
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure determination of microbial metabolites by the crystalline sponge method†
Yasuhide
Inokuma
ab,
Tomoya
Ukegawa
a,
Manabu
Hoshino
ac and
Makoto
Fujita
*ac
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: mfujita@appchem.t.u-tokyo.ac.jp
bJST PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cJST ACCEL, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 24th February 2016
Abstract
The structures of metabolites produced in microgram quantities by enzymatic reductions with baker's yeast were analyzed using the crystalline sponge method. The X-ray data provided reliable structures for trace metabolites including their relative and absolute stereochemistries that are not fully addressed by conventional NMR and LC-MS analyses. Technically, combining two or more chromatographic purification techniques is essential because, unlike abundant synthetic compounds, extracted metabolites contain many low level UV-silent impurities. The crystalline sponge method coupled with HPLC purification (LC-SCD) would thus be a useful method for metabolic analysis and drug discovery.
Introduction
The structural information of metabolites is key to understanding enigmatic cellular processes and for drug design.1 Important insights into active enzymes, gene expression of RNA sequences, and biochemical reactions can be extracted when the structures of metabolites are fully characterized. They are in most cases produced in minute quantities as low level components of a complex mixture. Combinations of spectroscopic measurements (e.g. NMR and MS) and separation techniques (e.g. GC and LC) have been frequently used and established to elucidate the structures of scarce metabolites from a mixture. However, full characterization of their structures, including their absolute stereochemistry, is still a laborious task because of limited sample supply.The crystalline sponge method2,3 is a recently developed technique to prepare single crystal samples for X-ray crystallographic analysis of trace and non-crystalline compounds. Given the low limit of the requisite sample amount (<0.1 μg), the method can be an innovative analytical tool for trace metabolites, but has only been used to examine abundant synthetic compounds in the past.4,5 The application of the method to bio-synthetically produced scarce metabolites is challenging because, unlike synthetic samples, extracted metabolites contain many unpredictable impurities. The major advantage of the crystalline sponge method is that a microgram level quantity of samples is sufficient to be analyzed. In this paper, we demonstrate that the microbial metabolites obtained only in microgram quantities are fully characterized using the crystalline sponge method coupled with HPLC separation (LC-SCD analysis).
Results and discussion
As a model case of metabolic analysis using a crystalline sponge [(ZnI2)3(tpt)2·(c-C6H12)x]n (1; tpt = tris(4-pyridyl)-1,3,5-triazine),3a,6 we conducted reductive dechlorination of 1,1-bis(4-chlorophenyl)-2,2,2-trichloroethane (DDT; 2) by reductases found in baker's yeast. When compound 2 (100 mg) was treated with fermenting baker's yeast (dry yeast (11.2 g) and sucrose (23.5 g) in 100 ml of water) at 30 °C for 1 week, a small amount of a metabolic product (∼1.3% yield based on UV spectroscopy) was detected in the ether extract obtained from the mixture. LDI-TOF MS spectrometry indicated the loss of one chlorine atom (m/z = 320.040; calcd for C14H10Cl4+ [2 − Cl + H]+: 319.9507) from 2, suggesting the formation of 1,1-bis(4-chlorophenyl)-2,2-dichloroethane (DDD; 3). The formation of 3 in a low yield is consistent with the previous report.7 However, another dechlorinated compound, 4, cannot be excluded as the product based solely on MS information.To unambiguously determine the structure of 3, the crude extract was subjected to the LC-SCD analysis. After pre-purification of the ether extract (ca. 15 μg) using PTLC, the analytical sample was further purified using HPLC with a narrow collection time window (Fig. S3†). This purification protocol is particularly important, or else the HPLC-separated sample was considerably contaminated with many UV-silent impurities. In fact, no satisfactory results were obtained in our earlier attempts in the crystalline sponge analysis when the crude extract was directly purified using HPLC with a wide collection window (Fig. S5†).
The HPLC separation system was slightly modified so that fraction collection and subsequent guest soaking could be done in the same microvial (Fig. S1†). The fraction was directly received by a microvial and, after the evaporation of the eluent (hexane), one crystal of crystalline sponge 1 and a solvent were added therein and the vial was allowed to stand at room temperature for 3 d for guest soaking.
