
Connecting structure, dynamics and viscosity in sheared soft colloidal liquids: a medley of anisotropic fluctuations†
Fabian
Westermeier
*a,
David
Pennicard
b,
Helmut
Hirsemann
b,
Ulrich H.
Wagner
c,
Christoph
Rau‡§
c,
Heinz
Graafsma¶
b,
Peter
Schall
d,
M.
Paul Lettinga||
*e and
Bernd
Struth
b
aMax Planck Institute for the Structure and Dynamics of Matter, CFEL, Luruper Chaussee 149, 22761 Hamburg, Germany. E-mail: fabian.westermeier@mpsd.mpg.de
bCenter for Free-Electron Laser Science, DESY, Notkestrasse 85, 22607 Hamburg, Germany
cDiamond Light Source Ltd, Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, UK
dVan der Waals-Zeeman Institute, University of Amsterdam, POSTBUS 94485, 1090 GL Amsterdam, The Netherlands
eForschungszentrum Jülich, Institute of Complex Systems (ICS-3), 52425 Jülich, Germany. E-mail: p.lettinga@fz-juelich.de
First published on 9th October 2015
Abstract
Structural distortion and relaxation are central to any liquid flow. Their full understanding requires simultaneous probing of the mechanical as well as structural and dynamical response. We provide the first full dynamical measurement of the transient structure using combined coherent X-ray scattering and rheology on electrostatically interacting colloidal fluids. We find a stress overshoot during the start-up of shear which is due to the strong anisotropic overstretching and compression of nearest-neighbor distances. The rheological response is reflected in uncorrelated entropy-driven intensity fluctuations. While the structural distortion under steady shear is well described by Smoluchowski theory, we find an increase of the particle dynamics beyond the trivial contribution of flow. After the cessation of shear, the full fluid microstructure and dynamics are restored, both on the structural relaxation timescale. We thus find unique structure-dynamics relations in liquid flow, responsible for the macroscopic rheological behavior of the system.
1 Introduction
The flow behavior of liquids and complex fluids is intimately connected to the adaptation of their structure. It is well known that many complex fluids dramatically change their mechanical properties when they are subjected to flow. One of the great challenges in fluid dynamics is to connect this transient material response to the underlying microstructure and dynamics. Such a connection is difficult to establish in conventional atomic liquids due to the involved small lengths and short time scales; this would require the observation of the atomic structure and dynamics simultaneously during rheological measurements, and on time scales shorter than their flow relaxation time, a prohibitively difficult task for atomic liquids. We show here how probing the microscopic response of charged colloidal dispersions by high-flux coherent X-ray scattering during transient shear experiments gives unique insight into the mechanical response of liquids.Suspensions of colloidal particles provide simple model systems for condensed matter as they can form liquid and crystal phases as well as gel and glass states by varying the concentration and interaction potential of the particles.1–3 Each of these phases displays rich rheological behavior, covering wide ranges of non-Newtonian responses.4,5 The rheological behavior is central to applications of these suspensions in food, paints and cosmetics as well as to the function of biological systems. The ability to directly observe colloidal suspensions allows for a direct test of theoretical descriptions of their flow and transient behavior.6–8
Although the fluid phase of colloidal hard spheres is conceptually the simplest phase, its rheology is still very complex.9 In the regime of low shear rates, the viscosity decreases with increasing shear rate – the sample shows shear thinning – even at low volume fractions.10 Yet, at high shear rates, strong shear thickening occurs due to hydro-clustering.11–13 The distortion of the microstructure when convective forces dominate Brownian motion is thought to play a crucial role both in shear thinning and thickening. Steady-state shear distortion has been observed for a broad range of particle volume fractions and systems, in hard sphere systems14–21 as well as in charge-stabilized systems interacting via long range particle interactions.18,21–23 A variety of methods has been used for these investigations, among them static light scattering,14–16,23 neutron scattering17–19,24,25 and X-ray scattering.26–28 During the last years, scanning confocal microscopy13,20,21 enabled the access to the three-dimensional structure of liquids under shear. The same was recently achieved by a combination of different sample geometries using neutron scattering.29,30
The structural distortions affect the shear-induced diffusion of the colloidal particles, hence coupling back to the flow dynamics. This effect is pronounced for colloidal glasses,7,31 but is expected to be less pronounced for liquids, as the liquid phase is ergodic. On the other hand, a soft interaction potential may enhance structural distortions, as observed for soft glasses.32–34 Clearly, the underlying dynamics of the particles also plays an important role and therefore, a pronounced coupling between rheology, microstructure and particle dynamics is to be expected. Connecting transient rheological behavior with the dynamically changing structure remains challenging even for this conceptually simple model system, despite its importance to many applications, and fundamental understanding of liquid flow.
In this paper we elucidate just this relation between transient rheology and adapting liquid structure, connecting these by dynamic structure factor measurements during rheological experiments. By performing coherent X-ray scattering experiments during the start-up, steady shear and relaxation of a colloidal fluid, we reveal the dynamically changing microscopic structure and dynamics that underlie the complex rheological behavior. Using two different scattering geometries, we determine the full three-dimensional structure factor revealing the strong shear-induced distortion of the nearest-neighbor structure. We find that the initial stress overshoot is caused by an overstretching and compression of particle distances, which increases nearest-neighbor potentials by several kBT, with Boltzmann constant kB and absolute temperature T. The increase in stress is accompanied by a high-entropy state, signaled by enhanced intensity fluctuations. After the cessation of shear, the driving force is the restoration of the compressed liquid structure to its quiescent configuration. These measurements for the first time give a full time-resolved, three-dimensional picture of the distortion and relaxation of liquids driven far out of equilibrium.
2 Experimental and methods
2.1 Rheometer and beamline setup
To probe the mechanical response simultaneously with the microscopic structure and dynamics, we employ a novel combination of rheology and dynamic X-ray scattering. This setup provides instantaneous structure factor measurements of the sheared suspension as reported in ref. 35–37, and is extended here to allow for full structural and dynamical characterization in three dimensions. We exploit the high flux and high brilliance of third-generation synchrotron sources to record dynamically correlated data sets and determine the dynamic structure factor. To use a commercial rheometer (MARS III, Thermo Fischer, Germany), a partially coherent X-ray beam was deflected by Germanium crystal optics by 90°, resulting in a vertical X-ray beam, parallel to the rotation axis of the rheometer. This beam passes through the layer of sheared suspension at around 14 mm to the rheometer axis. This setup is equivalent to the pilot installation at beamline BW1, DORIS III, DESY.35 We employed two shear geometries: plate–plate and Couette geometries, both fabricated from polyimide (Vespel, DuPont) to reduce X-ray absorption at moderate X-ray energies. A gap opening of 0.8 mm was used in the plate–plate geometry. The Couette geometry consists of two concentric cylinders of, respectively, 30 and 28 mm, so that the gap is 1.0 mm, while the height of the cylinders is 3.0 mm.The measurements in Couette geometry were performed in the middle of the sample cell's gap. In the plate–plate geometry the beam is directed along the gradient-of-velocity direction (in short the gradient direction ∇v) so that structures that scatter in the flow direction (v) or the vorticity direction (∇ × v) are probed. In the Couette geometry the beam is directed along the vorticity direction so that the flow-gradient plane is probed. Please note that the rheometer is stress-controlled and thus always moving slightly when switched on.
The use of two rheological geometries, a plate–plate and a Couette geometry,38 allows probing the nearest-neighbor structure in three dimensions. We scale shear rates ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) by the measured translational diffusion coefficient of a colloidal sphere of mean radius R0 at infinite dilution D0 ≈ kBT/(6πη0R0) in a solvent with viscosity η0, to define the Péclet number Pe = R02/D0. Structural distortions are expected to become pronounced for Pe > 1, when shear-induced motion dominates the diffusion and thermal motion can no longer equilibrate the structure.
by the measured translational diffusion coefficient of a colloidal sphere of mean radius R0 at infinite dilution D0 ≈ kBT/(6πη0R0) in a solvent with viscosity η0, to define the Péclet number Pe = R02/D0. Structural distortions are expected to become pronounced for Pe > 1, when shear-induced motion dominates the diffusion and thermal motion can no longer equilibrate the structure.
The experiments were performed at beamline P10 of PETRA III at DESY, Germany,39 at a photon energy of 8.05 keV corresponding to a wavelength of λ = 0.15 nm. To obtain a transversally coherent X-ray beam and a sufficient small speckle size in the detector plane, a set of slits upstream of the rheometer reduced the beamsize to typically 20 × 20 μm2 to 30 × 30 μm2 for coherent X-ray scattering experiments. Additional experiments have been performed at beamline I13 of the Diamond Light Source (DLS), Didcot, UK, at the same photon energy and beamsize.
