
Probing dispersion and re-agglomeration phenomena upon melt-mixing of polymer-functionalized graphite nanoplates
R. M.
Santos
,
C.
Vilaverde
,
E.
Cunha
,
M. C.
Paiva
* and
J. A.
Covas
Institute for Polymers and Composites/I3N, University of Minho, Campus de Azurém, 4800-058 Guimarães, Portugal. E-mail: mcpaiva@dep.uminho.pt
First published on 25th September 2015
Abstract
A one-step melt-mixing method is proposed to study dispersion and re-agglomeration phenomena of the as-received and functionalized graphite nanoplates in polypropylene melts. Graphite nanoplates were chemically modified via 1,3-dipolar cycloaddition of an azomethine ylide and then grafted with polypropylene-graft-maleic anhydride. The effect of surface functionalization on the dispersion kinetics, nanoparticle re-agglomeration and interface bonding with the polymer is investigated. Nanocomposites with 2 or 10 wt% of as-received and functionalized graphite nanoplates were prepared in a small-scale prototype mixer coupled to a capillary rheometer. Samples were collected along the flow axis and characterized by optical microscopy, scanning electron microscopy and electrical conductivity measurements. The as-received graphite nanoplates tend to re-agglomerate upon stress relaxation of the polymer melt. The covalent attachment of a polymer to the nanoparticle surface enhances the stability of dispersion, delaying the re-agglomeration. Surface modification also improves interfacial interactions and the resulting composites presented improved electrical conductivity.
1. Introduction
Nanostructures based on carbon allotropes – graphite, fullerenes, carbon nanotubes and graphene – are a versatile class of materials that are attracting substantial scientific and technological interest especially due to their remarkable mechanical and electrical properties.1 Their potential applications include single molecule gas detection,2 actuators and supercapacitors,3 transparent conducting electrodes,4 corrosion prevention,5 drug delivery and tissue engineering,6 biomaterials7 and polymer nanocomposites.8–12Graphite, formed by stacks of 2-D layers of sp2 hybridized carbon atoms hexagonally bonded with open edges, is a promising low cost (naturally abundant) reinforcement for functional and smart nanocomposites.8,9,13 Its in-plane stiffness (Young's modulus) is approximately 1 TPa, and its electrical (≈106 Ω cm−1) and thermal conductivity (≈400 W m−1 K−1) are also exceptional.14 It has been theoretically predicted that graphite has an edge-inherited non-bonding π-state (combination of zigzag and armchair edges) at about the Fermi level, in addition to the π-bonding and π*-antibonding states, resulting in unconventional magnetic features.15
However, in practice, the incorporation and dispersion of natural flaky graphite in polymer matrices is challenging due to the strong interaction and small spacing between graphene layers (0.335 nm center-to-center, generating an interstitial space of much less than 0.3 nm)16 and the lack of functional groups at the surface and edges.17 The former hinders the polymer intercalation in-between layers and their exfoliation, while the latter deters the creation of strong interfacial bonding with the polymer.
Exfoliated graphite can be synthesized by various methods, including intercalation with an alkali metal, alkali earth metal, rare earth metal for donors and metal halides, halogen and acids for acceptors.8,18,19 Most of the graphite flakes obtained by these procedures, usually denoted graphite nanoplates (GnP),20 have a thickness from 0.34 to 100 nm.21 Intercalation increases the interlayer spacing and weakens the mutual interactions, facilitating exfoliation by mechanical, thermal or microwave techniques.16
Chemical modification of GnP has been accomplished by non-covalent or covalent approaches to overcome the limitations associated with efficient dispersion and interfacial bonding with polymers.2,22,23 Non-covalent modification through electrostatic interactions, van der Waals forces, hydrogen bonding or π–π stacking interactions enables the attachment and stabilization of diverse functional groups (e.g. proteins, DNA–protein complexes, organic supermolecules) at the surface of the graphitic lattice without disturbing the electronic network.24 Covalent functionalization generally encompasses two routes: addition of free radicals or dienophiles to carbon–carbon bonds and formation of covalent bonds between functional groups and oxygen groups present in graphite oxide. Some of the most attractive organic species for chemical reactions with sp2 carbons of graphite are highly reactive free radicals formed by the diazonium addition reaction and the 1,3-dipolar cycloaddition (DCA)23 of azomethine ylides. This versatile reaction has been successfully applied in the functionalization of fullerenes,25 carbon nanotubes,26 carbon nanofibers27 and graphene, as it accesses a variety of organic derivatives by selecting the appropriate aldehyde and α-amino acid as precursors, depending on the required applications (polymer composites, drug delivery and nanoelectronic devices).28 The covalent bond presents the advantage of generating stronger interfaces with the polymer, and better withstand the processing conditions in comparison with non-covalent functionalization approaches.29
An overview of literature reveals that functionalization of conductive carbon nanoparticles plays an important role in widespread applications and commercialization of nanocomposites.17 Chemical modification enhances the dispersion and interfacial interactions with the polymer30–32 and prevents re-agglomeration.32,33 In this work, graphite nanoplates were functionalized via 1,3-dipolar cycloaddition and then grafted with polypropylene-graft-maleic anhydride (PP-g-MA). The kinetics of their dispersion in a polypropylene (PP) matrix, as well as the stability of the morphology generated under quasi-quiescent conditions, were studied and compared with the equivalent behaviour of the as-received graphite. The experiments were performed using a prototype small-scale mixer that generates a strong extensional flow component34,35 and is fixed to a capillary rheometer in order to induce flow under well-controlled conditions. Moreover, it allows for material sampling along its axis, thus providing the possibility of monitoring the evolution of dispersion. Polypropylene nanocomposites with 2 and 10 wt% of as-received and chemically modified GnP were prepared under identical conditions. The progression of nanoparticle dispersion along the mixer length was evaluated by optical and scanning electron microscopy; the electrical conductivity of the composites was measured. GnP re-agglomeration effects were analyzed upon melt-relaxation.
