
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed incorporation of atmospheric CO2: efficient synthesis of functionalized oxazolidinones†
Patricia
García-Domínguez
,
Lorenz
Fehr
,
Giulia
Rusconi
and
Cristina
Nevado
*
Department of Chemistry, University of Zürich, Winterthurerstrasse 180, CH-8057, Switzerland. E-mail: cristina.nevado@chem.uzh.ch
First published on 10th March 2016
Abstract
Methods to incorporate atmospheric CO2 into organic molecules are on demand. Here we present two Pd-catalyzed multicomponent reactions that provide functionalized oxazolidinones from propargylamines, aryl halides and CO2 as starting materials. These transformations, devoid of high CO2 pressures, represent a streamlined stereocontrolled synthesis of previously inaccessible versions of these useful heterocycles in an atom-economic manner, as up to four new single bonds are formed in a single synthetic operation.
The large emissions of CO2 to the atmosphere represent an ever-growing problem1 that continues fostering the development of processes for capture and utilization (CCU)2 of this inexpensive and non-toxic C1 source towards valuable compounds within the chemical community.3 Multi-component reactions have emerged in recent years as powerful synthetic tools to assemble molecular complexity from either commercial or easily accessible starting materials.4 Among the value-added compounds that can be produced from CO2 activation and conversion, oxazolidinones are particularly appealing due to their broad application as chiral auxiliaries,5 intermediates in organic synthesis,6 as well as agrochemicals and antibacterial drugs.7 Strategies towards this attractive chemical blueprint employing simple starting materials combined with environmentally friendly CO2 as a C1 source are thus highly desirable. Several reports dealing with CO2 fixation by propargylamines or aminoalcohols to access these useful heterocycles have appeared during the last decade.8 Nevertheless, most of these methodologies involve harsh conditions including the use of supercritical CO2 (scCO2)9 or strong organic bases (super bases) at high temperatures and pressures of the gas.10 Very recently, milder protocols with protic ionic liquids and low CO2 pressures have been reported.11 π-Acid catalysts have also been employed for this purpose,12,13 with prevalence of coinage metals, as demonstrated by the seminal reports of Yamada and coworkers using silver14 and Ikariya's work employing gold(I) complexes.15 However, several limitations still hamper the broad synthetic applicability of these methods. The fact that C–C bond formation reactions have been scarcely coupled to the carboxylation of propargylamines prevented the formation of tetrasubstituted olefins,16 whereas commonly obtained trisubstituted alkenes could only be accessed in Z-selective fashion. Furthermore, due to CO2's high thermodynamic and kinetic stability, substantial activation (temperature, pressure, strong agents) is still needed in most cases (Scheme 1, top). Inspired by the recognized flexibility of palladium catalysts, we have designed a one pot, 3-component carboxylation–cyclization–cross-coupling reaction to produce functionalized oxazolidinones out of simple building blocks (CO2, propargylamines and aryl halides) under mild reaction conditions. Up to four new bonds can be formed in a single operation during the reaction of terminal propargylamines through an additional Sonogashira cross-coupling reaction in the presence of copper salts. Our protocol, utilizing CO2 at atmospheric pressure furnishes, for the first time, tetrasubstituted 5-methylene-1,3-oxazolidin-2-ones in a stereocontrolled fashion. Moreover, the methodology described herein also allows the E-selective preparation of trisubstituted derivatives as well as modifications at the allylic position or at the N atom, thus encompassing different substitution patterns commonly found in oxazolidinone products (Scheme 1, bottom).