2.2 X-ray detector: LAMBDA detector
For the detection of anisotropic scattering patterns, a 2D photon counting detector is ideally suited, such as the novel LAMBDA detector, developed at DESY, Hamburg.50 It is based on the Medipix3RX readout Application Specific Integrated Circuits,40 bump bonded to a 300 μm silicon sensor, which results in a high quantum efficiency close to 100%. The LAMBDA satisfies the three main needs of a coherent scattering experiment: a sufficient small pixel size of 55 × 55 μm2 to spatially resolve speckle patterns, a high framing speed of up to 2000 frames per s with no time gap between consecutive frames to measure fast changes of the sample and a low noise level which is especially required if the framing rates of the detector are fast and thus the actually detected photons per pixel and frame are accordingly small.The LAMBDA was placed at a distance Rdet ≈ 3.5 m (PETRA III) or 2.0 m (DLS) downstream of the sample position. The distance between the rheometer and the detector was bridged by a flight tube filled with Helium to minimize air scattering.
2.3 Sample
The colloidal fluid consists of almost monodisperse spherical silica particles suspended at a particle volume fraction of ϕ = 0.075 in poly-propylene glycol with an average molecular weight of 4000 g mol−1 (PPG 4000) and a dynamic viscosity of 0.98 Pa s at 25 °C.41 The SiO2-particles were prepared by poly-condensation of tetraethyl-orthosilicate (Si(OC2H5)4) according to a Stöber synthesis.42 In a second reaction step, the particles were coated with 3-trimethoxysilyl-propyl-methacrylate (C10H20O5Si). The mean particle radius was R0 = 105 nm with a particle size polydispersity of ΔR/R0 = 5.2%. The charged particles interact via long-range electrostatic repulsion. We determine an effective charge of Zeff = 130e− and a Debye screening length 1/κ = 75 nm from excellent fits of the scattering data with the recently introduced Modified Penetrating Background-Corrected Rescaled Mean Spherical Approximation (MPB-RMSA).43,44 Thus, the effective radius of the particles is Reff = 180 nm and the effective volume fraction equals ϕeff = 0.38, corresponding to an average particle–particle distance of 361 nm. Hence, this is a very soft system.2.4 X-ray scattering experiments
Steady shear rates in the range between 0.0005 s−1 and 20 s−1 have been probed. Scattering patterns were recorded with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 24000 frames per series. These series were always started 5 seconds before the rheometer started its motion and continued for 15 to 35 seconds after the rheometer stopped probing. To evaluate both the time evolution and the formation of anisotropy of the microstructure of the sample, the momentum transfer Q dependent scattering intensity I(Q) was extracted temporally and azimuthally resolved. 25 frames of 0.002 s and 0.01 s exposure time each (resulting in a time resolution of 0.05 s and 0.25 s, respectively) were summed up for enhanced statistics, and I(Q) was extracted for azimuthal slices of a width of 4° and 10°, respectively.
000 to 24000 frames per series. These series were always started 5 seconds before the rheometer started its motion and continued for 15 to 35 seconds after the rheometer stopped probing. To evaluate both the time evolution and the formation of anisotropy of the microstructure of the sample, the momentum transfer Q dependent scattering intensity I(Q) was extracted temporally and azimuthally resolved. 25 frames of 0.002 s and 0.01 s exposure time each (resulting in a time resolution of 0.05 s and 0.25 s, respectively) were summed up for enhanced statistics, and I(Q) was extracted for azimuthal slices of a width of 4° and 10°, respectively.
The scattering intensity I(Q) depends inter alia on the shape, size and size-distribution of the scatterers, given by the single particle form-factor P(Q). For systems with direct particle interactions, I(Q) is also modulated due to additional ordering of the particles, I(Q) ∝ P(Q) × S(Q), where S(Q) is the static structure factor of the sample, which was subsequently derived from the scattering data by division with a scaled particle form factor. These S(Q) were fitted using the MPB-RMSA model.
To evaluate overall intensity fluctuations in a region of interest (defined by a scattering vector segment ΔQ and an azimuthal angle segment Δθ), the standard deviation of all recorded intensity values of 25 frames (corresponding to a time interval of 0.05 s to 0.25 s) was calculated. This value, σ(Δt, Δθ, ΔQ), was normalized by its temporal mean:
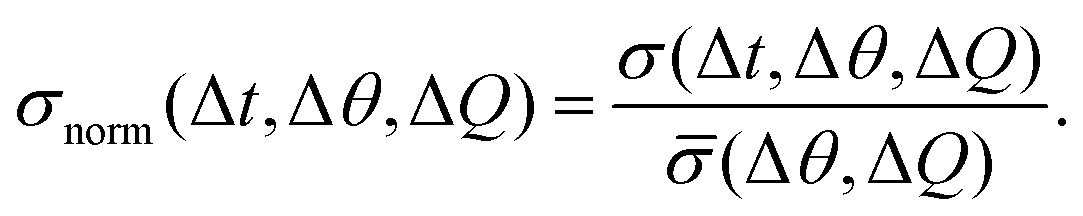 | (1) |
2.5 X-ray photon correlation spectroscopy in flow
In X-ray Photon Correlation Spectroscopy (XPCS),45 the motion of scatterers leads to fluctuations of the scattered light intensity which is thus directly related to the particle dynamics via the intensity auto-correlation function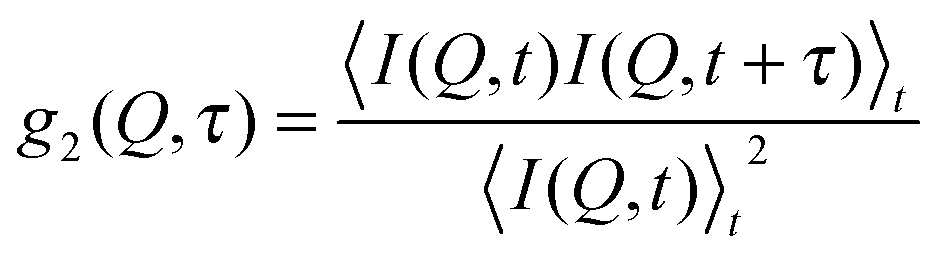 | (2) |
Due to the intrinsic assumption (and subsequent calculation) of time averages, XPCS as a technique can only give access to mean sample dynamics for a certain time period. Therefore, to access changes in the sample dynamics with respect to the experimental time, intensity autocorrelation functions were calculated for different time intervals. While the interval length was 2 s (corresponding to 1000 frames of 0.002 s exposure time), the time step between two subsequent intervals was 0.4 s (corresponding to 200 frames), thus resulting in a temporal overlap between two consecutive autocorrelation functions. Please note that due to the negligible scattering signal of the PPG 4000 polymers the scattering signal is strongly dominated by the colloidal particles and hence only the dynamics of the colloidal particles and not of the suspension medium PPG 4000 is accessed.
Recently XPCS has also been applied to measure local velocities in microfluidics,51 which is important since for complex fluids also complex responses like shear banding should be anticipated.52 In the case of a fluid in laminar flow, the autocorrelation functions are determined by three factors: (a) the diffusive motion of the scatterers due to thermal fluctuations in the sample, characterized by the relaxation rate Γ which is related to the short-time diffusion coefficient D(Q) by Γ(Q) = D(Q)Q2; (b) the transit time of the particles in the scattering volume, depending on the size of the scattering volume h and the average velocity of the particles νt ∝ v0/h; (c) a shear induced decorrelation depending on the velocity gradient of the scatterers, characterized by the shear relaxation rate Γs and depending on Q and the average flow velocity v0via Γs = Q∥v0.