2 Experimental section
2.1 Materials
A polypropylene copolymer, Icorene CO14RM® from ICO POLYMERS, with a melt flow index of 13.1 g/10 min (190 °C, 2.16 kg) and a density of 0.9 g cm−3 was used as a polymer matrix. PP-g-MA containing 8–10 wt% of maleic anhydride, with a weight-average molecular mass (Mw) of approximately 9.1 kg mol−1 and a density of 0.934 g cm−3 was supplied by Sigma-Aldrich. According to the manufacturer (XG Sciences, Inc.), graphite nanoplates xGnP® grade C-750 has a thickness of approximately 2 nm, an average diameter of less than 2 μm and a typical average surface area of 750 m2 g−1. They were supplied in powder form, and the powder particle projected area was assessed by dilution of the powder in an epoxy resin, spreading on glass slides and measuring by optical microscopy. The flakes of the two fillers (GnP and fGnP-PP) generate agglomerates whose particle size distribution is revealed in Fig. 1. It was computed from the analysis of at least 70 images of each, with a total measured area of agglomerates of 6.7 × 105 and 4.9 × 105 μm2 for as-received and fGnP-PP, respectively. While the larger agglomerate (maximum agglomerate area) was in the range of 2.5–3.0 × 104 μm2, the average agglomerate size was 1.1 × 104 and 7.2 × 103 μm2 for the as-received and functionalized GnP, respectively.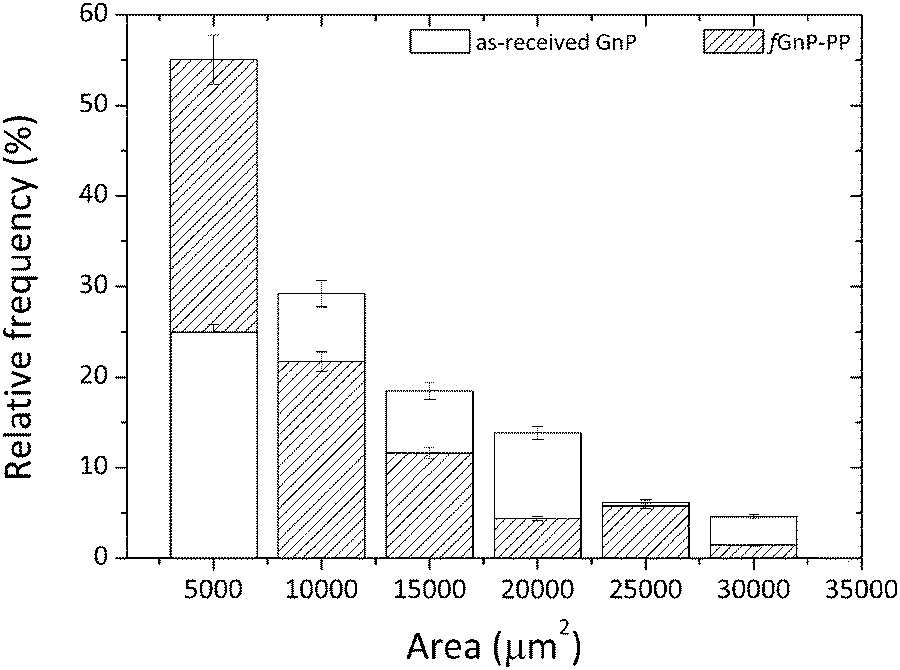 | ||
| Fig. 1 Particle size distribution of the as-received GnP and chemically modified fGnP-PP (powder form) before melt-mixing. | ||
2.2 Functionalization of graphite nanoplates
The as-received graphite nanoplates were chemically modified via 1,3-dipolar cycloaddition of azomethine ylides generated in situ by thermal decomposition of paraformaldehyde (analytical grade, from Sigma-Aldrich) and reaction with N-benzyloxycarbonylglycine (from Sigma-Aldrich) at 250 °C for 3 hours, yielding functionalized GnP (fGnP), using a procedure similar to that described for the functionalization of carbon nanotubes.36Fig. 2 summarizes the steps involved in the preparation of fGnP and fGnP-PP.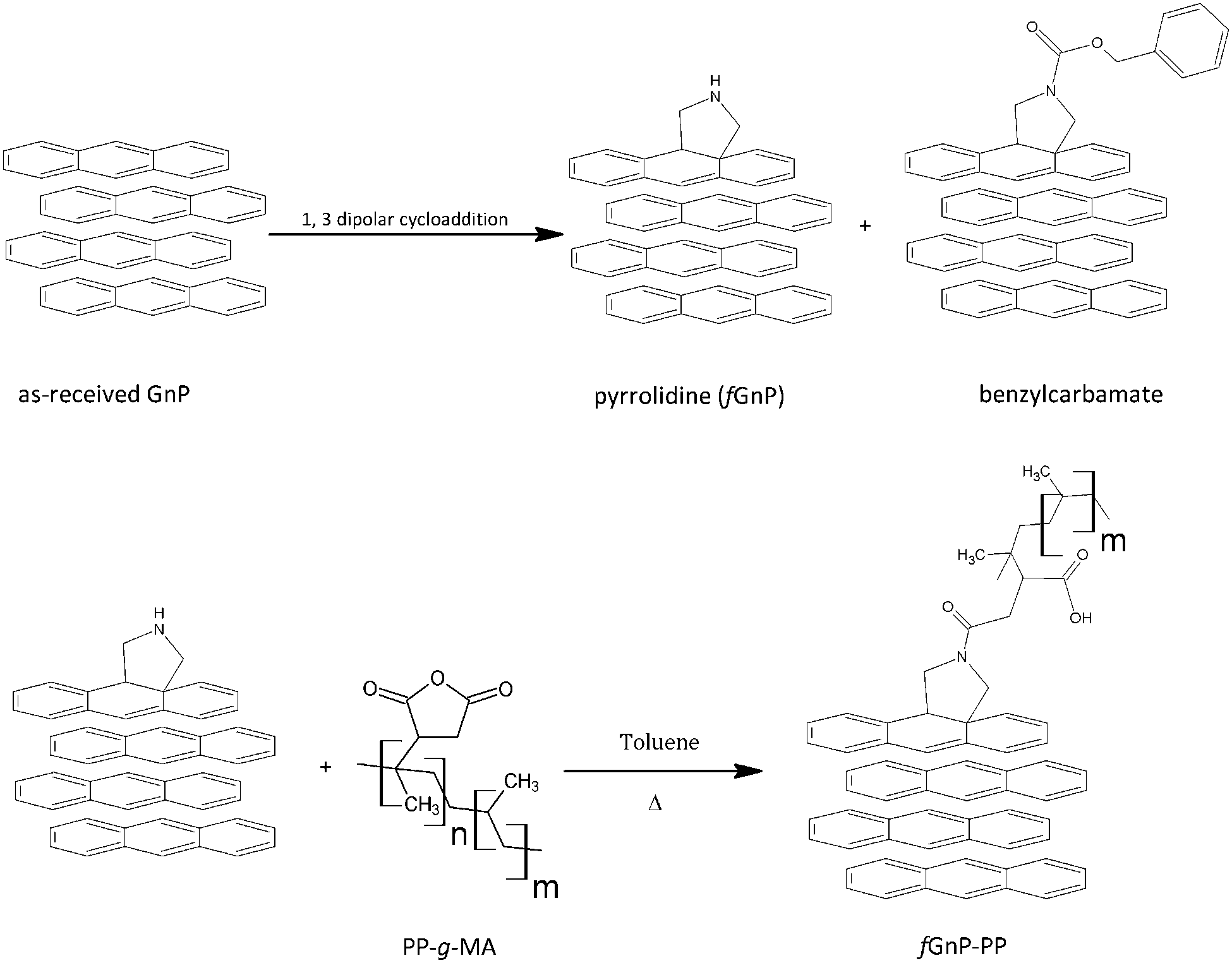 | ||
| Fig. 2 Functionalized graphite nanoplates after DCA reaction and grafting with PP-g-MA. | ||
PP-g-MA was then grafted onto the fGnP surface yielding PP-functionalized GnP (fGnP-PP). The reaction was carried out by refluxing the fGnP in a solution of PP-g-MA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) in toluene for 3 hours, under magnetic stirring. The resulting suspension was filtered, followed by repeated washing with hot toluene and dried overnight in a vacuum oven at 100 °C.
:2 molar ratio) in toluene for 3 hours, under magnetic stirring. The resulting suspension was filtered, followed by repeated washing with hot toluene and dried overnight in a vacuum oven at 100 °C.
2.3 Production of nanocomposites
The nanocomposites were manufactured by forcing a premix of polymer plus filler – by means of the descending ram of a capillary rheometer – through a mixer containing a series of circular channels with alternating small/large diameters, thus inducing repetitive convergent/divergent flows. It is well established that the normal stresses thus created, superimposed on the shear stress field, are particularly effective for dispersion.35,37,38 Changing the dimensions and/or the number of channels, the ram speed and temperature creates a wide range of flow conditions. Similarly to what has been observed for carbon nanotubes,35,39 graphite nanoplates seem to re-agglomerate once the thermomechanical stresses applied to achieve the dispersion are relieved.40 This morphology instability has obvious practical implications, considering the similitude to the usual industrial practice of manufacturing polymer-based nanocomposites by twin-screw extrusion, followed by processing of the final composite part, either by melt-processing in a single screw extruder or in a plasticising unit of an injection moulding equipment. The composite production step usually generates higher hydrodynamic stresses compared to the final processing step. Thus, the possible occurrence of this phenomenon was also investigated by appending a circular channel with a larger diameter after the mixer; in turn, the evolution of a second dispersion operation could also be studied by attaching a second series of circular channels with alternating diameters. The resulting layout is presented in Fig. 3.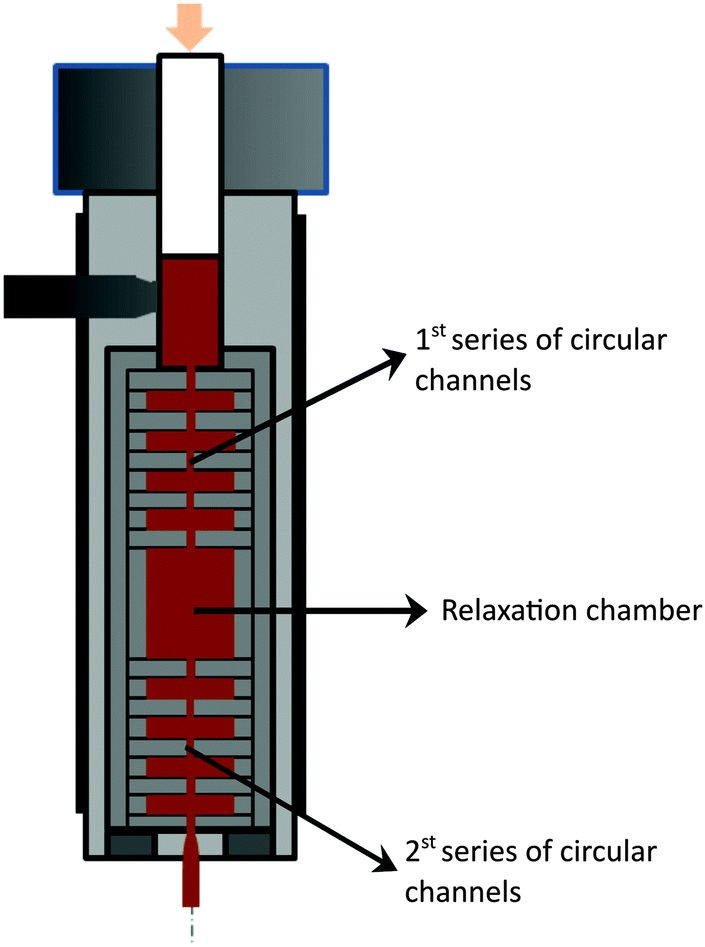 | ||
| Fig. 3 Schematics of the configuration of the prototype small-scale mixer used to prepare PP nanocomposites with the as-received GnP and chemically modified fGnP-PP. | ||
The mixer consists of a sequence of ten circular channels having different internal diameters, d (equal to 8 and 1 mm and lengths of 2 mm), inducing an 8:1 convergence, followed by a relaxation chamber (d of 18 mm and length of 24 mm) and another similar sequence of ten circular channels. Each individual channel is shaped by a circular ring, the entire flow configuration consisting of a vertical stack of rings assembled inside a sleeve. The latter can be quickly unscrewed from the main body of the mixer, in order to provide access to the material inside each channel. After pre-heating the mixed polymer–GnP powders at 200 °C for 5 min the melt was forced through the channels of the mixer at 50 mm min−1, corresponding to an average shear rate of approximately 1500 s−1 in the smaller capillary channel, 3.0 s−1 in the larger channel and 0.3 s−1 in the relaxation chamber. Once each experiment was concluded, the set-up was removed from the rheometer and samples were collected, cooled in N2 (l) and dried in air.
