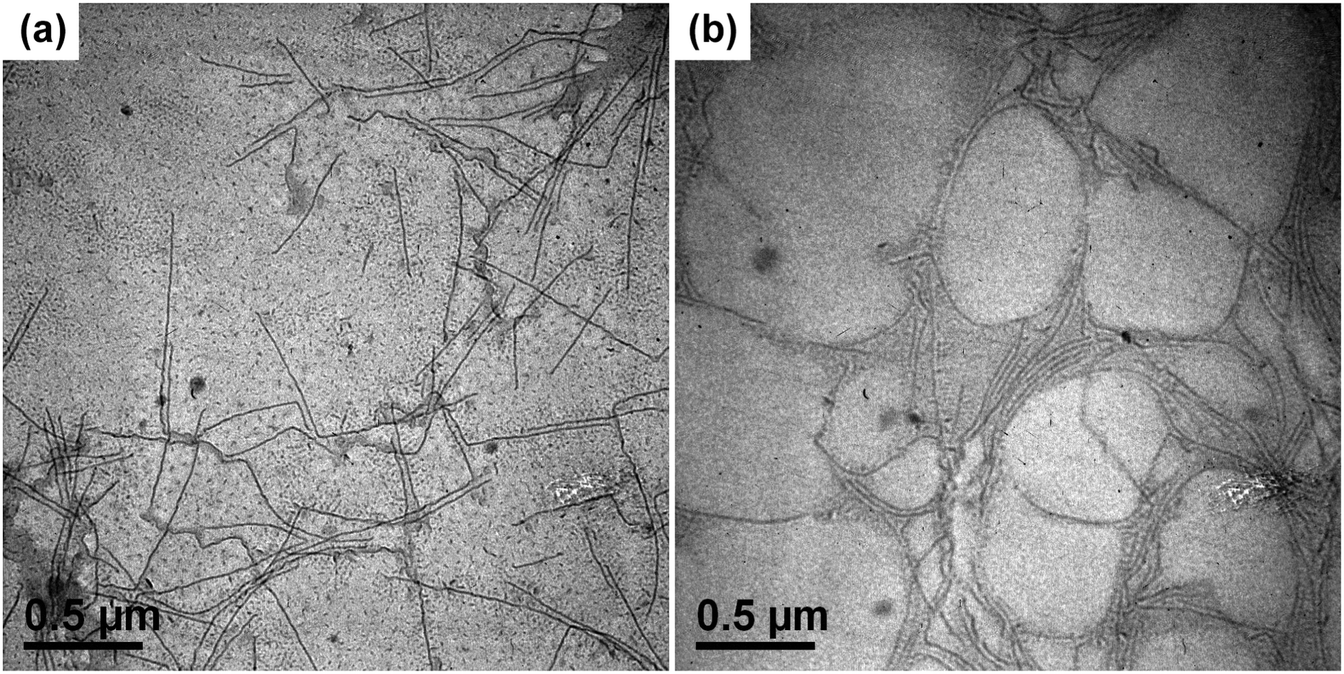 | ||
| Fig. 2 TEM micrographs of PE100-b-PtBA70/DMF (0.042 mmol L−1) after aging at 130 °C for 1 h and subsequent crystallization at 90 °C for 1 h. (a) Prepared by dropping the micellar solution onto carbon-coated copper grids and dried in air at room temperature; (b) prepared by freeze-drying method. | ||
In order to probe the formation mechanism of the long cylindrical micelles, the micellar morphology of PE100-b-PtBA70 after being held at 90 °C for different times was characterized, as illustrated in Fig. 3. Since the short cylinder micelles are produced by unimers during TEM sample preparation, only the long cylindrical micelles are discussed. One can see that, even at a very short crystallization time (5 min), the length of the long cylinders can reach 200–500 nm. With prolongation of holding time, the micellar length does not change too much, indicating a slow crystallization rate. The DSC result also confirms that the Tc of 90 °C is very close to the onset crystallization temperature of PE100-b-PtBA70 in DMF (Fig. S4 in ESI†). At such a high Tc, the crystallization rate of PE100-b-PtBA70 would be very slow. Consequently, we speculate that the instantaneous formation of the long cylindrical seed micelles at Tc = 90 °C is driven by micellization of the unimers, instead of crystallization. Nose and co-workers found that long cylindrical amorphous micelles of polystyrene-b-poly(dimethylsiloxane) (PS-b-PDMS) could be formed within 100 s by consuming large amounts of excess free unimers in the solution.52 They proposed that, because of the high immiscibility of the PS and PDMS blocks, there was a high free energy barrier for the PS block to go through the outer PDMS layer, leading to a slow growth rate of the spherical micelles.52 By contrast, the cylindrical micelles could exhibit a large growth rate via attachment of the unimers in the solution on the exposed ends of the cylindrical micelles. Similarly, the PE and PtBA blocks used in the present work are highly immiscible, thus long cylindrical micelles can also be produced quickly via micellization of the unimers. After micellization, nucleation may readily occur for the PE blocks aggregated in the core of the micelles due to the large dimension of the micelles in length and the Tc lower than the onset crystallization temperature of BCPs in DMF (Fig. S4 in ESI†). This means that the crystalline PE100-b-PtBA70 micelles at Tc = 90 °C may be formed via a stepwise micellization/crystallization process.