 | ||
| Scheme 1 Synthesis of oxazolidinones from propargylamines. | ||
Propargylamine 1 and iodobenzene (2a) were chosen as starting materials for the optimization of the reaction conditions. After a short preliminary screening,17 [PdCl2(dppf)] and NaOtBu were identified as suitable catalyst and base respectively for the fine tuning of the reaction conditions. With a sustained atmospheric pressure of CO2 at 40 °C, different solvents were tested. The reaction with THF produced the desired product 3a in 34% yield after 22 hours (Table 1, entry 1). A longer reaction time resulted in a limited increase of product yield (Table 1, entry 2). Different solvents were also screened (Table 1, entries 3–4) out of which DMSO was selected for the subsequent optimization as it delivered 3a in 89% yield (Table 1, entry 5). Decreasing the amount of base turned out to be beneficial (Table 1, entries 6–7). In contrast, a decrease in the reaction time or temperature resulted in lower yields (Table 1, entries 8–9). A compromise could be found when 1.5 equiv. of base were used, resulting in a 95% yield of 3a after 22 hours (Table 1, entry 10). Remarkably, no trace of 6-endo-dig cyclization products could be detected in these transformations.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | NaOtBu (equiv.) | T [°C] | t [h] | Yieldb [%] |
| a The reactions were performed in a Schlenk tube directly connected to a bottle of CO2. The pressure was fixed with a manometer. b Yields of isolated products after column chromatography on silica gel. c The same result was obtained when MeCN or MeNO2 were used as solvents. [PdCl2(dppf)] = dichloro[1,1′-bis-(diphenylphosphino)-ferrocene]palladium(II). | |||||
| 1 | THF | 3 | 40 | 22 | 34 |
| 2 | THF | 3 | 40 | 44 | 47 |
| 3 | DMF | 3 | 40 | 44 | 57 |
| 4 | DCE | 3 | 40 | 44 | Tracesc |
| 5 | DMSO | 3 | 40 | 44 | 89 |
| 6 | DMSO | 1.5 | 40 | 44 | 94 |
| 7 | DMSO | 1.1 | 40 | 44 | 97 |
| 8 | DMSO | 1.1 | 40 | 22 | 87 |
| 9 | DMSO | 1.1 | r.t. | 22 | 72 |
| 10 | DMSO | 1.5 | 40 | 22 | 95 |
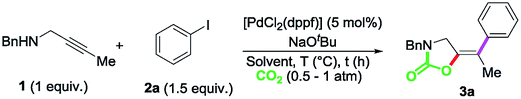
With the optimized reaction conditions in hand, we set out to explore the scope of this transformation (Scheme 2). The nature of the aryl iodide was explored first. Propargylamine 1, reacted with aryl iodides bearing electron-withdrawing groups to give the corresponding oxazolidinones 3b–e in excellent yields. Additional electron density in the aromatic ring appeared to be counterproductive given the reduced efficiency of the reaction to produce 3f. Single crystals of compound 3c allowed the unambiguous confirmation of the product structure and confirmed the E geometry of the exocyclic double bond.18
 | ||
| Scheme 2 Scope for the carboxylative cyclization and cross-coupling reaction of propargylamines and aryl iodides. Yields of isolated products after column chromatography on silica gel are given. aThe reaction was performed with 1.1 equiv. of base. bYields obtained using [Pd] 2.5 mol%. cThe reaction was run for 3 days. | ||
The reaction scope on the propargylamine substrates was explored next. Both alkyl (pentyl- and isopropyl-) as well as aryl-substituted alkynes were tolerated, delivering the corresponding cross coupling products 4–6a in excellent yields. Terminal alkynes were also accommodated as demonstrated by the reaction to produce 7a which could be isolated in 76% yield as a single isomer, as determined by X-ray diffraction analysis.18 Different substituents on the N-atom were also explored. While N-butyl, N-isopropyl and N-allyl propargylamines produced the corresponding oxazolidinones 8–10a in good yields, the more sterically demanding N-tert-butyl substrate required longer reaction times for a productive outcome (11a). Substituents at the propargylic position were also well tolerated as demonstrated by the reaction to produce 12a. Even commercially available propargylamine could be transformed into 13a in 51% yield. The exquisite reaction stereocontrol could also be confirmed by X-ray diffraction analysis of this compound.18