2.4 Characterization
X-ray photoelectron spectroscopy (XPS) was carried out using a Thermo Scientific K-Alpha ESCA instrument equipped with Al-Kα1,2 monochromatized radiation (1486.92 eV). The base pressure in the chamber was maintained at about 3 × 10−9 mbar. Photoelectrons were collected at an angle perpendicular to the sample surface. A constant analyzer energy mode (CAE) was used with a pass energy of 100 eV and 20 eV for the survey spectra and high resolution spectra, respectively. Charge referencing was done by setting the lower binding energy C 1s photopeak. The atomic percent (at%) compositions were determined from the survey XPS spectra considering the integrated peak areas using the Shirley background subtraction method and the Scofield sensitivity factors. Corel level curve fitting was performed using a convolution of a Lorentzian function (describing 1s hole lifetime effect) with a Gaussian function (temperature-dependent phonon broadening).
Optical microscopy of the as-received and chemically modified GnP (in powder form) was carried out on a BH2 Olympus microscope coupled to a Leica DFC 280 camera to characterize the initial particle size distribution before melt-compounding. For this purpose, 10.0 mg of GnP were gently dispersed in 4.25 mL of epoxy resin, then spread onto a glass slide and dried at room temperature for 24 h.
The as-received and chemically modified GnP were also dispersed in a butyl alcohol solution (1.2 g L−1) under sonication (CREST 230T ultrasonic bath, 80 W) for 2 hours at room temperature and then directly deposited onto a copper grid for subsequent observation by transmission electron microscopy (TEM) on a JEOL JEM1010, equipped with a CCD Orius camera and a tungsten filament as an electron source, at an acceleration voltage of 100 kV.
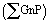 to the total composite area analyzed (AT):
to the total composite area analyzed (AT):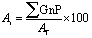 | (1) |
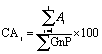 | (2) |
 and dividing by the total agglomerate area. The size of the larger agglomerate contained in 50% (Aχ50) and 90% (Aχ90) of the total agglomerate area was obtained from the cumulative distribution of agglomerate areas, CAj.
and dividing by the total agglomerate area. The size of the larger agglomerate contained in 50% (Aχ50) and 90% (Aχ90) of the total agglomerate area was obtained from the cumulative distribution of agglomerate areas, CAj.
The morphology of cryo-fractured nanocomposites sputtered with a gold/palladium mixture was examined by scanning electron microscopy (SEM) using a NanoSEM – FEI Nova 200, operating at an accelerating voltage of 5.00 kV in backscattering electroimage mode, coupled to an energy dispersive X-ray (EDAX-Pegasus X4M) spectrometer.
The dielectric properties of the nanocomposites were measured on the disk-shaped samples extracted from the mixer, using a Quadtech 1920 Precision LCR meter. Measurements were performed at a voltage of 1 V and a frequency ranging from 500 Hz to 1 MHz, at room temperature. The dielectric cell consisted of parallel plates having 3 mm diameter electrodes that were gold coated on the sample surface by magnetron sputtering. Four measurements were carried out for each sample, the average values being reported here. The real part of the electrical conductivity was calculated using:
| σ′(ω) = ωε0ε′′(ω) | (3) |
tanδ is the imaginary part of permittivity, where tanδ is the dissipation factor and ε′ is the real part of permittivity.41
3 Results and discussion
3.1 Graphite nanoplate functionalization
The thermal degradation of the as-received and chemically modified graphite nanoplates was compared by means of thermogravimetry, in order to confirm the presence of organic groups bonded through the DCA reaction. The variation of weight with temperature and its first derivative are shown in Fig. 4(a) and (b), respectively.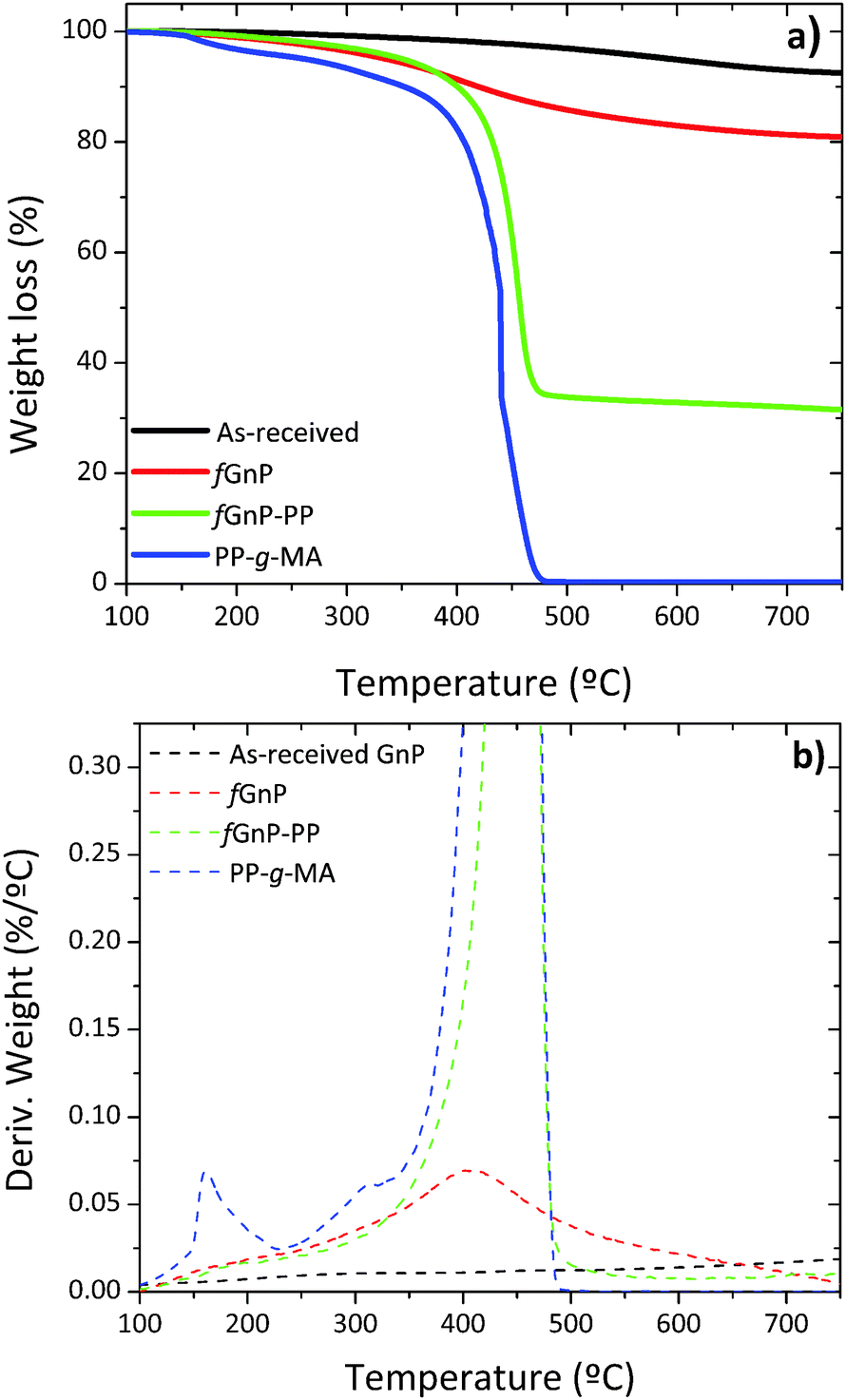 | ||
| Fig. 4 TGA of PP-g-MA and the as-received and chemically modified GnP; (a) TGA curves and (b) the first derivative of TGA curves. | ||
The thermal degradation of the as-received GnP shows a minor weight loss of approximately 3% up to 500 °C, due to the elimination of adsorbed compounds and residual oxidation. The first derivative evidences a broad band with low intensity that may be ascribed to the elimination of CO2 and CO (from 150 to 350 °C), hydroxylic or phenol functionalities (in the range of 350–500 °C) and carbonaceous impurities (at circa 600 °C).38,39 After carrying out the DCA reaction, fGnP is characterized by a weight loss of 14% from 150 to 500 °C, with a higher decomposition rate near 400 °C, assigned to the elimination of the covalently bonded organic groups (pyrrolidine or benzylcarbamate presented in Fig. 2). A further increase of weight loss is observed for fGnP-PP, close to 52.5%, indicating that a large uptake of PP-g-MA has taken place. The PP-g-MA shows two degradation steps near 150 and 300 °C, corresponding to the thermal decomposition of the grafted maleic anhydride (MA) groups. These degradation steps are absent in the TGA of fGnP-PP, confirming that most of the MA moieties grafted onto PP reacted with the pyrrolidine groups at the surface of fGnP, forming strong covalent bonds.