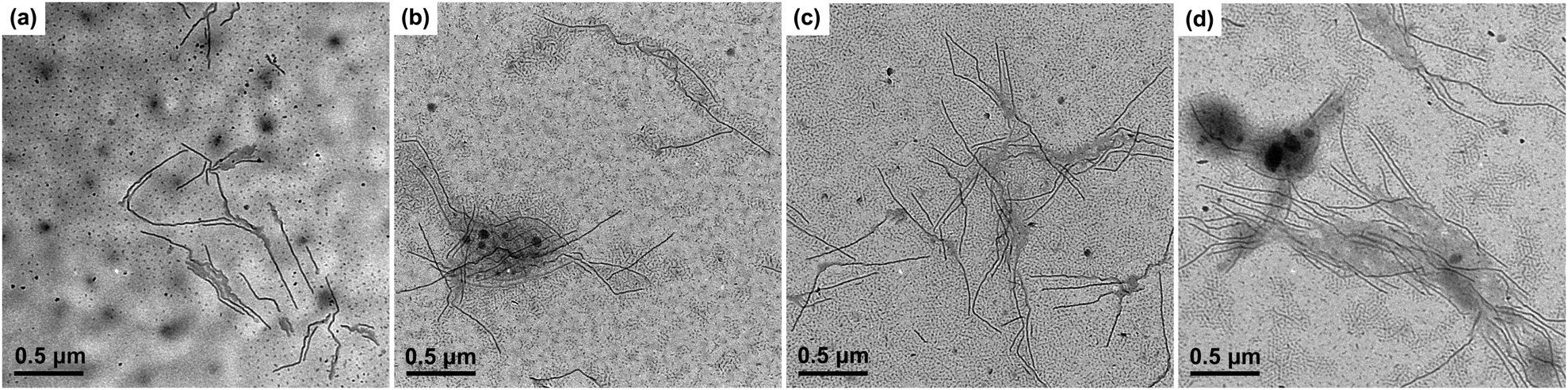 | ||
| Fig. 3 TEM micrographs of PE100-b-PtBA70/DMF (0.042 mmol L−1) micelles after crystallization at 90 °C for different times. (a) 5 min, (b) 10 min, (c) 15 min, (d) 25 min. The samples for TEM observation were prepared by dropping the micellar solution onto carbon-coated copper grids and dried in air at room temperature. | ||
On the other hand, the PE-b-PtBA unimers may also crystallize from the solution to form crystalline micelles directly (simultaneous crystallization/micellization) when the crystallization rate of the PE block is fast, which can compete with the stepwise micellization/crystallization process. The competition between these two processes is dependent on Tc and affected by the chain structure of the BCPs. As a result, we examined the effects of Tc and the corona-forming block on the formation of the seed micelles. Fig. 4 shows the TEM images of PE100-b-PtBA48, PE100-b-PtBA70 and PE100-b-PtBA30 micelles in DMF after being held at 90 °C or 50 °C for 5 min. Comparing Fig. 3a, Fig. 4a and b for different PE-b-PtBA BCPs at Tc = 90 °C, one can see that the length of the long cylindrical micelles is reduced upon decreasing the length of the PtBA block. Lowering the Tc has a similar effect with shortening the PtBA block, as shown by Fig. 3a and 4c. The effect of Tc is even stronger than that of the PtBA block length. Since the crystallizability of the PE-b-PtBA BCPs with a shorter PtBA block and at a lower Tc is stronger, the simultaneous crystallization/micellization process may overwhelm the stepwise micellization/crystallization process. It should be noted that, although we are sure about the occurrence of these two competitive processes based on the experimental phenomena, so far we cannot clearly distinguish the micelles formed by these two processes in a specific sample.
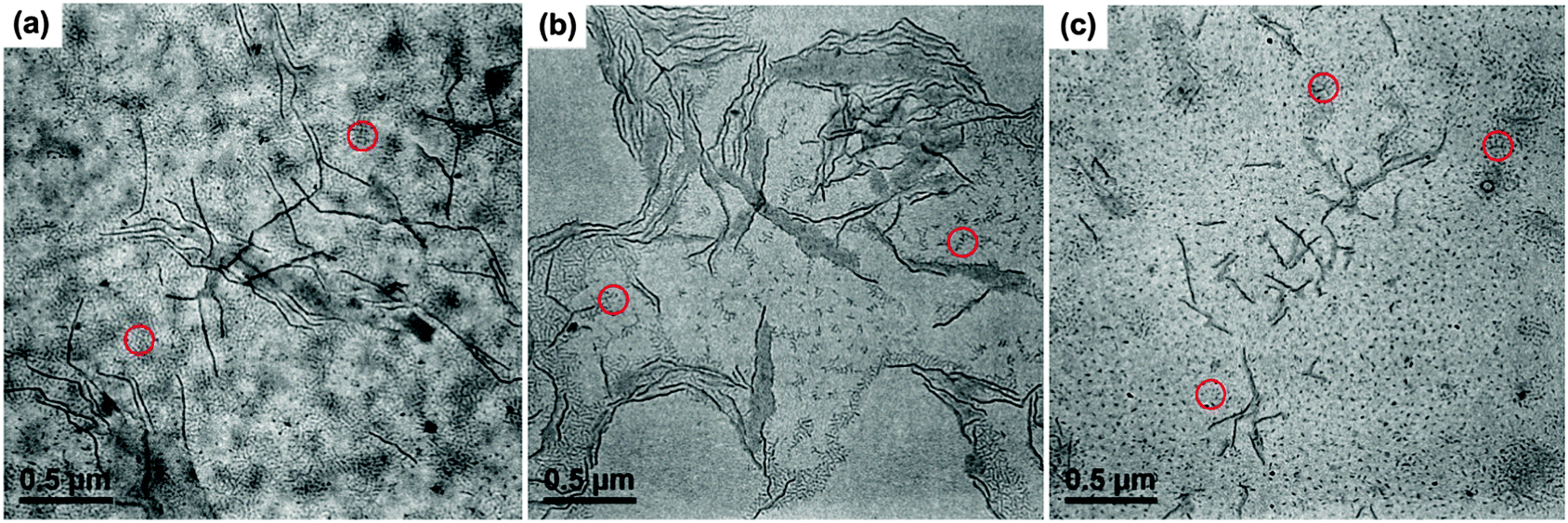 | ||
| Fig. 4 TEM images of PE-b-PtBA/DMF (0.042 mmol L−1) micelles in DMF after crystallization at Tc for 5 min. (a) PE100-b-PtBA48 at Tc = 90 °C; (b) PE100-b-PtBA30 at Tc = 90 °C; (c) PE100-b-PtBA70 at Tc = 50 °C. The short cylinders formed by un-consumed unimers during TEM sample preparation (highlighted by the red circles) are neglected in length analysis. | ||
As can be seen from Fig. 3 and 4, longer cylindrical micelles are formed for the BCP with a longer PtBA block at a higher Tc. Moreover, under such a condition, the solubility of the PE-b-PtBA BCPs is higher, meaning that fewer polymer chains form micelles. Based on these two facts, we can deduce that the number of the micelles is smaller for the BCP with a longer PtBA block at a higher Tc, which is hard to be seen from the TEM images due to the inhomogeneous location of the micelles on the grids.