As the three factors determining the intensity autocorrelation function depend on the scattering geometry, they lead to ref. 53–55
| g2,⊥(Q,τ) = β·exp[−2Γτ]·exp[−(νtτ)2] + 1 | (3) |
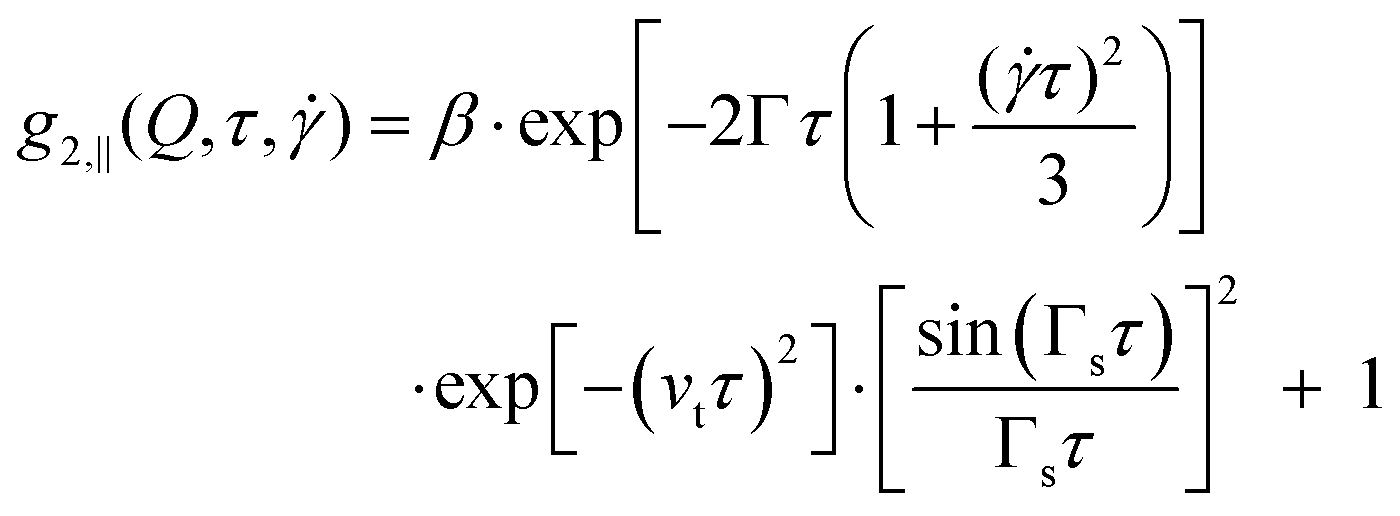 | (4) |
2.6 Liquid structure distortion theory
To analyze the distortion of the structure factor, we modeled S(Q) with generalized Smoluchowski theory on the two-particle level, which neglects hydrodynamic interactions.23,56 In particular, we followed eqn (3) of ref. 23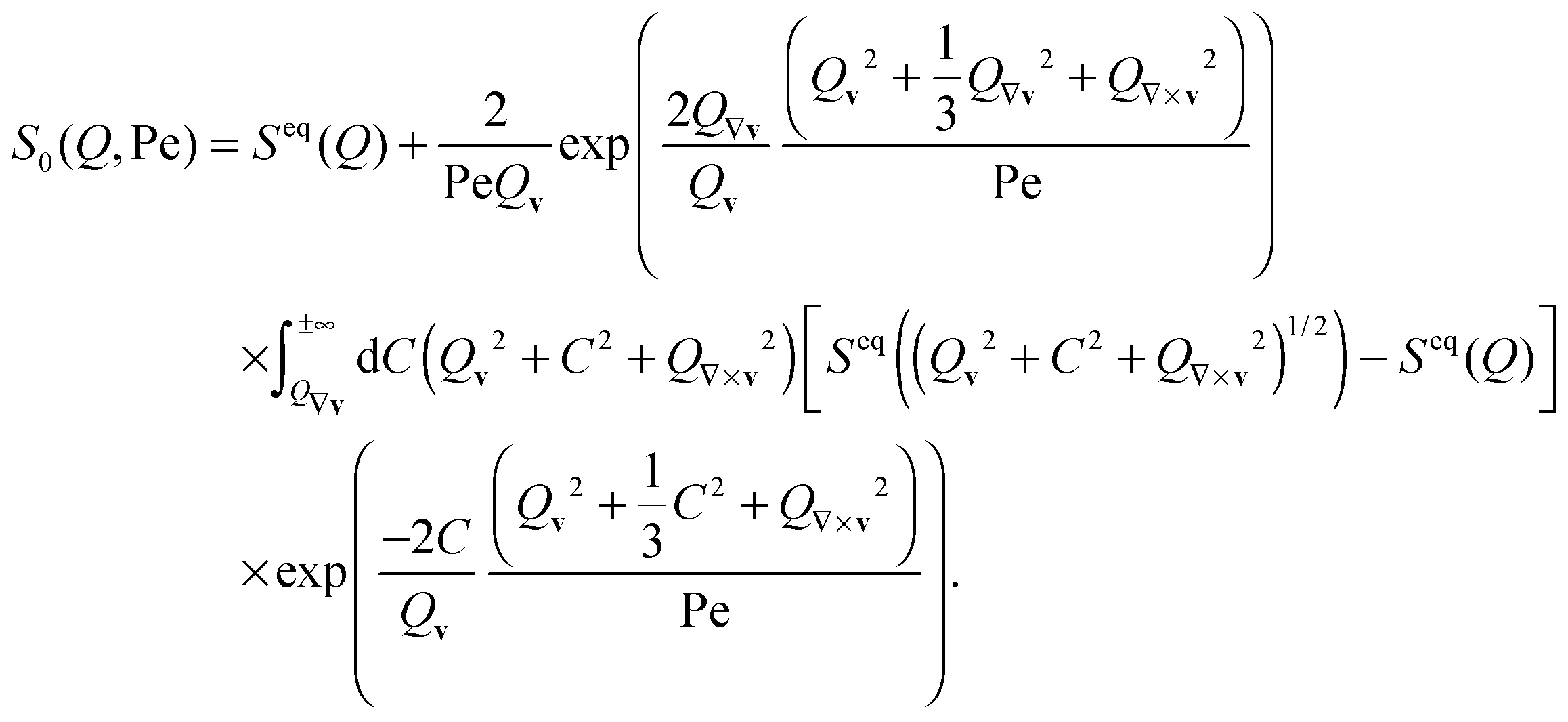 | (5) |
3 Results and discussion
3.1 Microstructure under steady shear flow
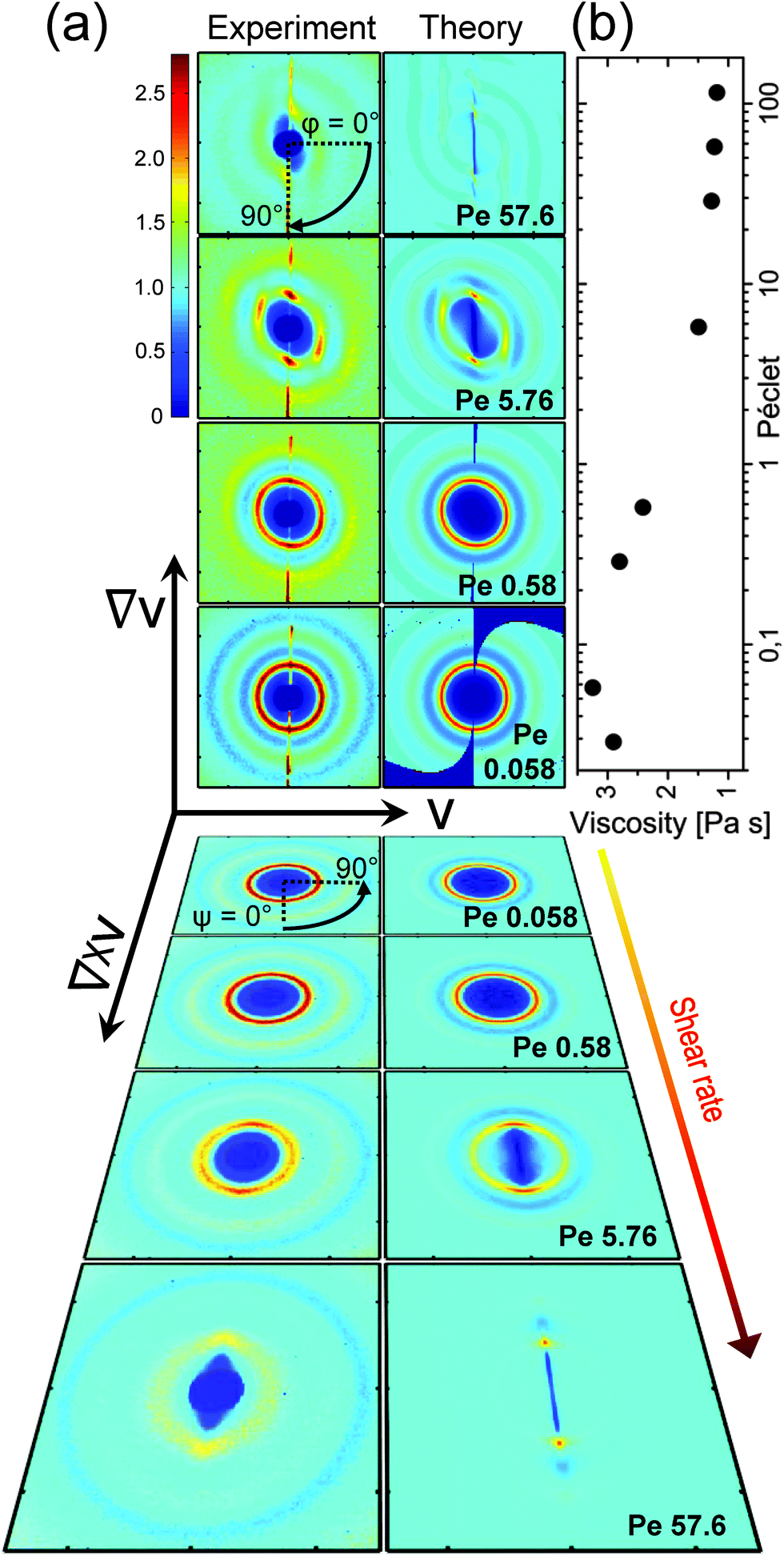 | ||
| Fig. 1 Viscosity and static structure factors at steady shear. (a) S(Q) as measured (left columns) and as calculated using eqn (5) (right columns) in the flow-gradient plane (Couette geometry, top 4 rows) and in the flow-vorticity plane (plate–plate geometry, bottom 4 rows, tilted) for Pe = 0.058, 0.58, 5.76 and 57.6 (bottom to top and in reverse order, respectively). The color-scale is identical for all images. Flow v, gradient ∇v and vorticity ∇ × v direction are indicated by the coordinate system. The definition of the angle in the flow-gradient plane, φ, and in the flow-vorticity plane, ψ, are indicated in the top and fifth row, respectively. (b) Viscosity as a function of Péclet number for values in between 0.0028 and 115.2 (corresponding to shear rates in between 0.005 s−1 and 20 s−1). | ||
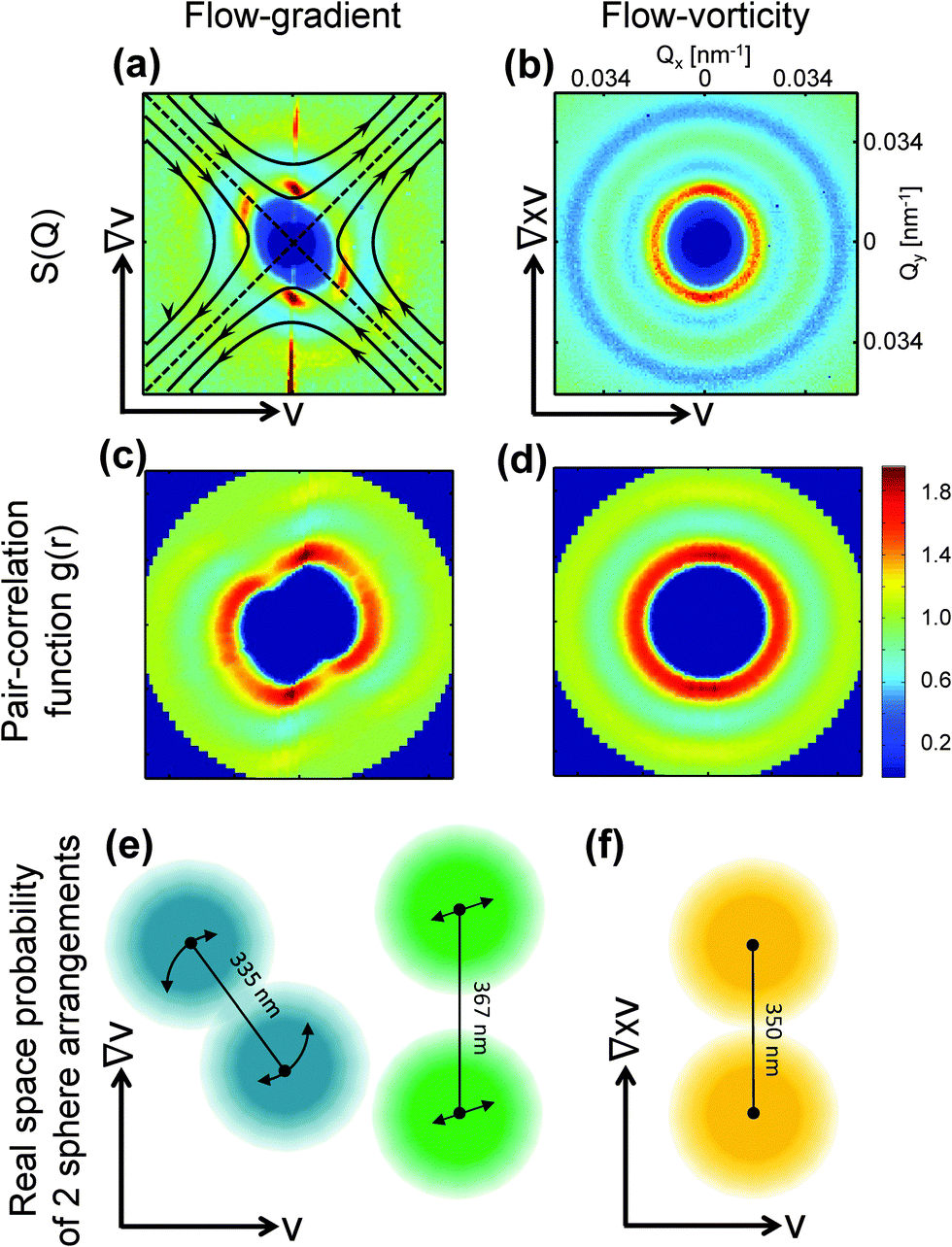 | ||
| Fig. 2 Three-dimensional structure factor, pair correlation function and real-space representation at Pe = 5.76. Left column: Flow-gradient plane (Couette geometry); Right column: Flow-vorticity plane (plate–plate geometry). (a and b) Measured S(Q) at Pe = 5.76, as also shown in Fig. 1, with a schematic representation of the extensional and compressional direction. (c and d) Corresponding pair-correlation functions. (e and f) Real space representation of the most probable two particle configurations in the particular plane. | ||