Additional evidence of functionalization of the GnP surface was obtained by XPS spectroscopy, as shown in Fig. 5. The atomic nitrogen and oxygen content at the surface of as-received and chemically modified GnP are presented in Table 1.
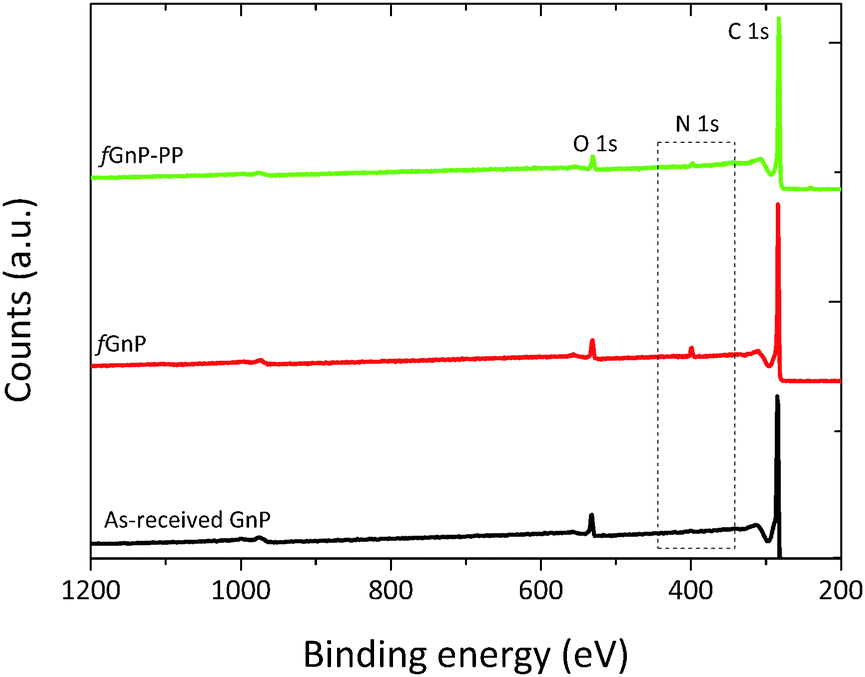 | ||
| Fig. 5 XPS survey spectra of the as-received and chemically modified graphite nanoplates. | ||
| Graphite nanoplatelets | Surface chemical elemental composition (at%) | ||||
|---|---|---|---|---|---|
| C | O | N | S | N/C ratio | |
| As-received GnP | 93.94 | 5.95 | 0.11 | — | 0.001 |
| fGnP | 90.84 | 5.50 | 3.31 | 0.30 | 0.040 |
| fGnP-PP | 94.72 | 4.14 | 1.14 | — | 0.012 |
The strong peak at 284.9 eV, common to the as-received and functionalized GnP, was attributed to the conjugated carbon atoms in the graphitic lattice (C 1s). The signal near 532 eV was ascribed to oxygen atoms (O 1s) incorporated during thermal expansion42 of the as-received GnP. After the DCA reaction, the N 1s peak appears at approximately 400 eV, confirming the presence of nitrogen-containing functional groups.22 The survey spectrum of fGnP provides a surface composition of 3.31 at% of N, and a similar oxygen content as the original GnP (Table 1). Besides confirming the bonding of N-containing functional groups to the GnP surface, this result indicates that these functional groups are mostly in the pyrrolidine form, as the presence of benzycarbamate would increase the overall oxygen content.
The analysis of the background-corrected high resolution C 1s core level XPS region of the as-received and chemically modified graphite nanoplates is presented in Fig. 6.
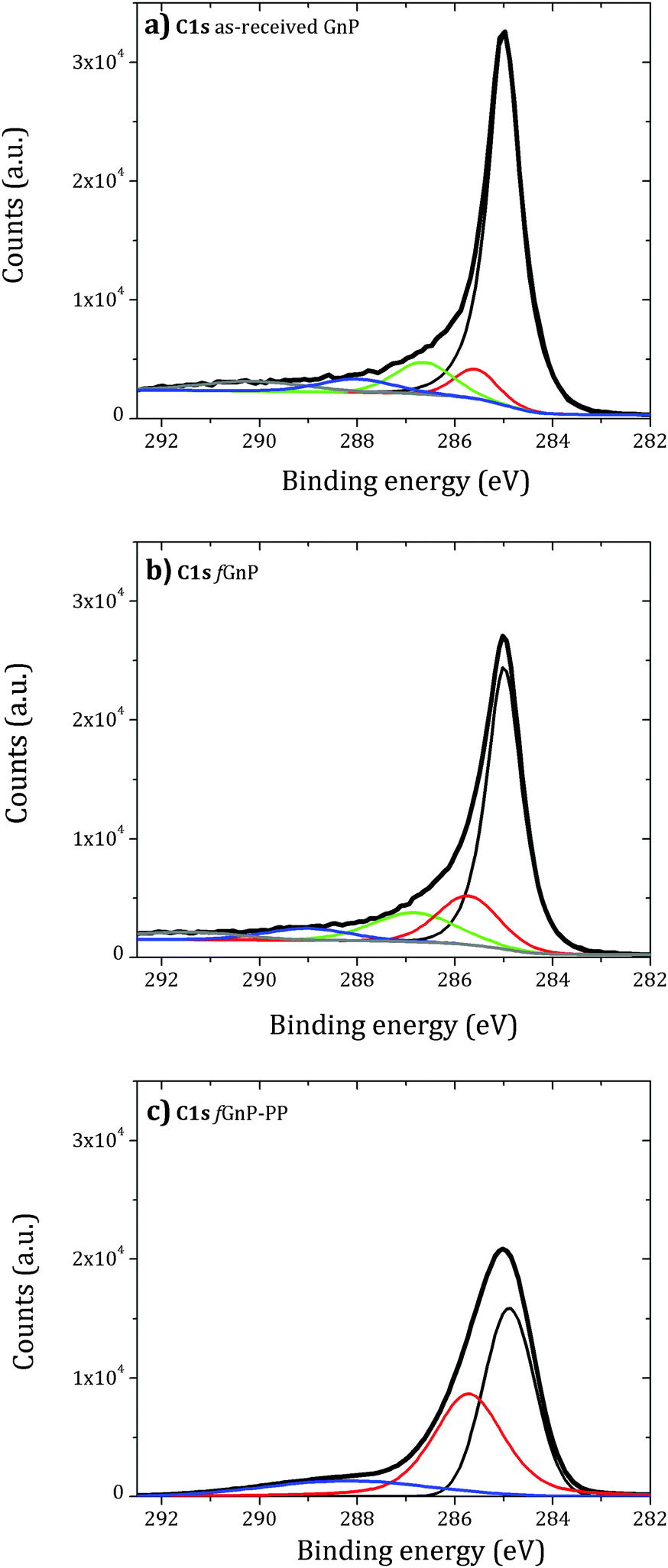 | ||
| Fig. 6 Background-corrected high resolution C 1s core level XPS region of the as-received (a) and functionalized (b) and (c) graphite nanoplates. | ||
The deconvolution of the C 1s peak of the as-received GnP shows two contributions above 286 eV (green) and near 288 eV (blue), attributed to carbon atoms singly bonded to an oxygen atom – alcohol or ether (−COR) – and carbonyl (−C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups, respectively.43 A small component peak shift of 0.5 eV to higher binding energy relative to sp2 C–C bonds (red) may be assigned to sp3 C–C bonds. An increase of this component peak area was observed for fGnP and may be associated with the presence of –CH2– and C–N of the cyclic pyrrolidine. After reaction with PP-g-MA, a large increase of this component peak area was observed. The C 1s peak becomes broad and is mainly composed of two contributions centered near 285 and 285.5 eV, with a ratio of approximately 6:5. Thus, either the grafted PP covers part of the fGnP surface, or the PP layer is thin and allows the detection of the GnP sp2 carbon. A new small component peak centered at 288.2 eV is observed, which may be assigned to amide or imide bonds (OC–N) formed by the reaction of maleic anhydride and pyrrolidine.