We also notice that small amounts of amorphous spherical micelles are formed at 130 °C. There are two possibilities for them after being held at Tc. Firstly, these micelles may crystallize to form crystalline micelles when the solution is cooled to Tc. However, due to the small micellar size and confinement, nucleation inside a spherical micelle at a high Tc is difficult,14 leading to a slow crystallization rate. Therefore, the crystalline micelles transformed from the amorphous spherical micelles can be neglected in the following discussion because of the small number and a slow crystallization rate. Secondly, these amorphous spherical micelles may be thermodynamically equilibrated and form cylindrical micelles at Tc, just like unimers. In this case, there is no need of additional consideration of them either.
Effects of crystallization temperature and the corona-forming block on the length of grown micelles
Above result shows that the crystalline seed micelles can be formed by either a stepwise micellization/crystallization or a simultaneous crystallization/micellization process at a Tc. However, there are still lots of dissolved unimers left in the solution after the crystalline seed micelles are produced, as indicated by the shorter cylindrical micelles formed upon sample preparation. The formed crystalline micelles can act as seed micelles and the unimers may epitaxially grow on them, leading to an increase in the length of the cylindrical micelles. In the previous section, we have revealed that a lower Tc and a shorter PtBA block result in more but shorter seed micelles. Here the effects of these two factors on the length of the grown micelles were further investigated. The TEM images of PE100-b-PtBA70 micelles after growth at four selected Tcs for 12 h and the TEM micrographs of PE100-b-PtBA70, PE100-b-PtBA48 and PE100-b-PtBA30 after growth at Tc = 90 °C for 60 h are shown in Fig. 5 and 6, respectively. It is found that all these three PE-b-PtBA BCPs can self-assemble into one-dimensional cylindrical micelles in DMF solution driven by crystallization of the PE block. Because of the strong crystallizability of PE, the PE-containing BCPs usually form disc-like or platelet crystalline micelles,5–8 whereas cylindrical micelles are seldom reported for PE-containing BCPs in the literature.9–12 Due to the good solubility of DMF toward PtBA as well as the slight solubility toward PE, the reduced crystallizability of PE and the highly swollen PtBA coils may be responsible for the formation of cylindrical crystalline micelles of PE-b-PtBA in DMF. This shows that the morphology of crystalline micelles of PE-containing BCPs can be regulated by solvent and the corona-forming block. | ||
| Fig. 5 TEM micrographs of PE100-b-PtBA70 micelles after growth at selected Tcs for 12 h. The samples for TEM observation were prepared by dropping the micellar solution onto carbon-coated copper grids and dried in air at room temperature. (a) Tc = 25 °C, (b) Tc = 50 °C, (c) Tc = 70 °C, (d) Tc = 90 °C. The short cylinders (like those highlighted by red circles) are neglected in later analysis. | ||
 | ||
| Fig. 6 TEM micrographs of PE-b-PtBA micelles after growth at Tc = 90 °C for 60 h. The samples for TEM observation were prepared by dropping the micellar solution onto carbon-coated copper grids and dried in air at room temperature. (a) PE100-b-PtBA30, (b) PE100-b-PtBA48, (c) PE100-b-PtBA70. The short cylinders (like those highlighted by red circles) are neglected in later analysis. | ||
Since the samples for TEM observation in Fig. 5 and 6 were prepared by drying at room temperature, both short and long cylinders are observed, which are generated by the un-consumed unimers in the solution during sample preparation and by growth of the unimers on the seed micelles, respectively. These short cylinders (20–40 nm) are neglected in our latter analysis. Because the amorphous micelles PE-b-PtBA in DMF at 130 °C are quite few, growth of the unimers on the short seed micelles is also ignored. One can see from Fig. 5 that the long cylindrical micelles become longer as Tc increases. On the other hand, as shown in Fig. 6, at the same growth time and Tc, the length of the long cylindrical micelles increases with increasing molecular weight of the PtBA block. These can be seen more clearly when the short cylinders formed by the unimers cooling from Tc to room temperature are screened out from the TEM images, which are shown in Fig. S5 and S6 of ESI.† Besides, comparing Fig. 5d and 6c for PE100-b-PtBA70 grown at Tc = 90 °C for different times, we can find that the length of the long cylindrical micelles increases with growth time.
The lengths of the crystalline cylindrical micelles after growth were further quantitatively and statistically measured from the TEM images. The number-average length (Ln) and mass-average length (Lw) of the cylindrical micelles can be calculated on the basis of the following equations:
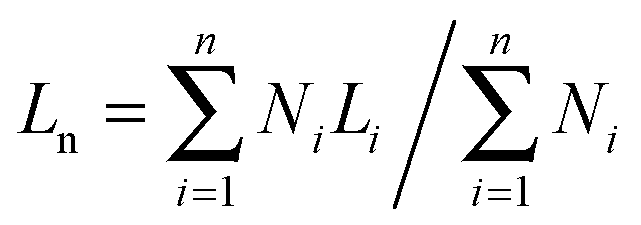 | (1) |
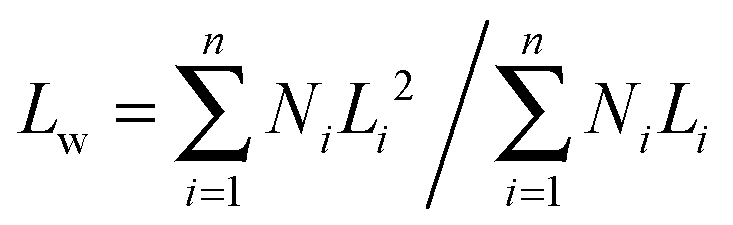 | (2) |
Fig. 7 shows the number-average contour lengths of three PE-b-PtBA BCPs at different Tcs. It is found that Tc has a great influence on the length of the grown cylindrical micelles. As Tc increases, Ln increases rapidly. The micelle length also depends on the length of the PtBA block. The BCP with a longer PtBA block tends to form longer micelles at Tc = 90 °C. Moreover, it is found that the contour length distributions (Lw/Ln) for the micelles of different PE-b-PtBA BCPs at various Tcs are smaller than 1.30.
