
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Persistent four-coordinate iron-centered radical stabilized by π-donation†
Yusuke
Sunada
*a,
Shintaro
Ishida
b,
Fumiya
Hirakawa
b,
Yoshihito
Shiota
c,
Kazunari
Yoshizawa
c,
Shinji
Kanegawa
c,
Osamu
Sato
c,
Hideo
Nagashima
ad and
Takeaki
Iwamoto
*b
aInstitute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasugakoen, Kasuga, Fukuoka 816-8580, Japan
bDepartment of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan
cInstitute for Materials Chemistry and Engineering, Kyushu University, Nishi-ku, Fukuoka 819-0395, Japan
dCREST, Japan Science and Technology Agency (JST), 6-1 Kasugakoen, Kasuga, Fukuoka 816-8580, Japan
First published on 25th September 2015
Abstract
Dinuclear iron carbonyl complex 2, which contains an elongated unsupported Fe–Fe bond, was synthesized by the reaction between Fe2(CO)9 and phosphinyl radical 1. Thermal Fe–Fe bond homolysis led to the generation of a four-coordinate carbonyl-based iron-centered radical, 3, which is stabilized by π-donation. Complex 3 exhibited high reactivity toward organic radicals to form diamagnetic five-coordinate Fe(II) complexes.
Introduction
The diverse reactivity of transition metal-centered radicals towards various organic substrates has garnered significant interest in both organometallic and catalytic synthetic chemistry over the past several decades.1,2 Among the transition metal complexes, iron carbonyl based five coordinate iron(I) species such as A–C shown in Chart 1 have received particular attention owing to their catalytic activity and unique properties.3–6 Thus, construction of coordinatively unsaturated iron(I) carbonyl complexes is expected to facilitate the development of novel organometallic and catalytic reactions. However, few examples of four or less coordinate iron(I) carbonyl complexes have been explored.7–9 Holland et al. described the synthesis of a four-coordinate Fe(I) dicarbonyl complex D in which the iron center adopts an S = 1/2 electronic configuration.8 Parkin et al. synthesized a four-coordinate Fe(I) monocarbonyl complex E using a tris(pyrazolyl)borate ligand.9 Chelating multidentate ligands bearing sterically hindered substituents are needed to stabilize these coordinatively unsaturated species.‡ We have attempted to construct a reactive, coordinatively unsaturated, iron carbonyl based iron-centered radical based on an alternative synthetic strategy involving stabilization by ligand-to-metal π-donation. Phosphinyl radical 1,10 shown in Scheme 1, was thought to be appropriate for this purpose, as it exhibits reactivity towards transition metal complexes, and the lone pair of electrons on the phosphorus center can effectively stabilize the low-coordinate metal complex via π-donation.10b In this paper, we report that 1 functions as an effective ligand to form the dinuclear iron carbonyl complex 2 with an unsupported Fe–Fe bond. Complex 2 was a suitable precursor for the generation of four-coordinate iron-centered radical 3via homolysis of the Fe–Fe bond. Reactions of 3 with organic radicals are also reported.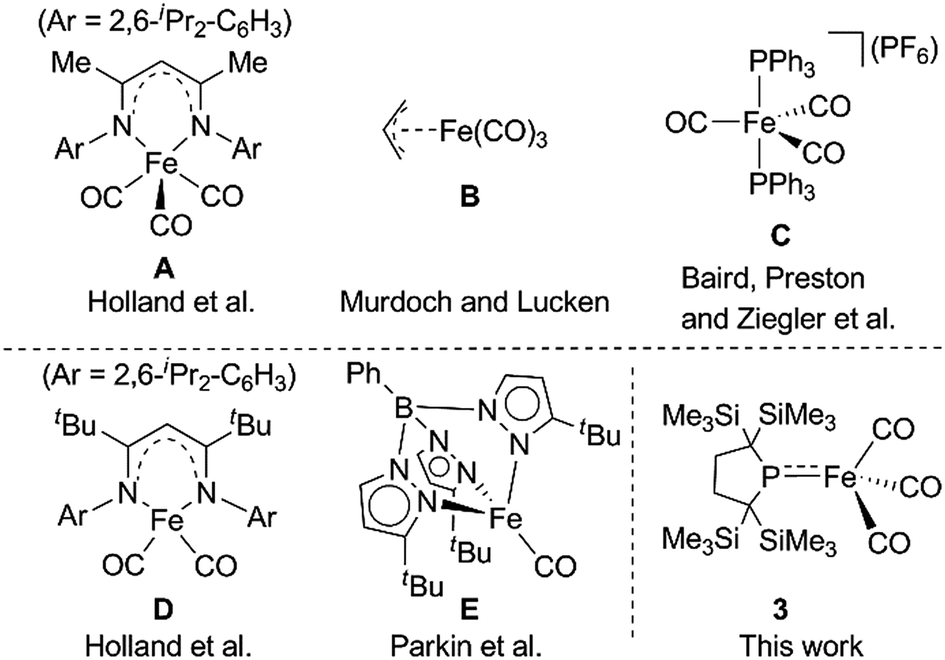 | ||
| Chart 1 Selected examples of five coordinate Fe(I) species (A–C) (upper) and previously reported four coordinate Fe(I) complexes D and E (lower). | ||
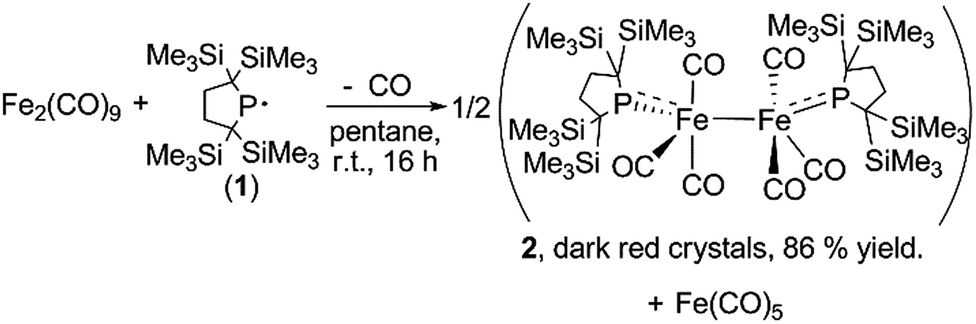 | ||
| Scheme 1 Synthesis of dinuclear iron carbonyl complex 2. | ||
A yellow suspension of Fe2(CO)9 in pentane gradually dissolved to give a dark red solution during the course of the reaction with 1 at room temperature for 16 h. From the dark red solution, complex 2 was obtained in 86% yield (based on 1) (Scheme 1). The reaction was accompanied by the formation of Fe(CO)5, which was confirmed by IR spectroscopy. In the course of the reaction, radical 1 formally underwent a one-electron reduction to form a monoanionic phosphido ligand. As a closely related example to this reaction, Cowley et al. prepared a five-coordinate iron-centered radical via the reaction between Fe2(CO)9 and [{(Me3Si)2CH}2P]˙. However, the molecular structure of this complex was not determined crystallographically.11