O) groups, respectively.43 A small component peak shift of 0.5 eV to higher binding energy relative to sp2 C–C bonds (red) may be assigned to sp3 C–C bonds. An increase of this component peak area was observed for fGnP and may be associated with the presence of –CH2– and C–N of the cyclic pyrrolidine. After reaction with PP-g-MA, a large increase of this component peak area was observed. The C 1s peak becomes broad and is mainly composed of two contributions centered near 285 and 285.5 eV, with a ratio of approximately 6:5. Thus, either the grafted PP covers part of the fGnP surface, or the PP layer is thin and allows the detection of the GnP sp2 carbon. A new small component peak centered at 288.2 eV is observed, which may be assigned to amide or imide bonds (OC–N) formed by the reaction of maleic anhydride and pyrrolidine.
TEM images (Fig. 7) depict the as-received GnP as thicker flakes compared to fGnP, demonstrating that the DCA reaction does not induce nanoparticle destruction, but may induce some exfoliation. After reaction with PP-g-Ma, the morphology is apparently maintained.
 | ||
| Fig. 7 TEM images of: (a) as-received GnP, (b) fGnP and (c) fGnP-PP. | ||
3.2 Dispersion of graphite nanoplates
Optical micrographs illustrating the morphology of PP nanocomposites with 2 and 10 wt% of the as-received GnP and fGnP-PP at various locations along the mixer are depicted in Fig. 8.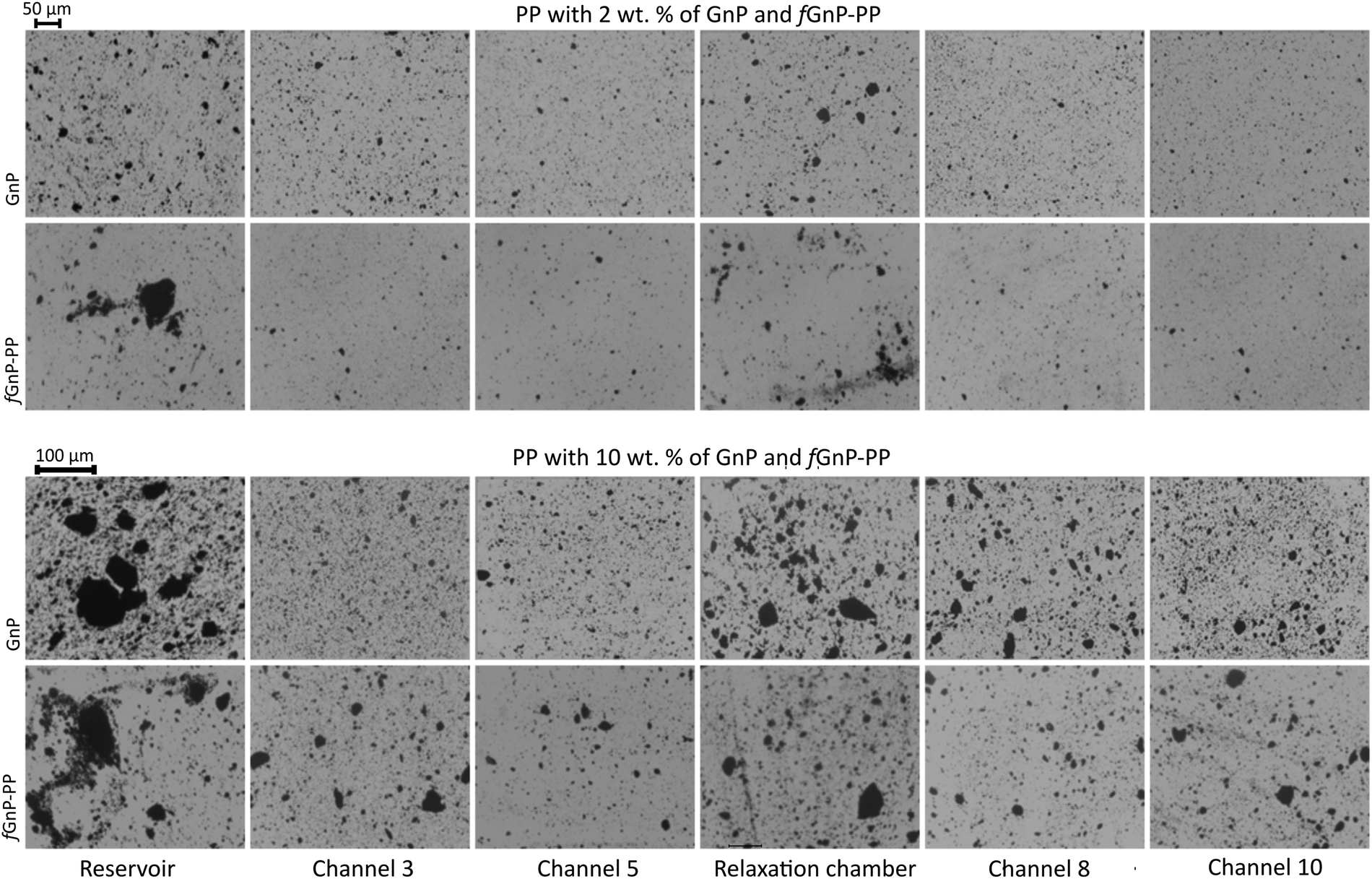 | ||
| Fig. 8 Morphology development of PP nanocomposites with 2 and 10 wt% of as-received GnP and fGnP-PP at various locations along the mixer. | ||
The agglomerate morphology was imaged at the reservoir of the capillary rheometer, channels 3 and 5 of the first mixing zone, the relaxation zone, and channels 8 and 10 of the second mixing zone (equivalent to channels 3 and 5 of the first mixing zone). Regardless of the filler content, a significant decrease of the GnP agglomerate size is revealed from the reservoir to channel 5, thus demonstrating the efficiency of the extensional flow to induce dispersion. In the relaxation chamber, where the applied shear stress is small (the shear rate is approximately 0.3 s−1), the area of the GnP agglomerates increases, suggesting that re-agglomeration has taken place. In the second mixing zone dispersion sets in again, the morphology at channel 10 approaching that at channel 5. Fig. 9 displays SEM images of the nanocomposites with 2 and 10 wt% of GnP and fGnP-PP removed from channel 5, illustrating the typical morphology of the agglomerates at the end of the first mixing section. In all cases, SEM shows that the agglomerates appear as composite areas of high GnP concentration, i.e., they are not constituted solely by GnP.