The crystallographic analysis confirmed the structure of the metabolite to be 3 (Fig. 1). The crystal structure revealed that there are two major binding sites for guest 3 in the pores of the crystalline sponge 1.‡ One is located near a tpt ligand where guest 3 was observed with 100% occupancy (guest A; Fig. S5†). Another one is located on a crystallographic twofold axis where guest 3 was statistically disordered (guest B). A better resolution was obtained for A, and the electron density map F0 clearly shows the structure of 3 (Fig. 1b). Slightly high R1 and wR2 values (0.0780 and 0.2736, respectively) were obtained presumably due to very minor disorder in 3, which cannot be modeled yet contributes to the residual electron density significantly because of the heavy atom (Cl).
 | ||
| Fig. 1 (a) HPLC chromatogram of the organic extract from the baker's yeast reaction. (b) The electron density map F0 (contoured at the 0.8σ level) around the binding site of metabolite 3 (guest A) in the pores of crystalline sponge 1. | ||
When a prochiral functional group is enzymatically reduced to a chiral one, the stereochemical issues are of major concern for the structural characterization. Moreover, with the product(s) isolated only in microgram quantities, NMR analyses (particularly, 13C or 2D NMR) become difficult even for determination of the relative stereochemistry. The absolute stereochemistry can hardly be addressed by any spectroscopic methods unless empirical rules or data for authentic compounds are available. Thus, we applied the LC-SCD analysis for the full structural characterization (including absolute stereochemistry) of a metabolite from tetralone 5. This compound exists as a racemate due to rapid enolization at C1 and can give four possible stereoisomers (including enantiomers) upon reduction of the carbonyl group at C2.
Treating tetralone 5 (100 mg) with baker's yeast (11.2 g) for 18 h gave analytically pure chiral alcohol 6 in 10 μg quantity after isolation using HPLC. The relative stereochemistry of 6 was confirmed to be cis by comparison with both an authentic cis–trans mixture obtained by the NaBH4 reduction of 5 and literature reported spectroscopic data.8 Chiral HPLC analysis indicated >98% ee for the isolated cis isomer 6. An inclusion crystal 1·6 was prepared by soaking a crystal of 1 in a cyclohexane/1,2-dichloroethane (v/v = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution of 6. The crystal structure was solved with a non-centrosymmetric space group C2.§ A reasonable Parsons' Flack parameter value of 0.010(4) was obtained and the final R1 and wR2 values were 0.0511 and 0.1396, respectively. In an asymmetric unit, three independent guest molecules 6 (G1–G3 in Fig. 2) were clearly observed with ∼100% occupancy. All of the observed molecules 6 (G1–G3) show a 1R,2S absolute configuration, consistent with a previous report,8 in which an absolute configuration was speculated based on the absolute stereochemistry of analogous compounds.8 Our crystallographic study provided unquestionable proof for the absolute configuration of 6. From this, a plausible reaction mechanism to give 6 is suggested to involve reduction from the Re face of a 1R isomer of 5 that is kinetically resolved within the enzyme pocket.
:1) solution of 6. The crystal structure was solved with a non-centrosymmetric space group C2.§ A reasonable Parsons' Flack parameter value of 0.010(4) was obtained and the final R1 and wR2 values were 0.0511 and 0.1396, respectively. In an asymmetric unit, three independent guest molecules 6 (G1–G3 in Fig. 2) were clearly observed with ∼100% occupancy. All of the observed molecules 6 (G1–G3) show a 1R,2S absolute configuration, consistent with a previous report,8 in which an absolute configuration was speculated based on the absolute stereochemistry of analogous compounds.8 Our crystallographic study provided unquestionable proof for the absolute configuration of 6. From this, a plausible reaction mechanism to give 6 is suggested to involve reduction from the Re face of a 1R isomer of 5 that is kinetically resolved within the enzyme pocket.
 | ||
| Fig. 2 (a) Reduction of tetralone 5 by baker's yeast. (b) Crystal structure of metabolite 6-incorporated crystalline sponge 1 viewing along the b-axis (metabolite 6: CPK model, others: stick model, disordered guests/solvents were omitted for clarity). (c) ORTEP drawings of G1–G3 at the 50% probability level. | ||
Conformational analysis of the observed guests G1–G3 is worthy of additional discussion. The methoxycarbonyl groups in G1 and G3 are located at the pseudo-axial position, while that in G2 is at the pseudo-equatorial position. For G2, intramolecular hydrogen bonding between the OH and COOMe groups was indicated by the short OO–H–OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O distance (∼2.8 Å) that may stabilize this conformation. Presumably, this conformer exists in solution and the crystalline sponge traps both conformers at different binding sites, enabling the concomitant observation of both conformers.