The two pairs of distinct peaks indicate two co-existing orientational states: first, maxima and minima along the compressional and extensional axis form as the particle distance is compressed from an equilibrium distance of 350 nm to 335 nm due to the compressional component of shear flow, and stretched to 367 nm due to the dilatational component. Second, density fluctuations in the flow direction are mixed by convection, while this is not the case in the other two principal directions. Hence, the structure peak in the flow direction disappears while it remains mainly unaffected in the gradient and vorticity direction.
These trends are largely confirmed in theoretical modeling of the shear-distorted structure factor. We modeled the distortion of the structure factor with generalized Smoluchowski theory on the two-particle level, neglecting hydrodynamic interactions following ref. 23 and 56 (see Experimental and methods for details). The theoretical results are compared with the experimental structure factor in Fig. 1a (right column). Generally, the theory and experiments show good agreement. In hard sphere systems, the thermodynamic, Brownian stresses dominate the hydrodynamic stresses in the shear thinning regime,17,57 which seems to hold for strongly charged systems mainly interacting via long range repulsive interactions.
We can understand the compression of the particles quantitatively: assuming an isotropic charge distribution, we estimate that the osmotically compressed particle distance gives rise to a force of 0.15kBT nm−1 in the compression direction at Pe = 5.8. With the assumption of a linear flow profile between the two rheometer walls, this force is indeed of the same order as the Stokes drag between these two particles which we estimate to be 0.20kBT nm−1, thus balancing the electrostatic force driving back the particles to their quiescent configuration.