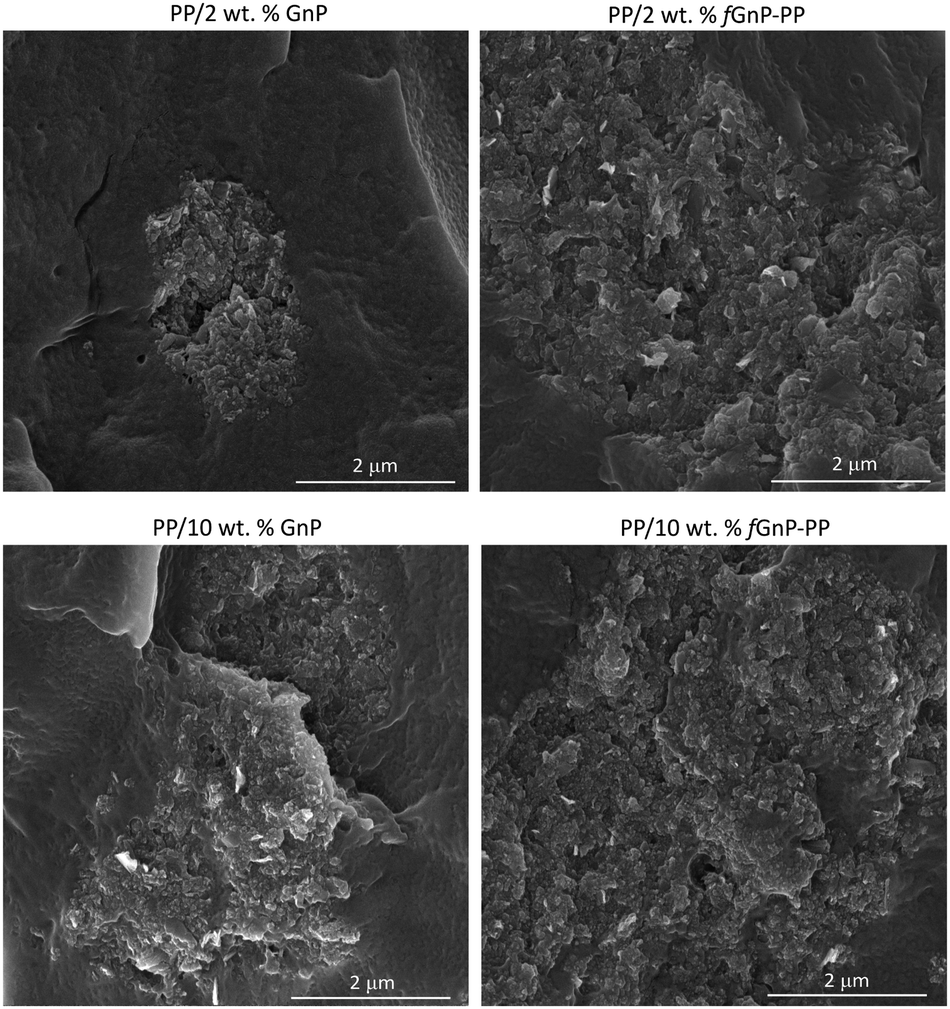 | ||
| Fig. 9 SEM micrographs of PP nanocomposites containing 2 and 10 wt% of as-received GnP and chemically modified fGnP-PP removed from channel 5. | ||
A quantitative assessment of the evolution of GnP and fGnP-PP dispersion in the PP matrix along the mixer, as described by the relevant statistical parameters, is provided in Fig. 10 and Table 2. Aχ50 and Aχ90 represent the area of the largest agglomerate in the subset of smaller agglomerates that constitute 50% or 90% of total 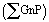 , respectively.
, respectively.
 | ||
| Fig. 10 Evolution of Ar along the extensional mixer for PP nanocomposites with (a) 2 and (b) 10 wt% of as-received GnP and chemically modified fGnP-PP, respectively. | ||
| Set-up | PP/2 wt% GnP | PP/2 wt% fGnP-PP | ||||
|---|---|---|---|---|---|---|
| A χ50 (μm2) | A χ90 (μm2) | N per unit area (mm−2) | A χ50 (μm2) | A χ90 (μm2) | N per unit area (mm−2) | |
| Reservoir | 26.0 | 240.7 | 4106 | 173.9 | 2000.3 | 1749 |
| Channel 3 | 22.6 | 152.2 | 2199 | 38.9 | 273.38 | 1648 |
| Channel 5 | 15.9 | 74.90 | 1057 | 29.4 | 131.5 | 1183 |
| Relaxation chamber | 25.5 | 167.4 | 3819 | 70.4 | 634.0 | 1120 |
| Channel 8 | 24.4 | 139.5 | 1617 | 40.0 | 279.1 | 1375 |
| Channel 10 | 17.8 | 88.5 | 1766 | 29.7 | 191.3 | 1278 |
| Set-up | PP/10 wt% GnP | PP/10 wt% fGnP-PP | ||||
|---|---|---|---|---|---|---|
| A χ50 (μm2) | A χ90 (μm2) | N per unit area (mm−2) | A χ50 (μm2) | A χ90 (μm2) | N per unit area (mm−2) | |
| Reservoir | 236.3 | 6328.0 | 6418 | 1940.9 | 8729.5 | 3026 |
| Channel 3 | 29.8 | 515.5 | 6774 | 131.9 | 997.4 | 4379 |
| Channel 5 | 26.3 | 220.3 | 5456 | 88.5 | 886.2 | 3230 |
| Relaxation chamber | 244.2 | 1585.7 | 2108 | 149.3 | 1977.0 | 3641 |
| Channel 8 | 59.5 | 651.8 | 4586 | 171.1 | 1222.3 | 3026 |
| Channel 10 | 44.6 | 297.6 | 4439 | 179.7 | 701.8 | 1763 |
Their evolution along the mixer shows a consistent dispersion of GnP agglomerates along the first mixing zone, a re-agglomeration stage in the relaxation chamber and a subsequent dispersion sequence in the second mixing zone. It should be noted that the material collected at the reservoir upstream of the mixer already evidenced some dispersion, the average particle size reducing by approximately one order of magnitude relative to the original value.
Indeed, for the as-received GnP the average size of 6.7 × 105 μm2 decreased to 1.5 × 104 and 5.2 × 104 μm2 for the composite concentrations of 2 and 10 wt% GnP, respectively, whilst for the functionalized filler the corresponding values are 4.9 × 105 μm2, 1.2 × 104 and 4.8 × 104 μm2, respectively. At a constant filler concentration, Table 2 shows that the number of agglomerates per unit area (mm−2), N, is typically smaller for the composite comprising fGnP-PP. The final N for the nanocomposites with 2 and 10 wt% of fGnP-PP was 1278 and 1763, respectively, while the nanocomposites with 2 and 10 wt% of as-received GnP contained 1766 and 4439, respectively.
Fig. 10 analyzes the evolution along every channel of the mixer of Ar, a dispersion parameter commonly used in the literature,17,39,44,45 showing consistently higher values for the as-received GnP compared to fGnP-PP. Hence, the amount of individually dispersed nanoplates (not detectable by optical microscopy) must be larger for the composite with fGnP-PP. The composites demonstrate a nearly linear progression of dispersion along the first mixing zone, indicating that the process is influenced by the residence time.
Manas-Zloczower and co-workers46,47 postulated that the mechanism of dispersion (rupture versus erosion) of agglomerated particles in a melt depends on the magnitude of the fragmentation number, which is defined as the ratio between hydrodynamic stresses and cohesive strength of the agglomerate, as well as on a finite probability for break-up, which is proportional to the residence time and the agglomerate surface area. Thus, at constant stress, the smaller the agglomerate the longer the exposure time required to break it. As for the rate of dispersion, it should increase with the intensity of the hydrodynamic stresses generated. This is confirmed in Fig. 10, which shows that the slope of the Ar variation along the mixer in the first mixing zone is almost 3 times greater for the system containing 10 wt% GnP compared to 2 wt% GnP, which may be explained by the higher viscosity expected for the composite with 10 wt% GnP.