O distance (∼2.8 Å) that may stabilize this conformation. Presumably, this conformer exists in solution and the crystalline sponge traps both conformers at different binding sites, enabling the concomitant observation of both conformers.
The structural and stereochemical analysis of a metabolite from adrenosterone (7) provided a more challenging task because three prochiral carbonyl carbons at C3, C11, and C17 can in principle generate 26 different compounds upon reduction of the carbonyl group(s). When steroid 7 was metabolized by baker's yeast on a 200 μg scale, HPLC analysis revealed the exclusive formation of a single metabolic product (hereafter denoted as 8).
The parent ion peak of metabolite 8 was observed at m/z = 325.1781 (calcd for C19H26NaO3+: 325.1780). The increase in mass unit by 2.0 Da from 7 suggests a mono-hydrogenation of 7 at either a carbonyl or CC group. The 1H NMR spectrum of 8 was, unfortunately, not conclusively able to determine its structure and stereochemistry because of overlapping signals. The unequivocal structure determination of 8 can be achieved, given that 8 is obtained only in microgram quantity, most reliably by the LC-SCD analysis.
Thus, ca. 20 μg of a crude mixture obtained from the ether extract was purified using HPLC and subjected to guest soaking with a crystal of 1. After guest soaking at 50 °C for 2 d, the crystallographic analysis revealed three independent molecules of 8 (H1–H3 in Fig. 3a) in an asymmetric unit with occupancies of 100, 80.5, and 100%, respectively.¶ All the structures revealed that the carbonyl group at C17 in 7 is stereoselectively reduced to a hydroxy group with an S configuration. The two carbonyl groups at C3 and C11 remain intact: typical CO distances (1.19(3) and 1.20(3)) were observed at C3 and C11, respectively, and these carbon atoms still adopt a trigonal planar geometry. In contrast, elongation of the C–O bond length of 1.41(3) Å and a tetrahedral geometry at C17 were observed, indicating that the carbonyl reduction took place only at C17. Notably, favorable host–guest interactions were observed. For example, guest H1 is trapped by the host framework of 1 with CO⋯H–C or C–H⋯I hydrogen bond interactions.
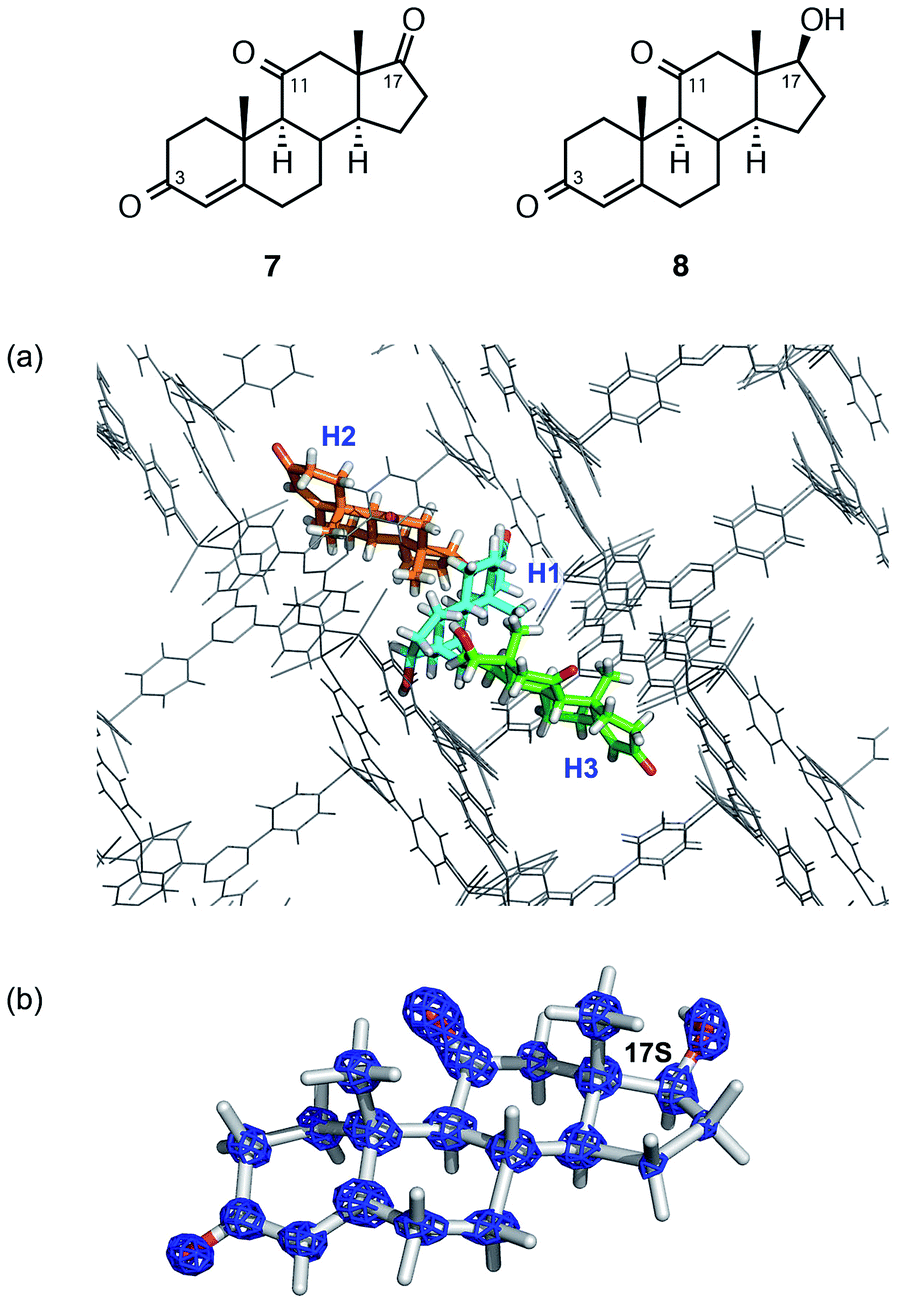 | ||