Given the broad functional group tolerance observed in this carboxylative cyclization/cross-coupling reaction, we questioned whether an additional Csp–Csp2 cross coupling reaction could be incorporated in these multicomponent transformations to introduce, in situ, a substituent on the terminal position of the propargylamine substrates. To this end, we set out to explore the reaction between N-benzyl propargylamine 14 and three equivalents of iodobenzene (2a) in the presence of an additional Cu-cocatalyst. The reaction required a re-optimization of the reaction conditions as summarized in Table 2.17 To our delight, the reaction in the presence of 10 mol% of CuI at 60 °C delivered the desired product 6a in 37% yield, although substantial amounts of the direct carboxylative cyclization and cross-coupling reaction (7a) could also be observed in this transformation (Table 2, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Cu cat. (mol%) | Base (equiv.) | 6a, yieldb [%] |
| a The reactions were performed in a Schlenk tube directly connected to a bottle of CO2. The pressure was fixed with a manometer. b Yield determined by 1H-NMR spectroscopy using 1,2-dibromoethane as the internal standard. In brackets, isolated yields after column chromatography on silica gel. c The by-product 7a could be isolated in a significant amount. d Concentration = 0.1 M. DABCO = 1,4-diazabicyclo[2.2.2]octane. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. BEMP = 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine. | ||||
| 1 | DMSO | CuI (10) | NaOtBu (3) | 37c |
| 2 | DMSO | CuI (10) | DBU (3) | — |
| 3 | DMSO | CuI (10) | Quinuclid. (3) | 63 |
| 4 | DMSO | CuI (10) | BEMP (3) | 70 (65) |
| 5 | DMSO | CuI (10) | DABCO (3) | 70 (64) |
| 6 | DMSO | CuI (5) | DABCO (2.6) | 67 (65) |
| 7 | DMSO | CuTC (5) | DABCO (2.6) | 68 |
| 8 | DMSO | CuOAc (5) | DABCO (2.6) | 70 |
| 9 | DMSO | Cu-3-Me-salicyl (5) | DABCO (2.6) | 72 |
| 10 | DMSO | CuI (5) | DABCO (2.6) | 73 (71) |

Different bases were explored (Table 2, entries 2–5) of which DABCO turned out to be the most suitable and cost-effective one providing 6a in a remarkable 64% isolated yield (Table 2, entry 5). A comparable efficiency was displayed when the Cu catalyst load was reduced to 5 mol% and the amount of base was adjusted to 2.6 equivalents (Table 2, entry 6). Copper salts such as CuTC, CuOAc and Cu-3-methylsalicylate offered comparable performances (Table 2, entries 7–9). Final adjustment of concentration and reaction time afforded compound 6a in 71% isolated yield providing the optimal reaction conditions for this transformation (Table 2, entry 10).
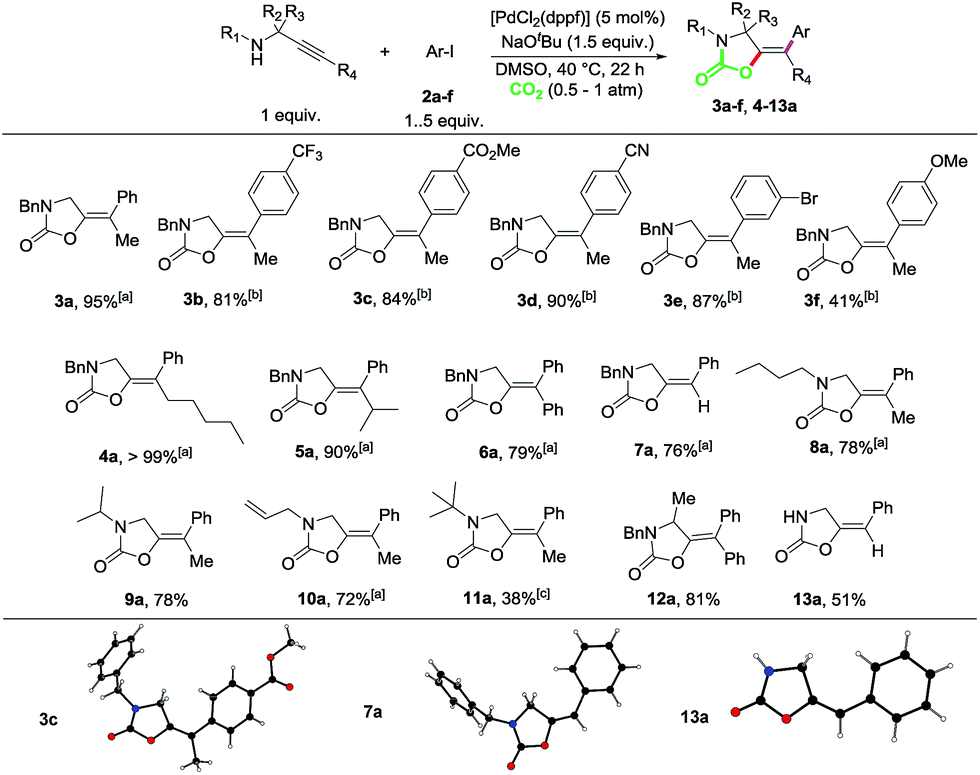
Next, we set out to explore the scope of this multicomponent reaction (Scheme 3). Propargylamine substrates modified at both the C-backbone and the N-protecting group were well tolerated as shown by the reactions to produce compounds 6a, 12a, 15 and 16. Taking 14 as a benchmark propargyl amine, different aryl iodides were explored. meta-Substituted-OMe, -OMOM, -OBn and -Me substrates could be efficiently coupled as shown by compounds 17–20. The reaction is highly chemoselective, as demonstrated by the reaction to produce 21, in which the Csp2–Br bond did not participate in the cross-coupling process. Di- and trimethoxy as well as 4-OMe and 4-methyl substituted 1-iodobenzenes could also be efficiently incorporated as demonstrated by the competent reactions to produce oxazolidinones 22–25. Heteroaromatic rings could also be incorporated, as shown by the reaction to obtain bis-thiophene derivative 26.