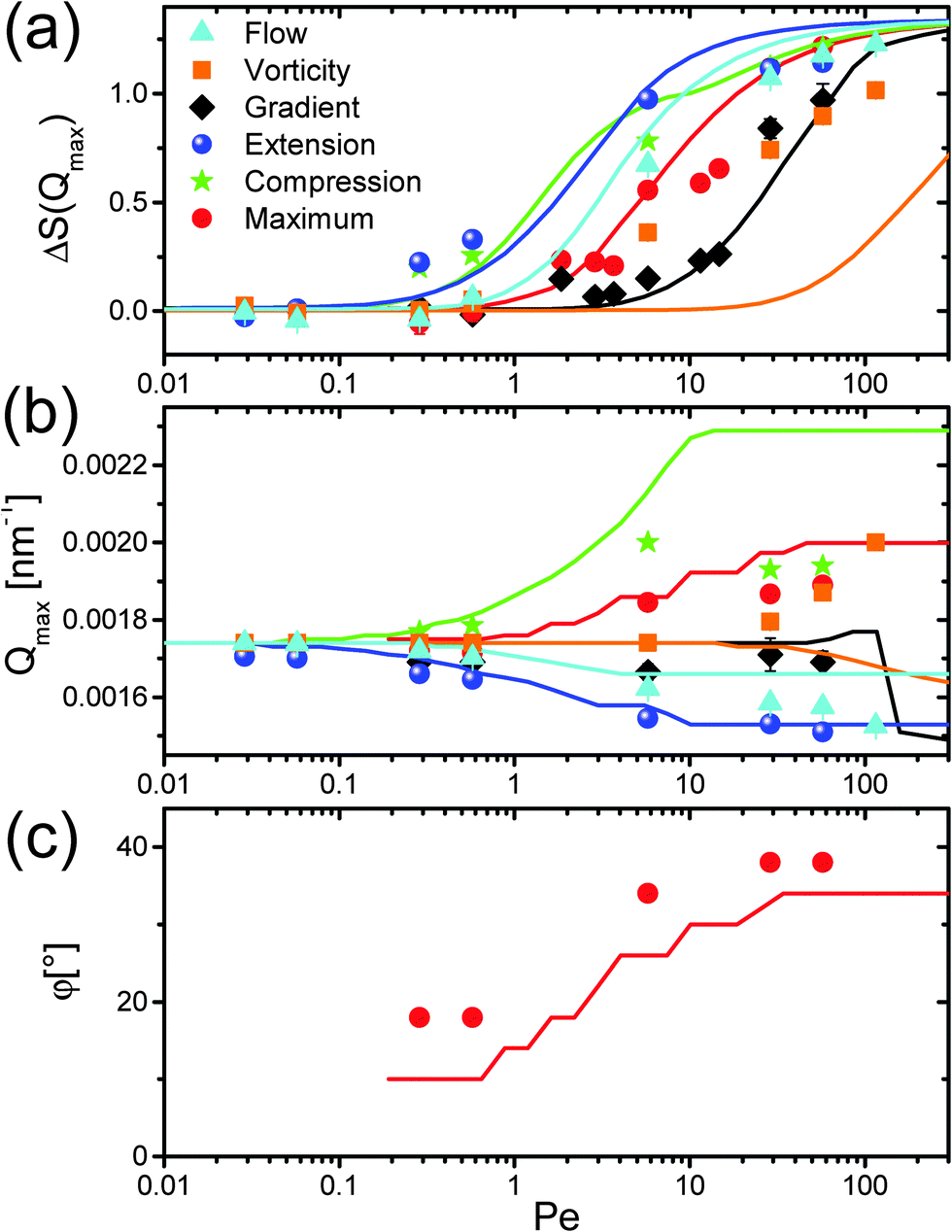 | ||
| Fig. 3 Shear-rate dependence of the structural distortions. (a) Relative height ΔS(Qmax) = Seq(Qmax) − Sshear(Qmax); (b) position Qmax; and (c) angular position φ of the first maximum of the static structure factor S(Q) for flow, gradient and vorticity direction, the direction of extensional and compressional flow and the maximum close to the compressional direction. The angular position in flow-gradient plane is defined in Fig. 1a, with respect to the flow direction. | ||
In contrast to concentrated hard sphere systems, we do not detect an increase of ΔS(Qmax) in the flow direction.15,25,36 Apparently, in the probed electrostatically interacting systems a compression does not lead to an enhancement of the structure. This can be attributed to the fact that in hard sphere systems structure arises due to the collision of nano-particles with each other, while this is strongly diminished in electrostatically interacting systems where the particles tend to maximize the distance between each other. As the potential is softer and particles have more room to adjust, one expects more pronounced flow-induced changes from the equilibrium structure factor. Indeed, around medium Pe in between 1 to 10 the structure is strongly diminished, especially in comparison to hard sphere systems.19,30 This could also explain the relatively low Péclet numbers where we first observe structural changes as compared to hard sphere experiments.21,30,60 Note also that the theory is only valid for low Péclet numbers, so that the correspondence of theory and experiments for Pe ≫ 1, such as the merging of the maxima in the flow-gradient plane with increasing Péclet numbers, may be fortuitous.
3.2 Transient behavior
Our measurements hence reveal the pronounced distortion of the nearest-neighbor structure underlying the rheological shear thinning. We can now use the full time resolution to follow dynamic behavior during the transients. To elucidate the build-up and relaxation of the distorted structure, we follow the system during the start-up, steady-state, and after the cessation of shear, as shown in Fig. 4.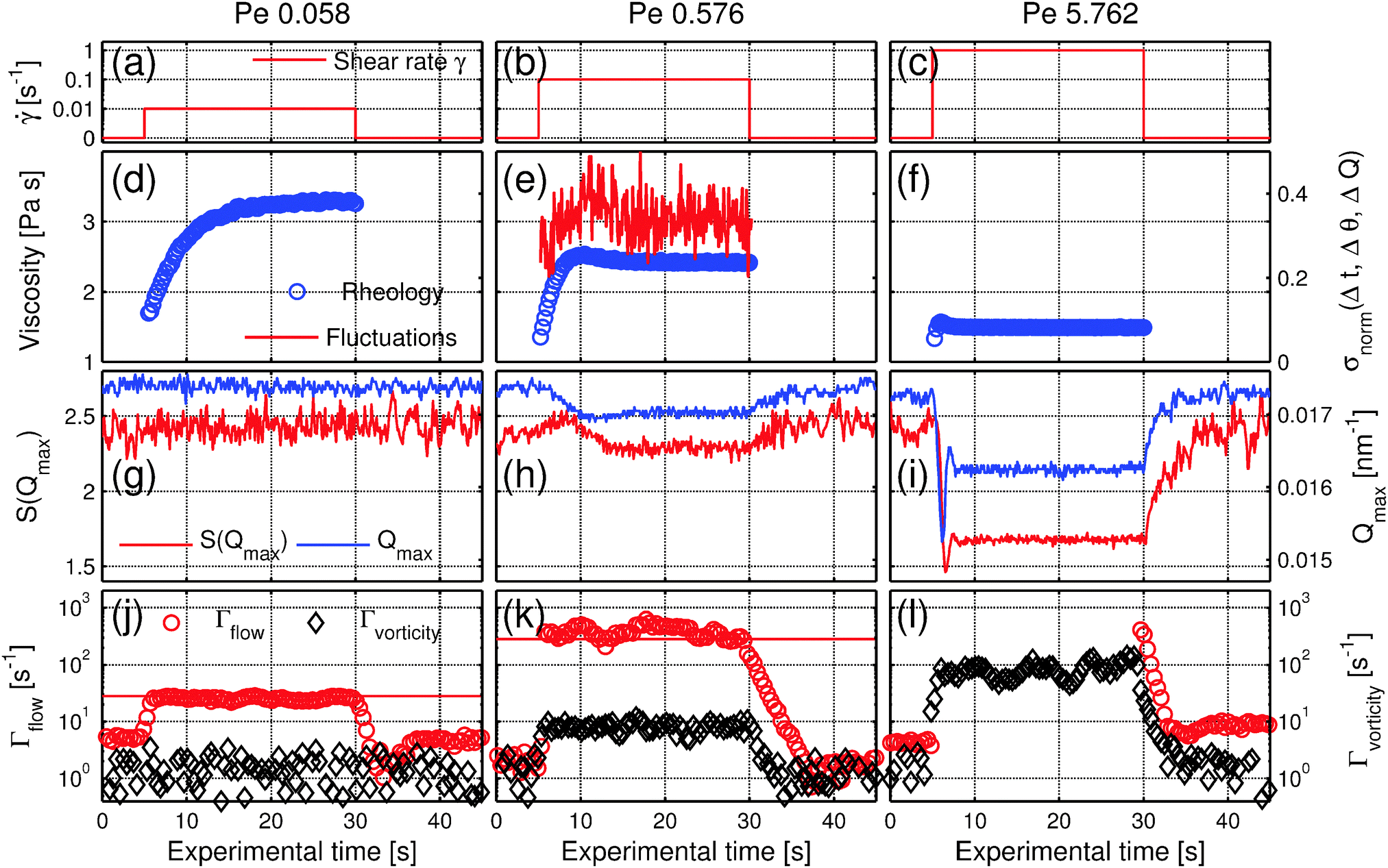 | ||
| Fig. 4 Transients at Pe = 0.058, 0.576 and 5.762. (a–c) Applied shear-rate . (d–f) Temporal evolution of the viscosity (blue circles) and for Pe = 0.576 fluctuations of the scattering intensity I(Q) (red solid line) at Q = 0.015 nm−1. (g–i) Temporal evolution of S(Qmax) (red line) and the position of the first maximum Qmax (blue line) at an angle φ = 0°, ψ = 90° in flow direction. (j–l) Relaxation rate in the flow direction Γflow (red circles) and in the vorticity direction Γvorticity (black diamonds). At Pe = 5.762, Γflow is shown only before and after shearing, as during shear the intensity was decorrelating too fast for reliable dynamics measurements. The solid red lines correspond to the expected relaxation rates assuming a linear flow profile in between the two rheometer plates. The columns correspond to Péclet numbers Pe = 0.0576, 0.576, 5.762, respectively. For all measurements the rheometer started after 5.0 s and sheared for 25 s. | ||
A new steady-state structure with decreased inter-particle correlations is established about 10 s after the start of shear. Interestingly, the transient behavior is also reflected in the fluctuations of the scattered intensity σnorm(Δt, Δθ, ΔQ) defined by eqn (1) (see Experimental and methods), as shown in Fig. 4e. The fluctuations were recorded at Q = 0.015 nm−1, near Qmax at the slope of the first maximum of S(Q). These enhanced fluctuations indicate higher entropy of the system: in equilibrium, the system is isotropic, while out of equilibrium, when stress is applied, the symmetry is broken as the sample evolves into an anisotropic oriented state. The more pronounced intensity fluctuations during the transient can be seen as the footprint of a different entropic state during the reorganization of the colloidal system. For Pe = 5.8 all responses are more pronounced, showing damped oscillations reminiscent of an elastic response, where the system swings around its new steady state structure, while for Pe < 0.1, where Brownian motion is dominating, no deformation is observed as the system can counteract deformation.