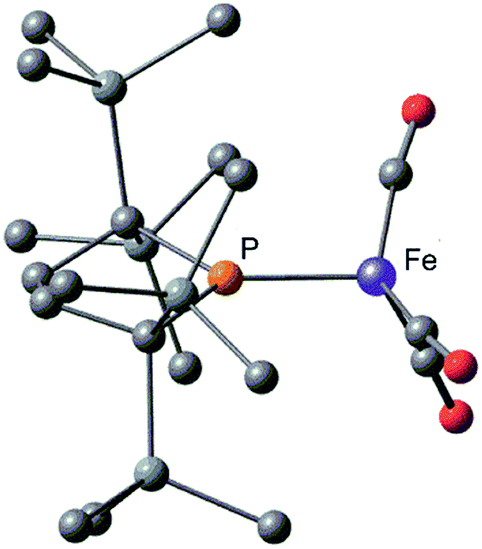
Complex 2 consists of two trigonal-bipyramidal iron centers with six CO ligands (Fig. 1). The distances between the iron and the carbon atoms of the CO ligands coordinated to the other iron center were within 2.932(4)–3.825(6) Å, which was too long for bonding interactions. Thus, complex 2 has an unsupported Fe–Fe bond. There are only two examples in the literature of structurally characterized coordinatively unsaturated neutral dinuclear iron complexes,12i.e., [Fe(tim)]2 (tim = 2,3,9,10-tetramethyl-1,4,8,11-tetraazacyclotetradeca-1,3,8,10-tetraene)12a and [(3,5-iPr2–Ar*)Fe–FeCp(CO)2] ((3,5-iPr2–Ar*) = C6H–2,6–(C6H2–2,4,6-iPr3)2–3,5–iPr2–Ar*).12b The Fe–Fe bond distance in these complexes (2.6869(6)12a and 2.3931(8)12b Å) are significantly shorter than that of 2 (2.7374(10) Å), which reflects the weak bonding interaction between the two iron centers in 2 (vide infra). The sum of the three angles around P(1) and P(2) are 358.6° and 358.1°, respectively, indicating planarity at P, which is consistent with π-bonding. The short Fe–P bond lengths, 2.0934(12) Å for Fe–P(1) and 2.1047(13) Å for Fe(2)–P(2), also support the multiple bond character of the Fe–P bond (vide infra).14
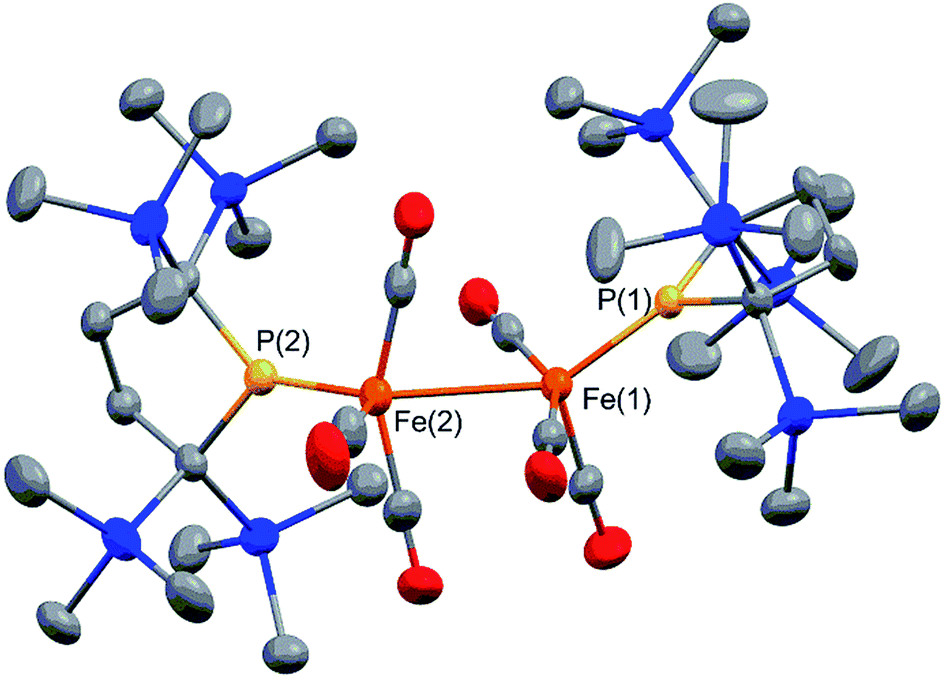 | ||
| Fig. 1 Molecular structure of 2 with 50% probability ellipsoids. | ||
Complex 2 is diamagnetic, and the 1H NMR of 2 confirms the molecular structure. The formal oxidation state of the two iron centers in 2 can be considered as Fe(I), and an intramolecular antiferromagnetic coupling between two adjacent iron centers makes complex 2 diamagnetic. A sharp singlet appears at 0.35 ppm in toluene-d8 at 293 K, whereas four slightly broad singlets are observed at 0.31, 0.35, 0.37, and 0.48 ppm with an integral ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 at 193 K. These signals are attributed to the magnetically inequivalent SiMe3 moieties in 2. Notably, the 31P NMR spectrum shows one significantly downfield shifted peak at 425.9 ppm, which is typically seen for planar sp2-phosphorus species.15 This 31P peak, as well as the planar geometry around P and the short Fe–P bond distances observed in X-ray crystallography, strongly suggest that the phosphido ligands formally function as LX-type ligands with the aid of π-donation.
:1:1:1 at 193 K. These signals are attributed to the magnetically inequivalent SiMe3 moieties in 2. Notably, the 31P NMR spectrum shows one significantly downfield shifted peak at 425.9 ppm, which is typically seen for planar sp2-phosphorus species.15 This 31P peak, as well as the planar geometry around P and the short Fe–P bond distances observed in X-ray crystallography, strongly suggest that the phosphido ligands formally function as LX-type ligands with the aid of π-donation.
At higher temperatures (293–353 K), one sharp singlet for the SiMe3 group and one doublet due to the CH2 moiety of the backbone of the phosphido ligand are observed in the 1H NMR of 2. Additionally, it should be noted that the appearance of a broad signal is confirmed at ∼3–5 ppm. The intensity of this broad peak increases with temperature (Fig. S5-2 in ESI†). Considering the elongated Fe–Fe bond in 2, we hypothesized that the broad peak observed at higher temperatures could be ascribed to the generation of paramagnetic species 3via the homolytic cleavage of the Fe–Fe bond, as shown in Scheme 2. The generation of a radical species is supported by ESR spectroscopy; the ESR spectrum of 2 in toluene exhibits one broad signal at g = 2.0519 (A(31P) = 3.43 mT) at 293 K. The intensity of this peak gradually increases upon heating, indicating that the concentration of the ESR-active species significantly increases with temperature (Fig. S6-1†). The observed g-value is comparable to those reported for iron carbonyl-based five-coordinate seventeen electron radicals.1a,3b,3c In addition, the ESR spectrum of a flash-frozen toluene solution measured at 77 K showed a rhombic signal (g = 2.0838, 2.0573, 2.0409), suggesting that the iron species generated in situ has an S = 1/2 ground state (Fig. S6-2†).