| Fig. 3 (a) Guest binding sites for metabolite 8 (H1–H3) in the network of 1 (viewing along the b-axis). (b) Electron density map F0 (blue mesh) contoured at the 1.0σ level overlaid on the structure of guest H1. | ||
Conclusions
In conclusion, LC-SCD analysis has been demonstrated as a useful tool for structural analysis of trace microbial metabolites. Some important lessons are learned in this study working with trace microbial metabolites, which we have not noticed in the previous analysis of abundant synthetic compounds. First, a key to the success of the analysis is to obtain high purity samples of the microbial metabolites by HPLC separation. As the crystalline sponge 1 often preferentially absorbs minor components, even low-level impurities may disturb efficient guest soaking. Given that trace metabolites separated using HPLC are normally contaminated with many impurities, great care should be taken in the purification steps. Therefore, pre-purification with PTLC or LC–LC is highly recommended. Second, pre-analysis of the abundant parent compounds would be beneficial because soaking conditions optimized for the parent compounds can usually be applied to the analysis of their metabolites. Third, having successful results in this study, we are more convinced that the crystalline sponge method, coupled with HPLC separation, will innovate the structural analysis of scarce amounts of microbial products in natural product chemistry as well as in drug discovery.Acknowledgements
This research was promoted as a part of the JST-PRESTO and JST-ACCEL projects in which Y. I. and M. F. are principle investigators, respectively. We thank Hiroyasu Sato and Akihito Yamano (Rigaku Corporation) for the support of the X-ray measurement of compound 1·8.Notes and references
- (a) G. Evans, A Handbook of Bioanalysis and Drug Metabolism, CRC Press, 2004 Search PubMed; (b) N. Bodor and P. Buchwald, Retrometabolic Drug Design and Targeting, Wiley, 2012 Search PubMed.
- (a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed; (b) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 501, 262 CrossRef CAS.
- For practical guidelines, see: (a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246–252 CrossRef CAS PubMed; (b) T. R. Ramadhar, S. Zheng, Y. Chen and J. Clardy, Acta Crystallogr., 2015, A71, 46–58 Search PubMed; (c) M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139–151 CAS.