In fact, Table 3 shows an increase in the total pressure measured for the composite with 10 wt% GnP relative to the composite with 2 wt% GnP, consistent with an increase in viscosity. Pressure is directly proportional to shear stress and thus to viscosity, since the shear rate remains constant. For the composites with fGnP-PP, Table 3 shows a decrease in the total pressure relative to GnP and even to the neat polymer (for 10 wt%), which is indicative of lower viscosity. This could be a consequence of slippage of the polymer melt on the surface of the polymer-grafted graphite nanoplates.48
| Nanoparticle | Nanoparticle content in the composite (wt%) | Pressure (MPa) |
|---|---|---|
| — | 0 | 17.1 |
| GnP | 2 | 18.3 |
| 10 | 24.3 | |
| fGnP-PP | 2 | 17.2 |
| 10 | 14.1 |
Thus, considering again the definition of the fragmentation number, and given the fact that the smaller hydrodynamic stresses are generated at a lower viscosity, the improvement in dispersion (lower Ar) observed for the composites with fGnP-PP can only be attributed to a significant decrease in the agglomerate cohesion. In fact, the PP molecules chemically bonded to the surface of the fGnP-PP increase the equilibrium distance between neighbouring GnP, thus considerably decreasing the van der Waals interactions responsible for the cohesion of their agglomerates.
Once the composites enter the relaxation chamber, the hydrodynamic (shear) stresses become lower than the cohesive forces and enough residence time is provided to allow progressive re-agglomeration, eventually yielding agglomerates with an Ar approaching the value measured at the reservoir. This type of phenomenon was reported before for the melt-mixing of carbon nanotubes with polymers35,39 and remains a major practical difficulty to their widespread use, since practical manufacturing sequences often require re-melting of the melt-mixed composites and processing by another method. GnP functionalization had limited success in stabilizing the morphology of the nanoplates in the relaxation chamber, although the data show a slower re-agglomeration pace.
Dispersion of the two types of fillers regains its approximate linear progression in the second mixing zone, albeit at a smaller pace and after a more abrupt reduction of Ar at the 6th channel. The values of Ar for the composites with a chemically modified filler remain consistently smaller than those for composites with the as-received filler. For the composite with 2 wt% of as-received GnP, the Ar measured after the 10th channel (exit of the second mixing section) is larger than that after the 5th channel (exit of the first mixing section). All the remaining composites reached similar Ar values after the 5th and 10th channels. It should be noticed that, for all composites, the slope of Ar variation in the second mixing section is considerably smaller than that measured along the first mixing section, which is surprising since the first and second mixing sections have exactly the same design, generating the same residence time and the same average shear rate. In terms of fragmentation number, this means that lower hydrodynamic stresses were generated in the second mixing section and/or the cohesive strength of the agglomerates increased from the first to the second mixing sections. While the former is not obvious to account for, the morphology and/or cohesion of the agglomerates formed upon re-agglomeration in the relaxation chamber could be different from that of the agglomerates at the reservoir of the capillary. Agglomerates consisting of smaller particles loosely attached to each other, but appearing as large agglomerates under the optical microscope, may be generated. In the second mixing zone, the small agglomerates will quickly detach from each other, but the probability of a further rupture is lower (under similar flow conditions). This effect is schematically depicted in Fig. 11. The differences between the as-received and chemically functionalized GnP should again be attributed to the differences in nanoplate–nanoplate interactions and slippage of the polymer melt at the nanoparticle polymer-grafted surface. Further studies need to be carried out to extensively characterize the evolution of agglomerate morphology along the dispersion system.
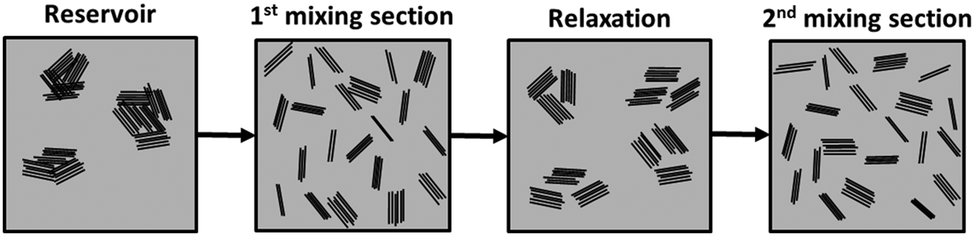 | ||
| Fig. 11 Schematic representation of a possible evolution of GnP agglomerates in PP (grey area), along the mixer. | ||
The electrical conductivity of polymer-based nanocomposites is known to be strongly dependent on the filler size, loading, shape, surface modification and degree of dispersion.49 The variation of AC conductivity with frequency for neat PP and its nanocomposites is presented in Fig. 12.
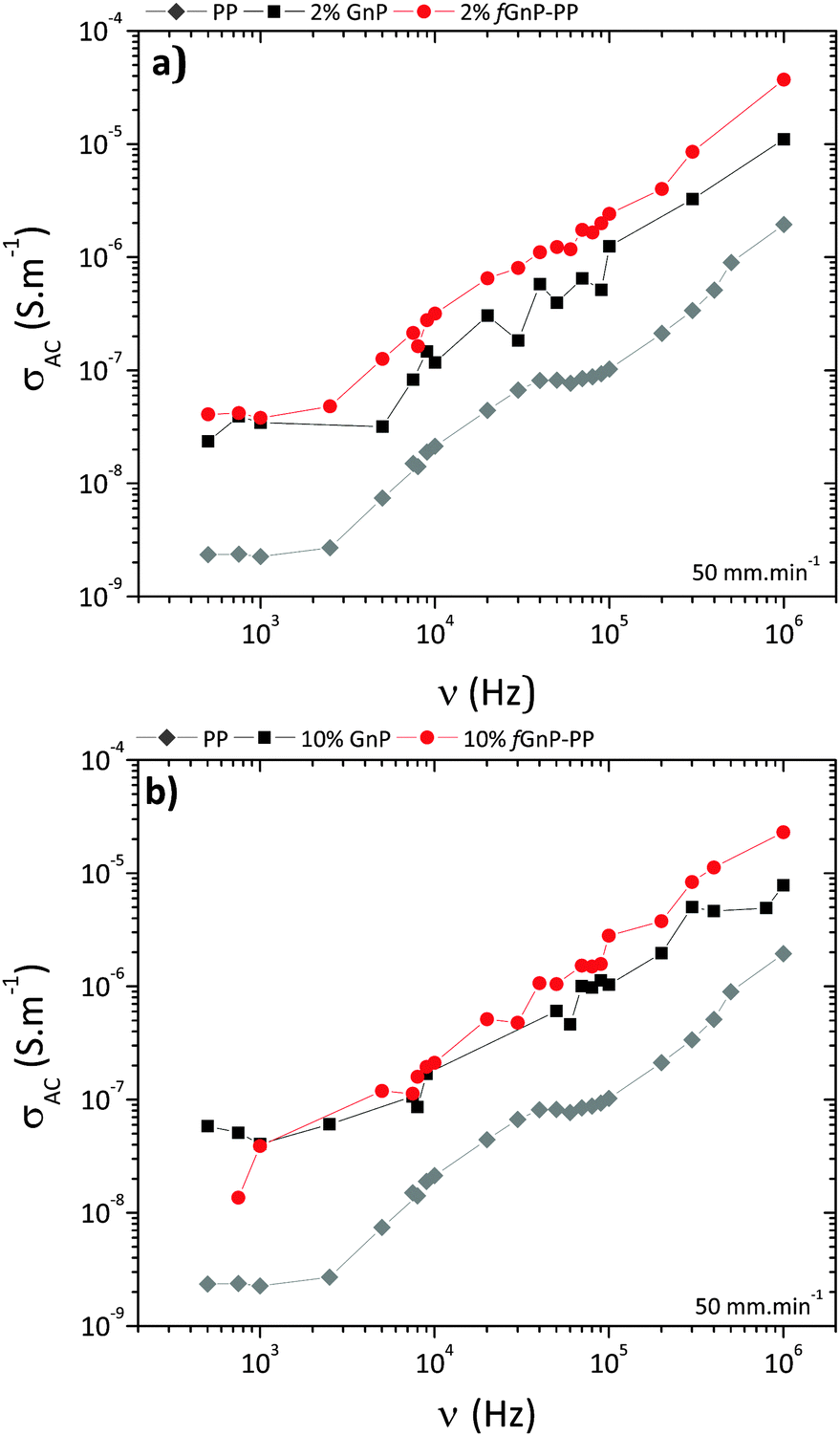 | ||
| Fig. 12 AC conductivity vs. frequency for neat PP and its nanocomposites with (a) 2 and (b) 10 wt% of GnP and fGnP-PP. | ||
The results illustrate the electrically insulating nature of PP, displaying increasing conductivity with increasing frequency, and a conductivity value of approximately 10−9 S m−1 at 1 kHz.50 After incorporation of 2 or 10 wt% of as-received GnP (Fig. 12a), the conductivity of nanocomposites at 1 kHz increases to approximately 10−8 S m−1 and still displays a frequency-dependent behaviour, suggesting that the formation of a three-dimensional interconnecting network was not achieved. Nanocomposites with fGnP-PP (Fig. 12b) show slightly higher electrical conductivity relative to the composites with the as-received GnP.
