
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli-responsive colorimetric and NIR fluorescence combination probe for selective reporting of cellular hydrogen peroxide†
Nagarjun
Narayanaswamy
a,
Sivakrishna
Narra
b,
Raji R.
Nair
b,
Deepak Kumar
Saini
b,
Paturu
Kondaiah
b and
T.
Govindaraju
*a
aBioorganic Chemistry Laboratory, New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bengaluru 560064, India. E-mail: tgraju@jncasr.ac.in
bDepartment of Molecular Reproduction, Development and Genetics, Indian Institute of Science, Bengaluru 560012, India
First published on 6th January 2016
Abstract
Hydrogen peroxide (H2O2) is a key reactive oxygen species and a messenger in cellular signal transduction apart from playing a vital role in many biological processes in living organisms. In this article, we present phenyl boronic acid-functionalized quinone-cyanine (QCy-BA) in combination with AT-rich DNA (exogenous or endogenous cellular DNA), i.e., QCy-BA⊂DNA as a stimuli-responsive NIR fluorescence probe for measuring in vitro levels of H2O2. In response to cellular H2O2 stimulus, QCy-BA converts into QCy-DT, a one-donor-two-acceptor (D2A) system that exhibits switch-on NIR fluorescence upon binding to the DNA minor groove. Fluorescence studies on the combination probe QCy-BA⊂DNA showed strong NIR fluorescence selectively in the presence of H2O2. Furthermore, glucose oxidase (GOx) assay confirmed the high efficiency of the combination probe QCy-BA⊂DNA for probing H2O2 generated in situ through GOx-mediated glucose oxidation. Quantitative analysis through fluorescence plate reader, flow cytometry and live imaging approaches showed that QCy-BA is a promising probe to detect the normal as well as elevated levels of H2O2 produced by EGF/Nox pathways and post-genotoxic stress in both primary and senescent cells. Overall, QCy-BA, in combination with exogenous or cellular DNA, is a versatile probe to quantify and image H2O2 in normal and disease-associated cells.
Introduction
The regulation of redox homeostasis is essential for maintaining normal cellular functions such as signaling, growth, survival, and death.1 Anomalous behavior of redox homeostasis adversely affects the normal physiological functions, and in turn is responsible for numerous pathological conditions.1 Normally, cells in the disease state exhibit high levels of aerobic glycolysis (Warburg effect), which result in oxidative stress.2 For example, the oxidative stress in cancer cells results in the accumulation of high levels of reactive oxygen species (ROS).3 ROS constitute an important class of chemically reactive species that are essential for normal cellular functions including cell proliferation and differentiation.4 The optimum levels of ROS are controlled by various cellular redox homeostasis mechanisms, and an abrupt increase in their concentration levels is directly linked to oxidative stress-related disorders. Abnormally high levels of ROS are generated in response to adverse environmental and physiological stresses, exposure to ultraviolet (UV) light, and ionizing and heat radiations.3c It is crucial to monitor the levels of intracellular ROS for maintaining effective cellular homeostasis. Notably, different levels of ROS are responsible for different biological responses.5 The cell maintains different levels of ROS by activating the ROS-scavenging systems such as superoxide dismutases, glutathione peroxidase, redox enzymes (peroxiredoxins, glutaredoxin, and thioredoxin) and catalase. Misregulation in any of these ROS-scavenging processes leads to the generation of excess amounts of ROS. Accumulation of high levels of ROS causes oxidative damage to cellular components such as proteins, lipids and nucleic acids, which is responsible for aging and many pathological conditions including cancer, cardiovascular, inflammatory and neurodegenerative diseases.6,7 It is known that cellular aging, also called cellular senescence, is a permanent cell cycle arrest state that results in increased production of ROS species.7a This increased ROS production is critical for maintaining the viability of the senescent cell.7b Therefore, it is necessary to develop molecular tools that are highly sensitive and can be activated by high levels of ROS to distinguish aged or disease-associated cells from normal cells.ROS mainly comprise free radicals, such as hydroxyl radical (OH˙) and superoxide (O2˙−), and reactive molecular species such as H2O2. H2O2 is one of the most prominent and essential ROS in biological systems, and its significantly higher levels are generated in aged and cancer cells than in normal cells.8 In fact, H2O2 is a small molecular metabolite and plays a vital role in the regulation of various physiological processes in living organisms.9 Most importantly, H2O2 serves as a messenger in normal cellular signal transduction and is also a known marker for oxidative damage in many disease-associated cells.9 In cells, H2O2 is generated through the tyrosine kinase receptor-mediated NADPH oxidase (Nox) activation, which affects the functioning of signaling proteins that control cell signaling, proliferation, senescence, and death.10 The biological significance of H2O2 in human physiology and pathology has generated immense interest in understanding the mechanistic details of its generation, partition and role in cellular function and signaling pathways. In comparison to other ROS, relatively higher stability and diffusion rates of H2O2 through the plasma membrane makes it an attractive candidate to study its signaling pathways in living cells.10 However, studies in the spatiotemporal dynamics of H2O2 as a messenger in cellular signal transduction have been limited on account of its chemical reactivity and instability. Molecular imaging of H2O2 using fluorescence probes is a highly attractive tool for studying its generation, accumulation,11 trafficking, and role in biological processes in a spatiotemporal manner in living cells.12 Recently, a new class of organochalcogens such as selenium- and tellurium-based fluorescence redox probes have been developed and explored for the in vivo imaging and real-time monitoring of ROS in cells.13
In recent years, stimuli-responsive fluorescence probes have gained momentum due to their flexibility in introducing diversity through chemical modification and liberation of biologically active probes at the site of target cellular organelles, in response to biological analytes of interest.14 Moreover, targeting specific subcellular organelles (mitochondria) and biomolecules, such as DNA and proteins, using stimuli-responsive fluorescence probes is an emerging and powerful imaging technique that presents enormous potential in biomedical applications related to diagnostics and therapeutics.15 In this context, we envisaged the functionalization of a DNA-binding fluorescence dye with a stimuli-responsive appendage as a promising and unconventional but efficient method for the in situ generation of an active probe in response to H2O2. Our group has been actively involved in the development of novel red fluorescence probes for biologically relevant thiols, metal ions and nucleic acids.16 Recently, we developed a sequence-specific DNA minor groove probe QCy-DT, which shows switch-on NIR fluorescence specifically in the presence of AT-rich DNA (exogenous or endogenous cellular DNA).17 Structurally, QCy-DT has a free hydroxyl group readily available for functionalization with a large number of chemically or enzymatically cleavable appendages to make it a versatile and promising stimuli-responsive probe. In response to a specific stimulus (chemical or enzyme), the appendage functionality is cleaved to release an NIR fluorescence-ready QCy-DT probe, which upon binding with the minor groove of DNA fluoresces strongly, and thus aids the imaging and quantification of the stimulus. We anticipated that functionalizing the DNA binding fluorescence probe QCy-DT with aryl boronates would be an attractive strategy for the development of a stimuli-responsive fluorescence probe for H2O2, because it provides sensitivity and built-in-correction to the probe against background signals from the cellular environment. Therefore, the QCy-DT hydroxyl group was functionalized to obtain phenyl boronic acid-conjugated quinone-cyanine (QCy-BA, Fig. 1a), which reacts selectively with H2O2 to release the parent DNA binding dye. We selected phenyl boronate as the preferred appendage owing to the fact that the reaction between H2O2 and boronic acid or ester is highly chemospecific, bioorthogonal and biocompatible, while the byproducts are non-toxic to living cells.11,18,19 Thus, we envision that probe QCy-BA, in combination with exogenous or endogenous cellular DNA, will be a promising, stimuli-responsive fluorescence probe for investigating H2O2 production and concentration levels in living cells, which will further facilitate the imaging and diagnosis of disease-associated cells.
 | ||
| Fig. 1 (a) Schematic representation for the conversion of QCy-BA to p-quinone-methide and QCy-DT, a DNA minor groove binder, in the presence of H2O2. (b) Time-dependent 1H NMR spectral monitoring of slicing of phenyl boronic acid of QCy-BA in the presence of H2O2. Red circles highlight the appearance of new signals for the newly-formed quinone system and QCy-DT. Ha and Hb represent the O–CH2 (C–Ha) bearing phenyl boronic acid group and newly-formed exocyclic (C–Hb) protons of p-quinone-methide, respectively. (c) The change in solution color upon addition of H2O2 to QCy-BA, as visualized after 2 h. | ||
Results and discussion
Synthesis and design principle of H2O2-triggered release of DNA minor groove binder
Aryl boronates are unique chemical moieties with selective reactivity towards the ambiphilic H2O2, a desirable property to achieve specificity and selectivity over other biologically relevant ROS.11 Initially, the boronate functionality acts as an electrophilic center and reacts with the nucleophile to generate the tetrahedral-boronate complex.11e Subsequently, the carbon–boron (C–B) bond becomes labile and acts as a nucleophile towards the electrophilic oxygen center of H2O2. In particular, the aryl boronate functionality becomes a specific reorganization center for H2O2 among all other biological oxygen metabolites and ROS, which operates through one electron transfer or electrophilic oxidation pathways.11Herein, we present a stimuli-responsive probe QCy-BA, a DNA minor groove binder (QCy-DT) functionalized with phenyl boronic acid. Although QCy-BA shares the backbone of Cy7 dyes, the major structural difference comes from two positively charged, nitrogen atom-containing benzothiazoles with distinct conjugation patterns around the central phenolic moiety derivatized with phenyl boronic acid functionality.17,20 Furthermore, the electron delocalization in QCy-BA is disrupted as a consequence of masking the central phenolic hydroxyl with phenyl boronic acid functionality. Upon slicing the phenyl boronic acid functionality, in response to the H2O2 stimulus, QCy-BA transforms into the negatively charged phenolate of QCy-DT. The generation of a phenolate restores the electron transfer towards one of the positively charged nitrogen atoms of the benzothiazole acceptor. This restores internal charge transfer (ICT) to generate a highly electron delocalized π-system similar to Cy-7 dye with NIR-fluorescence in the presence of DNA (as shown in Fig. 1a).17
Synthesis of QCy-BA was achieved by treating 4-(hydroxymethyl)phenyl boronic acid with a pinacol in the presence of magnesium sulfate in acetonitrile to obtain 4-(hydroxymethyl)phenyl boronic ester (1) (ESI Scheme 1†). The phenyl boronic ester 1 was treated with NaI and trimethyl silyl chloride in acetonitrile at 0 °C to obtain 4-(iodomethyl)phenyl boronic ester (2).20a The 4-(iodomethyl)phenyl boronic ester (2) was coupled to 4-hydroxy isophthaldehyde using potassium carbonate as a base in dimethylformamide (DMF) at room temperature to obtain phenyl boronic ester dialdehyde (3) in good yield. Finally, the dialdehyde (3) was coupled with N-methylated benzothiazole in the presence of piperidine to yield the probe QCy-BA. All the intermediates and probe QCy-BA were characterized by NMR and high-resolution mass spectrometry (HRMS).
NMR-analysis of H2O2-triggered release of DNA minor groove binder
In a preliminary study, we carried out time-dependent NMR spectroscopy analysis of QCy-BA in the presence of H2O2 to assess the stimuli-responsive slicing of phenyl boronic acid functionality. The 1H NMR spectrum of QCy-BA (2 mM) alone in D2O (0.5 mL) showed a single peak at 5.10 ppm corresponding to the O–CH2 (C–Ha)-bearing phenyl boronic acid group and peaks at 8.2–7.2 ppm corresponding to aromatic protons of the parent QCy-DT. The chemical shifts of O–CH2, aromatic region of QCy-BA and appearance of possible new peaks for p-quinone methide (sliced by-product corresponding to phenyl boronic acid functionality) upon sequential addition of H2O2 was monitored. After 1 h of H2O2 (10 mM, 5 μL from the stock 1 M H2O2 solution) addition, the peak intensity at 5.10 ppm, i.e., C–Ha (O–CH2) gradually decreased and new peaks appeared at 5.20 ppm and in the 6.5–7.0 ppm region, suggesting the coexistence of both phenyl boronic acid protected and deprotected forms of QCy-BA. The peaks at 5.20 ppm and aromatic region at 6.5–7.0 ppm corresponds to the newly-formed exocyclic C–Hb protons of p-quinone-methide and QCy-DT moieties, respectively. After 2 h, we observed a single peak at 5.20 ppm and prominent new peaks at 6.5–7.0 ppm, indicating the complete conversion of QCy-BA to QCy-DT and p-quinone methide (Fig. 1b). This study confirmed the H2O2 stimulus-triggered slicing of phenyl boronic acid functionality from QCy-BA to release QCy-DT, a DNA minor groove binding probe. Interestingly, the color of the solution changed from yellow to brown after the addition of H2O2 to QCy-BA, which facilitated naked eye detection of the formation of p-quinone-methide and QCy-DT (Fig. 1c).Photophysical properties of QCy-BA in the presence of H2O2
Next, we studied the photophysical properties of QCy-BA in the absence and presence of H2O2 using UV-vis absorption and emission studies in a PBS-buffer solution (10 mM, pH = 7.4) under ambient conditions. The UV-vis absorption spectrum of QCy-BA (5 μM) showed broad absorbance in the 300–500 nm region with an absorption maximum (λmax) at 400 nm. Upon excitation at 400 nm, the emission spectrum of QCy-BA (5 μM) showed weak fluorescence with emission maximum (Emax) at 565 nm (Fig. S1b†). As expected, QCy-BA did not emit in the NIR region due to the phenyl boronic acid protection of the backbone-phenolic hydroxyl moiety. Interestingly, the absorption spectrum of QCy-BA (5 μM) showed a gradual decrease in absorption maxima at 400 nm in the presence of H2O2 (1 mM); this was accompanied by the appearance of a new absorption band at 465 nm with a shoulder at 530 nm and an isosbestic point at 442 nm (Fig. S1a†). The new absorption bands at 465 nm and 530 nm revealed the transformation of QCy-BA to the phenolate form of QCy-DT. In agreement with the NMR study (Fig. 1b), UV-vis absorption data confirmed the generation of QCy-DT through H2O2-assisted oxidation of boronic acid in QCy-BA followed by the hydrolysis and 1,6-elimination of the p-quinone-methide group (Fig. S2†).11e,20a Evidently, UV-vis absorption spectral characteristics clearly support the observed change in solution color from yellow to brown, as a result of the newly-formed QCy-DT (λmax at 465 and 530 nm) from QCy-BA (λmax = 400 nm) (Fig. 1c). The emission spectra of QCy-BA (5 μM) in the presence of H2O2 (1 mM) displayed a gradual decrease in fluorescence intensity at 565 nm and a weak basal level fluorescence band centered around 680 nm, with a large Stokes shift (Δλmax = ∼280 nm) upon excitation at 400 nm (Fig. S1b†). Therefore, H2O2-triggered slicing of phenyl boronic acid functionality from QCy-BA is a highly useful transformation for the generation of a stimuli-responsive switch-on DNA binding fluorescence probe, QCy-DT, owing to its large Stokes shift and non-fluorescence in the unbound state.Furthermore, we performed a concentration-dependent fluorescence study on slicing of the phenyl boronic acid functionality from QCy-BA (5 μM) in response to the sequential addition of H2O2 (5–100 μM). The fluorescence intensity of QCy-BA at 565 nm was decreased in response to the addition of H2O2 in the concentration range from 5 to 50 μM and subsequently reached saturation at 100 μM. A linear relationship (R2 = 0.9877) was observed with increasing concentration of H2O2 in the concentration range of 5–20 μM. Based on 3σ/slope, the limit of detection (LOD) of H2O2 was found to be 5.3 μM, using the decrease in the fluorescence of QCy-BA at 565 nm (Fig. S3†).21
H2O2 is one of the many ROS present in biological systems, and it is necessary to test the probe QCy-BA against all of them to assess its selectivity and specificity. Therefore, we examined the response of QCy-BA towards H2O2 (100 μM) in the presence of other ROS (100 μM), including tert-butyl hydroperoxide (TBHP), superoxide (O2−), hydroxyl radical (HO˙), tert-butoxy radical (tBuO˙), hypochlorite (OCl−), peroxynitrite (ONOO−) and nitric oxide (NO). Remarkably, only H2O2 efficiently decreased the fluorescence emission at 565 nm owing to selective slicing of phenyl boronic acid functionality from QCy-BA (5 μM). On the other hand, we observed very minimal or no effect on the probe response in the presence of O2−, HO˙, tBuO˙, OCl−, ONOO− and NO (Fig. 2). These results are in agreement with the selective 1,6-elimination of the phenyl boronic acid functionality from QCy-BA only in the presence of H2O2 to liberate the p-quinone-methide moiety and QCy-DT.19,20
 | ||
| Fig. 2 Fluorescence response of QCy-BA (5 μM) to various reactive oxygen species (ROS) at an individual concentration of 100 μM, where F0 and F are the fluorescence intensities of QCy-BA in the absence and presence of ROS, respectively. | ||
Photophysical properties of combination probe QCy-BA⊂Drew-AT in the presence of H2O2
To further validate the H2O2-stimulated conversion of QCy-BA to QCy-DT, a DNA minor groove binder, the transformation was monitored using UV-vis absorption and emission studies in the presence of an AT-rich DNA strand (Drew-AT: 5′-GCGCAAATTTGCGC-3′). QCy-DT binds AT-rich DNA minor groove with high sequence-specificity (5′-AAATTT-3′), which reflects in the strong NIR-fluorescence.17 Thus, we chose Drew-AT, a self-complementary 14-base pair (bp) sequence containing a central 5′-AAATTT-3′ sequence,22 for fluorescence reporting of the QCy-DT released in response to H2O2 stimulus, by means of strong emission in the NIR region. The absorption spectrum of QCy-BA (2 μM) in the presence of Drew-AT (2 μM) duplex showed an increase in absorption maxima at 416 nm with a bathochromic shift (Δλmax = 16 nm) (Fig. S4a†). On the other hand, the fluorescence spectrum of QCy-BA (2 μM) in the presence of Drew-AT showed emission maxima at 500 nm with a hypsochromic shift (Δλmax = ∼50 nm) (Fig. S4b†). These changes in absorption and emission spectra are attributed to weak interactions between QCy-BA and Drew-AT duplex through electrostatic and hydrophobic interactions. Subsequently, absorption and emission spectra of QCy-BA were recorded in the presence of Drew-AT duplex and H2O2 (100 μM). The absorption spectrum showed a gradual decrease in absorption at 416 nm with corresponding increase in the absorption at 564 nm with an isosbestic point at 456 nm, which is in agreement with the absorption characteristics observed for QCy-DT/Drew-AT complex (Fig. 3a).17 Similarly, the emission spectrum of QCy-BA (excitation at λmax = 400 nm) in the presence of Drew-AT duplex and H2O2 showed fluorescence decrease at 500 nm and a corresponding increase at 650 nm (Fig. 3b). This remarkable ratiometric emission at 500 nm and 650 nm (Δλmax = ∼150 nm) is a desirable property of a fluorescence probe for increasing signal-to-noise ratio; measurement at low wavelengths minimizes the error arising from various environmental factors. Upon excitation with a wavelength corresponding to the isosbestic point (456 nm), probe QCy-BA in combination with Drew-AT showed gradual increase in I650/I500 ratio as a function of time (0 to 80 min) and concentration of H2O2 (0 to 200 μM) (Fig. S5†). Furthermore, upon excitation at 564 nm (λmax of QCy-DT bound to Drew-AT duplex), strong fluorescence enhancement at 650 nm was observed (Fig. 3c). Furthermore, probe QCy-BA in combination with Drew-AT showed selective fluorescence enhancement at 650 nm in the presence of H2O2 over other ROS (Fig. S6†). These results reiterated that the H2O2-triggered conversion of QCy-BA to a DNA minor groove binder QCy-DT is a promising ratiometric fluorescence platform for H2O2 in the presence of exogenous DNA (Drew-AT). | ||
| Fig. 3 (a) Absorption spectra of combination probe QCy-BA⊂Drew-AT (2 μM) in the presence of H2O2 (100 μM) in a PBS-buffer solution as a function of time. (b) Normalized fluorescence spectra of QCy-BA (2 μM) in the presence of Drew-AT (2 μM) upon excitation at 400 nm. (c) Fluorescence spectra of QCy-BA (2 μM) in the presence of Drew-AT (2 μM) upon excitation at 564 nm. All the spectra are acquired in the presence of H2O2 (100 μM). (d) Time-dependent fluorescence spectra of QCy-BA (2 μM) in the presence of Drew-AT (2 μM) after the addition of H2O2 (100 μM) upon excitation at 400 nm. (e) Schematic view of the conversion of QCy-BA to p-quinone-methide and a DNA minor groove binder (QCy-DT) with turn-on NIR fluorescence, in the presence of H2O2. | ||
Time-dependent fluorescence study was carried out to evaluate the release kinetics of QCy-BA (2 μM) to QCy-DT in response to H2O2 (100 μM) stimulus, in the presence of Drew-AT duplex. The change in fluorescence intensities at 500 nm and 650 nm, corresponding to emission maxima (Emax) of QCy-BA and QCy-DT, respectively, was monitored in the presence of Drew-AT. Upon excitation at 400 nm, the fluorescence intensity of QCy-BA gradually decreased at 500 nm, while that of QCy-DT increased at 650 nm (Fig. 3d). Similarly, the fluorescence spectra recorded upon excitation at 564 nm showed an exponential increase in emission intensity at 650 nm as a function of time and reached saturation within 4 h (Fig. S7†). The calculation of the kinetics parameter using pseudo-first-order conditions for the conversion of QCy-BA (2 μM) to QCy-DT in the presence of H2O2 (1 mM) and Drew-AT (2 μM) exhibited the rate constant of kobs = 1.0 × 10−3 s−1 (Fig. S8†).21 Overall, photophysical (absorption and emission) studies demonstrated that H2O2 triggers the slicing of phenyl boronic acid functionality from QCy-BA to generate QCy-DT, a DNA minor binding probe that shows switch-on NIR fluorescence in the presence of Drew-AT duplex (Fig. 3e).
Probing of in situ generated H2O2 using combination probe QCy-BA⊂Drew-AT
In biological systems, enzymes such as oxidases generate H2O2 by the oxidation of numerous biochemicals. Glucose oxidase (GOx) is one of the most important enzymes known to selectively catalyze the oxidation of glucose to gluconic acid in the presence of oxygen to generate H2O2. In this context, we set out to probe the in situ generation of H2O2 by the oxidation of glucose in the presence of GOx using our combination probe QCy-BA⊂Drew-AT (Fig. 4a). To monitor the in situ generation of H2O2, glucose was added to a PBS buffer (10 mM, pH = 7.4) containing GOx (4 U mL−1) and QCy-BA⊂Drew-AT (2 μM). The reaction mixture showed a gradual decrease in fluorescence at 500 nm (λex = 400 nm) and a corresponding increase in fluorescence intensity at 650 nm (Fig. S9a†). Similarly, upon excitation at 564 nm, the fluorescence spectra showed a strong enhancement in fluorescence emission at 650 nm, which may be attributed to the release and binding of QCy-DT to Drew-AT (Fig. 4b). Subsequently, the reaction kinetics of the in situ generation of H2O2 through the oxidation of glucose by GOx was investigated using the combination probe upon excitation at 564 nm. The fluorescence intensity at 650 nm was plotted as a function of time after the addition of glucose (Fig. 4c). Upon the addition of glucose (1 mM) in the presence of GOx, QCy-BA⊂Drew-AT showed a gradual increase in fluorescence intensity at 650 nm and reached saturation at 1 h. However, in the absence of glucose, GOx and QCy-BA⊂Drew-AT did not show such increase in fluorescence intensity. Furthermore, the fluorescence was monitored with the addition of increasing concentrations of glucose (0–1 mM) to the mixture of GOx and QCy-BA⊂Drew-AT. The fluorescence emission at 650 nm increased and showed a linear relationship in the concentration range of 0–0.2 mM (Fig. 4d and S9b†). Based on 3σ/slope, the LOD of H2O2 was found to be 6.11 μM (from the concentration of glucose) and is in good agreement with LOD of H2O2 (5.33 μM) using the combination probe (Fig. S10†). From the pseudo-first-order calculations, the combination probe QCy-BA⊂Drew-AT (2 μM) showed the rate constant of kobs = 6.87 × 10−4 s−1 in the presence of GOx (4 U mL−1) and glucose (1 mM) (Fig. S11†).21 Overall, the GOx assay demonstrated the in situ monitoring of H2O2 generated from the oxidation of glucose.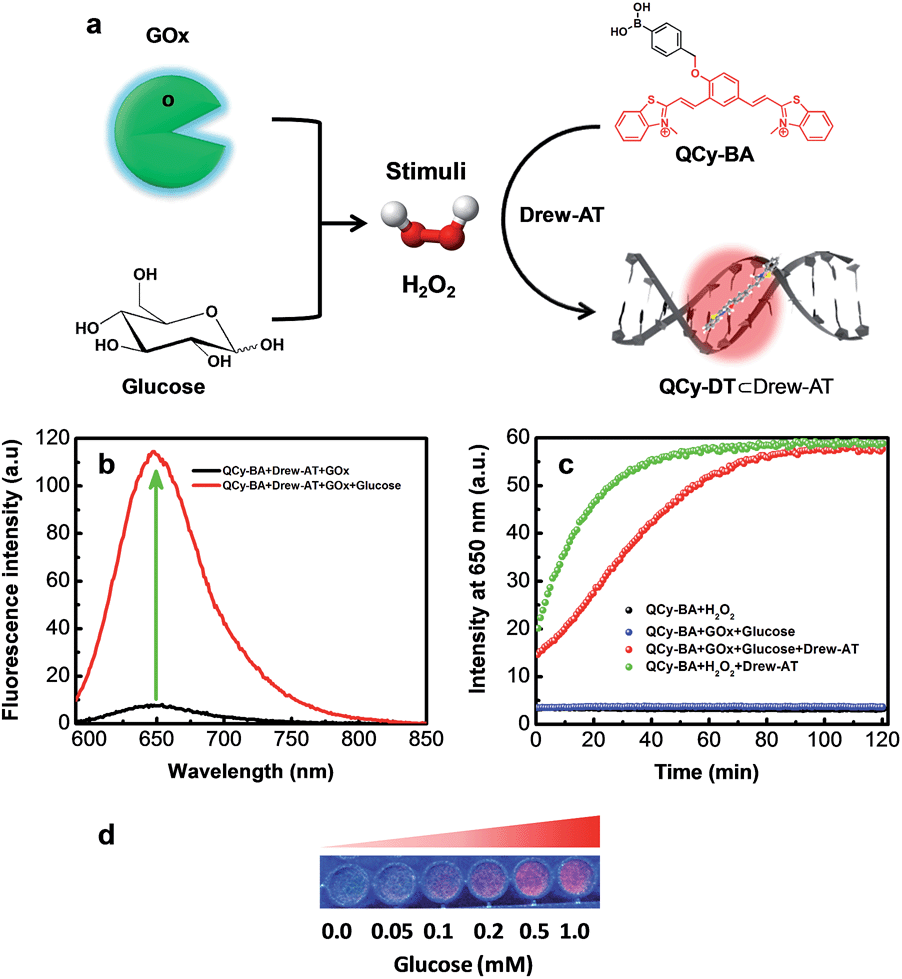 | ||
| Fig. 4 (a) Schematic diagram showing the GOx-assay, where GOx oxidizes glucose to gluconic acid, generating H2O2, followed by fluorescence reporting by the combination probe QCy-BA⊂Drew-AT. (b) Fluorescence spectra of combination probe QCy-BA⊂Drew-AT in the presence of GOx (4 U mL−1) and upon addition of glucose (1 mM). (c) Time-dependent fluorescence of combination probe QCy-BA⊂Drew-AT in the presence and absence of GOx (4 U mL−1) upon addition of glucose (1 mM). (d) Photographs of QCy-BA⊂Drew-AT complex under UV-light in the presence of GOx (4 U mL−1) with increasing glucose concentration from 0.0 to 1.0 mM. | ||
Subsequently, we studied the effect of an enzyme that spontaneously decomposes H2O2 on the conversion of QCy-BA to QCy-DT by the action of H2O2. Catalase is one of the most efficient enzymes that convert H2O2 to water and oxygen to protect cells from oxidative damage and ROS. Catalase exhibits highest turnover number for H2O2 and is capable of decomposing almost 106 molecules per second to water and oxygen. Interestingly, the fluorescence emission was not observed at 650 nm upon addition of H2O2 (1 mM) to a solution of QCy-BA⊂Drew-AT (2 μM) containing catalase (4 U mL−1). The lack of fluorescence emission at 650 nm can be attributed to the prevention of the QCy-BA to QCy-DT conversion, because the added H2O2 was used as a substrate by the catalase (Fig. S12†). These results validate that our combination probe QCy-BA⊂Drew-AT is a promising molecular tool for monitoring the in situ turnover of H2O2 involving oxidase and catalase.
Fluorescence imaging and cytotoxicity studies of QCy-BA in the presence of H2O2
Remarkable selectivity of QCy-BA towards H2O2 and its detection through DNA-assisted switch-on NIR fluorescence inspired us to evaluate the uptake and application of the probe to detect H2O2 in cells. For this purpose, confocal fluorescence imaging of HeLa cells treated with H2O2 (exogenous) was carried out. First, the HeLa cells were incubated with QCy-BA (5 μM) for 30 min and imaged under a confocal microscope. Confocal fluorescence images of these HeLa cells did not show any emission in the red channel (Fig. 5a and b). HeLa cells containing QCy-BA were then treated with H2O2 (100 μM) for 15 min, after which the cells were again scanned under a confocal microscope. Confocal images of these cells showed strong fluorescence in the red channel with maximum localization in the cell nucleus (Fig. 5c and d). Interestingly, cells also showed the pattern of black nucleoli, a characteristic feature of specific DNA minor groove binders over single-strand DNA and RNAs.23 Cell viability assay was performed in HeLa cells to check the cytotoxicity of probe QCy-BA. Upon incubation with QCy-BA, more than 80% of the cells were viable even at 25 μM concentration after 24 h (Fig. S13†). In general, the abovementioned results confirm the permeability and non-toxicity (at standard working concentration and time of 5 μM and 24 h, respectively) of QCy-BA and the detection of exogenously added H2O2 in HeLa cells through selective fluorescence staining of the cell nucleus. | ||
| Fig. 5 (a and b) Fluorescence microscope and differential interference contrast (DIC) images of HeLa cells incubated with QCy-BA (5 μM) in the absence of H2O2. (c and d) Fluorescence microscope and differential interference contrast (DIC) images of HeLa cells incubated with QCy-BA (5 μM) in the presence of H2O2 (100 μM). Fluorescence images were collected in the range of 600–800 nm upon excitation at 400 nm. (e and f) FACS/flow cytometry analysis shows the PerCP mean fluorescence intensity in HeLa cells. (e) Fluorescence intensity of QCy-BA (5 μM) in HeLa cells upon addition of H2O2 (100 μM) and N-acetyl-L-cysteine (NAC) (8 mM). (f) Fluorescence intensity of QCy-BA (5 μM) in HeLa cells upon addition of epidermal growth factor (EGF) (50 ng mL−1) and N-acetyl-L-cysteine (NAC) (8 mM). Error bars represent ± standard deviation. PerCP channel: λex = 482 nm and λem = 675 nm. | ||
Monitoring of in situ generated H2O2 levels by EGF/Nox pathways and post-genotoxic stress in live cells
The NMR, photophysical study, GOx assay and confocal fluorescence imaging of HeLa cells showed the detection of exogenously added H2O2 using QCy-BA. Subsequently, we employed QCy-BA for probing cellular (physiologically generated) H2O2 levels in live cells. HeLa cells were incubated with QCy-BA (5 μM) for 30 min in the absence and presence of N-acetyl-L-cysteine (NAC), a well-known H2O2 scavenger.24 In the absence of NAC, flow cytometry analysis of cells treated with the probe (5 μM) showed an increase in mean fluorescence intensity of PerCP as compared to control cells (Fig. S14†). Upon addition of NAC (8 mM), the fluorescence intensity of PerCP decreased significantly (Fig. 5e and S15a†). In a control experiment, flow cytometry analysis of live HeLa cells treated with QCy-BA and H2O2 (100 μM) for 30 min at 37 °C showed an increase in the mean fluorescence intensity of PerCP (Fig. 5e and S15a†). Thus, probe QCy-BA is also capable of detecting the cellular H2O2 levels in live cells.Furthermore, our study was extended to visualize the in situ H2O2 generation by a known signaling pathway in live cells. We selected the well-known epidermal growth factor (EGF) binding to epidermal growth factor receptor (EGFR) signaling pathway, which stimulates the production of H2O2 in cells by activating the NOx/PI3K pathways.10d In this experiment, live HeLa cells were incubated with the epidermal growth factor (EGF) (50 ng mL−1) for 40 min under physiological conditions (37 °C, pH = 7.4). EGF-treated live HeLa cells were incubated with QCy-BA (5 μM) for 30 min and flow cytometry analysis of these cells showed strong fluorescence intensity in the PerCP region (Fig. 5f and S15b†). On the other hand, the control experiment performed on live HeLa cells without EGF stimulation showed modest fluorescence due to the presence of cellular H2O2 level. In contrast, NAC-treated cells showed a decrease in fluorescence even in the presence of EGF (Fig. 5f and S15b†). FACS analysis showed a 2-fold higher NIR-fluorescence response in cells incubated with EGF (50 ng mL−1) compared to the cells treated with external H2O2 (100 μM). In the cells treated with an external source of H2O2, antioxidant enzymes rapidly consume and decrease its intracellular concentration by ∼7 to 10 fold.25 Consequently, the relative intracellular concentration of H2O2 available to react with probe QCy-BA is ∼7 to 10 fold lower than the original concentration of externally added H2O2. On the other hand, EGF binds to EGFR and induces the production of intracellular H2O2 in cells. Therefore, the concentration of H2O2 readily available to react with QCy-BA is more in the case of EGF treated cells, which in turn results in a higher fluorescence response. These results provide concrete evidence that QCy-BA is a versatile and practically viable molecular probe for monitoring concentration levels of H2O2 in live cells.
To detect the in situ generated H2O2 in other physiological conditions, we performed fluorescent plate reader-based studies for cellular senescence in primary and cancer cells using probe QCy-BA. First, we performed the fluorescence imaging of primary cells using probe QCy-BA in the presence of H2O2. Live cell imaging of MRC5 cells showed NIR fluorescence in the nucleus compared to control cells incubated with probe QCy-BA (5 μM) for 30 min after treating with H2O2 (100 μM) (Fig. 6a–d). It is well-established that genotoxic stress causes DNA damage in cells that can trigger the generation and accumulation of H2O2 inside the cells.26 Recently, it has been shown that DNA damage induces cell cycle arrest or cellular senescence, where ROS plays an integral role.27 To measure ROS generated concomitant to the dose of the DNA damage, HeLa cells were treated with increasing doses (0 to 200 μM) of 5-bromo-2′-deoxyuridine (BrdU). BrdU is a thymidine analog that gets directly incorporated into DNA and triggers a DNA damage response. From previously reported studies, it is well known that 48 h of treatment with BrdU (100 μM) or another DNA damaging agent, such as doxorubicin, at a concentration of 0.1 μM can lead to the induction of cellular senescence.7b,27b After 48 h of treatment with BrdU (100 μM), HeLa cells showed a 3-fold increase in fluorescence of 2′,7′-dichlorofluorescin diacetate (DCFDA) compared to the control cells; DCFDA is a known ROS probe for live cells (Fig. S16†). Interestingly, probe QCy-BA showed an almost 10-fold increase in fluorescence compared to control cells unlike DCFDA that showed only a 3 to 4-fold change, suggesting that QCy-BA dye has a much better dynamic range than DCFDA (Fig. 6e).
 | ||
| Fig. 6 (a and b) Fluorescence microscopy and differential interference contrast (DIC) images of MRC5 cells incubated with QCy-BA (5 μM) in the absence of H2O2. (c and d) Fluorescence microscope and differential interference contrast (DIC) images of MRC5 cells incubated with QCy-BA (5 μM) in the presence of H2O2 (100 μM). Scale bar = 5 μm. (e) H2O2 detection in attached live HeLa cells using QCy-BA (5 μM) after treatment with BrdU from 0 to 200 μM or doxorubicin (0.1 μM) for 48 h. Fold change of fluorescence per cell is normalized to 1 for control cells (n = 3). (f) MRC5 cells were treated with BrdU from 0 to 200 μM or doxorubicin (0.1 μM) for 72 h. H2O2 levels were estimated using QCy-BA (5 μM) dye and fold change of fluorescence per cell is normalized to 1 for control cells (n = 3). | ||
Furthermore, similar experiments were performed in primary MRC5 cells, which are human lung primary fibroblasts. To induce DNA damage, MRC5 cells were similarly treated with various doses of BrdU and doxorubicin (0.1 μM) for 72 h. After 72 h, the probe QCy-BA showed increase in fluorescence compared to control cells in a dose-dependent manner, indicating that the probe can be used to monitor the in situ generated H2O2 in primary cells as well (Fig. 6f). Therefore, the abovementioned results reveal that QCy-BA is a versatile probe to monitor the elevated levels of H2O2 in both primary and cancer cells in the senescence state.
Conclusions
In conclusion, we developed a stimuli-responsive, colorimetric and switch-on NIR fluorescence combination probe (QCy-BA in combination with AT-rich exogenous or endogenous nuclear DNA) for H2O2. In QCy-BA, the phenyl boronic acid functionality effectively suppressed the NIR fluorescence of QCy-DT, a DNA minor groove binder, and restored it selectively in the presence of H2O2. NMR and UV-vis absorption study showed the selective conversion of QCy-BA to QCy-DT and quinine methide in response to H2O2 while the solution color changed from yellow to brown for naked eye detection of H2O2 over other ROS. The fluorescence study demonstrated the selective conversion of QCy-BA to QCy-DT in response to H2O2 stimulus, which showed NIR fluorescence in the presence of AT-rich DNA duplex (Drew-AT). Furthermore, glucose oxidase assay confirmed the use of the combination probe QCy-BA⊂DNA for probing in situ generated H2O2 by the oxidation of glucose to gluconic acid. Cell viability and confocal fluorescence imaging of HeLa cells showed the cell permeability, non-toxicity and preferential nuclear staining of the probe in the presence of H2O2. Furthermore, QCy-BA is a sensitive probe to detect normal and in situ generated levels of H2O2 by EGF/Nox pathways in live cells. Probe QCy-BA was also found to be effective in the detection of H2O2 in primary cells as well as senescent cancer cells. Therefore, ease of synthesis, large Stokes shift, cell permeability and the ability to detect normal and elevated levels of H2O2 in primary as well as cancer cells makes QCy-BA a superior combination probe with a NIR fluorescence response (Table S1†). We anticipate that our approach of conjugating DNA fluorescence probes with stimuli-responsive appendages (combination probes) will open up new avenues in the development of DNA targeting theranostic prodrugs for targeting disease-associated cells. This approach can be further extended to create new stimuli-responsive probes for various biochemical processes including enzymatic activities.Experimental section
General information
All the chemicals, reagents, self-complementary Drew-AT, Hoechst 33258, phosphate buffer saline (PBS), 2′,7′-dichlorofluorescein (DCFDA), 5-bromo-2′-deoxyuridine (BrdU), doxorubicin (dox), hydrogen peroxide (H2O2), tert-butyl hydroperoxide (TBHP), potassium superoxide (KO2), sodium hypochlorite (NaOCl), diethylamine NONOate sodium salt hydrate, sodium nitrite (NaNO2) and N-acetyl-L-cysteine (NAC) were purchased from Sigma-Aldrich. All the synthesized compounds were purified by column chromatography using Rankem silica gel (60–120 mesh). 1H and 13C NMR spectra were recorded on a Bruker AV-400 MHz spectrometer with chemical shifts reported as parts per million (ppm) (in CDCl3, DMSO-d6, tetramethylsilane as an internal standard) at 20 °C. High resolution mass spectra (HRMS) were obtained on an Agilent Technologies 6538 UHD Accurate-Mass Q-TOF LC/MS spectrometer. The UV-vis absorption and emission spectra were recorded on Agilent Technologies Cary series UV-vis-NIR absorbance and Cary Eclipse fluorescence spectrophotometers, respectively. UV-vis absorption and emission spectra were obtained in quartz cuvettes with a path length of 1 cm.Sample preparation for UV-vis and fluorescence measurements
A stock solution of QCy-BA probe was prepared in millimolar concentration in Milli-Q water (MQ-water) and stored at −10 °C. DNA stock solutions were prepared by dissolving oligos in double distilled water in the order of 10−4 M. Double-stranded DNA samples were prepared in PBS (10 mM, pH = 7.4) buffer solution and subjected to annealing by heating up to 85 °C for 15 min, followed by subsequent cooling to room temperature for 7 h and storing in a refrigerator for 4 h.16d Peroxynitrite (ONOO−) solution was prepared according to the reported literature.28Maintenance of HeLa cells
Human cervix carcinoma cell line (HeLa) was cultured in DMEM (Dulbecco's modified Eagle's medium) with 10% FBS (fetal bovine serum). The antibiotics penicillin and streptomycin (1%) were mixed with 10% FBS medium. The cells were incubated at 37 °C temperature in a 5% CO2 humidified chamber. All the cell culture investigations were carried out under laminar flow hoods.Cytotoxicity studies on HeLa cells (MTT assay)
MTT [3-(4,5-dimethylthiozol-2yl)-2,5-diphenyltetrazolium bromide] assay was carried out with the QCy-BA probe on HeLa cells to determine the cytotoxicity effect. In a tissue culture 96-well plate, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well were plated and grown for 24 h. Cells were treated with various concentrations (25 μM, 12.5 μM, 6.25 μM, 3.125 μM and 0 μM) of QCy-BA probe for 24 h. All the treatments were carried out in triplicate. The required concentrations of QCy-BA were prepared from a 1 mg mL−1 aqueous stock solution in 0.2% DMEM. Four hours before the stipulated time of the experiment, MTT solution (5 mg mL−1 of 20 μL) was added to each well and incubated to form formazan crystals. The culture medium was completely removed by a 1 mL pipette, and 200 μL of DMSO was added to dissolve the formazan crystals. The purple-colored formazan was estimated by determining the absorbance at 590 nm using a spectrophotometer (Bio-RAD model 1680, Microplate reader). The results were represented as bar graphs (concentration of QCy-BAvs. % cell viability).
000 cells per well were plated and grown for 24 h. Cells were treated with various concentrations (25 μM, 12.5 μM, 6.25 μM, 3.125 μM and 0 μM) of QCy-BA probe for 24 h. All the treatments were carried out in triplicate. The required concentrations of QCy-BA were prepared from a 1 mg mL−1 aqueous stock solution in 0.2% DMEM. Four hours before the stipulated time of the experiment, MTT solution (5 mg mL−1 of 20 μL) was added to each well and incubated to form formazan crystals. The culture medium was completely removed by a 1 mL pipette, and 200 μL of DMSO was added to dissolve the formazan crystals. The purple-colored formazan was estimated by determining the absorbance at 590 nm using a spectrophotometer (Bio-RAD model 1680, Microplate reader). The results were represented as bar graphs (concentration of QCy-BAvs. % cell viability).
Exogenous and endogenous detection of H2O2 in HeLa cells by QCy-BA
For the detection of endogenous H2O2 in HeLa cells by flow cytometric analysis, 3 × 105 HeLa cells were plated in each well of 6-well tissue culture plates and grown for 24 h. Cells were serum deprived for 1 h. In addition, the serum-deprived cells of the 6-well tissue culture plates were treated with N-acetyl-L-cysteine (NAC) (8 mM) and incubated for 1 h. Subsequently, the cells were treated with the QCy-BA probe (5 μM) and incubated for 30 min. After 30 min incubation, the cells were washed with DPBS (Dulbecco's phosphate buffer saline) to remove the excess of QCy-BA. These cells were harvested after trypsinization. Exogenously, H2O2 (100 μM) was added to the QCy-BA- and NAC + QCy-BA-treated cells and incubated for 15 min. These samples were subjected to FACS analysis in the PerCP (λex = 482 nm and λem = 675 nm) channel.Epidermal growth factor (EGF)-generated H2O2 detection by QCy-BA in HeLa cells
In a 12-well plate, 3 × 105 HeLa cells were plated in each well and grown for 24 h. Cells were serum-deprived for 1 h. Three wells of serum-deprived cells were treated with EGF (50 ng mL−1) for 40 min and NAC (8 mM) for 1 h. Then, the cells were treated with QCy-BA (5 μM) for 30 min. After 30 min incubation of cells with QCy-BA, cells were washed with DPBS (Dulbecco's phosphate buffer saline) to remove the excess of QCy-BA. The cells were harvested after trypsinization and a single cell suspension was obtained. The distribution of QCy-BA-stained HeLa cells was determined by flow cytometry in the PerCP channel.Immunofluorescence studies with QCy-BA for the detection of H2O2 in HeLa cells
Immunofluorescence studies were carried out in HeLa cells to validate exogenous and endogenous detection of H2O2 by QCy-BA. The HeLa cells (10000 cells) were grown on cover slips. These cells were treated with 5 μM concentration of QCy-BA for 30 min. The cells were washed several times with DPBS to remove the excess of QCy-BA. The cells were treated with H2O2 (100 μM) for 30 min. These samples were subjected to confocal microscopy for immunofluorescence and images were collected from 600–800 nm upon excitation at 400 nm. Fluorescence images were obtained by a Carl Zeiss laser scanning microscope (LSM510 META).
Detection of ROS using a fluorescence plate reader
The cells were incubated with QCy-BA (5 μM) for 30 min in the dark, washed with PBS and analyzed to detect QCy-BA dye fluorescence using an Infinite M1000 Pro (Tecan, Austria). Wavelengths used for excitation and emission for QCy-BA dye were 400 nm and 650 nm, respectively. The ROS measurement assays were conducted using a plate reader and after fluorescence measurements, cells were washed, trypsinized and counted to estimate fluorescence per cell values.Live cell imaging of MRC5 cells
MRC5 PDL 23 cells were seeded overnight and treated with H2O2 for live cell imaging after the addition of QCy-BA (5 μM) for 30 min. Images were acquired using an Olympus IX 83 inverted epifluorescence microscope using a 20× objective from 600–800 nm upon excitation at 400 nm.Acknowledgements
We thank Prof. C. N. R. Rao FRS for constant support and encouragement; the Council of Scientific and Industrial Research (CSIR), New Delhi [grant no. 02/(0128)/13/EMR-II]; Innovative Young Biotechnologist Award (IYBA) and the Department of Biotechnology (DBT), India (BT/03/IYBA/2010), for financial support; Alexander von Humboldt Foundation, Germany, for special equipment donation to T. G; ICMS-JNCASR for awarding Sheikh Saqr Career Award Fellowship to T. G.; CSIR for SRF fellowship to N. N.Notes and references
- (a) C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed; (b) P. D. Ray, B.-W. Huang and Y. Tsuji, Cell. Signalling, 2012, 24, 981–990 CrossRef CAS PubMed.
- (a) R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed; (b) C. Gorrini, I. S. Harris and T. W. Mak, Nat. Rev. Drug Discovery, 2013, 12, 931–947 CrossRef CAS PubMed.
- (a) T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794–798 CAS; (b) S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1–3 CrossRef CAS PubMed; (c) K. P. Mishra, J. Environ. Pathol., Toxicol. Oncol., 2004, 23, 61–66 CrossRef CAS; (d) S. Kawanishi, Y. Hiraku, S. Pinlaor and N. Ma, Biol. Chem., 2006, 387, 365–372 CrossRef CAS PubMed.
- (a) F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS; (b) J. Boonstra and J. A. Post, Gene, 2004, 337, 1–13 CrossRef CAS PubMed; (c) M. P. Murphy, A. Holmgren, N.-G. Larsson, B. Halliwell, C. J. Chang, B. Kalyanaraman, S. G. Rhee, P. J. Thornalley, D. Gems, T. Nyström, V. Beluosov, P. T. Schumacker and C. C. Winterbourn, Cell Metab., 2011, 13, 361–366 CrossRef CAS PubMed.
- (a) G. Perry, K. A. Raine, A. Nunomura, T. Watayc, L. M. Sayre and M. A. Smith, Free Radical Biol. Med., 2000, 28, 831–834 CrossRef CAS; (b) B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine 1–677, Oxford University Press, Oxford, 2007 Search PubMed.
- (a) M. T. Lin and M. F. Beal, Nature, 2006, 443, 787–795 CrossRef CAS PubMed; (b) N. Houstis, E. D. Rosen and E. S. Lander, Nature, 2006, 440, 944–948 CrossRef CAS PubMed; (c) D. Jay, H. Hitomi and K. K. Griendliing, Free Radical Biol. Med., 2006, 40, 183–192 CrossRef CAS PubMed; (d) J. P. Fruehauf and F. L. Meysken Jr, Clin. Cancer Res., 2007, 13, 789–794 CrossRef CAS PubMed; (e) T. Finkel, M. Serrano and M. A. Blasco, Nature, 2007, 448, 767–774 CrossRef CAS PubMed; (f) D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- (a) R. Colavitti and T. Finkel, IUBMB Life, 2005, 57, 277–281 CrossRef CAS PubMed; (b) R. R. Nair, M. Bagheri and D. K. Saini, J. Cell Sci., 2015, 128, 342–353 CrossRef CAS PubMed.
- (a) B. Chance, H. Sies and A. Boveris, Physiol. Rev., 1979, 59, 527–605 CAS; (b) M. Lopez-Lazaro, Cancer Lett., 2007, 252, 1–8 CrossRef CAS PubMed.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef PubMed.
- (a) K. N. Schmidt, P. Amstad, P. Cerutti and P. A. Baeuerle, Chem. Biol., 1995, 2, 13–22 CrossRef CAS PubMed; (b) M. Sundaresan, Z. X. Yu, V. J. Ferrans, K. Irani and T. Finkel, Science, 1995, 270, 296–299 CAS; (c) K. Z. Guyton, Y. Liu, M. Gorospe, Q. Xu and N. J. Holbrook, J. Biol. Chem., 1996, 271, 4138–4142 CrossRef CAS PubMed; (d) Y. S. Bae, S. W. Kang, M. S. Seo, I. C. Baines, E. Tekle, P. B. Chock and S. G. Rhee, J. Biol. Chem., 1997, 272, 217–221 CrossRef CAS PubMed; (e) S. R. Lee, K. S. Kwon, S. R. Kim and S. G. Rhee, J. Biol. Chem., 1998, 273, 15366–15372 CrossRef CAS PubMed; (f) M. V. Avshalumov and M. E. Rice, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11729–11734 CrossRef CAS PubMed; (g) Z. A. Wood, L. B. Poole and P. A. Karplus, Science, 2003, 300, 650–653 CrossRef CAS PubMed.
- (a) E. W. Miller, A. E. Albers, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2005, 127, 16652–16659 CrossRef CAS PubMed; (b) D. Lee, S. Khaja, J. C. Velasquez-Castano, M. Dasari, C. Sun, J. Petros, W. R. Taylor and N. Murthy, Nat. Mater., 2007, 6, 765–769 CrossRef CAS PubMed; (c) A. E. Albers, B. C. Dickinson, E. W. Miller and C. J. Chang, Bioorg. Med. Chem. Lett., 2008, 18, 5948–5950 CrossRef CAS PubMed; (d) B. C. Dickinson, Y. Tang, Z. Chang and C. J. Chang, Chem. Biol., 2011, 18, 943–948 CrossRef CAS PubMed; (e) A. R. Lippert, G. C. Van de Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed; (f) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed; (g) Y. Wen, K. Liu, H. Yang, Y. Li, H. Lan, Y. Liu, X. Zhang and T. Yi, Anal. Chem., 2014, 86, 9970–9976 CrossRef CAS PubMed.
- (a) E. W. Miller, O. Tulyathan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263–267 CrossRef CAS PubMed; (b) G. C. Van de Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783–1795 CrossRef CAS PubMed; (c) R. Weinstain, E. N. Savariar, C. N. Felsen and R. Y. Tsien, J. Am. Chem. Soc., 2014, 136, 874–877 CrossRef CAS PubMed.
- (a) F. B. Yu, P. Li, P. Song, B. S. Wang, J. Z. Zhao and K. Han, Chem. Commun., 2012, 48, 4980–4982 RSC; (b) Z. Lou, P. Li, X. Sun, S. Yang, B. Wang and K. Han, Chem. Commun., 2013, 49, 391–393 RSC; (c) Z. Lou, P. Li and K. Han, Acc. Chem. Res., 2015, 48, 1358–1368 CrossRef CAS PubMed.
- (a) A. M. Caamaño, M. E. Vázquez, J. Martínez-Costas, L. Castedo and J. L. Mascarenas, Angew. Chem., Int. Ed., 2000, 39, 3104–3107 CrossRef; (b) J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvine and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed; (c) H. M. Lee, D. R. Larson and D. S. Lawrence, ACS Chem. Biol., 2009, 4, 409–427 CrossRef CAS PubMed; (d) A. Deiters, ChemBioChem, 2010, 11, 47–53 CrossRef CAS PubMed.
- (a) M. I. Sanchez, J. M. Costas, F. Gonzalez, M. A. Bermudez, M. E. Vazquez and J. L. Mascareñas, ACS Chem. Biol., 2012, 7, 1276–1280 CrossRef CAS PubMed; (b) P. Murat, M. V. Gormally, D. Sanders, M. D. Antonio and S. Balasubramanian, Chem. Commun., 2013, 49, 8453–8455 RSC; (c) M. I. Sanchez, C. Penas, M. E. Vazquez and J. L. Mascareñas, Chem. Sci., 2014, 5, 1901–1907 RSC; (d) Q. Hu, M. Gao, G. Feng and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 14225–14229 CrossRef CAS PubMed; (e) R. Kumar, J. Han, H. J. Lim, W. X. Ren, J. Y. Lim, J. H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836–17843 CrossRef CAS PubMed; (f) E.-J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- (a) D. Maity, A. Raj, D. Karthigeyan, T. K. Kundu and T. Govindaraju, RSC Adv., 2013, 3, 16788–16794 RSC; (b) D. Maity and T. Govindaraju, Org. Biomol. Chem., 2013, 11, 2098–2104 RSC; (c) D. Maity, A. Raj, P. K. Samanta, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, RSC Adv., 2014, 4, 11147–11151 RSC; (d) N. Narayanaswamy, M. Kumar, S. Das, R. Sharma, P. K. Samanta, S. K. Pati, S. K. Dhar, T. K. Kundu and T. Govindaraju, Sci. Rep., 2014, 4, 6476 CrossRef CAS PubMed.
- N. Narayanaswamy, S. Das, P. K. Samanta, K. Banu, G. P. Sharma, N. Mondal, S. K. Dhar, S. K. Pati and T. Govindaraju, Nucleic Acids Res., 2015, 43, 8651–8663 CrossRef CAS PubMed.
- H. G. Kuivila and A. G. Armour, J. Am. Chem. Soc., 1957, 79, 5659–5662 CrossRef CAS.
- R. B. Greenwald, A. Pendri, C. D. Conover, H. Zhao, Y. H. Choe, A. Martinez, K. Shum and S. Guan, J. Med. Chem., 1999, 42, 3657–3667 CrossRef CAS PubMed.
- (a) N. Karton-Lifshin, E. Segal, L. Omer, M. Portnoy, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2011, 133, 10960–10965 CrossRef CAS PubMed; (b) S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970–2984 CrossRef CAS PubMed.
- C. Chang, D. Srikun, C. S. Lim, C. J. Chang and B. R. Cho, Chem. Commun., 2011, 47, 9618–9620 RSC.
- (a) M. Coll, C. A. Frederick, A. H. J. Wang and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8385–8389 CrossRef CAS PubMed; (b) N. Spink, D. G. Brown, J. V. Skelly and S. Neidle, Nucleic Acids Res., 1994, 22, 1607–1612 CrossRef CAS PubMed.
- G. M. Spitzer, J. E. Fuchs, P. Markt, J. Kirchmair, B. Wellenzohn, T. Langer and K. R. Liedl, ChemPhysChem, 2008, 9, 2766–2771 CrossRef CAS PubMed.
- C. C. Winterbourn and D. Metodiewa, Free Radical Biol. Med., 1999, 27, 322–328 CrossRef CAS PubMed.
- (a) F. Antunes and E. Cadenas, FEBS Lett., 2000, 475, 121–126 CrossRef CAS PubMed; (b) B. K. Huang and H. D. Sikes, Redox Biol., 2014, 2, 955–962 CrossRef CAS PubMed.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS PubMed.
- (a) F. d'Adda di Fagagna, Nat. Rev. Cancer, 2008, 8, 512–522 CrossRef PubMed; (b) K. M. Tewey, T. C. Rowe, L. Yang, B. D. Halligan and L. F. Liu, Science, 1984, 226, 466–468 CAS.
- J. W. Reed, H. H. Ho and W. L. Jolly, J. Am. Chem. Soc., 1974, 96, 1248–1249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, UV-vis absorption, emission, catalase assay, cell viability and FACS data. See DOI: 10.1039/c5sc03488d |
| This journal is © The Royal Society of Chemistry 2016 |