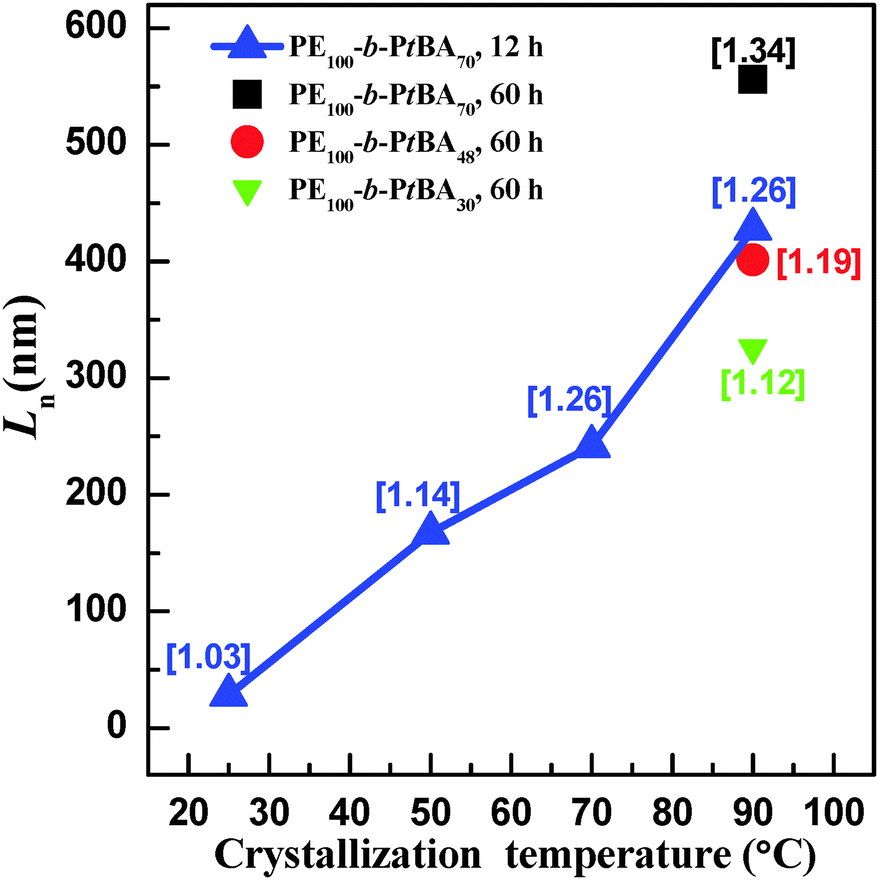 | ||
| Fig. 7 Dependence of the number-average contour length (Ln) on crystallization temperature (Tc) for three different PE-b-PtBA BCPs. The contour length distributions (Lw/Ln) are indicated in the brackets. Short cylinders formed during sample preparation are neglected in this statistics. | ||
We also used DLS to characterize the micellar solutions of PE-b-PtBA BCPs in DMF. It should be noted that, for anisotropic particles, such as cylinders, the size obtained by DLS is not the real one of the micelles, which cannot be compared with that measured by TEM. As larger particles generally exhibit a longer relaxation time and smaller particles exhibit a shorter relaxation time, the intensity correlation function of micelles may be fitted with a double-relaxation mode.53 We can compare the length and composition of the cylindrical micelles in solution in terms of the DLS parameters obtained by fitting. The DLS analysis indicates that longer micelles are formed and more unimers are left in the solution when the PtBA block is longer or Tc is higher, which is in accordance with the TEM results (please see details in the ESI†).
Both TEM and DLS results show that longer cylindrical micelles are formed for PE-b-PtBA with a longer corona-forming block at a higher Tc in DMF. A similar phenomenon was observed by O'Reilly and Dove for the micelles of poly(L-lactide)-b-poly(acrylic acid) (PLLA-b-PAA) BCPs in the THF/H2O mixture.27 The PLLA-b-PAA BCPs with a longer PAA block generated longer cylindrical micelles. The larger dimension of the cylindrical micelles at a higher Tc and for a longer corona-forming block can be interpreted from the concentrations of the seed micelles and the dissolved unimers. A higher Tc and a longer corona-forming block lead to good solubility of the BCPs, thus fewer seed crystalline micelles are produced and more unimers are left in the solution. Therefore, the dimension of the PE-b-PtBA cylindrical micelles is also determined by munimers/mseeds, which is similar to the living growth of the crystalline micelles of PFDMS-containing BCPs.37 Moreover, the seed micelles are longer at a higher Tc and for a longer corona-forming block, which may also be partially responsible for the larger size of the grown micelles.