- For applications in organic chemistry, see: (a) D. Kamimura, D. Urabe, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 5122–5125 CrossRef CAS PubMed; (b) E. V. Vinogradova, P. Müller and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 126, 3189–3192 CrossRef; (c) A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed; (d) N. Zigon, M. Hoshino, S. Yoshioka, Y. Inokuma and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 9033–9037 CrossRef CAS PubMed; (e) S. Yoshioka, Y. Inokuma, M. Hoshino, T. Sato and M. Fujita, Chem. Sci., 2015, 6, 3765–3768 RSC; (f) S. Takizawa, K. Kishi, Y. Yoshida, S. Mader, F. Arteaga, S. Lee, M. Hoshino, M. Rueping, M. Fujita and H. Sasai, Angew. Chem., Int. Ed., 2015, 54, 15511–15515 CrossRef CAS PubMed; (g) S. Urban, R. Brkljača, M. Hoshino, S. Lee and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 2678–2682 CrossRef CAS PubMed.
- (a) T. R. Ramadhar, S. Zheng, Y. Chen and J. Clardy, Chem. Commun., 2015, 51, 11252–11255 RSC; (b) E. Sanna, E. C. Escudero-Adán, A. Bauzá, P. Ballester, A. Frontera, C. Rotger and A. Costa, Chem. Sci., 2015, 6, 5466–5472 RSC.
- (a) K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3392–3395 CrossRef CAS; (b) O. Ohmori, M. Kawano and M. Fujita, J. Am. Chem. Soc., 2004, 126, 16292–16295 CrossRef CAS PubMed.
- B. J. Kallman and A. K. Andrews, Science, 1963, 141, 1050–1051 CAS.
- D. Seebach, S. Roggo, T. Maetzke, H. Braunschweiger, J. Cercus and M. Krieger, Helv. Chim. Acta, 1987, 70, 1605–1615 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of sample preparation and crystallographic analysis. CCDC 1451761–1451763. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc00594b |
| ‡ Crystallographic data for 1·3: C36H24N12Zn3I6·(C14H10Cl4)·(C7H4Cl2)·2.5(C3H6), M = 2230.02, colorless block, 0.23 × 0.11 × 0.09 mm3, monoclinic, space group C2/c, a = 35.7365(13) Å, b = 15.0610(5) Å, c = 30.9345(11) Å, β = 104.141(7)°, V = 16145.2(11) Å3, Z = 8, Dc = 1.835 g cm−3, T = 93(2) K, 2.989° < θ < 27.468°, 17652 unique reflections out of 88524 with I > 2σ(I), GoF = 1.063, final R factors R1 = 0.0780, and wR2 = 0.2736 for all data, CCDC deposit number 1451761. |
| § Crystallographic data for 1·6: C72H48I12N24Zn6·3.79(C13H16O4)·1.93(C6H12), M = 4220.24, colorless rod, 0.18 × 0.11 × 0.06 mm3, monoclinic, space group C2, a = 36.3783(8) Å, b = 14.6755(2) Å, c = 31.2278(6) Å, β = 103.184(2)°, V = 16232.2(5) Å3, Z = 4, Dc = 1.727 g cm−3, T = 100(2) K, 4.1720° < θ < 74.0140°, 31961 unique reflections out of 69980 with I > 2σ(I), GoF = 1.031, final R factors R1 = 0.0511, and wR2 = 0.1396 for all data, Flack parameter (Parsons) = 0.010(4), CCDC deposit number 1451762. |
| ¶ Crystallographic data for 1·8: C72H48I12N24Zn6·2.8(C19H26O3)·1.5(C6H12), M = 4147.41, colorless plate, 0.28 × 0.08 × 0.07 mm3, monoclinic, space group C2, a = 34.7707(6) Å, b = 14.8406(3) Å, c = 31.2152(5) Å, β = 102.601(2)°, V = 15719.6(5) Å3, Z = 4, Dc = 1.749 g cm−3, T = 93(2) K, 2.9030° < θ < 73.3970°, 29574 unique reflections out of 106306 with I > 2σ(I), GoF = 1.005, final R factors R1 = 0.0788, and wR2 = 0.2259 for all data, Flack parameter (Parsons) = −0.008(8), CCDC deposit number 1451763. |
| This journal is © The Royal Society of Chemistry 2016 |