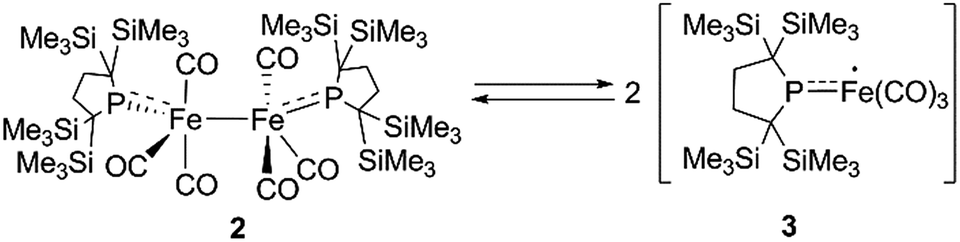 | ||
| Scheme 2 Equilibrium between 2 and 3. | ||
IR spectroscopy provided more detailed information about the species generated at higher temperatures. The solid-state IR (ATR) spectrum of 2 features bands at 2014, 1963, 1932, and 1916 cm−1, which are attributed to terminal CO ligands (Fig. S7-1†). In the IR spectrum of 2 in n-octane at 293 K, four νCO absorptions appear at 2017, 1968, 1940, and 1921 cm−1, indicating the presence of dinuclear complex 2 in solution at this temperature (Fig. 2). An absorption band assignable to the bridging CO ligand is not observed in the solid or solution states. The solution spectrum at 293 K includes a shoulder at around 1938 cm−1 together with the four aforementioned νCO absorption bands. At 353 K in n-octane in the dark, this band represents the major absorption and is accompanied by a strong band at 2015 cm−1, and the four CO bands observed at 293 K are almost completely absent (Fig. 2). If we assume that three CO ligands are maintained on the iron center during heating, this spectral change could be explained by the formation of an iron tricarbonyl species with C3v symmetry (vide infra).
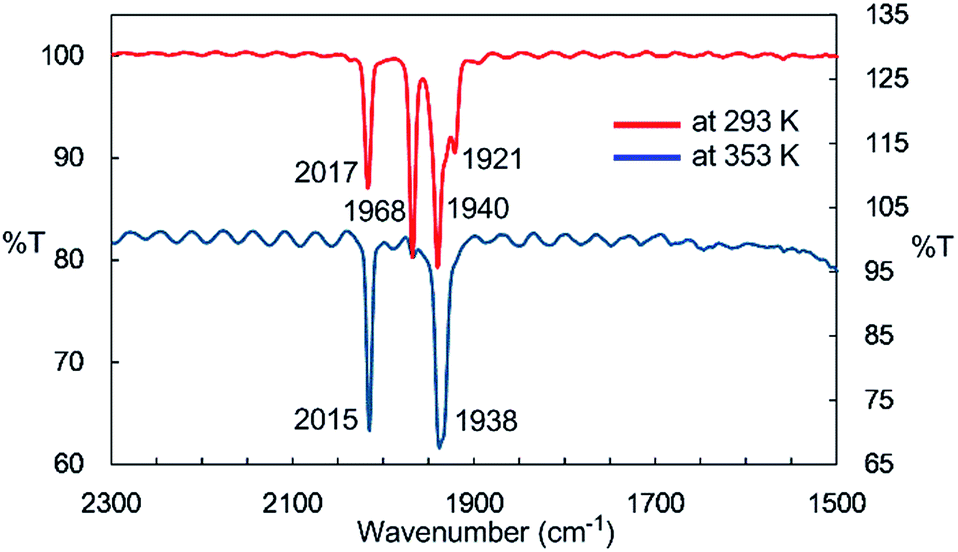 | ||
| Fig. 2 IR spectra of the n-octane solution of 2 at 293 K and 353 K. | ||
The UV-Vis-NIR spectrum of an n-octane solution of 2 at 293 K displays an intense band (A) at 380 nm and two relatively weak bands (B and C) at 502 and 720 nm, respectively (Fig. 3, upper). Their intensity progressively decreases with heating, and three new bands, namely A′ (362 nm), B′ (496 nm), and C′ (818 nm) are exclusively observed at 353 K (Fig. 3, lower). After cooling the solution to 293 K, the original bands, A, B, and C, gradually reappear over time, and equilibrate after 5 h. Notably, an isosbestic point is observed at 810 nm, providing good evidence for an equilibrium between two species (Fig. S8-3†).
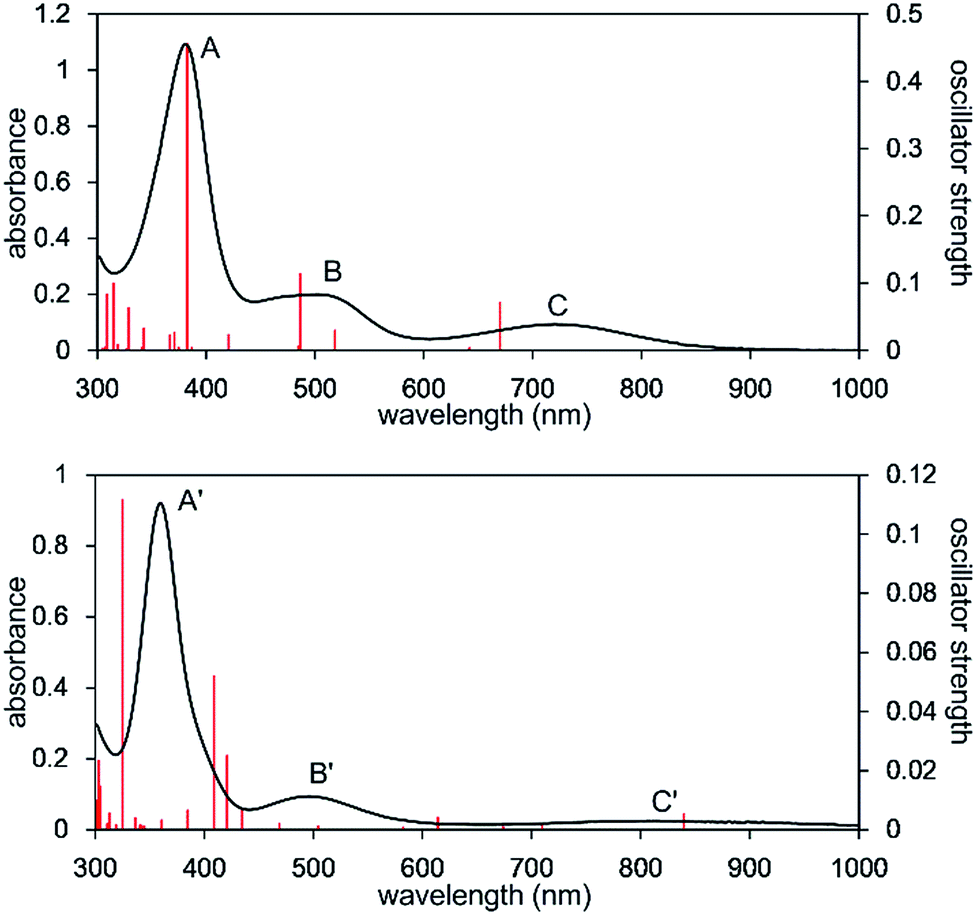 | ||
| Fig. 3 UV-Vis-NIR spectra of the n-octane solution of 2 measured at 293 K (upper) and 353 K (lower). Solid red bars indicate the calculated transition of the 2opt (upper) and 3opt (lower). | ||
In order to gain further insight into the electronic structure and bonding in the iron complexes, a theoretical investigation of 2 was undertaken. Geometry optimization and time-dependent density functional theory (TD-DFT) spectral calculation were carried out at the PBE0, CAM-B3LYP, and B3LYP levels, and the PBE0 functional was found to give the most satisfactory results.§
The calculated spectrum of 2opt is given in Fig. 3 (upper). Although a low energy absorption band is predicted to appear at 670 nm, which is blue-shifted by 50 nm as compared to the actual spectrum presumably due to the overestimation of the low-energy gap, the TD-DFT analysis of 2opt is in reasonable agreement with the actual spectrum. The Mayer Fe–Fe bond order is calculated to be 0.33. The bond order between Fe and P is calculated to be 1.4, indicating this bond to have a double bond nature. Molecular orbital analysis of HOMO and HOMO − 1 of 2opt highlights the π-bonding nature of the Fe–P bond (Fig. 4). This π-bonding nature was also confirmed by density distribution analysis in HOMO and HOMO − 1 (Fig. S17†).
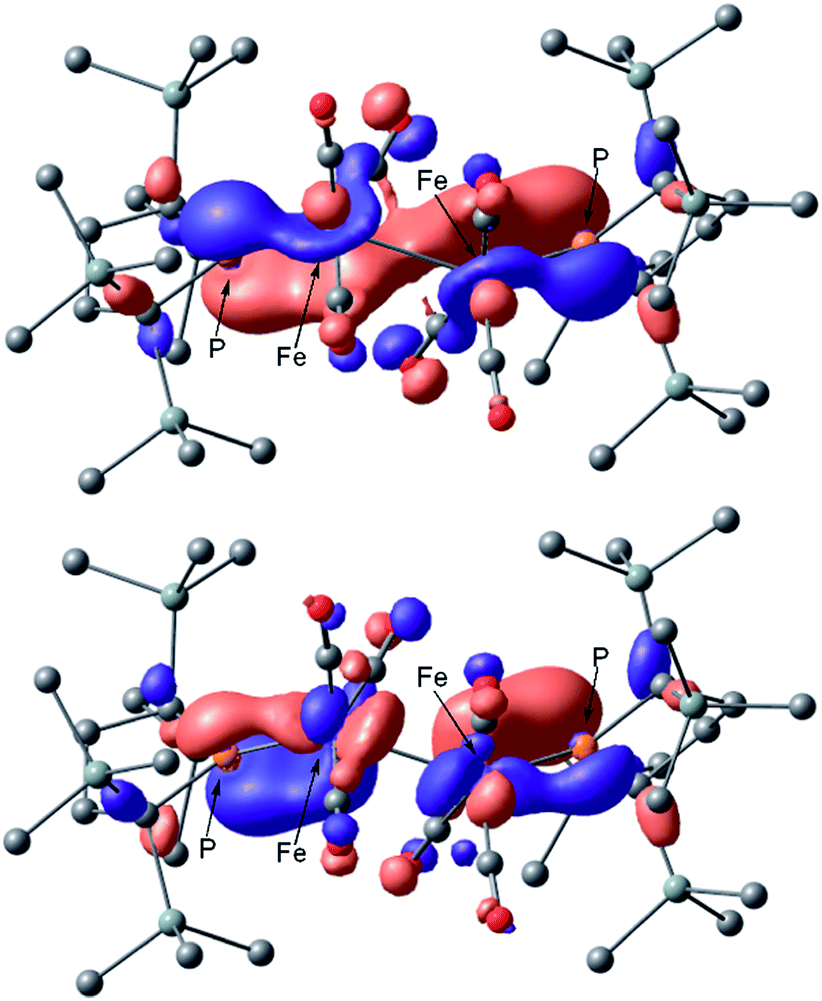 | ||
| Fig. 4 Graphical representations of the DFT(PBE0)-derived HOMO (upper) and HOMO − 1 (lower) for 2opt. | ||
Next, we calculated the molecular and electronic structures of 3 at the PBE0 level. We found that 3opt possesses a slightly distorted C3v tetrahedral coordination geometry in the open-shell doublet ground state (Fig. 5). Interestingly, DFT studies of 3opt indicate that the spin densities around Fe and P are 1.50 and −0.52, respectively. The bonding orbital in 3 was carefully analysed, and the orbital interactions in 3opt were depicted in Fig. S20.† We found that βHOMO consists of the dyz orbital in iron and py orbital in phosphorus, and π-bonding character was confirmed in the βHOMO by molecular orbital analysis of 3opt (Fig. 6). In contrast, αHOMO consists of dx2−y2 orbital in iron leading to a non-bonding interaction (molecular orbital details are given in the ESI, Fig. S19 and S20†). This non-bonding nature was also confirmed by density distribution in αHOMO (Fig. S21†). This partial Fe–P π-bonding is consistent with the estimated Mayer bond order for Fe–P (1.2). These results indicate that electron donation from the P atom to Fe found in the βHOMO electronically stabilizes the four-coordinate iron-centered radical. TD-DFT calculations based on 3opt give the absorption bands shown in Fig. 3 (lower). Although the accuracy of the TD-DFT calculation of 3opt appears to be limited due to the open-shell nature of 3opt,16 the obtained predicted spectrum roughly approximates the actual spectrum. The two remarkable peaks in the range of 300–550 nm in the predicted spectrum of 3opt are blue-shifted compared with those of 2opt. A similar feature is also observed in the actual spectrum of 3. It should be noted that one peak appears at 839 nm in the predicted spectrum of 3opt, which effectively reproduces the actual band observed at 818 nm (band C′ in Fig. 3 (lower)). The electron density difference map of 2opt and 3opt indicates that these absorptions originate from the metal-to-ligand charge transfer (MLCT) transitions (Fig. S22 and S23†). It should be mentioned that Lappert et al. have reported the synthesis of four coordinated Co(I) tricarbonyl, [{(Me3Si)2N}(iPr2N)P]Co(CO)3, by the reaction of the in situ generated phosphinyl radical [{(Me3Si)2N}(iPr2N)P]˙ with Co2(CO)8.17 This cobalt(I) phosphido complex also adopts tetrahedral coordination geometry, as shown by X-ray diffraction analysis. Thus, complex 3 can be considered analogous to Lappert's cobalt(I) phosphido complex minus one electron.
 | ||
| Fig. 5 DFT(PBE0) optimized structure of 3opt. | ||
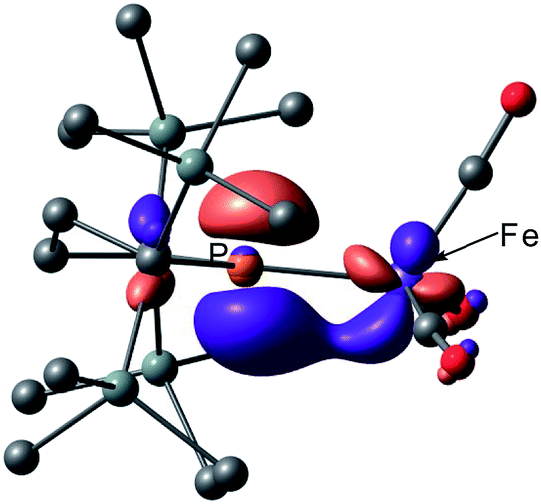 | ||
| Fig. 6 Graphical representations of the DFT(PBE0)-derived βHOMO for 3opt. | ||
Considering the experimental and theoretical results, it is highly probable that the generation of four-coordinate iron-centered radical 3 takes place via the homolysis of the unsupported Fe–Fe bond of 2 in solution. The coordination geometry of 3 was predicted to be slightly distorted tetrahedral, and the local symmetry to resemble C3v. This is consistent with the IR spectrum of 2 in n-octane at high temperatures. As mentioned above, two strong bands are observed at 2015 and 1938 cm−1 at 353 K. This spectral feature is characteristic of tetrahedral C3v-symmetrical tricarbonyl species; the former can be assigned as the symmetric A1 stretch, and the latter can be assigned as the asymmetric E stretch. These CO stretching vibrations are blue-shifted relative to those found in Holland's four coordinate Fe(I) dicarbonyl complex, (NacNactBu)Fe(CO)2 (1992 and 1908 cm−1),8a but are significantly shifted to lower wavenumbers compared with those of previously reported five coordinate Fe(I)-tricarbonyl complexes.6a,8a,8b
The equilibrium constant (Keq) between 2 and 3 was determined by variable temperature NMR spectroscopy. The concentration of 3 in solution was estimated by the Evans method,18 and the thermodynamic parameters for the homolytic cleavage of the Fe–Fe bond in 2 were calculated to be ΔH = 18.8 ± 0.5 kJ mol−1 and ΔS = 46.1 ± 1.8 J mol−1 K−1, respectively. The thermodynamic parameters were also estimated based on the concentration of 2 (ΔH = 15.8 ± 0.2 kJ mol−1 and ΔS = 36.7 ± 0.4 J mol−1 K−1), which are roughly consistent with those obtained by the Evans method.¶ The large positive ΔS value is reasonable for the homolysis of Fe–Fe bond.
It is well known that metalloradicals can be captured by reaction with radical traps, such as stable organic radicals, metal-hydride species including HSnR3, and group 15 based cage compounds (e.g., P4). As a recent representative example, Scheer et al. have described that the sterically hindered iron dimer [CpBIGFe(CO)2]2 (CpBIG = C5(4-nBuC6H4)5), readily dissociates in solution to afford a monomeric iron-centered radical, which can be easily trapped by reaction with P4 or As4.19 In manganese chemistry, Figueroa has synthesized an isolable mononuclear manganese-centered radical by introduction of sterically bulky isocyanide ligands, the reactivity of which was clearly indicated by its reaction with HSnR3 and some organic substrates.6k Because 3 consists of only one bulky phosphido and three less hindered CO ligands, the iron center of 3 is expected to have enough room to react with certain organic substrates. Thus, we performed reactions of 3 with organic radicals. First, treatment of 3 (generated in situ from 2) with 9-azanoradamantane N-oxyl (nor-AZADO) was examined. The reaction proceeds smoothly, even at room temperature, to afford diamagnetic five-coordinate mononuclear iron(II) complex 4 in 74% isolated yield (Scheme 3).|| Identification of 4 was carried out by IR and NMR spectroscopy, and X-ray diffraction analysis. The signals due to the SiMe3 groups of 4 appear as two singlets at 0.36 and 0.47 ppm in the 1H NMR spectrum in C6D6. One phosphorus resonance is observed at 282.16 ppm in the 31P NMR spectrum. It is shifted to a higher field as compared to 1, presumably due to the coordination of one σ-donating nitrogen instead of a π-accepting CO ligand. Two strong absorption bands derived from the Fe–CO moiety appear at 1958 and 1892 cm−1 in the IR spectrum of 4. The ORTEP representation of 4 is depicted in Fig. 7. Two CO ligands are bound to the iron center, and the O–N bond of nor-AZADO is coordinated to the iron center in a κ2-fashion with a bond distance of 1.3851(14) Å. The sum of the three angles around P is 358.1°, indicating the contribution of π-bonding. Although 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and its analogues are known to coordinate to the metal center in a κ2-(ON) coordination mode in the reaction with some transition metal complexes,20 to the best of our knowledge, complex 4 is the first example of a structurally characterized nor-AZADO complex having a κ2-(ON) moiety. The overall reaction can be explained as follows: radical recombination between in situ-generated iron-centered radical 3 and the oxygen-centered radical takes place to create the Fe–O bond, followed by the liberation of one CO ligand from the iron center concomitant with the formation of the Fe–N bond. In the course of this reaction, nor-AZADO formally undergoes one electron reduction to coordinate to the iron(II) center.
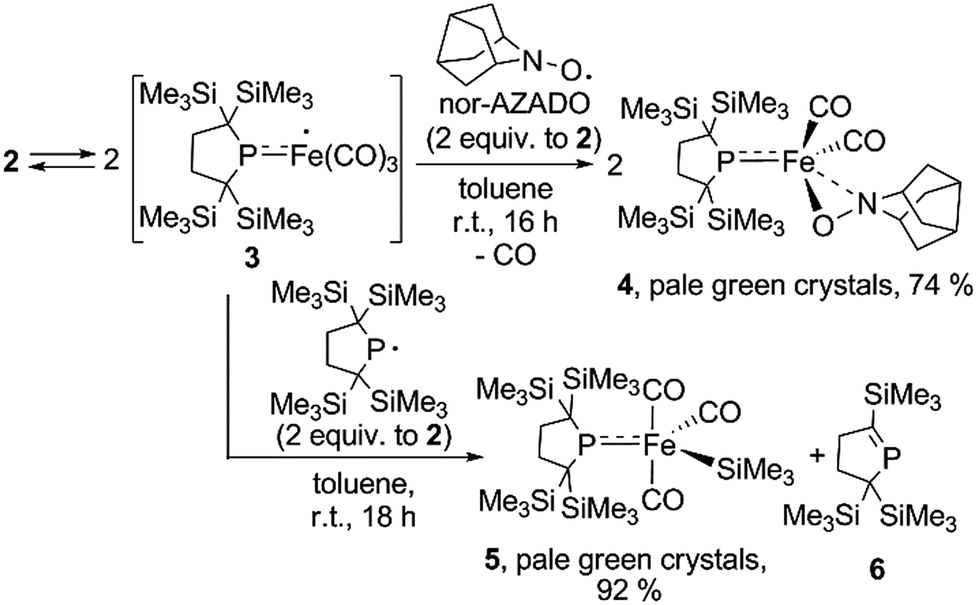 | ||
| Scheme 3 Reactions of in situ generated 3 with organic radicals. | ||
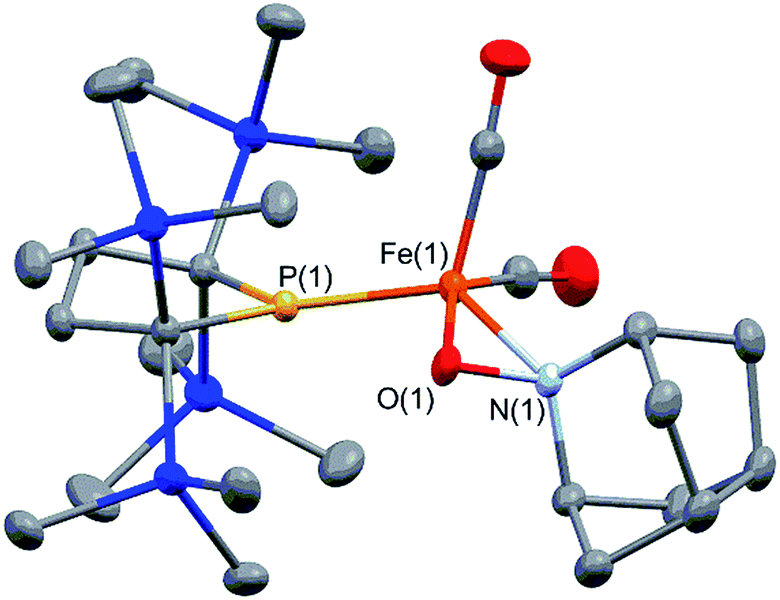 | ||
| Fig. 7 Molecular structure of 4 with 50% probability ellipsoids. | ||
Next, we performed the reaction between 3 and phosphinyl radical 1, from which formation of five-coordinate iron-silyl complex 5 and phosphaalkene 6 in a 1:1 ratio is confirmed. The presence of π-donation in 5 was ascertained by 31P NMR, as one significantly downfield shifted singlet is observed at 461.4 ppm. The molecular structure of 5 was unequivocally determined by X-ray diffraction analysis (Fig. S27†). The relatively short Fe–P bond length (2.1009(7) Å) as well as the planar geometry around the P atom (the sum of the three angles around P was found to be 359.92°) indicate the contribution of π-donation to the Fe–P interaction. This reaction can also be considered the result of the radical behaviour of complex 3; the iron-centered radical 3 abstracts one of four SiMe3 groups from 1via homolytic substitution (SH2) to form a Fe–SiMe3 bond concomitant with the formation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond to afford 6 (detailed reaction mechanism is shown in Scheme S1 in ESI†). Given the fact that diamagnetic closed shell organoiron(II) complexes tend to adopt coordinatively saturated octahedral coordination geometry, the results described here could contribute to the development of new synthetic methodologies towards novel iron complexes.
P bond to afford 6 (detailed reaction mechanism is shown in Scheme S1 in ESI†). Given the fact that diamagnetic closed shell organoiron(II) complexes tend to adopt coordinatively saturated octahedral coordination geometry, the results described here could contribute to the development of new synthetic methodologies towards novel iron complexes.
Reactions of in situ generated 3 with HSnR3 (R = Ph, Bu) were also performed. Reaction of 2 with HSnPh3 was monitored by 1H and 31P NMR spectra in C6D6 at room temperature. In the 31P NMR spectrum of the crude product, three peaks were observed at −33.6, 29.3 and 464.9 ppm, respectively, in an intensity ratio of 10:4:1. The peak at −33.6 ppm was assigned as free phosphine 7,10a whereas the other two peaks can be assigned as complexes 8 and 9. Although 9 could not be obtained in pure form (see ESI† for detail), complex 8 can be isolated in 26% yield, and the molecular structure of 8 was determined by X-ray diffraction analysis (Fig. S28†). The relative ratio of 7, 8 and 9 was determined from the 1H NMR spectrum of the crude product using an internal standard, and was estimated to be 60:37:3 based on iron. Formation of trace amount of Ph3Sn–SnPh3 (ca. 2%) was detected in the 1H NMR spectrum of the crude product.** These results strongly suggest that the reaction of 3 with HSnPh3 gave a complex mixture, and the major reaction pathway is the formation of free phosphine 7. At the first stage of this reaction, iron-centered radical 3 would abstract a H atom from HSnPh3 to generate five coordinate “(phosphido)Fe(H)(CO)3” species. Because such Fe–H species is expected to be unstable, following disproportionation would afford 7 and iron species including 8 and 9. This possible reaction sequence is shown in Scheme S2 in ESI.† In a similar manner, reaction of 2 with HSnBu3 was also performed. The 31P NMR spectrum of the crude product suggests the formation of 7 as the major product concomitant with the formation of 8′ which is analogous to 8. The slightly broadened 1H NMR signals are presumably due to the partial formation of paramagnetic species and prevented further detailed analysis. Formation of 7 was also confirmed in the crude 1H NMR spectrum (Fig. S13†). Formation of Bu3Sn–SnBu3 was observed in ca. 10% yield in the GC-MS analysis, suggesting that the partial radical recombination took place after the abstraction of a H atom from the Sn center (Scheme 4).
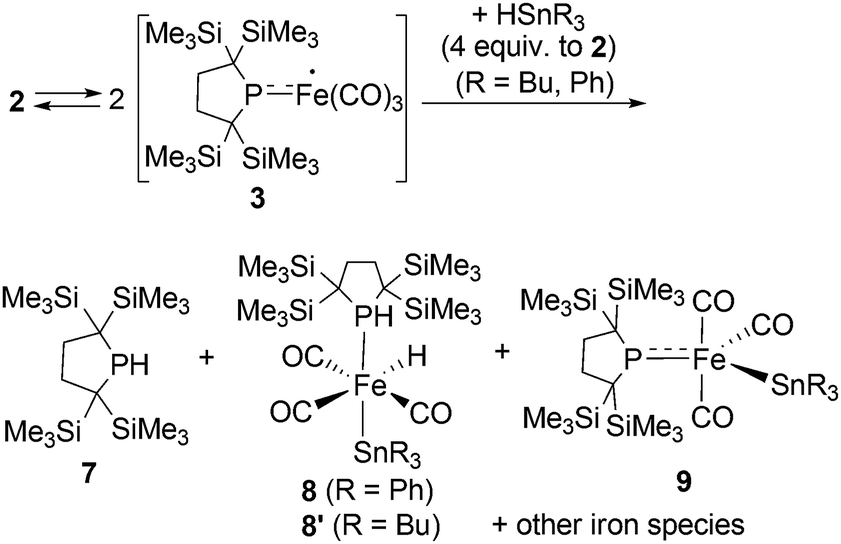 | ||
| Scheme 4 Reactions of in situ generated 3 with HSnR3 (R = Ph, Bu). | ||
The reactions shown above clearly indicate the radical nature of 3. Next, we have performed the reaction of 3 (generated in situ from 2) with 9,10-dihydroanthracene or 1,4-cyclohexadiene. However, no reaction took place below 333 K, and partial decomposition of 2 was confirmed by 1H and 31P NMR in the reaction at 353 K for 24 h. Considering the difference in the bond dissociation energy for the C–H bond of 1,4-cyclohexadiene (73 kcal mol−1)21a and the Fe–H bond (58.5 ± 5 kcal mol−1; determined in the gas phase),21b the results obtained here may be reasonable. Further research to elucidate the bond dissociation energy of Fe–H derived from complex 3 will be carried out in our laboratory.
Conclusions
In conclusion, we succeeded in preparing coordinatively unsaturated dinuclear iron carbonyl complex 2 by the reaction of Fe2(CO)9 with phosphinyl radical 1. The generation of four-coordinate iron-centered radical 3 was realized by the thermal homolysis of the unsupported Fe–Fe bond of 2. Experimental analysis and theoretical calculation revealed that π-donation from the phosphido ligand to the iron center electronically stabilizes the four-coordinate iron-centered radical 3. In both complex 2 and 3, π-bonding electrons in the p-orbital of phosphorus lies on the HOMO and HOMO − 1 for 2 and βHOMO for 3, which are effectively involved in π-donation from phosphorus to the iron center. The orbital interactions in 3opt shown in Fig. S20† clearly suggest that the 3py orbital of phosphorus and the dyz orbital of iron can effectively interact to create the π-bonding interaction. Complex 3 effectively reacted with organic radicals to afford diamagnetic five-coordinate organoiron(II) species. These results may facilitate the development of new synthetic methodologies towards the design and construction of highly reactive metal-centered radicals. Efforts to develop novel fundamental and catalytic reactions realized by coordinatively unsaturated iron-centered radicals are now underway in our laboratory.Acknowledgements
This work was supported by Grant in Aid for Young Scientist (A) (No. 24685011), and Scientific Research on Innovative Areas “Stimuli-responsive Chemical Species” (No. 25109534, 24109004, 24109014 and 25109533) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. This work was also supported by the Cooperative Research Program of the “Network Joint Research Center for Materials and Devices”.Notes and references
- (a) M. C. Baird, Chem. Rev., 1988, 88, 1217–1227 CrossRef CAS; (b) T. J. Meyer and J. V. Caspar, Chem. Rev., 1985, 85, 187–218 CrossRef CAS; (c) A. E. Stiegman and D. R. Tyler, Coord. Chem. Rev., 1985, 63, 217–240 CrossRef CAS; (d) D. R. Tyler, Acc. Chem. Res., 1991, 24, 325–331 CrossRef CAS.