Growth kinetics of PE-b-PtBA crystalline cylindrical micelles in DMF
Besides the micelle size, the growth kinetics of the crystalline micelles is also important, through which the growth mechanism can be inferred and the micelle size may be regulated. We monitored the micelle size of PE-b-PtBA BCPs in DMF after growth at 90 °C for different times. The TEM images of the PE100-b-PtBA70 micelles at selected times are illustrated in Fig. 8. The detailed data for three PE-b-PtBA BCPs (PE100-b-PtBA70, PE100-b-PtBA48 and PE100-b-PtBA30) are shown in Fig. S9–S11 of ESI,† respectively. The variations in the number-average length (Ln) of the cylindrical micelles measured by TEM with growth time for the different PE-b-PtBA micelles in DMF solution are plotted in Fig. 9. One can see that, the length of the cylindrical micelles increases with growth time rapidly in the initial period and reaches a constant after a certain time due to consumption of the unimers in the solution. As the PtBA block becomes longer, the time required for the micelles to reach a constant length increases. The final length of the cylindrical micelles increases with increasing PtBA block length as well, which agrees with the result in the previous section.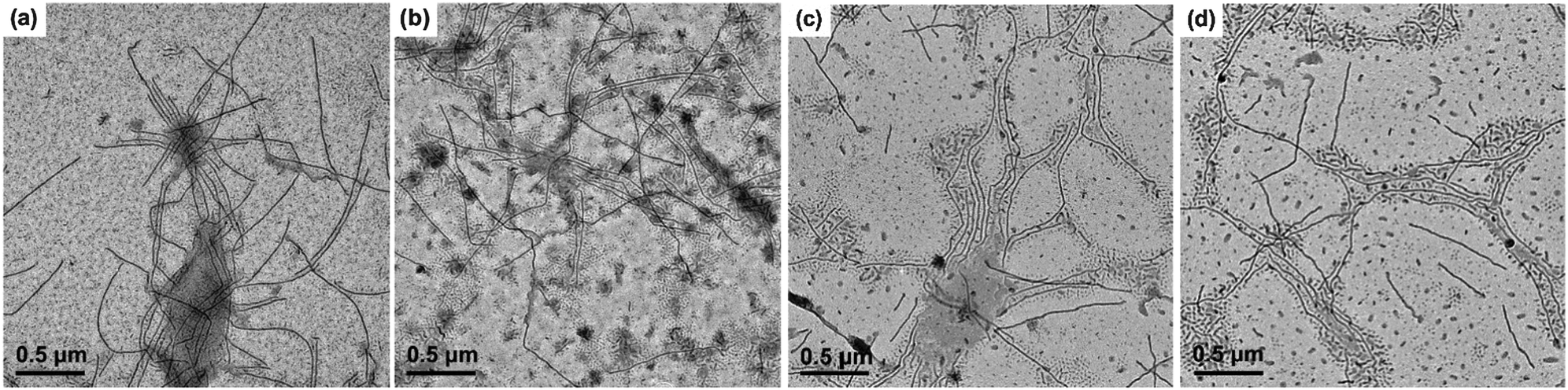 | ||
| Fig. 8 TEM micrographs of the cylindrical micelles for the PE100-b-PtBA70 micelles in DMF solution after annealing at 130 °C for an hour and then growth at 90 °C for different times. (a) 0.83 h, (b) 23.5 h, (c) 60 h, (d) 120 h. | ||
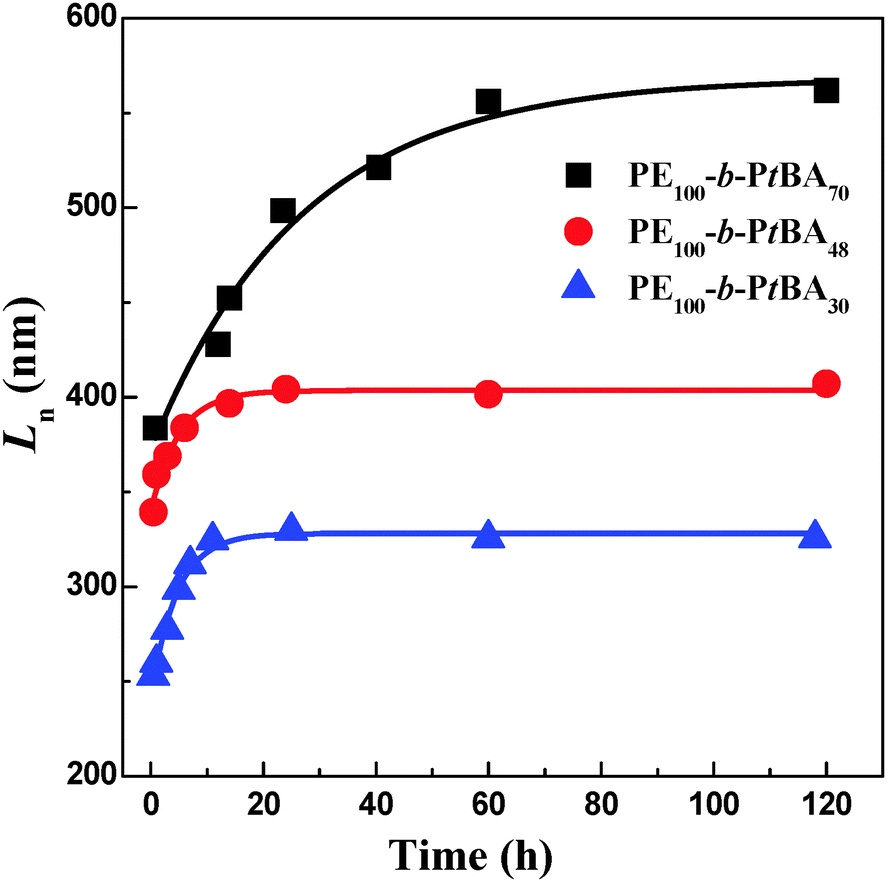 | ||
| Fig. 9 Variations of the number-average length (Ln) of the cylindrical micelles measured by TEM with growth time for the different PE-b-PtBA micelles in DMF solution. The scattering symbols represent the experimental data and the lines are the fitting results using eqn (8). | ||
In the present work, the end-to-end coupling between two crystalline cylindrical micelles is not observed,54 thus crystallization-driven growth of the cylindrical PE-b-PtBA micelles can be viewed as epitaxial attachment of unimers in the solution on both the ends of the crystalline seed micelles. Such a process is quite similar to addition polymerization if we suppose the semicrystalline seed micelles and the unimers in the solution as the active species and monomer, respectively.54 Thus, we have:
| d[M]/dt = −k1[ns][M] | (3) |
| ln[M] = ln[M0] − k1[ns]t | (4) |
Eqn (4) can be transformed into:
| [M] = [M0] exp(−k[ns]t) | (5) |
| Ln = L0 + Q([M0] − [M])/[ns] | (6) |
Then, we have:
| Ln = L0 + Q[M0](1 − exp(−k[ns]t))/[ns] | (7) |
| Ln = L0 + B(1 − exp(−Ct)) | (8) |
The product of the parameters B and C is kQ[M0]. Since Q and [M0] are the same for different PE-b-PtBA BCPs, the product of B and C, i.e. kQ[M0], can be used to evaluate the growth rate of different PE-b-PtBA micelles. The fitting results using eqn (8) are shown in Fig. 9 and the obtained values for different parameters are summarized in Table 2.