4. Conclusions
Nanocomposites containing 2 or 10 wt% of graphite nanoplates were prepared by melt-mixing using a small scale intensive mixer coupled to a capillary rheometer. Regardless of filler loading, a significant decrease of the agglomerate size takes place, showing that extensional flow efficiently stimulates the dispersion of graphite nanoplates. In the relaxation chamber, where the shear rate is approximately 0.3 s−1, a prominent increase of the agglomerate area occurs, suggesting that re-agglomeration has taken place. The morphology and/or cohesion of these re-formed agglomerates seem to be different from that of the initial agglomerates at the reservoir of the capillary rheometer, affecting its subsequent dispersion rate in the second mixing zone. Surface modification of GnP with the polymer enhances the stability of dispersion and delays the re-agglomeration. A better GnP/polymer interface and enhanced electrical conductivity could also be noticed for nanocomposites containing functionalized graphite nanoplates.Further advances in surface functionalization of GnP may contribute to the stability of the dispersed nanoparticles in the polymer melt. The magnitude of GnP re-agglomeration may be minimized by merging the steps of nanocomposite manufacturing and processing into one single operation, avoiding its negative effects on the performance.
Acknowledgements
The authors acknowledge the financial support to Project Matepro – Optimizing Materials and Processes, with reference NORTE-07-0124-FEDER-000037 by the Programa Operacional Regional do Norte (ON.2) and Portuguese Foundation for the Science and Technology (FCT) for PEst-C/CTM/LA0025/2013. EC acknowledges FCT for a PhD grant SFRH/BD/87214/2012.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- U. K. Sur, Int. J. Electrochem., 2012, 2012, 12 Search PubMed.
- P. Avouris, Nano Lett., 2010, 10, 4285–4294 CrossRef CAS PubMed.
- E. P. Randviir, D. A. C. Brownson and C. E. Banks, Mater. Today, 2014, 17, 426–432 CrossRef CAS.
- S. Goenka, V. Sant and S. Sant, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- W. G. Zheng, S. C. Wong and H. Sue, J. Polym., 2002, 43, 6767 CrossRef CAS.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5–25 CrossRef CAS.
- R. J. Young, I. A. Kinloch, L. Gong and K. S. Novoselov, Compos. Sci. Technol., 2012, 72, 1459–1476 CrossRef CAS.
- T. K. Das and S. Prusty, Polym.-Plast. Technol. Eng., 2013, 52, 319–331 CrossRef CAS.
- V. Dhand, K. Y. Rhee, H. Ju Kim and D. Ho Jung, J. Nanomater., 2013, 2013, 14 Search PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- R. Sengupta, M. Bhattacharya, S. Bandyopadhyay and A. K. Bhowmick, Prog. Polym. Sci., 2011, 36, 638–670 CrossRef CAS.
- H. Kim and C. W. Macosko, Macromolecules, 2008, 41, 3317–3327 CrossRef CAS.
- T. Enoki and Y. Kobayashi, J. Mater. Chem., 2005, 15, 3999–4002 RSC.
- B. Z. Jang and A. Zhamu, J. Mater. Sci., 2008, 43, 5092–5101 CrossRef CAS.
- P.-C. Ma, N. A. Siddiqui, G. Marom and J.-K. Kim, Composites, Part A, 2010, 41, 1345–1367 CrossRef.
- H. Selig and L. B. Ebert, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1980, vol. 23, pp. 281–327 Search PubMed.
- F. L. Vogel, US Pat., 4565649, 1986 Search PubMed.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- L. R. Bunnell, US Pat., 5186919, 1993 Search PubMed.
- M. Quintana, K. Spyrou, M. Grzelczak, W. R. Browne, P. Rudolf and M. Prato, ACS Nano, 2010, 4, 3527–3533 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- J. Liu, J. Tang and J. J. Gooding, J. Mater. Chem., 2012, 22, 12435–12452 RSC.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- N. Tagmatarchis and M. Prato, J. Mater. Chem., 2004, 14, 437–439 RSC.
- R. Araújo, F. Fernandes, M. Proença, C. Silva and M. Paiva, J. Nanosci. Nanotechnol., 2007, 7, 3441–3445 CrossRef.
- P. A. Denis and F. Iribarne, Int. J. Quantum Chem., 2010, 110, 1764–1771 CAS.
- C. Klumpp, K. Kostarelos, M. Prato and A. Bianco, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 404–412 CrossRef CAS PubMed.
- C. A. Mitchell, J. L. Bahr, S. Arepalli, J. M. Tour and R. Krishnamoorti, Macromolecules, 2002, 35, 8825–8830 CrossRef CAS.
- J. Zhu, J. Kim, H. Peng, J. L. Margrave, V. N. Khabashesku and E. V. Barrera, Nano Lett., 2003, 3, 1107–1113 CrossRef CAS.
- Y.-J. Wan, L.-C. Tang, D. Yan, L. Zhao, Y.-B. Li, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, Compos. Sci. Technol., 2013, 82, 60–68 CrossRef CAS.
- P.-C. Ma, S.-Y. Mo, B.-Z. Tang and J.-K. Kim, Carbon, 2010, 48, 1824–1834 CrossRef CAS.
- R. M. Novais, J. A. Covas and M. C. Paiva, Composites, Part A, 2012, 43, 833–841 CrossRef CAS.
- S. Jamali, M. C. Paiva and J. A. Covas, Polym. Test., 2013, 32, 701–707 CrossRef CAS.
- M. Conceição Paiva, F. Simon, R. M. Novais, T. Ferreira, M. Fernanda Proença, W. Xu and F. Besenbacher, ACS Nano, 2010, 4, 7379–7386 CrossRef PubMed.
- X. Q. Nguyen and L. A. Utracki, US Pat., 5451106, 1995, to Nacional Research Council Canada Search PubMed.
- L. A. Utracki, A. Luciani and D. J. J. Bourry, US Pat., 6550956, B1, 2003 Search PubMed.
- I. Alig, P. Pötschke, D. Lellinger, T. Skipa, S. Pegel, G. R. Kasaliwal and T. Villmow, Polymer, 2012, 53, 4–28 CrossRef CAS.
- C. Vilaverde, R. M. Santos, M. C. Paiva and J. A. Covas, Composites, Part A, 2015, 78, 143–151 CrossRef CAS.
- D. S. McLachlan, C. Chiteme, C. Park, K. E. Wise, S. E. Lowther, P. T. Lillehei, E. J. Siochi and J. S. Harrison, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3273–3287 CrossRef CAS.
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS.
- T. I. T. Okpalugo, P. Papakonstantinou, H. Murphy, J. McLaughlin and N. M. D. Brown, Carbon, 2005, 43, 153–161 CrossRef CAS.
- S. Pegel, P. Pötschke, G. Petzold, I. Alig, S. M. Dudkin and D. Lellinger, Polymer, 2008, 49, 974–984 CrossRef CAS.
- G. R. Kasaliwal, S. Pegel, A. Göldel, P. Pötschke and G. Heinrich, Polymer, 2010, 51, 2708–2720 CrossRef CAS.
- A. Scurati, D. L. Feke and I. Manas-Zloczower, Chem. Eng. Sci., 2005, 60, 6564–6573 CrossRef CAS.
- N. Domingues, A. Gaspar-Cunha, J. A. Covas, M. Camesasca, M. Kaufman and I. Manas-Zloczower, Int. Polym. Process., 2010, 25, 188–198 CrossRef CAS.
- F. Brochard-Wyart, C. Gay and P. G. de Gennes, Macromolecules, 1996, 29, 377–382 CrossRef CAS.
- Y. Pan, L. Li, S. H. Chan and J. Zhao, Composites, Part A, 2010, 41, 419–426 CrossRef.
- C.-R. Yu, D.-M. Wu, Y. Liu, Z.-Z. Yu, A. Dasari, X.-S. Du and Y.-M. Mai, Compos. Sci. Technol., 2011, 71, 1706–1712 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |