
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-objective active machine learning rapidly improves structure–activity models and reveals new protein–protein interaction inhibitors†
D.
Reker
 ,
P.
Schneider
and
G.
Schneider
*
,
P.
Schneider
and
G.
Schneider
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog Weg 4, 8093 Zürich, Switzerland. E-mail: gisbert.schneider@pharma.ethz.ch
First published on 10th March 2016
Abstract
Active machine learning puts artificial intelligence in charge of a sequential, feedback-driven discovery process. We present the application of a multi-objective active learning scheme for identifying small molecules that inhibit the protein–protein interaction between the anti-cancer target CXC chemokine receptor 4 (CXCR4) and its endogenous ligand CXCL-12 (SDF-1). Experimental design by active learning was used to retrieve informative active compounds that continuously improved the adaptive structure–activity model. The balanced character of the compound selection function rapidly delivered new molecular structures with the desired inhibitory activity and at the same time allowed us to focus on informative compounds for model adjustment. The results of our study validate active learning for prospective ligand finding by adaptive, focused screening of large compound repositories and virtual compound libraries.
Introduction
Active machine learning implements an automated, feedback-driven discovery process.1,2 We showcase this concept for the rapid identification of bioactive compounds with desired properties by the iterative adaptation of the underlying structure–activity relationship (SAR) model.3–6 In each iteration, the machine learning SAR model selects a compound set for biochemical testing, and the additional structure–activity data obtained from these experiments serves to refine the model for the subsequent iterations. Thereby, the best use is made of the bioactivity data, while limiting the overall number of assays performed.7 New compounds are either selected with a focus on maximal information content and diversity of the molecular reference structures (explorative strategy)8,9 or with a focus on improved bioactivity (exploitive/greedy strategy).10–12 Until now, this concept has essentially been studied only theoretically.3 Two noteworthy exceptions are prospective applications to lead discovery, in which a greedy strategy was combined with exploration through biased sampling via scaffold-centric filtering10 or pre-sampling by genetic algorithms.11 Here, we present full-fledged prospective active learning that implements a multi-objective selection function for balancing the exploration of chemical space and the exploitation of the SAR model. We achieved rapid model improvement while retrieving novel active compounds. Furthermore, we propose a technique for the informed batch-wise selection of compounds, which is of particular practical relevance for the application of active learning in the context of biological studies where many assays are effectively performed in batches.9,13We selected the CXC chemokine receptor 4 (CXCR4, “fusin”) as our prospective drug target.14 CXCR4 and its endogenous ligand CXCL-12 (SDF-1) are both part of a phylogenetically conserved inter-cellular signaling system15 that controls chemotaxis and plays an important role in brain development and intestinal morphogenesis14 but is also relevant for the pathobiology of various diseases.15,16 In 1996, CXCR4 was identified as the co-receptor used by T-tropic human immunodeficiency virus (HIV) for internalization by CD4-positive T cells,14 and this has been associated with late-stage infection and disease progression to immunodeficiency.15 The CXCR4 gene is up-regulated in several cancers and serves as a diagnostic and prognostic marker17,18 due to its association with cell survival and metastatic behavior.19,20 Despite the suggested pharmacological relevance of the CXCR4–CXCL-12 interaction, only a modest number (287 curated ligands in ChEMBL19; Fig. S1†) of CXCR4-modulating small molecules have been identified to date,16 which might be explained in part by the difficulty of finding low molecular weight inhibitors of such protein–protein interactions.21 Consequently, CXCR4 is an attractive target for active machine learning because sufficient data for the initial model construction is available and there is ample opportunity for the discovery of new chemical entities as CXCR4 modulators.
Results and discussion
Balanced learning is a new strategy for compound selection
For the estimation of ligand affinity (pAffinity) to CXCR4, we used random forest prediction technology,22 which we had already studied in the context of active learning3 and validated prospectively.23 For ligand selection, we pursued a strategy that balanced exploitation and exploration simultaneously to identify informative actives for improving the machine learning SAR model. To capture novelty, we used the uncertainty of the prediction (variance of the predicted ligand affinity, “query-by-committee”)3,9 and the similarity of a newly picked compound to the existing training data.24 For the latter, we used the random forest similarity metric, which ensures compound novelty in terms of the model architecture,25 rather than relying on chemical compound similarity.10,11 Accordingly, two compounds are deemed similar when they are predicted by the same leaf of a regression tree, and the sum over all of the trees that make up the random forest ensemble yields the final similarity value.The two measures of novelty (uncertainty and similarity) enable different compound selection strategies: (i) the uncertainty of a molecule is high for a compound containing features of both active and inactive training examples and will help to focus on compounds that lie at the interface between the active and the inactive molecules in chemical space. (ii) The similarity strives to pick compounds that differ from the training data as a whole in terms of the model architecture. Retrospective evaluation demonstrated that, while the individual strategies focus solely on novelty or potency and perform like random selection for the other objective, combining the selection scores using weighted averages26 results in a balanced selection strategy that actively enriches compound sets with potent and structurally new molecules for a broad range of different drug targets (Fig. S2†).
Based on these preliminary results, we decided to optimize the weights of the multi-objective selection function for the CXCR4 application. To this end, we retrospectively simulated the identification of CXCR4 inhibitors by “time-split cross-validation”.27 Using a non-redundant, grid-based search, we were able to test a broad range of weight settings. A balanced weighting of novelty and affinity resulted in good performance on different evaluation criteria that captured the activity of the selected compounds as well as model improvement (Table S1†), suggesting that considering all three selection measures simultaneously might be key to active learning. To compare the performance of active learning with the annual improvement of CXCR4 ligands deposited in ChEMBL19,28 we performed our optimized selection beginning with ligands that were published before 2003 (6% of all available CXCR4 ligands). We observed that the active learning approach rapidly identified ligand-efficient29 compounds and turned out to be more explorative than historical medicinal chemistry (Fig. 1). Importantly, active learning was robust against the distraction caused by 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 randomly added, presumably inactive, decoy compounds to choose from.
000 randomly added, presumably inactive, decoy compounds to choose from.