It can be seen from Fig. 9 that the experimental data are well fitted with eqn (8), verifying the proposed mechanism for the growth of crystalline PE-b-PtBA cylindrical micelles in DMF. It should be noted that eqn (8) corresponds to a living growth model. The applicability of the living growth model to the present system may lie in following two aspects. Firstly, the seed micelles produced by crystallization of the original amorphous micelles can be ignored due to the small number of the amorphous spherical micelles at 130 °C and the difficulty in nucleation inside the micelles. Secondly, the formation of the crystalline seed micelles at Tc may be completed in a short period, and then the concentration of the seed micelles, [ns], can be viewed as a constant. As compared with the long growth period due to the slow growth rate, the time for the formation of the seed micelles is much shorter. This is the reason why we choose a higher Tc (90 °C) for the growth kinetics study. At a lower Tc, the growth rate is so fast that the periods of forming seed micelles and the growth of the micelles cannot be well separated and the growth kinetics is no longer quasi-living. However, due to the low concentration of the unimers in the late stage of growth and the slow growth rate at high Tc, longer times is needed for the complete incorporation of unimers into micelles. As shown in Fig. S12 of ESI,† the short cylindrical micelles formed by the un-consumed unimers obviously become fewer after the PE100-b-PtBA30 micelles grow at 90 °C for 71 h. This shows that there should be very few unimers left in the solution and the unimers can be almost completely incorporated into the crystalline micelles with sufficient growth time. As a result, the growth of the crystalline micelles is not a process of thermodynamic equilibrium. The kinetics study shows that the growth of the crystalline PE-b-PtBA cylindrical micelles in DMF at Tc = 90 °C can be viewed as quasi-living. The achievement of quasi-living growth in the PE-b-PtBA/DMF system means that the length of the crystalline cylindrical micelles can be well controlled by growth time. Such a result may also be applicable to other BCP crystalline micelles, in which the CMC of the BCPs varies greatly with temperature. However, two preconditions should be satisfied: the formation of seed micelles should be completed in a short period and the growth rate of the unimers on the seed micelles should be slow.
It is found that both the initial length of the seed micelles (L0) and the limit length of the cylindrical micelles after growth (Lend) increase as the PtBA block becomes longer (Table 2), as previously discussed. On the other hand, it is observed that the product of B and C decreases with increasing length of the PtBA block, showing that the cylindrical micelles of the PE-b-PtBA with a longer PtBA block have a smaller growth rate. This can be well explained in terms of the reduced tethering density in the crystalline micelles.18 The reduced tethering density, ![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10d.gif) , is defined as σπRg2, where σ is the tethered chain density and is equal to the reciprocal of the area occupied by each soluble chain and Rg is the radius of gyration of the tethered chain in its end-free state under the same conditions. The reduced tethering density reflects the crowding of the corona-forming block. The length of the corona-forming block is an important factor affecting . When the crystalline block is fixed, a longer corona-forming block leads to a larger . At a large , the corona-forming block is more crowded and then tends to bend and cover the lateral surfaces of the crystals formed by the core-forming block. This will hinder the epitaxial attachment of the unimers to the active ends of the cylindrical micelles, i.e. slow the growth of the crystalline micelles.
, is defined as σπRg2, where σ is the tethered chain density and is equal to the reciprocal of the area occupied by each soluble chain and Rg is the radius of gyration of the tethered chain in its end-free state under the same conditions. The reduced tethering density reflects the crowding of the corona-forming block. The length of the corona-forming block is an important factor affecting . When the crystalline block is fixed, a longer corona-forming block leads to a larger . At a large , the corona-forming block is more crowded and then tends to bend and cover the lateral surfaces of the crystals formed by the core-forming block. This will hinder the epitaxial attachment of the unimers to the active ends of the cylindrical micelles, i.e. slow the growth of the crystalline micelles.
The effects of Tc and the corona-forming block on the formation of the seed micelles and growth of the crystalline cylindrical micelles of PE-b-PtBA BCPs in DMF can be schematically depicted in Scheme 1. PE-b-PtBA BCPs are dispersed as unimers and a small amount of amorphous micelles in DMF at 130 °C. When the BCP solution is cooled to Tc, unimers tend to form cylindrical seed micelles via two competitive processes: stepwise micellization/crystallization and simultaneous crystallization/micellization. The former prevails at a higher Tc or for the BCPs with a longer PtBA block, forming fewer but longer seed micelles. By contrast, the latter dominates at a lower Tc or for the BCP containing a shorter PtBA block, leading more but shorter seed micelles. Besides, a small amount of seed micelles is also generated through the crystallization of the amorphous micelles. The seed micelles can grow via epitaxial crystallization of the dissolved unimers on the ends of the cylindrical micelles, leading to a larger dimension in length. A quasi-living growth may be achieved at a higher Tc. A longer corona-forming block may retard the attachment of the unimers. However, the final length of the grown micelles at a higher Tc is determined by the number of the seed micelles over the mass of unimers in solution (mseeds/munimers), thus longer cylindrical micelles are yielded for the BCP with a longer PtBA block at a higher Tc.