After the cessation of flow, all parameters relax to their equilibrium value within about 10 seconds both for Pe = 0.576 and Pe = 5.76. This relaxation time is on the order of the structural relaxation time τS = 4R02/D0 = 20 s. The relaxation time is independent of the previous applied shear rate, the only difference being the amplitude of relaxation, which is highest for Pe = 5.76 where the sheared state is furthest from the quiescent state. At a lower Péclet number these transient structural effects vanish, as shown for Pe = 0.0576, for which the smooth evolution of viscosity is reflected in an unaffected micro-structure. This decrease of the stress overshoot with a decreasing shear rate has been also observed in hard sphere systems.61–63
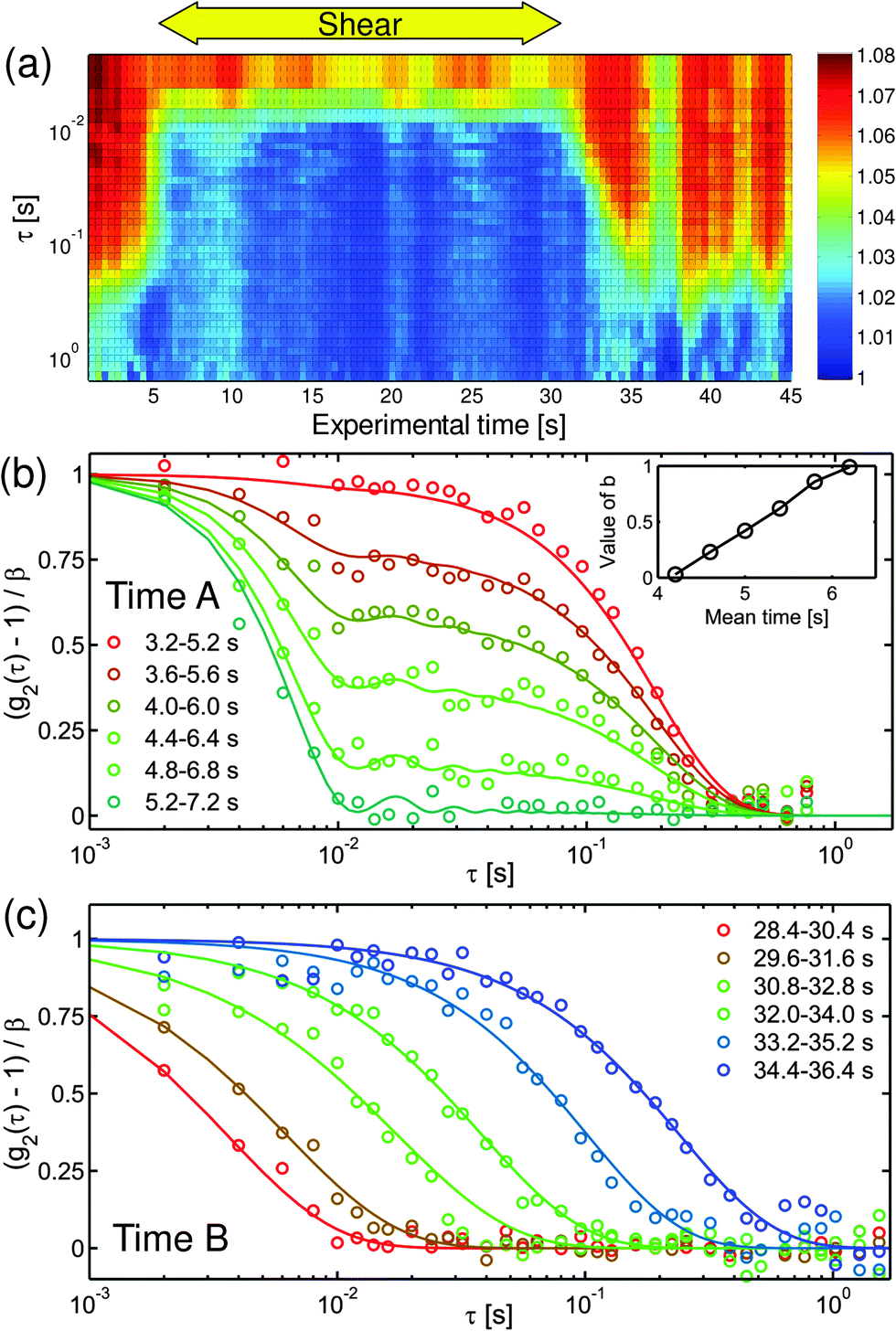 | ||
| Fig. 5 Time evolution of the intensity autocorrelation functions at Pe = 0.288. (a) Intensity autocorrelation functions g2(τ) for different experimental times at Q = 0.021 nm−1 in flow direction. The time interval for the calculation of each autocorrelation function is 2 s while the gap between two consecutive autocorrelation function is 0.4 s. The rheometer started to shear with = 0.05 s−1, corresponding to Pe = 0.288 at experimental time 5.0 s and stopped at 30.0 s. A and B correspond to the experimental times shown in (b) and (c) during the start-up and cessation of shear. The loss of visibility of the autocorrelation functions between 35 s and 40 s is possibly due to some fluctuations of the experimental setup during this period. (b) Normalized intensity autocorrelation functions (g2(τ) − 1)/β in flow direction of 6 time intervals Q = 0.021 nm−1 at the start-up of shear. The solid lines show a fit of eqn (6) to the data. The inset shows the weighting factor b as a function of mean experimental time. (c) Normalized intensity autocorrelation functions (g2(τ) − 1)/β in flow direction for 6 time intervals after the cessation of flow at Q = 0.021 nm−1. The solid lines show a modelization using a stretched exponential decay. | ||
During the start-up, the autocorrelation function shows two distinct decays as depicted in Fig. 5b, indicating the presence of two relaxation processes, thermal (before the onset of shear) and shear induced (after the onset of shear). Assuming that shear-induced relaxation with rate Γs superimposes on the thermal relaxation of the quiescent suspension with rate Γ, the decay of the autocorrelation function can be described by
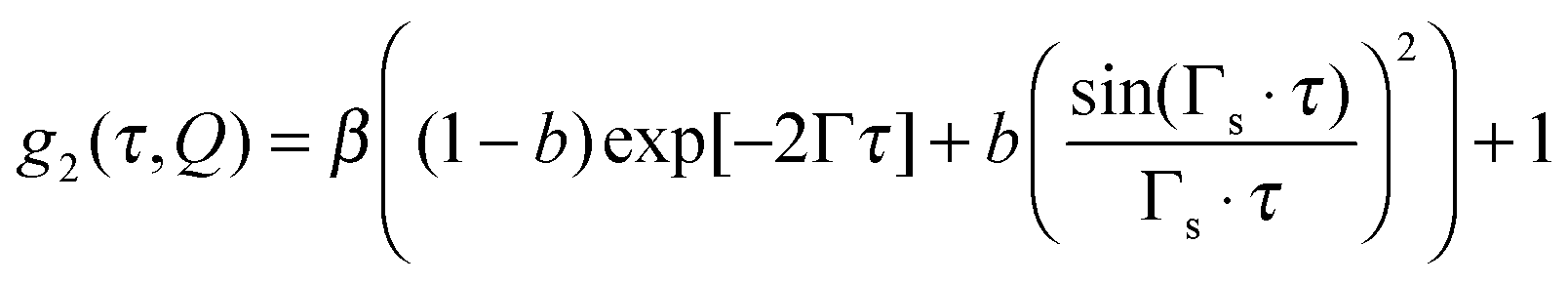 | (6) |
The particle relaxation after the cessation of flow is characterized by a gradual shift of the autocorrelation functions towards a slower decay, as is apparent for t > 30 s in Fig. 5a. To compare the autocorrelation functions at all experimental times, we modeled them with the Kohlrausch–Williams–Watts function,64g2(Q) = β·exp(−2·(Γτ)γ) + 1, with γ being the exponent of the exponential decay. The fits nicely agree with the experimental autocorrelation functions during the cessation of shear, indicating a broad distribution of decay times and a gradual recovery of the equilibrium relaxation time, as depicted in Fig. 5c. The extracted relaxation rates Γ in flow and vorticity directions are depicted in Fig. 4j–l, together with the structural changes in panels Fig. 4g–i. For a low Péclet number of 0.0576, no changes in Γ can be detected perpendicular to the flow direction during the start up or after the cessation of shear, indicating that there is almost no contribution of shear to the thermal relaxation rate due to diffusion. This situation changes for increasing shear rates Pe > 0.1, where the relaxation rate increases due to the applied shear. The relaxation process after the cessation of shear takes place for approximately 10 s, corresponding to the timescale of the relaxation of the microstructure, which is on the order of the structural relaxation time τS. Hence, the time scale of the dynamic relaxation coincides nicely with the time scale of structural relaxation, confirming the tight relationship between the microstructure and dynamics. Because of this coupling, the dynamics continues to be fast, even after the complete cessation of shear, while the original quiescent structure has not reformed, until the original structure has re-established, and concomitantly, the dynamics have decayed to its original rate. The initial decay rate during the recovery of the equilibrium relaxation time is about one to two orders of magnitude higher than the quiescent diffusion, indicating thus that there is a driving force for the reestablishment of the equilibrium microstructure. This driving force is given by the difference between the potential at the non-equilibrium compressed state and the potential in the liquid quiescent state, where the colloidal system balances the forces between individual particles and thus restores the initial microstructure. Moreover, the decay of overall fluctuations, comprising both correlated and uncorrelated ones, can be seen as an indication of energy release from the system.
4 Summary and conclusion
Our combined measurements reveal the three-dimensional structure and dynamics underlying the rheology during the start-up, steady shear and relaxation of charged colloidal liquids. The shear thinning of the colloidal fluid can be attributed to the overall distortion of the structure factor. We confirm predictions by generalized Smoluchowski theory56 for the distortion of the full three-dimensional structure factor in steady shear flow which leads to shear thinning. In particular, the flow gradient plane gives us access to the compressional and extensional direction where the structure is compressed and stretched, respectively. This allows us to estimate the forces to compress the particles. We show that the compression force is sufficient to overcome the electrostatic potential inducing the microstructure in the quiescent liquid.The compression forces form the basis for understanding the transient behavior. The structural restoration after the cessation of shear flow is accompanied by a pronounced slowing down of the colloidal dynamics which is initially up to two orders of magnitude higher than the quiescent diffusion. This anomalous flow-enhanced diffusion manifests directly the electrostatic driven restoring force: it tends to equilibrate the particle concentration in the compression and vorticity direction, while filling the depleted space in the extensional and flow direction on microscopic length scales corresponding to Qmax. We suggest that this is in analogy to collective diffusion in equilibrium, where the osmotic compressibility 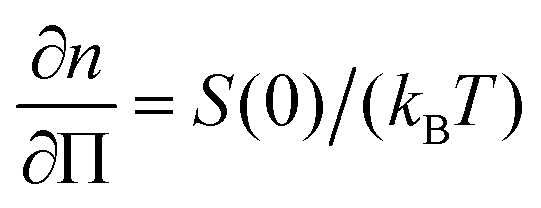 drives the diffusion for Q → 0 for high-density gradients. It is shown theoretically that this effect is enhanced by shear flow,65–67 which is confirmed by simulations of fluidized soft glassy systems,32,34 where unbalanced contact forces drive the system back to its equilibrium state. In analogy, the long-range repulsive interactions between the charged particles likely enhance this process and make it occur more pronounced than is found for short-ranged repulsive systems, an effect which prompts for more theoretical work.
drives the diffusion for Q → 0 for high-density gradients. It is shown theoretically that this effect is enhanced by shear flow,65–67 which is confirmed by simulations of fluidized soft glassy systems,32,34 where unbalanced contact forces drive the system back to its equilibrium state. In analogy, the long-range repulsive interactions between the charged particles likely enhance this process and make it occur more pronounced than is found for short-ranged repulsive systems, an effect which prompts for more theoretical work.
Stress relaxation after the cessation of shear flow was also observed for soft glasses and related to the restoration of structure in simulations.33 We show here experimentally that indeed the stress relaxation is governed by the structural and dynamical relaxation in the fluid phase, which takes place on the timescales of the structural relaxation time.
In addition, entropic intensity fluctuations, quantified by σnorm(Δt, Δθ, ΔQ), show a similar transient behavior to both the stress and the microstructure. Due to the breaking of symmetry when shear is applied, the system can evolve towards more possible configurations, pointing towards higher entropy of the system reflected by the increased fluctuations. The events that cause the fluctuations persist in these soft systems, because of their regenerative character. This is consistent with the enhanced diffusion as found by XPCS.
In conclusion, the combination of rheology and high-flux coherent X-ray scattering on soft colloidal dispersion allowed us to identify stress overshoots in transient with overshoots in the structure deformation as well as in entropic intensity fluctuations. These observations reveal the connection between the microscopic structure and dynamics and the mechanical response of fluids. Moreover, we could show that shear flow strongly enhances the diffusion, which returns to the equilibrium behavior at the same time scale as the equilibrium structure is restored after the cessation of shear flow. We argue that the unique combination of experiments we put forward here opens up the road for the full understanding of complex flow behavior on all relevant length-scales.
Acknowledgements
We wish to acknowledge the support of the beamline staff of P10, in particular Michael Sprung, and the support of the beamline staff of I13, in particular Andrew Peach. We would like to thank Dmitry Denisov and Dang Minh Triet for fruitful discussions. For the LAMBDA development, we would like to thank Sergej Smoljanin. We are grateful to Jan Dhont and Gerhard Nägele for helpful suggestions and discussions.References
- P. N. Pusey and W. van Megen, Nature, 1986, 320, 340–342 CrossRef CAS
.
- P. J. Lu and D. A. Weitz, Annu. Rev. Condens. Matter Phys., 2013, 4, 217–233 CrossRef CAS
- V. J. Anderson and H. N. W. Lekkerkerker, Nature, 2002, 416, 811–815 CrossRef CAS PubMed
- J. Vermant and M. J. Solomon, J. Phys.: Condens. Matter, 2005, 17, R187–R216 CrossRef CAS
- D. Vlassopoulos and M. Cloitre, Curr. Opin. Colloid Interface Sci., 2014, 19, 561–574 CrossRef CAS
- J. M. Brader, J. Phys.: Condens. Matter, 2010, 22, 363101 CrossRef CAS PubMed
- M. Laurati, K. J. Mutch, N. Koumakis, J. Zausch, C. P. Amann, A. B. Schofield, G. Petekidis, J. F. Brady, J. Horbach, M. Fuchs and S. U. Egelhaaf, J. Phys.: Condens. Matter, 2012, 24, 464104 CrossRef CAS PubMed
- E. Nazockdast and J. F. Morris, Soft Matter, 2012, 8, 4223–4234 RSC
- H. M. Laun, Angew. Makromol. Chem., 1984, 123, 335–359 CrossRef
- D. R. Foss and J. F. Brady, J. Rheol., 2000, 44, 629–651 CrossRef CAS
- D. R. Foss and J. F. Brady, J. Fluid Mech., 2000, 407, 167–200 CrossRef CAS
- N. J. Wagner and J. F. Brady, Phys. Today, 2009, 62, 27–32 CrossRef CAS
- X. Cheng, J. H. McCoy, J. N. Israelachvili and I. Cohen, Science, 2011, 333, 1276–1279 CrossRef CAS PubMed
- B. J. Ackerson, J. van der Werff and C. G. de Kruif, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 37, 4819–4827 CrossRef
- B. J. Ackerson, C. G. De Kruif, N. J. Wagner and W. B. Russel, J. Chem. Phys., 1989, 90, 3250–3253 CrossRef CAS
- N. J. Wagner and W. B. Russel, Phys. Fluids A, 1990, 2, 491–502 CrossRef CAS
- J. W. Bender and N. J. Wagner, J. Colloid Interface Sci., 1995, 172, 171–184 CrossRef CAS
- B. J. Maranzano and N. J. Wagner, J. Chem. Phys., 2002, 117, 10291–10302 CrossRef CAS
- D. P. Kalman and N. J. Wagner, Rheol. Acta, 2009, 48, 897–908 CrossRef CAS
- X. Cheng, X. Xu, S. A. Rice, A. R. Dinner and I. Cohen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 63–67 CrossRef CAS PubMed
- B. Xu and J. F. Gilchrist, J. Chem. Phys., 2014, 140, 204903 CrossRef PubMed
- B. J. Ackerson, Phys. A, 1991, 174, 15–30 CrossRef CAS
- Y. Yan and J. Dhont, Phys. A, 1993, 198, 78–107 CrossRef CAS
- B. J. Ackerson, J. B. Hayter, N. A. Clark and L. Cotter, J. Chem. Phys., 1986, 84, 2344–2349 CrossRef CAS
- H. Watanabe, M.-L. Yao, K. Osaki, T. Shikata, H. Niwa, Y. Morishima, N. P. Balsara and H. Wang, Rheol. Acta, 1998, 37, 1–6 CrossRef CAS
- H. Hoekstra, J. Mewis, T. Narayanan and J. Vermant, Langmuir, 2005, 21, 11017–11025 CrossRef CAS PubMed
- H. Versmold, S. Musa, C. Dux, P. Lindner and V. Urban, Langmuir, 2001, 17, 6812–6815 CrossRef CAS
- H. Versmold, S. Musa and A. Bierbaum, J. Chem. Phys., 2002, 116, 2658–2662 CrossRef CAS
- A. K. Gurnon, C. R. Lopez-Barron, A. P. R. Eberle, L. Porcar and N. J. Wagner, Soft Matter, 2014, 10, 2889–2898 RSC
- A. K. Gurnon and N. J. Wagner, J. Fluid Mech., 2015, 769, 242–276 CrossRef CAS
- V. Chikkadi, D. M. Miedema, M. T. Dang, B. Nienhuis and P. Schall, Phys. Rev. Lett., 2014, 113, 208301 CrossRef CAS PubMed
- J. R. Seth, L. Mohan, C. Locatelli-Champagne, M. Cloitre and R. T. Bonnecaze, Nat. Mater., 2011, 10, 838–843 CrossRef CAS PubMed
- L. Mohan, R. T. Bonnecaze and M. Cloitre, Phys. Rev. Lett., 2013, 111, 268301 CrossRef PubMed
- L. Mohan, C. Pellet, M. Cloitre and R. Bonnecaze, J. Rheol., 2013, 57, 1023–1046 CrossRef CAS
- B. Struth, K. Hyun, E. Kats, T. Meins, M. Walther, M. Wilhelm and G. Grübel, Langmuir, 2011, 27, 2880–2887 CrossRef CAS PubMed
- D. Denisov, M. Dang, B. Struth, G. Wegdam and P. Schall, Sci. Rep., 2013, 3, 1631 CAS
- M. P. Lettinga, P. Holmqvist, P. Ballesta, S. Rogers, D. Kleshchanok and B. Struth, Phys. Rev. Lett., 2012, 109, 246001 CrossRef CAS PubMed
- J. M. Kim, A. P. R. Eberle, A. K. Gurnon, L. Porcar and N. J. Wagner, J. Rheol., 2014, 58, 1301–1328 CrossRef
- A. V. Zozulya, S. Bondarenko, A. Schavkan, F. Westermeier, G. Grübel and M. Sprung, Opt. Express, 2012, 20, 18967–18976 CrossRef CAS PubMed
- R. Ballabriga, J. Alozy, G. Blaj, M. Campbell, M. Fiederle, E. Frojdh, E. H. M. Heijne, X. Llopart, M. Pichotka, S. Procz, L. Tlustos and W. Wong, J. Instrum., 2013, 8, C02016 Search PubMed
- M. G. McLin and C. A. Angell, J. Phys. Chem., 1991, 95, 9464–9469 CrossRef CAS
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef
- M. Heinen, P. Holmqvist, A. J. Banchio and G. Nägele, J. Chem. Phys., 2011, 134, 044532 CrossRef PubMed
- M. Heinen, P. Holmqvist, A. J. Banchio and G. Nägele, J. Chem. Phys., 2011, 134, 129901 CrossRef
- G. Grübel and F. Zontone, J. Alloys Compd., 2004, 362, 3–11 CrossRef
- M. Sutton, S. G. J. Mochrie, T. Greytak, S. E. Nagler, L. E. Berman, G. A. Held and G. B. Stephenson, Nature, 1991, 352, 608–610 CrossRef
- T. Thurn-Albrecht, W. Steffen, A. Patkowski, G. Meier, E. W. Fischer, G. Grübel and D. L. Abernathy, Phys. Rev. Lett., 1996, 77, 5437–5440 CrossRef CAS PubMed
- J. Gapinski, A. Patkowski, A. J. Banchio, J. Buitenhuis, P. Holmqvist, M. P. Lettinga, G. Meier and G. Nagele, J. Chem. Phys., 2009, 130, 084503 CrossRef CAS PubMed
- F. Westermeier, B. Fischer, W. Roseker, G. Grübel, G. Nägele and M. Heinen, J. Chem. Phys., 2012, 137, 114504 CrossRef PubMed
- D. Pennicard, S. Lange, S. Smoljanin, H. Hirsemann and H. Graafsma, J. Instrum., 2012, 7, C11009 CrossRef
- A. Fluerasu, P. Kwasniewski, C. Caronna, F. Destremaut, J.-B. Salmon and A. Madsen, New J. Phys., 2010, 12, 035023 CrossRef
- J. K. G. Dhont and W. J. Briels, Rheol. Acta, 2008, 47, 257–281 CrossRef CAS
- D. P. Chowdhury, C. M. Sorensen, T. W. Taylor, J. F. Merklin and T. W. Lester, Appl. Opt., 1984, 23, 4149–4154 CrossRef CAS PubMed
- S. Busch, T. Jensen, Y. Chushkin and A. Fluerasu, Eur. Phys. J. E: Soft Matter Biol. Phys., 2008, 26, 55–62 CrossRef CAS PubMed
- W. R. Burghardt, M. Sikorski, A. R. Sandy and S. Narayanan, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 021402 CrossRef PubMed
- J. K. G. Dhont, J. Fluid Mech., 1989, 204, 421–431 CrossRef CAS
- N. J. Wagner and B. J. Ackerson, J. Chem. Phys., 1992, 97, 1473–1483 CrossRef CAS
- M. P. Lettinga, H. Wang and J. K. G. Dhont, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 061405 CrossRef CAS PubMed
- J. Bergenholtz, J. F. Brady and M. Vicic, J. Fluid Mech., 2002, 456, 239–275 CAS
- M. C. Newstein, H. Wang, N. P. Balsara, A. A. Lefebvre, Y. Shnidman, H. Watanabe, K. Osaki, T. Shikata, H. Niwa and Y. Morishima, J. Chem. Phys., 1999, 111, 4827–4838 CrossRef CAS
- H. Watanabe, M.-L. Yao, A. Yamagishi, K. Osaki, T. Shitata, H. Niwa and Y. Morishima, Rheol. Acta, 1996, 35, 433–445 CrossRef CAS
- C. P. Amann, M. Siebenbürger, M. Krüger, F. Weysser, M. Ballauff and M. Fuchs, J. Rheol., 2013, 57, 149–175 CrossRef CAS
- N. Koumakis, A. Pamvouxoglou, A. S. Poulos and G. Petekidis, Soft Matter, 2012, 8, 4271–4284 RSC
- G. Williams and D. C. Watts, Trans. Faraday Soc., 1970, 66, 80–85 RSC
- M. Marchioro and A. Acrivos, J. Fluid Mech., 2001, 443, 101–128 Search PubMed
- A. M. Leshansky and J. F. Brady, J. Fluid Mech., 2005, 527, 141–169 CrossRef
- A. M. Leshansky, J. F. Morris and J. F. Brady, J. Fluid Mech., 2008, 597, 305–341 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information on the angular dependence of the intensity autocorrelation function and the shear dependent viscosity of PPG 4000. See DOI: 10.1039/c5sm01707f |
| ‡ Also at: Department of Otolaryngology, Northwestern University Feinberg School of Medicine, Chicago, Illinois 60611, USA. |
| § Also at: School of Materials, University of Manchester, Grosvenor St., Manchester M1 7HS, UK. |
| ¶ Also at: Mid Sweden University, 851 70 Sundsvall, Sweden. |
| || Also at: Laboratory for Soft Matter and Biophysics, KU Leuven, Celestijnenlaan 200D, B-3001 Leuven, Belgium. |
| This journal is © The Royal Society of Chemistry 2016 |