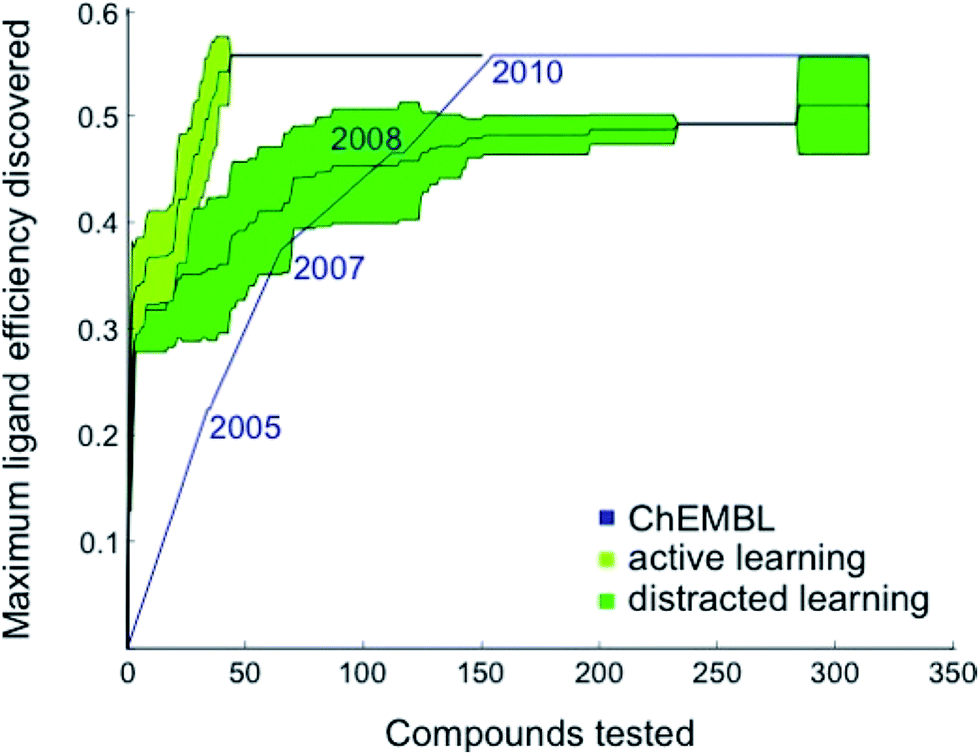 | ||
| Fig. 1 Retrospective comparison of active machine learning with historic data. The algorithm was provided with 6% learning data (21 ChEMBL28 compounds tested for CXCR4 binding and published before 2003) and had to pick from the remaining ChEMBL CXCR4 activity data (light green curve, “active learning”) or the remaining data plus 50000 compounds assumed to be inactive (dark green curve, “distracted learning”). The maximum ligand efficiency29 discovered using this method is compared to historic data (blue curve; 314 compounds tested). | ||
Active virtual screening retrieves potent and diverse hits
We applied the balanced active learning approach to the virtual screening of the Enamine HTS compound collection (version 201410;30 1465960 compounds). For this purpose, we trained a random forest model using all available CXCR4 ligand data (IC50, Ki) from ChEMBL19 and scored all compounds from the pool with the balanced selection function. The compound with the best score served as the seed for batch selection. We re-scored the n − 1 remaining pool compounds by random forest similarity to the compound with the best score. Visual comparison of the batch-selected compounds with naïvely picked top compounds revealed that greater structural diversity could be achieved through the re-scoring step (Table S2†). The average Tanimoto similarity for the top 10 selected compounds to their respective nearest neighbor differs about two-fold in favor of re-scoring (r = 2, 2048 bit, RDKit; Tc = 0.2 and 0.47 for batch-selected and naïvely selected compounds, respectively).
The first batch of 30 compounds was tested at a concentration of 10 μM for CXCR4 inhibition by monitoring intracellular arrestin recruitment (performed by DiscoverX, Fremont, CA, USA).31 We observed a near-normal distribution of efficacy values (Table S3†) with a maximal observed inhibition of 84% of the control (AMD3100) and six structurally new (Tc ≤ 0.23 for the ChEMBL19 CXCR4 ligand structures) compounds yielding an inhibition >50% (Fig. 2, compounds 1–6). Because the random forest SAR model was trained on predicting IC50 values, we needed to convert these inhibition values into approximate IC50 values as an input for the active machine learning step. To this end, we inverted the Hill equation assuming a Hill parameter of one: pIC50 ≐ log10(x) − log10[(100 − y)/y], where x is the ligand concentration, and y is the measured inhibition (percent of the control). While such values are approximations for the sake of model development only, the estimated IC50 value of 131 nM for positive control AMD3100 is in agreement with the experimentally measured value for AMD3100 competing with CXCL-12 (EC50 = 260 nM).32 For our six best hits, the equation computes approximate IC50 values between 2 and 10 μM. Importantly, none of these compounds has been investigated before (SciFinder; Chemical Abstract Service, Columbus, OH, USA), and they structurally differ from their nearest neighbors in known CXCR4 ligand space (Tc ≤ 0.23; Fig. S3†).
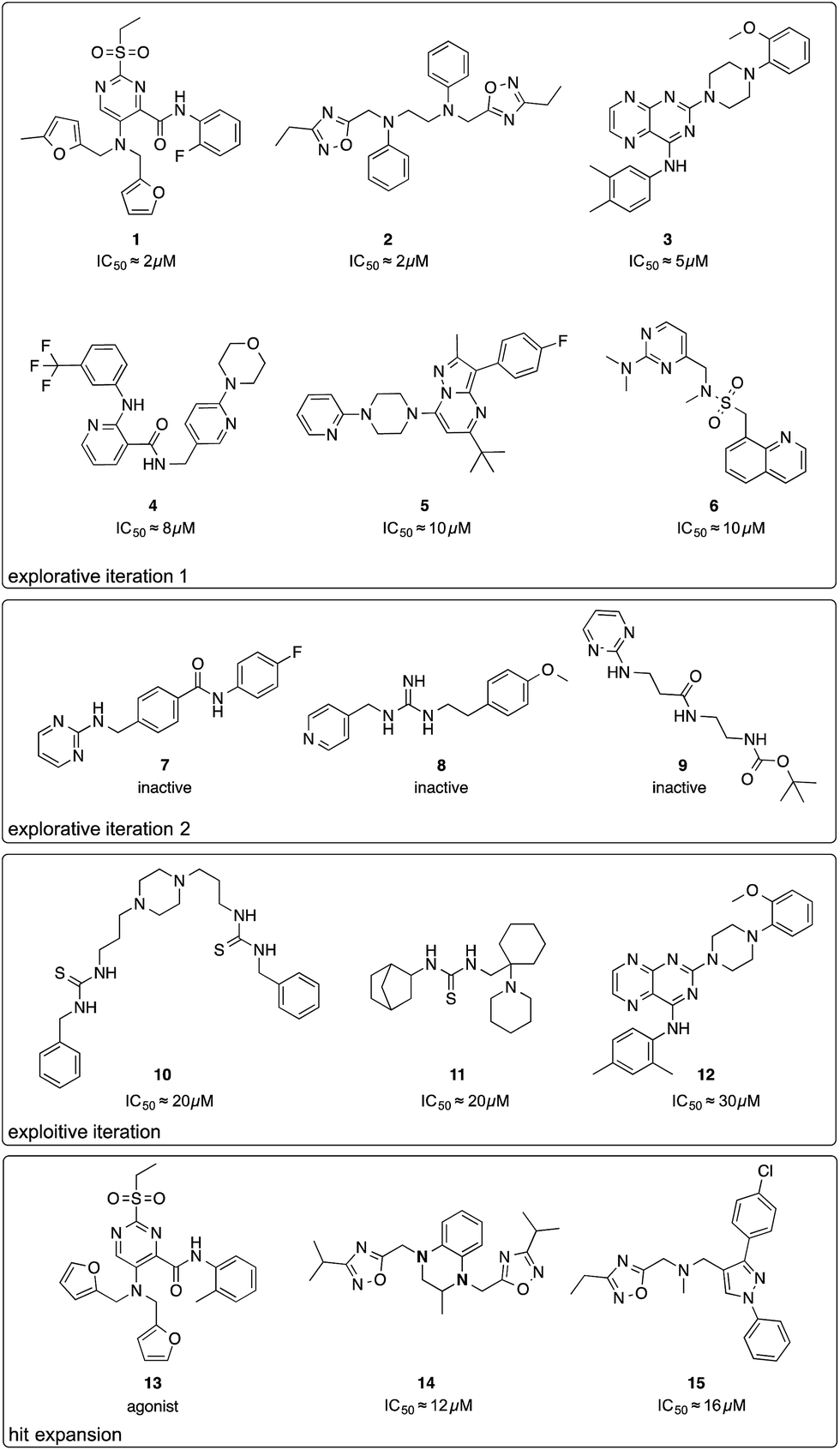 | ||
| Fig. 2 Chemical structures of compounds selected by the active learning algorithm. IC50 values were approximated from two single-dose measurements using an inverse of the Hill equation. | ||
We performed a second active learning cycle with the updated random forest model and the same balanced selection function (Table S4†). We observed that the selected compounds had approximately half a log unit lower predicted affinity compared to the compounds selected in the first iteration. This prediction was reflected in our screening results. At a concentration of 10 μM, the 30 selected compounds from round two showed only weak or no activity in the CXCR4–arrestin assays. Poor inhibition was confirmed by re-testing the compounds at a concentration of 30 μM in a secondary screen that measured cAMP signaling.33 Judging from the substructure similarity to known CXCR4 antagonists, one might have expected stronger activity for some of the selected compounds (Fig. 2, compounds 7–9). For example, compound 7 is a close structural analog of KRH-1636 (EC50 = 18 nM),34 compound 8 contains a presumable CXCR4-binding guanidine moiety,35 and compound 9 expresses a secondary amine pattern that is observed in structurally related potent CXCR4 antagonists.32 The random forest SAR model rightfully lacked confidence regarding the activity of these inactive analogues, which suggests that they are valuable for improving the understanding of the SAR.4
Active learning changes and improves the architecture of the SAR model
At this point, we decided to halt the balanced exploration of the screening compound pool because the second iteration suggested that we had arrived in chemical subspaces where we were unable to reliably enhance the compound activity (Fig. S4†). To evaluate whether active learning had actually improved the SAR model, we investigated the development of the predictive uncertainty for the screening compound pool over the learning cycles. We computed the standard deviation of the predictions made by the trees of the initial random forest model and the optimized models after the first and second active learning cycles.23,36 The predictive uncertainty was reduced after both learning cycles (Fig. 3A). The active learning process sampled the screening compound pool in such a way that the 2 × 30 = 60 added compounds helped to capture the SAR of the structurally diverse 1.5 million pool compounds.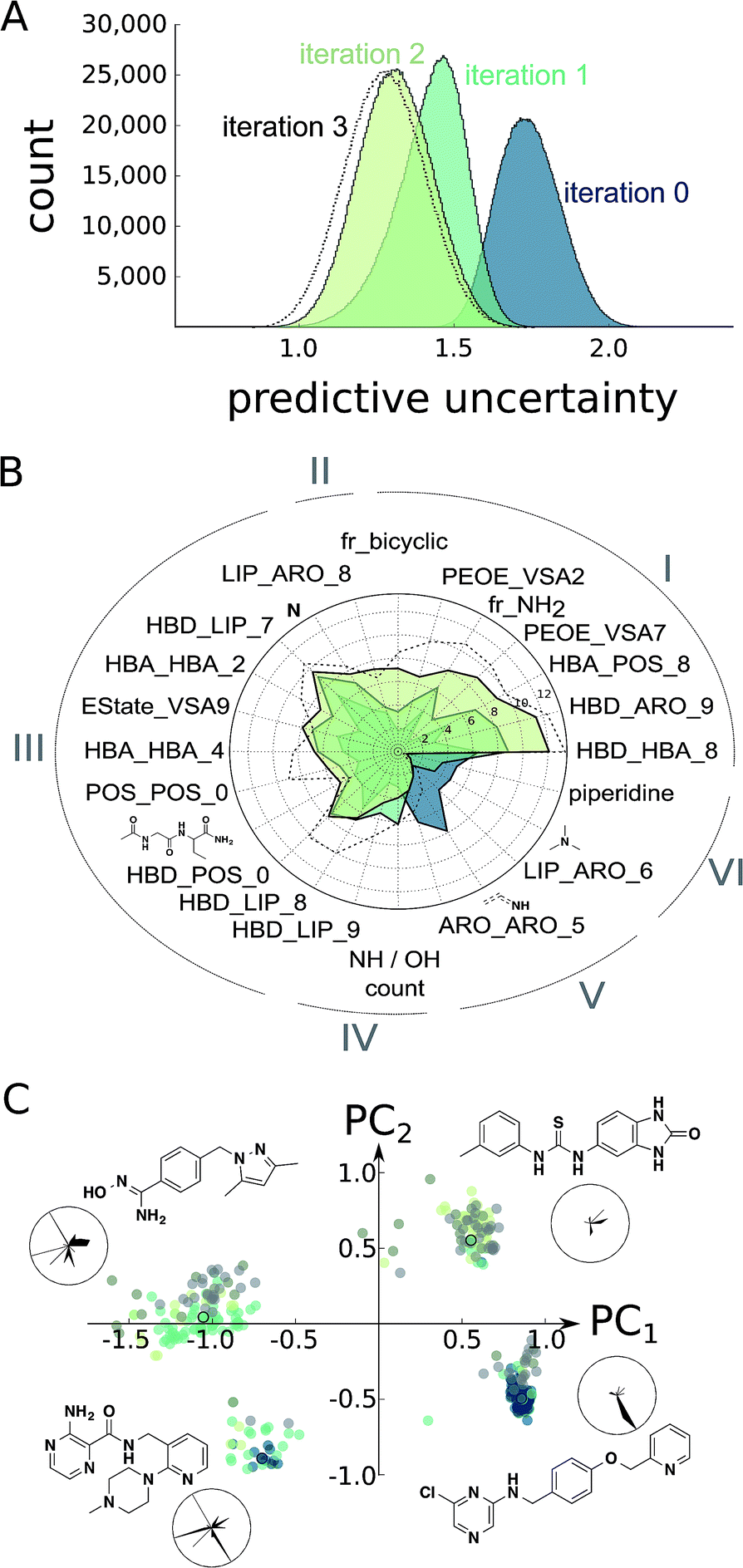 | ||
| Fig. 3 Estimation of model improvement and architecture change after the prospective active learning iterations. (A) Difference in predictive uncertainty (standard deviation of predictions of trees)23 for the Enamine screening collection30 using the random forest models. The individual random forest models were trained on the ChEMBL19 data (“iteration 0”),28 the ChEMBL19 data plus the first active learning iteration results (“iteration 1”), the ChEMBL19 data plus both active learning iteration results (“iteration 2”), or the ChEMBL19 data plus both learning and the exploitive and hit-expansion iterations (“iteration 3”). (B) Change in random forest feature importance25 for the top features of the models “iteration 0”, “iteration 1” and “iteration 2”. We can clearly observe the development of different classes of feature importance. For example, many features became consistently more or less important during learning (I and VI), while others seem to have converged after the first learning iteration (III and V). More interestingly, a few features have been only discovered (II) or have been disvalued (IV) during the second iteration. The importance values for the model “iteration 3” are shown for comparison. (C) Position of the top 100 predicted screening compounds from each model in feature space (colored dots). The feature space was generated as the first two principle components (PC1, PC2) of normalized features selected in (B). The cluster representatives (colored dots with black circles) are shown as chemical structures and their normalized feature values in radar charts. In these radar charts, the circle corresponds to the maximal feature values, and the black, filled areas correspond to the feature values for the respective chemical structure shown. The features are arranged as in (B). | ||
Furthermore, we calculated the random forest feature importance for all of the models. The summed feature importance increased during active learning, suggesting that the algorithm improved at explaining the underlying SAR using the molecular pharmacophore and substructure representations. The absolute and relative importance of the individual features dynamically changed with each iteration, with dozens of features temporarily considered relevant but discarded as learning proceeded (Fig. S5†). Visualizing the most relevant features extracted by the random forest approach illustrates this variation of model architectures (Fig. 3B). Overall, the models valued abstract descriptors over substructure fingerprints in spite of their much smaller number (386 vs. 2048 features). This observation might be explained in part by the known tendency of random forest classifiers to rate continuous descriptors higher than binary fingerprints.37
To asses whether the observed change of the most important features translates into an altered perception of compound potency, we plotted the position of the 100 most potently predicted (pAffinity) screening compounds from each individual model in the important feature space (Fig. 3C). While the initial ChEMBL-based model picked potent compounds exclusively from two clusters, our two active learning iterations discovered two additional clusters of promising compounds. Inspection of cluster representatives and their descriptor values suggest that the newly learned features can assist in navigating CXCR4 ligand space.
The improved machine learning model identifies a novel chemotype
Motivated by the observed model change and the improvement of prediction accuracy after the first two learning cycles, we decided to tweak our selection function to focus on the retrieval of actives (exploitation) in the third virtual screening round. We scored the compounds according to a conservative affinity estimate (pAffinity − uncertainty),23,26 purchased the 10 top-scoring compounds, and tested them in the arrestin assay at a concentration of 30 μM (Table S5†). This time, we observed a strong readout (inhibition below −80% or greater than 50% of the control) for six of the 10 compounds. Approximately half of the hits showed agonistic behavior in the assay, which suggests that the model was still unable to distinguish agonists from inverse agonists and antagonists.The active learning approach discovered thiourea derivatives as innovative CXCR4 ligands in the exploitive iteration (4/10 compounds, Table S5†). Novartis previously reported fully substituted isothiourea derivatives as CXCR4 antagonists.38 Crystallographic receptor–ligand complexes confirmed this substructure forms at least two relevant hydrogen-bond interactions,39 which we consistently observed for our thiourea compounds in hypothetical ligand–receptor complexes obtained by computational ligand docking (Fig. 4A and Fig. S6†). Importantly, the molecular descriptors employed do not perceive this substructure variation as a trivial modification, which is reflected in the low ranks (>5000) of these hits when predicting their activity with the initial random forest model that was trained on the ChEMBL CXCR4 data containing the isothiourea compounds. In fact, with their elongated shape and terminal aromatic rings, some of our hits seem to constitute hybrids of known CXCR4 ligands, suggesting that the model successfully generalized over the known SARs. We tested this hypothesis by investigating the reference compounds used for predicting the most potent thiourea compound 10, and found that the model coupled this chemical structure with distinct types of CXCR4 antagonists, including diamines,32 cyclam AMD3100 derivatives,16 isothiourea38 and guanidine-containing compounds35 (Table S6†). The notable chemical similarity to known antagonists is attractive for model interpretation. It originates from the greedy selection strategy that forces the algorithm to borrow from known actives to maintain high confidence in the predictions. The activities of the retrieved hits are fully in line with the prediction. The mean absolute difference between the conservative predictions and the observed effect is 0.47log units for the five compounds that elicited antagonistic assay readout (Table S5†).
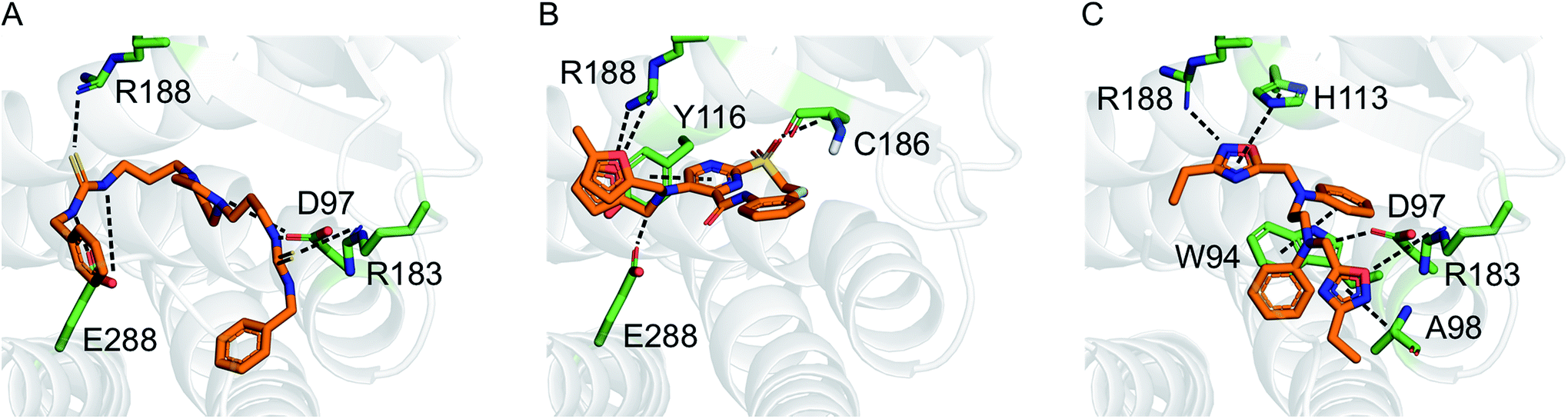 | ||
| Fig. 4 Hypothetical binding modes of three representative CXCR4 ligands predicted by GOLD (5.1) docking (PDB-ID: 3odu). Compounds 10 (A), 1 (B), and 2 (C) are shown as orange stick models. Dashed lines indicate potential polar interactions with the receptor atoms (green). | ||
We next posed the question of whether the newly added training data had an impact on the final predictions. Almost all of the compounds selected in the exploitive iteration were predicted using data from the two learning iterations (Table S7†). The only exception was integrilin, which seemed a reasonable choice of the greedy algorithm given the initial training SAR data for circular peptides with mono- or di-arginine groups.40 Such structures were not investigated in the active learning approach. Integrilin is the only macrocycle among the 61 pool compounds with a similar molecular weight (500 < MW < 1000 g mol−1) that contain an arginine residue. The lack of activity of integrilin adds to the SAR and promotes the utility of the actively added information for use in the final predictive model (Table S7†). For example, hit compound 12 is an analog of the compound 3 that was discovered here.
Hit expansion improves the understanding of novel CXCR4 inhibitors
Finally, we employed hit expansion of the most potent hits, compounds 1 and 2, through sampling via the random forest similarity. Almost all of the derivatives of compound 1 showed activity (Table S8†). Losing the halogen substituent seemed to be better tolerated compared with removing one of the two furan rings. This is consistent with the hypothetical binding mode of compound 1 where the two furan rings jointly interact with R188, while the phenyl substituent forms no interactions (Fig. 4B and S6†). The agonistic activity of compound 13 suggests a close relationship between agonists and antagonists in this compound class. Further biochemical evaluation of compound 13 will be necessary to ensure that this result is consistent with other assays (Fig. S7†). When performing hit expansion for compound 2, we did not find many structural analogs in the screening pool, which is reflected by low levels of similarity to compound 2 (Table S9†). This provides an explanation for why these molecules were largely inactive with only two exceptions. Compound 14 highlights the importance of correctly positioning the two oxadiazoles, while the aromatic, linear framework of compound 2 can be substituted by a tetrahydroquinoxaline. This is consistent with the binding-mode hypothesis for compound 2 in which both oxadiazoles form hydrogen bonds and arene interactions with the receptor, and the aromatic linker is involved in π–π stacking (Fig. 4C and S6†). In the asymmetric compound 15, a tricyclic ring system replaces one of the oxadiazoles while maintaining activity, suggesting possibilities for hit optimization.The learning strategy determines the information gain
As a final step of model evaluation, we repeated our analysis of prediction uncertainty and feature importance with a model trained on all the ChEMBL and screening data, including the final results from the greedy and the two hit expansion iterations. We observed only marginal improvement of the predictive uncertainty using the additional data points (Fig. 3A), suggesting that the active compounds retrieved in the last iteration did not add much information to the model. In line with this observation, the feature importance was similar to the previous learning exercise (Fig. 3B). The minor model improvement while retrieving actives contrasts the second learning iteration (“iteration 2”), which sampled multiple inactive compounds that strongly improved the model. These results further underline the impact of the learning strategy on the actual value of the retrieved compounds in terms of their activity and information content. Our balanced learning strategy aims at finding informative actives. Accordingly, the first learning iteration (“iteration 1”) led to numerous informative actives (e.g., 1–6). Several retrospective studies have proposed adaptive learning behavior to compromise between the identification of actives and the model improvement, for example by evolving stochastic combinations of learning functions,41 Pareto-optimization,42 or automated switching strategies.43 Jain and colleagues have shown that using several selection strategies in parallel can help identify novel inhibitors in subsequent iterations.44,45 As an extension to these studies, our balanced approach considers multiple objectives for each individual compound selection instead of performing parallel or alternating selections.Conclusions
We prospectively applied the emerging concept of active learning to the identification of inhibitors of the CXCR4–CXCL-12 protein–protein interaction. In contrast to other prospective active learning studies,10,11 we simultaneously considered both the activity and the novelty of the selected compounds. Analysis of the model architecture and the predictive uncertainty suggests that this multi-objective strategy enabled rapid model improvement while discovering structurally new inhibitors from previously uncharted compound clusters. Some of the hits ranked in the lower half of the screening pool when predicted with models that were trained exclusively on ChEMBL CXCR4 data, thereby endorsing active learning as a promising technique to exploratively sample compound libraries for finding actives. Importantly, we explicitly addressed batch selection by employing the random forest similarity metric and, consequently, observed only low structural redundancy among the selected compounds. At the same time, using the random forest similarity as a selection function for hit expansion allowed us to identify active derivatives of the original hits, further highlighting the value of the random forest similarity measure for virtual screening. From a hit finding perspective, our learning approach led to the discovery of new classes of CXCR4 inhibitors. A shortcoming of the current prediction model is its apparent inability to distinguish between agonists and antagonists. This might be attributed to our model relying in part on Ki data and to the structural similarities between known CXCR4 agonists and antagonists. Another observation is the lack of highly active ligands among the selected compounds. Similar results were observed for compounds identified by structure-based screening, suggesting a limitation of screening out-of-the-box compound libraries against CXCR4.21,46,47 Our study demonstrates the applicability of active machine learning for rapid hit retrieval against relevant drug targets with reduced consumption of materials. Recent success stories of machine learning models used for virtual compound screening9–11,26,48,49 suggest that a successful transfer of the active learning concept to different hit discovery projects is possible. Successful hit finding will critically depend on the performance of the SAR model, the amount and quality of the available data for model building, and a customized selection strategy to ensure the expected outcome in terms of desired molecular structure and model improvement. Our tunable selection approach prototypes the design of such learning functions. With the increasing availability of automated assay systems that enable rapid feedback loops, we expect active machine learning to become an important tool for hit and lead discovery.Methods and materials
Data and affinity prediction models
CXCR4 ligand data (log-transformed IC50, Ki, Kd values) were extracted from the ChEMBL database (version 19).28 We removed entries for which the comment field indicated inconclusive results (e.g., “Not Tested”, “Insoluble”, “Unstable”).48,49 In cases for which the annotation was a lower bound (“>”), we increased the annotated value by one log unit to avoid overestimating the activity of such compounds. Ki values were shifted by 0.4log units, which corresponds to the mean shift observed for all Ki and IC50 value pairs for the same compound and the same target found in our data. We excluded entries with pAffinity less than 3 or greater than 12. Annotated inactive compounds were annotated with a pAffinity of 3. Compounds with multiple affinity annotations were included once with the arithmetic mean when the standard deviation was smaller than one log unit; otherwise these compounds were excluded. We also extracted the year of publication of the molecules for the retrospective analysis. Molecules were described using an in-house CATS2 (http://www.cadd.ethz.ch/software/catslight2.html) implementation in Python (Version 2.7.3) with a maximum correlation distance of 10 bonds and type-sensitive scaling,50 RDKit physicochemical properties,51 and RDKit Morgan fingerprints (radius = 4, 2048 bits).52 This led to a 2434 dimensional descriptor vector for every molecule. Random forest models were fitted in Python (Version 2.7.3) using the scikit-learn (0.14.1) library,53 which we modified to use subagging instead of bagging in the random forest training54 as this had proven beneficial for active learning in preliminary investigations. We used 500 trees that were provided with molecular representations containing a maximum of 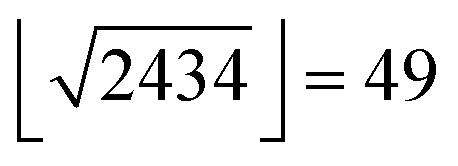 features per tree.25
features per tree.25
Compound scoring
Compounds were ranked according to their predicted affinity, the variance of the individual affinity predictions acquired with the 500 trees, and the random forest similarity to the training data. The random forest similarity was calculated as the number of common leafs that resulted when predicting two structures using the model. The final scoring function was constructed as a weighted average of normalized affinity (with weight w1), variance (with weight w2), and similarity values (with weight w3). Comparing the arccos of the dot product of the vectors of the different weighting parameters allowed the identification of parameter sets that would give equivalent rankings to reduce the necessary time for parameter optimization when using the grid-based search. The weightings (w1, w2, w3) for the model were set to (2, 1, −1) during the explorative learning, (1, −1, 0) for the exploitive/greedy iteration, and (0, 0, 1) for hit expansion to identify analogues of the hit compounds 1 and 2.Retrospective evaluation
The first 33% of the compounds sorted according to their year of publication served as the training set. The remaining 66% were randomly split into learning and test data. The active learning was run for 50 iterations on the learning set with different parameters while monitoring the area under the learning curve (ALC), the average activity of the 50 selected compounds, the reduction of the mean squared error on the test set, and the number of unique Murcko scaffolds55 of the 50 selected compounds (Table S1†). For comparing the optimized active learning model to the annual improvement in the ChEMBL data,28 we considered compounds published before 2003 for training (21 compounds, 6%) and calculated the maximum ligand efficiency29 of the selected compounds after every selection [maxLE = max(1.4pAffinity/nheavy_atoms)]. These values were compared to the maximum ligand efficiency for all compounds published up to a certain year after 2003. The initial training data (6%) that was not considered for the annual improvement visualization contained a few compounds with ligand efficiency of approximately 3.0 (e.g., CHEMBL1202231; LE = 0.32). For the distracted active learning, we supplied the algorithm with an additional 50000 compounds that we randomly sampled from the ChemDB.56 These molecules were annotated as assumed inactive (pAffinity = 3.0). Active learning was performed until the compound with the maximal ligand efficiency (ChEMBL237830; LE = 0.56) was discovered.
Compound selection
During the prospective study, compounds were selected by the algorithm in automated fashion. We performed pre-filtering of potentially insoluble compounds (clogS < 7) using the molecular operating environment (MOE; Version 2011.10).57 None of the active compounds was flagged according to PAINS substructural alerts58 using the publicly available KNIME59 workflow (http://myexperiment.org/workflows/2164.html).
Biochemical assay
CXCR4 inhibition was measured using DiscoverX's (Fremont, CA, USA) arrestin31 and cAMP33 assays on a fee-for-service basis.Feature importance and feature space visualization
Calculation of the feature importance per model was performed by first calculating the feature importance per tree as IMPORTANCE = MSE(P) − fLMSE(L) − fRMSE(R), where MSE is the mean squared error, P is the set of ligands in the node using the feature for classification, L is the set of ligands that do not fulfill the constraint given by the feature, R is the set of ligands that do fulfill the constraint given by the feature and fL = |L|/|P| and fR = |R|/|P| are the fractions of examples in L and R. Then, the importance values per tree were summed to yield the total importance values for the whole random forest model. For the estimation of the architectural changes induced by the data, we trained a total of 10 random forest models on each of the three data sets from active learning cycles 0, 1, 2, and 3. For each set, the feature importance was calculated as the average value given by the models trained. The set of 24 most important features is the union of the 15 most important features according to models 0, 1, and 2. To facilitate their chemical interpretation, the multi-dimensional Morgan fingerprints were represented by the most occurring substructure for each feature. For visualizing the feature space, we extracted the first two principle components (PC) on the normalized 24 most important features using the scikit-learn (0.14.1) library.53 Cluster representatives were selected manually according to their position in feature space. Calculation and visualization were performed in Python.Ligand docking
We retrieved the crystal structure of CXCR4 with the highest resolution (2.5 Å) from the Protein Data Bank (PDB-ID: 3odu).39 Chain B, waters, and ligands, except the copy of IT1t in chain A, were deleted in PyMOL.60 Computational docking was performed with GOLD (version 5.1)61 and the GoldScore function, which had proven beneficial for correctly identifying the binding poses for CXCR4 ligands.62 Ligands were preprocessed using the MOE (2011.10)57 “wash” and “energy minimize” functions and docked with 30 genetic algorithm runs per ligand. We defined the binding pocket as 10 Å around the reference ligand IT1t. Binding poses were selected by visual inspection of the structures forming potential interactions with receptor atoms that are known to interact with potent CXCR4 antagonists.39 Ligand and receptor conformations were relaxed using the MOE energy minimization with default parameters and the PFROSST force field.57 Interactions were analyzed in MOE and graphical models were generated with PyMOL.60Conflict-of-interest statement
P. S. and G. S. are the founders of inSili.com LLC, Zürich.Acknowledgements
We would like to thank Dr Tiago Rodrigues for commenting on the first subset selection. This research was supported by the Swiss National Science Foundation (grant no. CR32I2_159737 to G. S.). The Chemical Computing Group Inc. (Montreal, Canada) provided a research license for the Molecular Operating Environment.References
- T. M. Gureckis and D. B. Markant, Perspect Psychol. Sci., 2012, 7, 464–481 CrossRef PubMed
.
-
S. Burr, Active Learning, Synthesis Lectures on Artificial Intelligence and Machine Learning, Morgan & Claypool Publishers, San Rafael, CA, USA, 2012 Search PubMed
- D. Reker and G. Schneider, Drug Discovery Today, 2015, 20, 458–465 CrossRef PubMed
- R. F. Murphy, Nat. Chem. Biol., 2011, 7, 327–330 CrossRef CAS PubMed
- R. D. King, K. E. Whelan, F. M. Jones, P. G. K. Reiser, C. H. Bryant, S. H. Muggleton, D. B. Kell and S. G. Oliver, Nature, 2004, 427, 247–252 CrossRef CAS PubMed
- G. Schneider, M. Hartenfeller, M. Reutlinger, Y. Tanrikulu, E. Proschak and P. Schneider, Trends Biotechnol., 2009, 27, 18–26 CrossRef CAS PubMed
- A. Schüller and G. Schneider, J. Chem. Inf. Model., 2008, 48, 1473–1491 CrossRef PubMed
- M. K. Warmuth, J. Liao, G. Rätsch, M. Mathieson, S. Putta and C. Lemmen, J. Chem. Inf. Comput. Sci., 2003, 43, 667–673 CrossRef CAS PubMed
- Y. Fujiwara, Y. Yamashita, T. Osoda, M. Asogawa, C. Fukushima, M. Asao, H. Shimadzu, K. Nakao and R. Shimizu, J. Chem. Inf. Model., 2008, 48, 930–940 CrossRef CAS PubMed
- B. Desai, K. Dixon, E. Farran, Q. Feng, K. R. Gibson, W. P. van Hoom, J. Mills, T. Morgan, D. M. Parry, M. K. Ramjee, C. N. Selway, G. J. Tarver, G. Whitlock and A. G. Wright, J. Med. Chem., 2013, 56, 3033–3047 CrossRef CAS PubMed
- J. Besnard, G. F. Ruda, V. Setola, K. Abecassis, R. M. Rodriguiz, X.-P. Huang, S. Norval, M. F. Sassano, A. I. Shin, L. A. Webster, F. R. C. Simeons, L. Stojanovski, A. Prat, N. G. Seidah, D. B. Constam, G. R. Bickerton, K. D. Read, W. C. Wetsel, I. H. Gilbert, B. L. Roth and A. L. Hopkins, Nature, 2012, 492, 215–220 CrossRef CAS PubMed
- M. Ahmadi, M. Vogt, P. Iyer, J. Bajorath and H. Fröhlich, J. Chem. Inf. Model., 2013, 53, 553–559 CrossRef CAS PubMed
- A. W. Naik, J. D. Kangas, C. J. Langmead and R. F. Murphy, PLoS One, 2013, 8, e83996 Search PubMed
- C. Murdoch, Immunol. Rev., 2000, 177, 175–184 CAS
- K. L. Arnolds and J. V. Spencer, Infect., Genet. Evol., 2014, 25, 146–156 CrossRef CAS PubMed
- B. Debnath, S. Xu, F. Grande, A. Garofalo and N. Neamati, Theranostics, 2013, 3, 47–75 CrossRef CAS PubMed
- H. Liang, Y. Zhong, Z. Luo, Y. Huang, H. Lin, S. Zhan, K. Xie and Q. Q. Li, Anticancer Res., 2011, 31, 3433–3440 CAS
- B. K. Aravindan, J. Prabhakar, T. Somanathan and L. Subhadra, Ann. Transl. Med., 2015, 3, 23 Search PubMed
- X. Sun, G. Cheng, M. Hao, J. Zheng, X. Zhou, J. Zhang, R. S. Taichman, K. J. Pienta and J. Wang, Cancer Metastasis Rev., 2010, 29, 709–722 CrossRef CAS PubMed
- A. M. Roccaro, A. Sacco, W. G. Purschke, M. Moschetta, K. Buchner, C. Maasch, D. Zboralski, S. Zöllner, S. Vonhoff, Y. Mishima, P. Maiso, M. R. Reagan, S. Lonardi, M. Ungari, F. Facchetti, D. Eulberg, A. Kruschinski, A. Vater, G. Rossi, S. Klussmann and I. M. Ghobrial, Cell Rep., 2014, 9, 118–128 CrossRef CAS PubMed
- M. M. Mysinger, D. R. Weiss, J. J. Ziarek, S. Gravel, A. K. Doak, J. Karpiak, N. Heveker, B. K. Shoichet and B. F. Volkman, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5517–5522 CrossRef CAS PubMed
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef
- T. Rodrigues, D. Reker, M. Welin, M. Caldera, C. Brunner, G. Gabernet, P. Schneider, B. Walse and G. Schneider, Angew. Chem., Int. Ed., 2015, 54, 15079–15083 CrossRef CAS PubMed
- F. Yang, H.-Z. Wang, H. Mi, C.-D. Lin and W.-W. Cai, BMC Bioinf., 2009, 10, S22 CrossRef PubMed
- V. Svetnik, A. Liaw, C. Tong, J. C. Culberson, R. P. Sheridan and B. P. Feuston, J. Chem. Inf. Comput. Sci., 2003, 43, 1947–1958 CrossRef CAS PubMed
- M. Reutlinger, T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 4244–4248 CrossRef CAS PubMed
- R. P. Sheridan, J. Chem. Inf. Model., 2013, 53, 783–790 CrossRef CAS PubMed
- A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. Davies, F. A. Kruger, Y. Light, L. Mak, S. McGlinchey, M. Nowotka, G. Papadatos, R. Santos and J. P. Overington, Nucleic Acids Res., 2014, 42, D1083–D1090 CrossRef CAS PubMed
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed
- Enamine Ltd, 2014, HTS Collection, http://www.enamine.net, accessed October 2014.