 | ||
| Scheme 3 Scope for the Sonogashira-carboxylative cyclization and cross-coupling reaction of propargylamines and aryl iodides. Isolated yields after column chromatography are given. In brackets, yields determined by 1H-NMR spectroscopy using 1,2-dibromoethane as the internal standard. aThe reaction was performed with a 0.5 M concentration. | ||
Several control experiments were designed in order to interrogate the reaction mechanism (Scheme 4).17 The reaction of propargylamines 1 and 27 with one equivalent of [PhPd(dppf)I]19 (28) under the standard conditions produced the corresponding oxazolidinones 3a and 6a in good yields respectively (eqn (1), right). When 1 reacted in the presence of a catalytic amount of 28, a successful transformation into 3a was also observed (eqn (1), left). As shown in eqn (2), disubstituted oxazolidinones such as 29 are not produced via Heck-reaction of monosubstituted ones. The reactions of N-benzyl propargylamine 14 with 2a, 4-CF3– (2b) and 4-OMe-1-iodobenzene (2f) under the standard conditions from Table 2, entry 10 in the absence of CO2 delivered the corresponding substituted alkynes 27, 30 and 31 in a >99%, 79% and 69% yield, respectively, which confirms the feasibility of a Sonogashira cross-coupling under the reaction conditions (eqn (3A)). The reactions of 27, 30 and 31 with iodobenzene 2a under the same conditions but in the presence of CO2 delivered the corresponding oxazolidinones 6a, 32 and 33 in high yields (eqn (3B)). When 30 reacted with 4-CF3-1-iodobenzene 2b, symmetrically substituted oxazolidinone 34 could be isolated in 38% yield (eqn (3C)). With these experiments in hand, the following mechanistic proposal can be envisioned (Scheme 5).
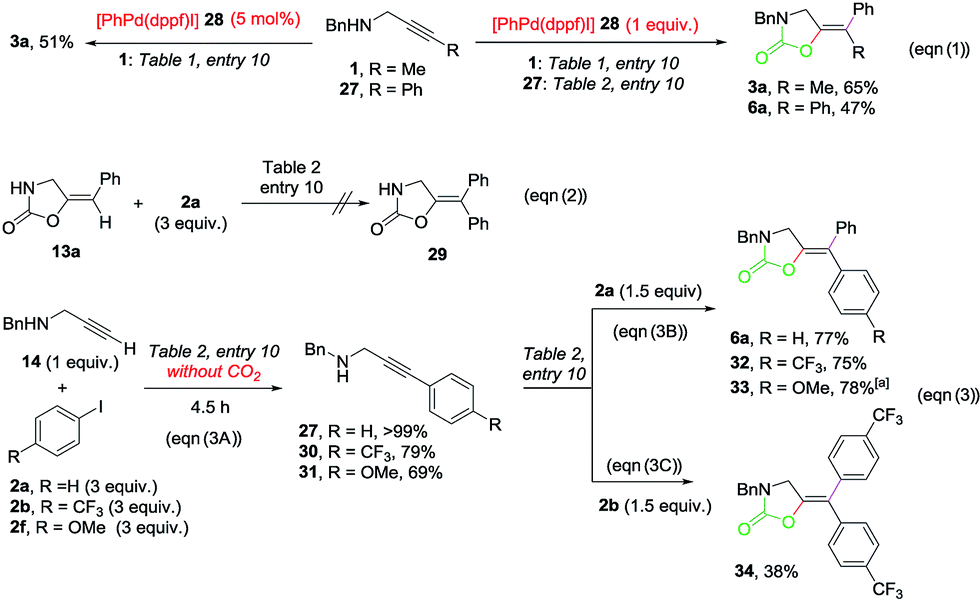 | ||
| Scheme 4 Control experiments. Isolated yields after column chromatography are given. aYield determined by 1H-NMR spectroscopy using 1,2-dibromoethane as the internal standard. | ||
 | ||
| Scheme 5 Mechanistic proposal. | ||
In situ generated Pd(0)20 undergoes oxidative addition with aryl iodide present in the reaction media to give [ArPd(dppf)I] (I) (Cycle B). In the case of terminal alkynes, and in the presence of a catalytic amount of copper, transmetalation of I with copper acetylide II (Cycle A) delivers Pd–alkynyl intermediate III, which upon reductive elimination produces substituted propargylamine IV, as experimentally confirmed by the results reported in eqn (3A). Amine IV reacts with the base and CO2 to produce V2,10c,21 which reacts with additional I present in the reaction media to give, upon π-activation of the triple bond, vinyl palladium complex VI. Reductive elimination in VI delivers the observed oxazolidinone products (Cycle C). For starting materials with internal alkynes (Scheme 2), only E isomers were obtained as confirmed by the X-ray diffraction analysis of 3c, 7a and 13a. Formation of I