- (a) T. L. Brwon, Ann. N. Y. Acad. Sci., 1980, 333, 80–89 CrossRef; (b) J. Halpern, Pure Appl. Chem., 1979, 51, 2171–2182 CrossRef CAS; (c) J. Halpern, Pure Appl. Chem., 1986, 58, 575–584 CrossRef CAS; (d) A. Lewandowska-Andralojc, D. C. Grills, J. Zhang, R. M. Bullock, A. Miyazawa, Y. Kawanishi and E. Fujita, J. Am. Chem. Soc., 2014, 136, 3572–3578 CrossRef CAS PubMed; (e) B. Giese and G. Thoma, Helv. Chim. Acta, 1991, 74, 1135–1142 CrossRef CAS; (f) B. Giese and G. Thoma, Helv. Chim. Acta, 1991, 74, 1143–1155 CrossRef CAS; (g) B. B. Wayland, A. E. Sherry, G. Poszmil and A. G. Bunn, J. Am. Chem. Soc., 1992, 114, 1673–1681 CrossRef CAS; (h) S. L. Scott, J. H. Espenson and Z. Zhu, J. Am. Chem. Soc., 1993, 115, 1789–1797 CrossRef CAS; (i) D. H. Hill, M. A. Parvez and A. Sen, J. Am. Chem. Soc., 1994, 116, 2889–2901 CrossRef CAS; (j) I. Kuksis, I. Kovács, M. C. Baird and K. F. Preston, Organometallics, 1996, 15, 4991–5002 CrossRef CAS; (k) P. A. Wegner and M. S. Delaney, Inorg. Chem., 1976, 15, 1918–1921 CrossRef CAS; (l) C. A. MacConnachie, J. M. Nelson and M. C. Baird, Organometallics, 1992, 11, 2521–2528 CrossRef CAS.
- (a) C. P. Putnik, J. J. Welter, G. D. Stucky, M. J. D'Aniello Jr, B. A. Sosinsky, J. F. Kirner and E. L. Muetterties, J. Am. Chem. Soc., 1978, 100, 4107–4116 CrossRef CAS; (b) E. L. Muetterties, B. A. Sosinsky and K. I. Zamaraev, J. Am. Chem. Soc., 1975, 97, 5299–5300 CrossRef CAS; (c) H. D. Murdoch and E. A. C. Lucken, Helv. Chim. Acta, 1964, 47, 1517–1524 CrossRef CAS.
- (a) K. Szaciłowski, W. Macyk, G. Stochel, Z. Stasicka, S. Sostero and O. Traverso, Coord. Chem. Rev., 2000, 208, 277–297 CrossRef; (b) T. E. Bitterwolf, Coord. Chem. Rev., 2000, 206–207, 419–450 CrossRef CAS; (c) H. B. Abrahamson, M. C. Palazzotto, C. L. Reichel and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 4123–4127 CrossRef CAS; (d) J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1980, 102, 7794–7795 CrossRef CAS; (e) I. Kuksis, I. Kovács, M. C. Baird and K. F. Preston, Organometallics, 1996, 15, 4991–5002 CrossRef CAS; (f) H. Sitzmann, T. Dezember, W. Kaim, F. Baumann, D. Stalke, J. Kärcher, E. Dormann, H. Winter, C. Wachter and M. Kelemen, Angew. Chem., Int. Ed. Engl., 1996, 35, 2872–2875 CrossRef CAS; (g) Y.-M. Wuu, C. Zou and M. S. Wrighton, J. Am. Chem. Soc., 1987, 109, 5861–5862 CrossRef CAS.
- (a) J. H. MacNeil, A. C. Chiverton, S. Fortier, M. C. Baird, R. C. Hynes, A. J. Williams, K. F. Preston and T. Ziegler, J. Am. Chem. Soc., 1991, 113, 9834–9842 CrossRef CAS; (b) M. Cory MacLeod, D. J. Vinyard and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 10226–10229 CrossRef PubMed; (c) S. D. Brown and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 1913–1923 CrossRef CAS PubMed; (d) Y. Lee and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 4438–4446 CrossRef CAS PubMed; (e) S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322–323 CrossRef CAS PubMed; (f) M. J. Ingleson, B. C. Fullmer, D. T. Buschhorn, H. Fan, M. Pink, J. C. Huffman and K. G. Caulton, Inorg. Chem., 2008, 47, 407–409 CrossRef CAS PubMed; (g) M. T. Mock, C. V. Popescu, G. P. A. Yap, W. G. Dougherty and C. G. Riordan, Inorg. Chem., 2008, 47, 1889–1891 CrossRef CAS PubMed; (h) A. M. Tondreau, C. Milsmann, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2011, 50, 9888–9895 CrossRef CAS PubMed; (i) C. E. MacBeth, S. B. Harkins and J. C. Peters, Can. J. Chem., 2005, 83, 332–340 CrossRef CAS; (j) L. Fohlmeister and C. Jones, Aust. J. Chem., 2014, 67, 1011–1016 CrossRef CAS; (k) C. G. Riordan, Coord. Chem. Rev., 2010, 254, 1815–1825 CrossRef CAS PubMed; (l) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS.
- Examples of transition metal carbonyl based metal centered radicals other than iron, see: (a) S. Fortier, M. C. Baird, K. F. Preston, J. R. Morton, T. Ziegler, T. J. Jaeger, W. C. Watkins, J. H. MacNeil, K. A. Watson, K. Hensel, Y. L. Page, J.-P. Charland and A. J. Williams, J. Am. Chem. Soc., 1991, 113, 542–551 CrossRef CAS; (b) E. F. van der Eide, M. L. Helm, E. D. Walter and R. M. Bullock, Inorg. Chem., 2013, 52, 1591–1603 CrossRef CAS PubMed; (c) J. A. S. Roberts, J. A. Franz, E. F. van der Eide, E. D. Walter, J. L. Petersen, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2011, 133, 14593–14603 CrossRef CAS PubMed; (d) E. F. van der Eide, T. Liu, D. M. Camaioni, E. D. Walter and R. M. Bullock, Organometallics, 2012, 31, 1775–1789 CrossRef CAS; (e) J. A. S. Roberts, D. L. DuBois and R. M. Bullock, Organometallics, 2011, 30, 4555–4563 CrossRef CAS; (f) S. J. McLain, J. Am. Chem. Soc., 1988, 110, 643–644 CrossRef CAS; (g) W. E. Lindsell and P. N. Preston, J. Chem. Soc., Dalton Trans., 1979, 1105–1108 RSC; (h) D. Chong, A. Nafady, P. J. Costa, M. J. Calhorda and W. E. Geiger, J. Am. Chem. Soc., 2005, 127, 15676–15677 CrossRef CAS PubMed; (i) J. H. Brownie, M. C. Baird, D. R. Laws and W. E. Geiger, Organometallics, 2007, 26, 5890–5901 CrossRef CAS; (j) W. Wang, X. Wang, Z. Zhang, N. Yuan and X. Wang, Chem. Commun., 2015, 51, 8410–8413 RSC; (k) D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201506498 in press.
- The generation of low coordinate iron carbonyl-based radicals have been achieved in the gas phase. Selected examples, see: (a) A. Massey, A. D. Bass and L. Sanche, J. Chem. Phys. C, 2015, 119, 12708–12719 CrossRef; (b) Y. Tamenori and I. Koyano, J. Chem. Phys. A, 1998, 102, 368–374 CrossRef CAS; (c) R. E. Winters and R. W. Kiser, Inorg. Chem., 1964, 3, 699–702 CrossRef CAS; (d) S. Hsieh and J. H. D. Eland, Int. J. Mass Spectrom. Ion Processes, 1997, 167/168, 415–424 CrossRef CAS; (e) M. N. Rocklein and D. P. Land, Int. J. Mass Spectrom., 1998, 177, 83–89 CrossRef CAS; (f) M. Zhou and L. Andrews, J. Chem. Phys., 1999, 110, 10370–10379 CrossRef CAS; (g) M. Zhou, G. V. Chertihin and L. Andrews, J. Chem. Phys., 1998, 109, 10893–10904 CrossRef CAS.