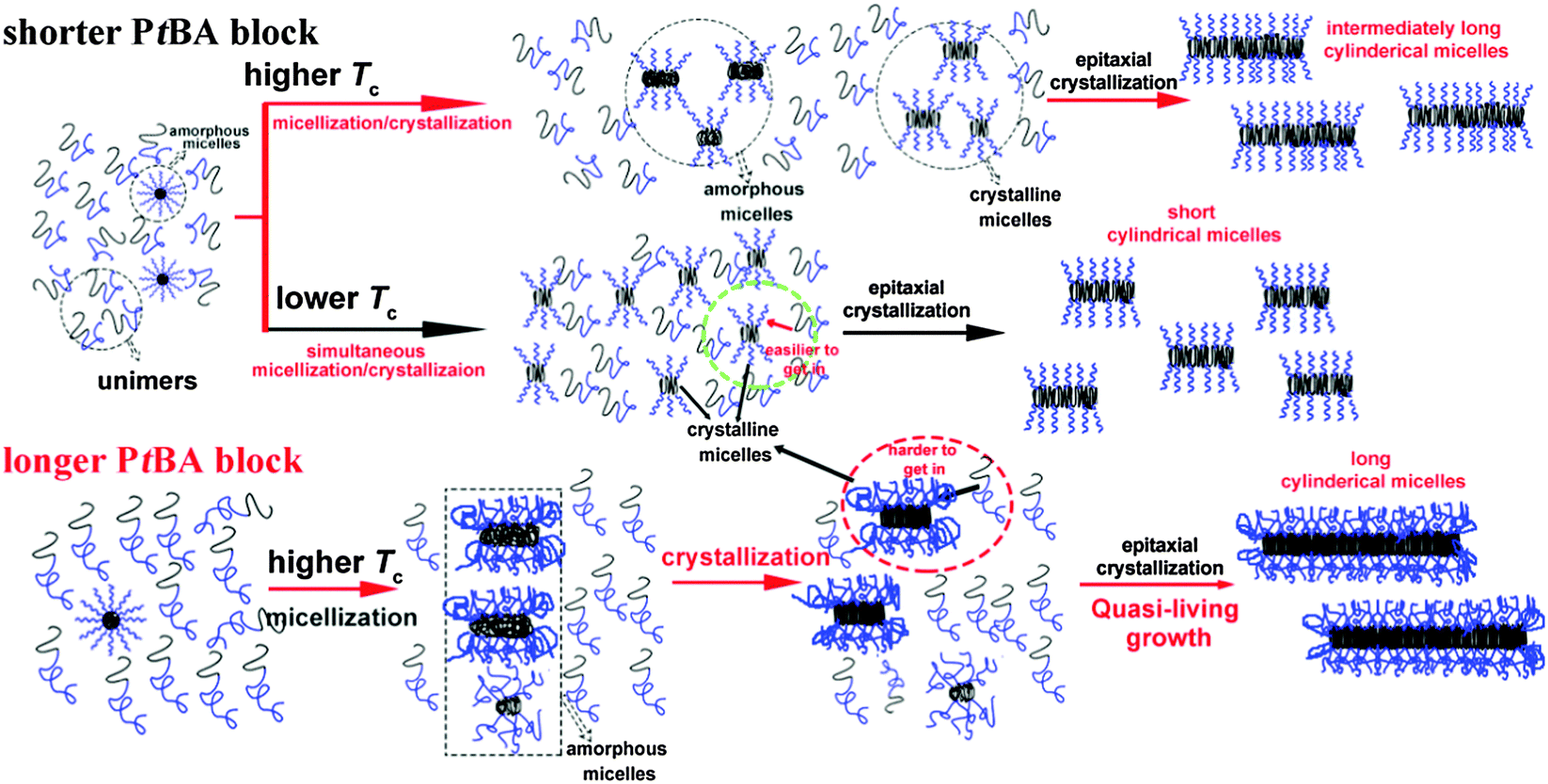 | ||
| Scheme 1 Scheme of the effects of Tc and the length of the corona-forming block on the formation of seed micelles and the growth of the cylindrical micelles of PE-b-PtBA BCPs in DMF. | ||
Conclusions
In summary, the PE-b-PtBA BCPs can self-assemble into one-dimensional crystalline cylindrical micelles driven by crystallization of the core-forming PE block. When the BCP solution is cooled to Tc, the micellization mechanism and solubility of the BCPs evidently vary with Tc and the core-forming block. As a result, both the “self-seeding” and stepwise micellization/crystallization methods are inapplicable to this system. The crystalline seed micelles are formed via two competitive processes: stepwise micellization/crystallization and simultaneous crystallization/micellization. The cylindrical micelles formed can act as seeds to allow epitaxial growth of the unimers left in the solution. The amount of the unimers left in the solution increases with increasing Tc and the PtBA block length, but the amount of the seed micelles exhibits an inverse tendency. This leads to the formation of longer cylindrical micelles at a higher Tc and for the PE-b-PtBA with a longer PtBA block. Because the formation of the seed micelles is much faster than the epitaxial growth and the amount of the seed micelles formed by the amorphous micelles is negligible, a quasi-living mode can be applied to the growth kinetics of PE-b-PtBA crystalline cylindrical micelles in DMF at a higher Tc. A longer PtBA block results in a smaller growth rate of the micelles.Acknowledgements
This work was supported by National Natural Science Foundation of China (21274130).Notes and references
- X. S. Wang, G. Guerin, H. Wang, Y. S. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- Y. Gao, H. B. Qiu, H. Zhou, X. Y. Li, R. Harniman, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2015, 137, 2203–2206 CrossRef CAS PubMed.
- Z. M. Hudson, J. S. Qian, C. E. Boott, M. A. Winnik and I. Manners, ACS Macro Lett., 2015, 4, 187–191 CrossRef CAS.
- H. Zhou, Y. J. Lu, H. B. Qiu, G. Guerin, I. Manners and M. A. Winnik, Macromolecules, 2015, 48, 2254–2262 CrossRef CAS.
- E. K. Lin and A. P. Gast, Macromolecules, 1996, 29, 4432–4441 CrossRef CAS.
- Z. Y. Li, R. Liu, B. Y. Mai, W. J. Wang, Q. Wu, G. D. Liang, H. Y. Gao and F. M. Zhu, Polymer, 2013, 54, 1663–1670 CrossRef CAS.
- L. G. Yin and M. A. Hillmyer, Macromolecules, 2011, 44, 3021–3028 CrossRef CAS.
- H. F. Wang, C. Wu, G. M. Xia, Z. Ma, G. Mo and R. Song, Soft Matter, 2015, 11, 1778–1787 RSC.
- J. Schmelz, M. Karg, T. Hellweg and H. Schmalz, ACS Nano, 2011, 5, 9523–9534 CrossRef CAS PubMed.
- J. Schmelz, A. E. Schedl, C. Steinlein, I. Manners and H. Schmalz, J. Am. Chem. Soc., 2012, 134, 14217–14225 CrossRef CAS PubMed.
- J. Schmelz, F. H. Schacher and H. Schmalz, Soft Matter, 2013, 9, 2101–2107 RSC.
- L. G. Yin, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2012, 45, 9460–9467 CrossRef CAS.
- J. T. Xu, J. P. A. Fairclough, S. M. Mai and A. J. Ryan, J. Mater. Chem., 2003, 13, 2740–2748 RSC.
- J. T. Xu, W. Jin, G. D. Liang and Z. Q. Fan, Polymer, 2005, 46, 1709–1716 CrossRef CAS.
- A. M. Mihut, A. Chiche, M. Drechsler, H. Schmalz, E. Di Cola, G. Krausch and M. Ballauff, Soft Matter, 2009, 5, 208–213 RSC.
- A. M. Mihut, J. J. Crassous, H. Schmalz, M. Drechsler and M. Ballauff, Soft Matter, 2012, 8, 3163–3173 RSC.