and its ability to catalyse the subsequent reaction steps is supported by experiments shown in eqn (1). The results collected in our control experiments provide evidence of the existence of a Csp–Csp2 coupling prior to the carboxylative-cyclization and Csp2–Csp2 cross-coupling, which indeed seems to be favourable with all kind of aryl iodides (neutral, electron-rich and electron-poor, eqn (3A)). In contrast, the cyclization process is sensitive to the electronic density on the active catalyst ([ArPd(dppf)I]), but not on the alkyne (carboxylative-cyclization & cross-coupling, eqn 3(B) and (C)). Together these results suggest that the cyclization process might be the responsible for the final outcome (yield) of this multicomponent reaction.
Conclusions
In summary, two Pd-catalyzed multicomponent reactions producing highly functionalized 5-methylene-1,3-oxazolidin-2-ones from propargylamines and aryl iodides under mild conditions have been developed. These transformations employ CO2 at atmospheric pressure and provide a streamlined access to previously inaccessible versions of these useful heterocycles in a stereoselective and atom-economic manner. Further applications of these methodologies are currently being explored and will be reported in due course.Acknowledgements
The European Research Council (ERC Starting grant agreement no. 307948) and the Xunta de Galicia are acknowledged for financial support. We thank Dr Anthony Linden for the X-ray crystal structure determination of 3c, 7a and 13a.Notes and references
- A. A. Lacis, G. A. Schmidt, D. Rind and R. A. Ruedy, Science, 2010, 330, 356–359 CrossRef CAS PubMed.
- Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 CAS.
- (a) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (c) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; (d) C. S. Yeung and V.-M. Dong, Top. Catal., 2014, 57, 1342–1350 CrossRef CAS.
- (a) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed; (b) A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- (a) D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS; (b) M. M. Heravi and V. Zadsirjan, Tetrahedron: Asymmetry, 2013, 24, 1149–1188 CrossRef CAS.
- L. Aurelio, R. T. C. Brownlee and A. B. Hughes, Chem. Rev., 2004, 104, 5823–5846 CrossRef CAS PubMed.
- (a) T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529–542 CrossRef CAS PubMed; (b) T. Andreou, A. M. Costa, L. Esteban, L. Gonzàlez, G. Mas and J. Vilarrasa, Org. Lett., 2005, 7, 4083–4086 CrossRef CAS PubMed; (c) M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010–2023 CrossRef CAS PubMed.
- (a) Y. Takada, S. W. Foo, Y. Yamazaki and S. Saito, RSC Adv., 2014, 4, 50851–50857 RSC; (b) S. Fujita, H. Kanamaru, H. Senboku and M. Arai, Int. J. Mol. Sci., 2006, 7, 438–450 CrossRef CAS; (c) Y. P. Patil, P. J. Tambade, S. R. Jagtap and B. M. Bhanage, J. Mol. Catal. A: Chem., 2008, 289, 14–21 CrossRef CAS.