- (a) A. R. Sadique, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2008, 47, 784–786 CrossRef CAS PubMed . See also; (b) J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756–769 CrossRef CAS PubMed; (c) R. E. Cowley, N. A. Eckert, J. Elhaïk and P. L. Holland, Chem. Commun., 2009, 1760–1762 RSC; (d) T. R. Dugan and P. L. Holland, J. Organomet. Chem., 2009, 694, 2825–2830 CrossRef CAS PubMed; (e) A. R. Sadique, W. W. Bremmessel and P. L. Holland, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m175–m176 Search PubMed.
- J. L. Kisko, T. Hascall and G. Parkin, J. Am. Chem. Soc., 1998, 120, 10561–10562 CrossRef CAS.
- (a) S. Ishida, F. Hirakawa and T. Iwamoto, J. Am. Chem. Soc., 2011, 133, 12968–12971 CrossRef CAS PubMed; (b) T. Iwamoto, F. Hirakawa and S. Ishida, Angew. Chem., Int. Ed., 2012, 51, 12111–12114 CrossRef CAS PubMed; (c) S. Ishida, F. Hirakawa and T. Iwamoto, Chem. Lett., 2015, 44, 94–96 CrossRef; (d) F. Hirakawa, H. Ichikawa, S. Ishida and T. Iwamoto, Organometallics, 2015, 34, 2714–2716 CrossRef CAS.
- A. H. Cowley, R. A. Kemp and J. C. Wilburn, J. Am. Chem. Soc., 1982, 104, 331–332 CrossRef CAS.
- A Cambridge Structural Database search (CSD version 5.36) was performed, and only two examples of coordinatively unsaturated neutral dinuclear iron complexes having unsupported Fe–Fe bond were found.13 (a) H. Lei, J.-D. Guo, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2010, 132, 17399–17401 CrossRef CAS PubMed; (b) C. R. Hess, T. Weyhermüller, E. Bill and K. Wieghardt, Angew. Chem., Int. Ed., 2009, 48, 3703–3706 CrossRef CAS PubMed.
- A trianionic iron dinuclear complex having a unsupported Fe–Fe bond, (NBu4)3[FeCl16Pc]2 (Cl16Pc = hexadecachlorophthalocyanine), has been synthesized by Konarev et al. The Fe–Fe bond distances is slightly elongated (2.899(4) Å) compared with that of 2, presumably due to electronic repulsion. See, D. V. Konarev, A. V. Kuzmin, M. Ishikawa, Y. Nakao, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2014, 3863–3870 CrossRef CAS.
- Complexes 2, 4, and 5 represent the first examples of crystallographically characterized iron phosphido complexes, with π-bond character in the Fe–P bond. The Fe–P bond distances in the mononuclear iron phosphido complexes without π-donation are in the range of 2.265(3)–2.359(2) Å. For selected examples, see: (a) W. Petz and F. Weller, Z. Naturforsch., B: Chem. Sci., 1996, 51, 715–721 CAS; (b) H. Brombach, E. Niecke and M. Nieger, Organometallics, 1991, 10, 3949–3951 CrossRef CAS.
- L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606–626 CrossRef CAS.
- A. Ipatov, F. Cordova, L. J. Doriol and M. E. Casida, J. Mol. Struct.: THEOCHEM, 2009, 914, 60–73 CrossRef CAS.
- J.-P. Bezombes, P. B. Hitchcock, M. F. Lappert and J. E. Nycz, Dalton Trans., 2004, 499–501 RSC.
- (a) E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS; (b) C. Piguet, J. Chem. Educ., 1997, 74, 815–816 CrossRef CAS; (c) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- S. Heini and M. Scheer, Chem. Sci., 2014, 5, 3221–3225 RSC.
- Selected examples, see; (a) D. Isrow, N. J. deYonker, A. Koppaka, P. J. Pellechia, C. E. Webster and B. Captain, Inorg. Chem., 2013, 52, 13882–13893 CrossRef CAS PubMed; (b) D. J. Mindiola, R. Waterman, D. M. Jenkins and G. L. Hillhouse, Inorg. Chem., 2003, 345, 299–308 CAS; (c) S. Pelties, D. Herrmann, B. de Bruin, F. Hartl and R. Wolf, Chem. Commun., 2014, 50, 7014–7016 RSC; (d) D. Isrow and B. Captain, Inorg. Chem., 2011, 50, 5864–5866 CrossRef CAS PubMed; (e) M. Ito, T. Matsumoto and K. Tatsumi, Inorg. Chem., 2009, 48, 2215–2223 CrossRef CAS PubMed; (f) P. Jaitner, W. Huber, A. Gieren and H. Betz, J. Organomet. Chem., 1986, 311, 379–385 CrossRef CAS; (g) P. Jaitner, W. Huber, G. Huttner and O. Scheidsteger, J. Organomet. Chem., 1983, 259, C1–C5 CrossRef CAS.
- (a) T. J. Burkey, M. Majewski and D. Griller, J. Am. Chem. Soc., 1986, 108, 2218–2221 CrossRef CAS PubMed; (b) M. L. Mandich, L. F. Halle and J. L. Beauchamp, J. Am. Chem. Soc., 1984, 106, 4403–4411 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic, computational details, and crystal data for 2, 4, 5 and 8. CCDC 1057111–1057113 and 1425703. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02601f |
| ‡ Although complex D in Chart 1 can capture atmospheric CO, no further reactions of four coordinated iron carbonyl based Fe(I) complexes have been reported. |
| § The ground state of 2opt was estimated to be the closed-shell singlet in all calculations. Calculated bond lengths and angles at the PBE0 and CAM-B3LYP levels are similar to experimental values; however, the structural parameters at the B3LYP level are inconsistent with the experimental results (see Table S2 in ESI†). The results of TD-DFT calculations at the CAM-B3LYP and B3LYP levels are also reported in ESI (Fig. S24†). |
| ¶ The thermodynamic parameters were calculated based on the concentration of 3, which was estimated by the Evans method,18 in which the number of unpaired electrons at the iron center was assumed to be 1. Because it is difficult to quantitate the exact concentration of 2 and 3 simultaneously in solution, the thermodynamic parameters were alternatively calculated based on the concentration of 2, as estimated from the relative ratio of the integral value in the 1H NMR spectra measured in C6D6 at various temperatures. Although the relative ratio of the integral values of sharp (diamagnetic) vs. broad (paramagnetic) peaks may include some experimental error, the obtained thermodynamic parameters were roughly consistent with those obtained by the Evans method. The details are given in ESI.† |
| || One of the reviewers pointed out that dinuclear complex 2 may have the possibility of directly reacting with nor-AZADO to form product 4. Because one broad peak assignable to 3 almost disappeared in 1H NMR spectrum of 2 at 253 K, we have performed the reaction of 2 with nor-AZADO in toluene-d8 at 253 K. We found that only ca. 8% of 2 was converted to product 4 after 25 days at 252 K. This result may indicate that in situ generated radical 3 can react with nor-AZADO, and that direct reaction of dinuclear complex with nor-AZADO can be ruled out. |
| ** Although powder formation was not observed after the reaction of 2 with HSnPh3, low solubility of Ph3Sn–SnPh3 toward C6D6 may hamper the accurate detection of the amount of Ph3Sn–SnPh3 formed in this reaction. |
| This journal is © The Royal Society of Chemistry 2016 |