- DiscoveRX Corporation, Fremont, CA, USA; #93-0203C7, URL: http://www.discoverx.com/product-data-sheets-3-tab/93-0203c7, accessed October 2015.
- L. Ros-Blanco, J. Anido, R. Bosser, J. Esté, B. Clotet, A. Kosoy, L. Ruíz-Ávila, J. Teixidó, J. Seoane and J. I. Borrell, J. Med. Chem., 2012, 55, 7560–7570 CrossRef CAS PubMed
- DiscoveRX Corporation, Fremont, CA, USA; #95-0081C2, URL: http://www.discoverx.com/product-data-sheets-3-tab/95-0081c2, accessed October 2015.
- K. Ichiyama, S. Yokoyama-Kumakura, Y. Tanaka, R. Tanaka, K. Hirose, K. Bannai, T. Edamatsu, M. Yanaka, Y. Niitani and N. Miyano-Kurosaki, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4185–4190 CrossRef CAS PubMed
- R. A. Wilkinson, S. H. Pincus, K. Song, J. B. Shepard, A. J. Weaver, M. E. Labib and M. Teintze, Bioorg. Med. Chem. Lett., 2013, 23, 2197–2201 CrossRef CAS PubMed
- N. Meinshausen, J. Mach. Learn. Res., 2006, 7, 983–999 Search PubMed
- C. Strobl, A.-L. Boulesteix, A. Zeileis and T. Hothorn, BMC Bioinf., 2007, 8, 25 CrossRef PubMed
- G. Thoma, M. B. Streiff, J. Kovarik, F. Glickman, T. Wagner, C. Beerli and H.-G. n. Zerwes, J. Med. Chem., 2008, 51, 7915–7920 CrossRef CAS PubMed
- B. Wu, E. Y. Chien, C. D. Mol, G. Fenalti, W. Liu, V. Katritch, R. Abagyan, A. Brooun, P. Wells and F. C. Bi, Science, 2010, 330, 1066–1071 CrossRef CAS PubMed
- S. Ueda, S. Oishi, Z.-x. Wang, T. Araki, H. Tamamura, J. Cluzeau, H. Ohno, S. Kusano, H. Nakashima and J. O. Trent, J. Med. Chem., 2007, 50, 192–198 CrossRef CAS PubMed
- Y. Baram, R. El-Yaniv and K. Luz, J. Mach. Learn. Res., 2004, 5, 255–291 Search PubMed
- M. Zuluga, A. Krause, G. Sergent and M. Püschel, JMLR Workshop Conf. Proc., 2013, 28, 462–470 Search PubMed
- P. Donmez, J. G. Carbonell and P. N. Bennett, Proceedings of the 18th European conference on Machine Learning, ECML 07, 2007, 116–127 Search PubMed
- R. Varela, W. P. Walters, B. B. Goldman and A. N. Jain, J. Med. Chem., 2012, 55, 8926–8942 CrossRef CAS PubMed
- A. Steudle, R. Varela and A. N. Jain, J. Cheminf., 2014, 6, 1 Search PubMed
- C. Castaldo, T. Benicchi, M. Otrocka, E. Mori, E. Pilli, P. Ferruzzi, S. Valensin, D. Diamanti, W. Fecke and M. Varrone, J. Biomol. Screening, 2014, 19, 659–859 CrossRef PubMed
- J. Kim, M. L. R. Yip, X. Shen, H. Li, L.-Y. C. Hsin, S. Labarge, E. L. Heinrich, W. Lee, J. Lu and N. Vaidehi, PLoS One, 2012, 7, e31004 CAS
- M. Reutlinger, T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 582–585 CrossRef CAS PubMed
- T. Rodrigues, N. Hauser, D. Reker, M. Reutlinger, T. Wunderlin, J. Hamon, G. Koch and G. Schneider, Angew. Chem., Int. Ed., 2015, 54, 1551–1555 CrossRef CAS PubMed
- M. Reutlinger, C. P. Koch, D. Reker, N. Todoroff, T. Rodrigues, P. Schneider and G. Schneider, Mol. Inf., 2013, 32, 133–138 CrossRef CAS PubMed
-
G. Landrum, RDKit: Open-source cheminformatics, 2015, http://www.rdkit.org, accessed October 2014 Search PubMed
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed
- P. Bühlmann and B. Yu, Ann. Stat., 2002, 30, 927–961 CrossRef
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed
- J. H. Chen, E. Linstead, S. J. Swamidass, D. Wang and P. Baldi, Bioinformatics, 2007, 23, 2348–2351 CrossRef CAS PubMed
- Chemical Computing Group CCG, Montreal, Canada, http://www.chemcomp.com.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed
-
M. R. Berthold, N. Cebron, F. Dill, T. Gabriel, T. Kötter, T. Meinl, P. Ohl, C. Sieb, K. Thiel and B. Wiswedel, in Data Analysis, Machine Learning and Applications, ed. C. Preisach, H. Burkhardt, L. Schmidt-Thieme and R. Decker, Springer, Berlin, Heidelberg, Germany, 2008, ch. 38, pp. 319–326 Search PubMed
-
The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC Search PubMed
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed
- J. M. Planesas, V. I. Pérez-Nueno, J. I. Borrell and J. Teixidó, J. Mol. Graphics Modell., 2012, 38, 123–136 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details about computational comparisons and all screening results. See DOI: 10.1039/c5sc04272k |
| This journal is © The Royal Society of Chemistry 2016 |