- (a) Y. Kayaki, M. Yamamoto, T. Suzuki and T. Ikariya, Green Chem., 2006, 8, 1019–1021 RSC; (b) R. Maggi, C. Bertolotti, E. Orlandini, C. Oro, G. Sartori and M. Selva, Tetrahedron Lett., 2007, 48, 2131–2134 CrossRef CAS.
- (a) M. Costa, G. P. Chiusoli and M. Rizzardi, Chem. Commun., 1996, 1699–1700 RSC; (b) M. Costa, G. Paolo Chiusoli, D. Taffurelli and G. Dalmonego, J. Chem. Soc., Perkin Trans. 1, 1998, 1541–1546 RSC; (c) R. Nicholls, S. Kaufhold and B. N. Nguyen, Catal. Sci. Technol., 2014, 4, 3458–3462 RSC; (d) M. Yoshida, T. Mizuguchi and K. Shishido, Chem.–Eur. J., 2012, 18, 15578–15581 CrossRef CAS PubMed.
- (a) J. Hu, J. Ma, Q. Zhu, Z. Zhang, C. Wu and B. Han, Angew. Chem., Int. Ed., 2015, 54, 5399–5403 CrossRef CAS PubMed; (b) M.-Y. Wang, Q.-W. Song, R. Ma, J.-N. Xie and L.-N. He, Green Chem., 2016, 18, 282–287 RSC.
- T.-A. Mitsudo, Y. Hori, Y. Yamakawa and Y. Watanabe, Tetrahedron Lett., 1987, 28, 4417–4418 CrossRef CAS.
- (a) A. Bacchi, A. P. Chiusoli, M. Costa, B. Gabriele, C. Righi and G. Salerno, Chem. Commun., 1997, 1209–1210 RSC; (b) L. B. Wolf, K. C. M. F. Tjen, H. T. ten Brink, R. H. Blaauw, H. Hiemstra, H. E. Schoemaker and F. P. J. T. Rutjes, Adv. Synth. Catal., 2002, 344, 70–83 CrossRef CAS.
- (a) S. Kikuchi, S. Yoshida, Y. Sugawara, W. Yamada, H.-M. Cheng, K. Fukui, K. Sekine, I. Iwakura, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2011, 84, 698–717 CrossRef CAS; (b) T. Ishida, S. Kikuchi, T. Tsubo and T. Yamada, Org. Lett., 2013, 15, 848–851 CrossRef CAS PubMed.
- S. Hase, Y. Kayaki and T. Ikariya, Organometallics, 2013, 32, 5285–5288 CrossRef CAS.
- A tandem reaction to afford cyclic carbonates using allyl chlorides has been developed: (a) K. Iritani, N. Yanagihara and K. Utimoto, J. Org. Chem., 1986, 51, 5499–5501 CrossRef CAS; (b) Y. Inoue, K. Ohuchi, I. F. Yen and S. Imaizumi, Bull. Chem. Soc. Jpn., 1989, 62, 3518–3522 CrossRef CAS; also with aryl iodides: (c) Y. Inoue, Y. Itoh, I.-F. Yen and S. Imaizumi, J. Mol. Catal., 1990, 60, L1–L3 CrossRef CAS , although the severe reaction conditions and low yields for a reduced substrate scope precluded the further impact and application of this method.
- For experimental procedures, characterization of new products and additional control experiments, please, see ESI.†.
- Crystallographic data for 3c (CCDC 1439789†), 7a (CCDC 1439720†) and 13a (CCDC 1439788†).
- T. L. Andersen, S. D. Friis, H. Audrain, P. Nordeman, G. Antoni and T. Skrydstrup, J. Am. Chem. Soc., 2015, 137, 1548–1555 CrossRef CAS PubMed.
- B. Das, P. Kundu and C. Chowdhury, Org. Biomol. Chem., 2014, 12, 741–748 CAS and ref. 13a.
- D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857–3898 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of new products and control experiments. CCDC 1439789, 1439720 and 1439788. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00419a |
| This journal is © The Royal Society of Chemistry 2016 |