- K. Rajagopal, A. Mahmud, D. A. Christian, J. D. Pajerowski, A. E. X. Brown, S. M. Loverde and D. E. Discher, Macromolecules, 2010, 43, 9736–9746 CrossRef CAS PubMed.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromolecules, 2007, 40, 7633–7637 CrossRef CAS.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromol. Rapid Commun., 2008, 29, 467–471 CrossRef CAS.
- W. N. He, J. T. Xu, B. Y. Du, Z. Q. Fan and X. S. Wang, Macromol. Chem. Phys., 2010, 211, 1909–1916 CrossRef CAS.
- W. N. He, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromol. Chem. Phys., 2012, 213, 952–964 CrossRef CAS.
- J. X. Yang, W. N. He, J. T. Xu, B. Y. Du and Z. Q. Fan, Chin. J. Polym. Sci., 2014, 32, 1128–1138 CrossRef CAS.
- M. Su, H. Y. Huang, X. J. Ma, Q. Wang and Z. H. Su, Macromol. Rapid Commun., 2013, 34, 1067–1071 CrossRef CAS PubMed.
- J. Wang, W. Zhu, B. Peng and Y. M. Chen, Polymer, 2013, 54, 6760–6767 CrossRef CAS.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- N. Petzetakis, D. Walker, A. P. Dove and R. K. O'Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013, 46, 9074–9082 CrossRef CAS.
- A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2014, 5, 1427–1436 RSC.
- L. Sun, A. Pitto-Barry, N. Kirby, T. L. Schiller, A. M. Sanchez, M. A. Dyson, J. Sloan, N. R. Wilson, R. K. O'Reilly and A. P. Dove, Nat. Commun., 2014, 5, 5746 CrossRef CAS PubMed.
- M. Lazzari and M. A. Lopez-Quintela, Macromol. Rapid Commun., 2009, 30, 1785–1791 CrossRef CAS PubMed.
- H. Wang, C. L. Liu, G. Wu, S. C. Chen, F. Song and Y. Z. Wang, Soft Matter, 2013, 9, 8712–8722 RSC.
- M. J. Wang, H. Wang, S. C. Chen, C. Chen and Y. Liu, Langmuir, 2015, 31, 6971–6980 CrossRef CAS PubMed.
- J. Gwyther, J. B. Gilroy, P. A. Rupar, D. J. Lunn, E. Kynaston, S. K. Patra, G. R. Whittell, M. A. Winnik and I. Manners, Chem. – Eur. J., 2013, 19, 9186–9197 CrossRef CAS PubMed.
- J. S. Qian, X. Y. Li, D. J. Lunn, J. Gwyther, Z. M. Hudson, E. Kynaston, P. A. Rupar, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2014, 136, 4121–4124 CrossRef CAS PubMed.
- T. Vilgis and A. Halperin, Macromolecules, 1991, 24, 2090–2095 CrossRef CAS.
- L. Glavas, P. Olsen, K. Odelius and A. C. Albertsson, Biomacromolecules, 2013, 14, 4150–4156 CrossRef CAS PubMed.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS PubMed.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef CAS PubMed.
- W. N. He and J. T. Xu, Prog. Polym. Sci., 2012, 37, 1350–1400 CrossRef CAS.
- J. X. Yang, L. Liu and J. T. Xu, Prog. Chem., 2014, 26, 1811–1820 Search PubMed.
- J. J. Crassous, P. Schurtenberger, M. Ballauff and A. M. Mihut, Polymer, 2015, 62, A1–A13 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- J. M. Dean, N. E. Verghese, H. Q. Pham and F. S. Bates, Macromolecules, 2003, 36, 9267–9270 CrossRef CAS.
- J. S. Qian, G. Guerin, Y. J. Lu, G. Cambridge, I. Manners and M. A. Winnik, Angew. Chem., Int. Ed., 2011, 50, 1622–1625 CrossRef CAS PubMed.
- J. Qian, Y. Lu, A. Chia, M. Zhang, P. A. Rupar, N. Gunari, G. C. Walker, G. Cambridge, F. He, G. Guerin, I. Manners and M. A. Winnik, ACS Nano, 2013, 7, 3754–3766 CrossRef CAS PubMed.
- J. S. Qian, G. Guerin, G. Cambridge, I. Manners and M. A. Winnik, Macromol. Rapid Commun., 2010, 31, 928–933 CrossRef CAS PubMed.
- Y. Xue, S. S. Zhang, K. Cui, J. Huang, Q. L. Zhao, P. Lan, S. K. Cao and Z. Ma, RSC Adv., 2015, 5, 7090–7097 RSC.
- B. B. Busch, M. M. Paz, K. J. Shea, C. L. Staiger, J. M. Stoddard, J. R. Walker, X. Z. Zhou and H. D. Zhu, J. Am. Chem. Soc., 2002, 124, 3636–3646 CrossRef CAS PubMed.
- Z. Z. Tong, R. Y. Wang, J. Huang, J. T. Xu and Z. Q. Fan, Polym. Chem., 2015, 6, 2214–2225 RSC.
- X. X. Ke, J. T. Xu, B. Y. Du and Z. Q. Fan, Chin. J. Polym. Sci., 2015, 33, 1038–1047 CrossRef CAS.
- Z. Z. Tong, B. Zhou, J. Huang, J. T. Xu and Z. Q. Fan, Macromolecules, 2014, 47, 333–346 CrossRef CAS.
- K. Iyama and T. Nose, Macromolecules, 1998, 31, 7356–7364 CrossRef CAS.
- X. X. Ke, L. Wang, J. T. Xu, B. Y. Du, Y. F. Tu and Z. Q. Fan, Soft Matter, 2014, 10, 5201–5211 RSC.
- W. N. He, B. Zhou, J. T. Xu, B. Y. Du and Z. Q. Fan, Macromolecules, 2012, 45, 9768–9778 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PE-b-PtBA BCPs, WAXD pattern of dried PE100-b-PtBA30 micelles, DSC crystallization curves of PE-b-PtBA BCPs, outlines of the long PE-b-PtBA micelles at Tc, DLS size distributions of PE-b-PtBA micelles, DLS correlation functions at various Tcs, DLS fitting results and analysis, TEM images of PE100-PtBA70, PE100-PtBA48 and PE100-b-PtBA30 micelles at different growth times. See DOI: 10.1039/c5sm02226f |
| This journal is © The Royal Society of Chemistry 2016 |